
Nickel–platinum nanoparticles immobilized on graphitic carbon nitride as highly efficient catalyst for hydrogen release from hydrous hydrazine†
Lixin
Xu
a,
Na
Liu
a,
Bing
Hong
a,
Ping
Cui
a,
Dang-guo
Cheng
b,
Fengqiu
Chen
b,
Yue
An
b and
Chao
Wan
*a
aCollege of Chemistry and Chemical Engineering, Anhui University of Technology, 59 Hudong Road, Ma'anshan 243002, China. E-mail: wanchao1219@hotmail.com
bCollege of Chemical and Biological Engineering, Zhejiang University, 38 Zheda Road, Hangzhou 310027, China
First published on 24th March 2016
Abstract
Here we demonstrate that the combination of NiPt alloy nanoparticles with a graphitic carbon nitride (g-C3N4) support facilitates H2 production from hydrous hydrazine in an alkaline solution under moderate conditions. Of all the heterogeneous catalysts tested, Ni37Pt63/g-C3N4 shows superior catalytic performance with a maximum initial turnover frequency (TOF) of 570 h−1 at 323 K.
In light of the upcoming energy crisis and the increasingly severity of environmental pollution it is desirable to use sustainable, renewable and clean energies.1 Hydrogen, whose only by-product is water, is an environmentally attractive and ideal energy carrier, especially for electricity generation in low-temperature fuel cells.2–5 However, despite the numerous studies conducted over several decades, the hydrogen economy has not been fulfilled due to the challenges in safely and efficiently storing hydrogen.6–9 Chemical hydrogen storage is a promising solution because it can be conveniently transported and has a considerable hydrogen content.10–15 Hydrous hydrazine, such as hydrazine monohydrate (H2NNH2·H2O), is considered a promising candidate for storing hydrogen. It is advantageous due to its high hydrogen storage capacity (8.0 wt%), easy recharging as a liquid (the current infrastructure for liquid fuel cells can be utilized), and its environment-friendliness, i.e. the only byproduct is nitrogen via a complete decomposition reaction: H2NNH2 → N2(g) + 2H2(g).16–21 Nevertheless, to efficiently liberate hydrogen, the undesired reaction 3H2NNH2 → 4NH3(g) + N2(g) must be avoided.22–27 To solve this problem, one must develop highly efficient, selective, and economic catalysts for producing hydrogen from hydrous hydrazine in practical applications.
For implementing this target, much attention has been devoted to synthesize metal catalysts for improving the hydrogen release from H2NNH2·H2O, among which noble metals, e.g., Pt, Pd, Rh, or their bimetallic alloy catalysts28–34 have proved to be efficient. In light of this, many studies have been devoted to developing monometallic and polymetallic nanoparticles (NPs), where materials such as Al2O3, CeO2, graphene oxide, carbon black, etc. act as the support35–44 and metals such as Pt,45–50 Pd,33,51 and other elements19–21,52,53 are the active ingredients. In particular, binary NiPt alloy NPs are efficient catalysts for hydrous hydrazine decomposition in alkaline solutions. The dispersion of the metal NPs throughout the solution significantly influences the performance of the catalysts;45–49 therefore, to improve their activity one must reduce the clustering of NPs via the use of proper supports.
Recently, graphitic carbon nitride (g-C3N4) has received a great deal of attention as a metal-free candidate to supplement traditional carbon materials due to its many appealing properties: low cost, semiconductivity, high chemical and physical stability, basicity, energy-storage capacity, and nontoxicity.54,55 g-C3N4 has been comprehensively investigated for use in gas storage, fuel cells, photocatalysis, and heterogeneous catalysis.54–57 It has emerged as one of the more promising supports for metal NPs because the nitrogen-rich structure of its amine-bridged heptazine units provides many adsorption sites for grafting metal ions on its surface.58 Furthermore, to the best of our knowledge, no NPs have yet been immobilized on g-C3N4 supports for the catalyzing H2NNH2·H2O dehydrogenation. Here we present the first instance of NiPt alloy nanoparticles (NPs) supported on g-C3N4 and show their excellent catalytic performance in the dehydrogenation of H2NNH2·H2O under moderate conditions.
g-C3N4 was prepared via thermal condensation of melamine powder.54 Melamine powder (5 g) was put in a covered alumina crucible, heated to 823 K at a rate of 5 K min−1 and maintained at 823 K for 4 h in a nitrogen flow (30 mL min−1). The final yellow product was collected and ground into powder with an agate mortar.
For the preparation of NiPt/g-C3N4 catalysts with variable Ni/Pt ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 1:4, 2:3, 3:2, 4:1, 0:1), 0.2 g of g-C3N4 was impregnated by a mixed solution containing 0.4 mmol of metal ions with variable molar ratios of Ni2+ and Pt4+, and the solution was diluted with distilled water to 20 mL. After stirring for 24 h at 298 K, a solution containing sodium borohydride (NaBH4) and sodium hydroxide (NaOH) was added to the above-mentioned mixture while stirring vigorously at 273 K. The catalysts were extracted by centrifugation, washed with ethanol, and dried in vacuum at 373 K for 12 h.
:0, 1:4, 2:3, 3:2, 4:1, 0:1), 0.2 g of g-C3N4 was impregnated by a mixed solution containing 0.4 mmol of metal ions with variable molar ratios of Ni2+ and Pt4+, and the solution was diluted with distilled water to 20 mL. After stirring for 24 h at 298 K, a solution containing sodium borohydride (NaBH4) and sodium hydroxide (NaOH) was added to the above-mentioned mixture while stirring vigorously at 273 K. The catalysts were extracted by centrifugation, washed with ethanol, and dried in vacuum at 373 K for 12 h.
The synthesis of the NiPt/g-C3N4 is displayed in Scheme 1. The NiPt/g-C3N4 catalysts were reproducibly prepared by this simple impregnation method, followed by reduction by the NaBH4 and NaOH solution at 273 K.49 After centrifugation and washing with copious amounts of water, NiPt/g-C3N4 was obtained, characterized using various techniques, and was used for the dehydrogenation of hydrous hydrazine. The molar ratio of the NiPt NPs in the catalysts was modified by tuning the initial composition of NiCl2 and H2PtCl6, and determined using inductively coupled plasma atomic emission spectroscopy (ICP-AES). As shown in Table S1 (ESI†), the Ni/g-C3N4, Ni24Pt76/g-C3N4, Ni37Pt63/g-C3N4, Ni58Pt42/g-C3N4, Ni82Pt18/g-C3N4, and Pt/g-C3N4 catalysts were synthesized using metal precursors at Ni/Pt molar ratios of 1:0, 1:4, 2:3, 3:2, 4:1, and 0:1, respectively. The catalytic reactions were performed in alkaline solutions of hydrazine at 323 K, as shown in Fig. 1 and Table S2 (ESI†). The performances of the catalysts were distinctly improved when Pt was alloyed to Ni. Ni37Pt63/g-C3N4 showed the best catalytic performance and a maximum turnover frequency (TOF) of 570 h−1 at 323 K, which is much higher than most of literature values for NiPt catalysts (Table 1).45–49 The 100% hydrogen selectivity of the decomposition reaction of hydrous hydrazine over Ni37Pt63/g-C3N4 was verified using mass spectrometry (Fig. S1, ESI†). Pt/g-C3N4 was nearly catalytically inactive, but catalytic activity increased by alloying and increasing the composition of Ni, which highlights the existence of synergistic effects on the molecular level from alloying NiPt NPs.
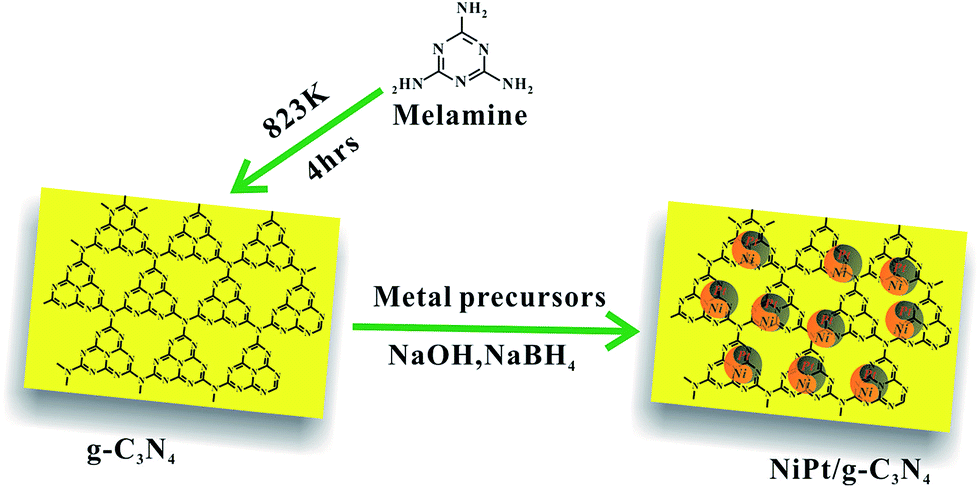 | ||
| Scheme 1 Schematic illustration of the synthesis of the NiPt/g-C3N4 catalysts. | ||
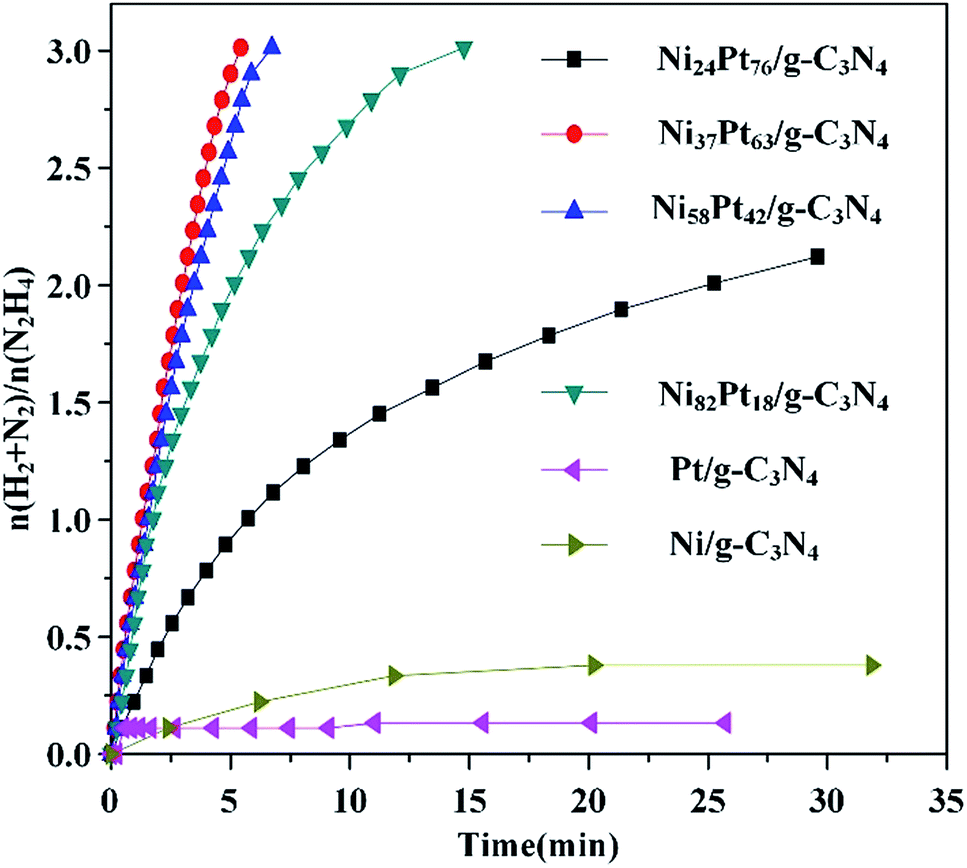 | ||
| Fig. 1 Time course plots for the decomposition of hydrous hydrazine over NiPt/g-C3N4 with NaOH (0.5 M) at 323 K (catalyst = 0.100 g; N2H4·H2O = 0.1 mL). | ||
| Catalyst | T (K) | TOF (h−1) | E a (kJ mol−1) | Reference |
|---|---|---|---|---|
| Ni37Pt63/g-C3N4 | 323 | 570 | 36.6 | This work |
| Ni0.99Pt0.01 | 323 | 12 | 49.95 | 59 |
| Ni0.9Pt0.1/Ce2O3 | 298 | 28.1 | 42.3 | 36 |
| Ni84Pt16/graphene | 323 | 415 | 40 | 44 |
| Ni88Pt12@MIL-101 | 323 | 375.1 | 51.29 | 43 |
| Ni3Pt7/graphene | 323 | 416 | 49.36 | 45 |
| Ni60Pt40/CeO2 | 303 | 293 | 58.2 | 39 |
| Ni64.1Mo11.5B24.4–La(OH)3 | 323 | 30 | 55.1 | 19 |
| Ni–Al2O3-HT | 303 | 3.9 | 49.3 | 17 |
| NiIr0.059/Al2O3 | 303 | 12.4 | 38.6 | 24 |
| NiPt0.057/Al2O3 | 303 | 16.5 | 34 | 30 |
| Ni30Fe30Pd40 NPs | 323 | 21.5 | 40.0 | 33 |
| Ni85Ir15@MIL-101 | 323 | 464 | 66.9 | 53 |
| Rh58Ni42@MIL-101 | 323 | 344 | 33 | 44 |
| Ni3Rh7/NPC-900 | 323 | 156 | — | 40 |
| Pt0.6Ni0.4/PDA-rGO | 323 | 2056 | 33.39 | 48 |
The dehydrogenation of hydrous hydrazine catalyzed by the Ni37Pt63/g-C3N4 was conducted at different temperatures (298–343 K) to acquire the activation energy (Ea) of this reaction. The values of the rate constant (k) were acquired from the slope of the linear part of the graph of dehydrogenation reaction versus time, at different temperatures (T) in Fig. 2(a). The Arrhenius plot (lnk vs. 1/T) for this catalyst is displayed in Fig. 2(b). Ea is determined to be 36.6 kJ mol−1, which is smaller than most of literature value for this reaction over NiPt catalysts (Table 1). Furthermore, the recyclability of the Ni37Pt63/g-C3N4 catalyst was investigated during the decomposition of hydrazine hydrate at 323 K (Fig. S2, ESI†). After the four times cycle run, the catalytic activity of catalyst decreased slightly and hydrogen selectivity remained stable. As shown in Fig. S3,† the reason for the decrease in the dehydrogenation performance can be attributed to agglomeration of the particles, which is consistent with the literature findings.47,49
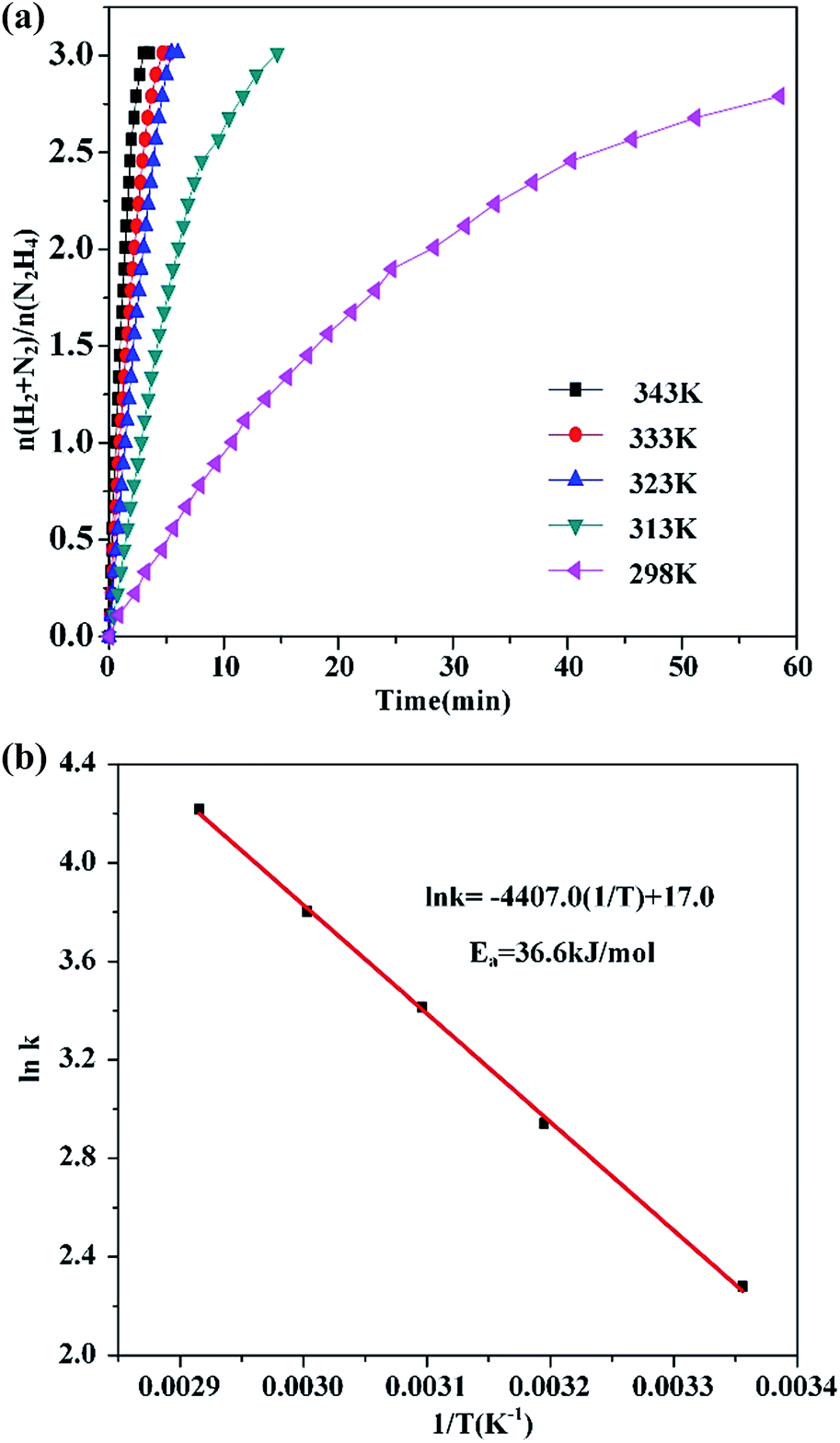 | ||
| Fig. 2 (a) Time course plots for hydrogen generation by the decomposition of hydrous hydrazine by Ni37Pt63/g-C3N4 at, 298 K, 313 K, 323 K, 333 K and 343 K. (b) Plot of lnk versus 1/T during the hydrous hydrazine decomposition over Ni37Pt63/g-C3N4 at different temperatures (catalyst = 0.100 g; N2H4·H2O = 0.1 mL). | ||
The X-ray diffraction (XRD) patterns of the g-C3N4, Ni/g-C3N4, Pt/g-C3N4, Ni24Pt76/g-C3N4, Ni37Pt63/g-C3N4, Ni58Pt42/g-C3N4 and Ni82Pt18/g-C3N4 are shown in Fig. S4 (ESI†). A distinct peak centered at 2θ = 27.5° is attributed to the XRD pattern of the g-C3N4.57 The XRD patterns of the Ni/g-C3N4 and Pt/g-C3N4 catalysts (Fig. S4(b) and (c), ESI†) are consistent with the literature values (JCPDS, PDF 65-0380 for Ni and PDF 65-2868 for Pt), and confirm the presence of pure Ni and Pt NPs.36 No peaks characteristic of pure Ni and Pt were observed in the XRD patterns of NiPt/g-C3N4 with different Ni/Pt molar ratios (Fig. S4(d–g), ESI†); this reveals the existence of a bimetallic phase rather than a mixture of monometallic Ni and Pt NPs.34–36,43,45–49 As seen in the literature,34–36,45–49 the diffraction peaks of all the NiPt nanocatalysts are partially shifted compared to those of pure Ni and Pt NPs; this highlights that an alloy has formed by expanding the Ni crystal lattice through the substitution of larger Pt atoms for the smaller Ni atoms. Additionally, the morphologies of the as-synthesized Ni37Pt63/g-C3N4 catalysts were observed using transmission electron microscopy (TEM). The high-resolution TEM (HRTEM) image in Fig. 3(a–d) demonstrates the crystalline nature of the Ni37Pt63 NPs. The lattice spacing is calculated to be 0.210 nm, which lies between the (111) plane of face-centered cubic (fcc) Ni (0.204 nm) and fcc Pt (0.227 nm), further demonstrating the formation of an alloy structure of NiPt NPs.34–36,43,45–49 Fig. S5 (ESI†) shows that the NiPt NPs have a narrow size distribution with a mean particle size of 3.2 ± 0.2 nm.
 | ||
| Fig. 3 (a–d) TEM images of Ni37Pt63/g-C3N4 with different magnification. | ||
The elemental chemical states of the Ni37Pt63/g-C3N4 were elucidated using X-ray photoelectron spectroscopy (XPS). The high-resolution XPS spectra for C 1s, N 1s, Ni 2p, and Pt 4f in Ni37Pt63/g-C3N4 are shown in Fig. S6 (ESI†). The XPS spectra of C 1s are centered at 287.8 and 284.6 eV. The first peak at 287.8 eV is attributed to the sp2-hybridized carbon atoms in the aromatic rings of g-C3N4, while the other peak at 284.6 eV is assigned to fortuitous carbon impurities. The N 1s spectrum of Ni37Pt63/g-C3N4 shows two distinct peaks at binding energies of 399.8 and 398.1 eV that are attributed to amino groups (C–N–H) and the sp2-hybridized nitrogen atoms of triazine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C) units, respectively. These assignments are in good agreement with the reported values for g-C3N4.57 Furthermore, the XPS results (Fig. S6, ESI†) of Ni37Pt63/g-C3N4 demonstrate the presence of metallic Ni0 with Ni 2p3/2 at 855.2 eV and Ni 2p1/2 at 872.7 eV, as well as metallic Pt0 with Pt 4f7/2 71.5 eV and Pt 4f5/2 74.7 eV.34–36,43,45–49 The binding energies for Ni 2p and Pt 4f in Ni37Pt63 NPs were different from those of pure Ni and Pt NPs; thus, the XPS results confirm the alloy structure of Ni37Pt63 NPs in Ni37Pt63/g-C3N4. The formation of oxidized Ni centered at 861.1 and 879.3 eV is a result of the reported sample preparation process for XPS measurements.43,46,49 The results obtained from XRD, HRTEM, and XPS all demonstrate that NiPt NPs form an alloy when deposited on the surface of g-C3N4.
N–C) units, respectively. These assignments are in good agreement with the reported values for g-C3N4.57 Furthermore, the XPS results (Fig. S6, ESI†) of Ni37Pt63/g-C3N4 demonstrate the presence of metallic Ni0 with Ni 2p3/2 at 855.2 eV and Ni 2p1/2 at 872.7 eV, as well as metallic Pt0 with Pt 4f7/2 71.5 eV and Pt 4f5/2 74.7 eV.34–36,43,45–49 The binding energies for Ni 2p and Pt 4f in Ni37Pt63 NPs were different from those of pure Ni and Pt NPs; thus, the XPS results confirm the alloy structure of Ni37Pt63 NPs in Ni37Pt63/g-C3N4. The formation of oxidized Ni centered at 861.1 and 879.3 eV is a result of the reported sample preparation process for XPS measurements.43,46,49 The results obtained from XRD, HRTEM, and XPS all demonstrate that NiPt NPs form an alloy when deposited on the surface of g-C3N4.
In conclusion, well-dispersed NiPt NPs have been successfully immobilized on g-C3N4 using a simple co-reduction method, which catalyze the dehydrogenation of hydrous hydrazine in an alkaline solution. The dehydrogenation performance of the catalysts depends on the composition of NiPt in the as-synthesized catalysts, indicating a synergistic molecular-scale alloy effect in bimetallic NiPt NPs. The Ni37Pt63/g-C3N4 catalysts demonstrate 100% hydrogen selectivity and remarkable catalytic performance with a maximum TOF value of 570 h−1 at 323 K, which is much higher than most of literature values for NiPt catalysts (Table 1). Designing and developing these high-performance catalysts using g-C3N4 as a support may accelerate the use of hydrous hydrazine as a hydrogen storage material.
Acknowledgements
Financial supports from National Natural Science Foundation of China (21376005, 21476001) and the Program for Zhejiang Leading Team of S&T Innovation (2013TD07) are gratefully appreciated.Notes and references
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- A. Züttel, A. Borgschulte and L. Schlapbach, Hydrogen as a Future Energy Carrier, Wiley-VCH, Weinheim, 2008 Search PubMed.
- D. A. J. Rand and R. M. Dell, Hydrogen Energy: Challenges and Prospects, RSC, Cambridge, 2008 Search PubMed.
- J. Graetz, Chem. Soc. Rev., 2009, 38, 73–82 RSC.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS.
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 CAS.
- A. W. C. Van de Berg and C. O. Areán, Chem. Commun., 2008, 668–681 RSC.
- C. W. Hamilton, R. T. Baker, A. Staubitzc and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- U. Eberle, M. Felderhoff and F. Schuth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- S. I. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS PubMed.
- Q. L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 CAS.
- D. G. Tong, D. M. Tang, W. Chu, G. F. Gu and P. Wu, J. Mater. Chem. A, 2013, 1, 6425–6432 CAS.
- L. He, Y. Q. Huang, A. Q. Wang, X. D. Wang, X. W. Chen, J. J. Delgado and T. Zhang, Angew. Chem., Int. Ed., 2012, 51, 6191–6194 CrossRef CAS PubMed.
- M. Y. Zheng, X. W. Chen, R. H. Cheng, N. Li, J. Sun, X. D. Wang and T. Zhang, Catal. Commun., 2006, 7, 187–191 CrossRef CAS.
- J. J. Zhang, Q. Kang, Z. Q. Yang, H. B. Dai, D. W. Zhuang and P. Wang, J. Mater. Chem. A, 2013, 1, 11623–11628 CAS.
- S. K. Singh, A. K. Singh, K. Aranishi and Q. Xu, J. Am. Chem. Soc., 2011, 133, 19638–19641 CrossRef CAS PubMed.
- H. L. Wang, J. M. Yan, S. J. Li, X. W. Zhang and Q. Jiang, J. Mater. Chem. A, 2015, 3, 121–124 CAS.
- J. Wang, Y. Li and Y. Zhang, Adv. Funct. Mater., 2014, 24, 7073–7077 CAS.
- X. W. Chen, T. Zhang, M. Y. Zheng, Z. L. Wu, W. C. Wu and C. Li, J. Catal., 2004, 224, 473–478 CrossRef CAS.
- L. He, Y. Q. Huang, X. Y. Liu, L. Li, A. Q. Wang, X. D. Wang, C. Y. Mou and T. Zhang, Appl. Catal., B, 2014, 147, 779–788 CrossRef CAS.
- D. G. Tong, X. L. Zeng, W. Chu, D. Wang and P. Wu, Mater. Res. Bull., 2010, 45, 442–447 CrossRef CAS.
- J. B. Yoo, H. S. Kim, S. H. Kang, B. Lee and N. H. Hur, J. Mater. Chem. A, 2014, 2, 18929–18937 CAS.
- S. K. Singh, X. B. Zhang and Q. Xu, J. Am. Chem. Soc., 2009, 131, 9894–9895 CrossRef CAS PubMed.
- Z. J. Zhang, Z. H. Lu, H. L. Tan, X. S. Chen and Q. L. Yao, J. Mater. Chem. A, 2015, 3, 23520–23529 CAS.
- O. Song-II, J. M. Yan, H. L. Wang, Z. L. Wang and Q. Jiang, Int. J. Hydrogen Energy, 2014, 39, 3755–3761 CrossRef.
- L. He, Y. Q. Huang, A. Q. Wang, Y. Liu, X. Y. Liu, X. W. Chen, J. J. Delgado, X. D. Wang and T. Zhang, J. Catal., 2013, 298, 1–9 CrossRef CAS.
- S. K. Singh and Q. Xu, J. Am. Chem. Soc., 2009, 131(50), 18032–18033 CrossRef CAS PubMed.
- J. Wang, W. Li, Y. R. Wen, L. Gu and Y. Zhang, Adv. Energy Mater., 2015, 1401879 Search PubMed.
- D. Bhattacharjee and S. Dasgupta, J. Mater. Chem. A, 2015, 3, 24371–24378 CAS.
- A. K. Singh and Q. Xu, Int. J. Hydrogen Energy, 2014, 39, 9128–9134 CrossRef CAS.
- Y. Y. Jiang, Q. Kang, J. J. Zhang, H. B. Dai and P. Wang, J. Power Sources, 2015, 273, 554–560 CrossRef CAS.
- H. L. Wang, J. M. Yan, Z. L. Wang, O. Song-II and Q. Jiang, J. Mater. Chem. A, 2013, 1, 14957–14962 CAS.
- J. Lin, J. Chen and W. P. Su, Adv. Synth. Catal., 2013, 355, 41–46 CrossRef CAS.
- B. Q. Xia, N. Cao, H. M. Dai, J. Su, X. J. Wu, W. Luo and G. Z. Cheng, ChemCatChem, 2014, 6, 2549–2552 CrossRef CAS.
- Y. Y. Jiang, H. B. Dai, Y. J. Zhong, D. M. Chen and P. Wang, Chem.–Eur. J., 2015, 21, 15439–15445 CrossRef CAS PubMed.
- B. Q. Xia, K. Chen, W. Luo and G. Z. Cheng, Nano Res., 2015, 8, 3472–3479 CrossRef CAS.
- J. Wang, X. B. Zhang, Z. L. Wang, L. M. Wang and Y. Zhang, Energy Environ. Sci., 2012, 5, 6885–6888 CAS.
- J. K. Sun and Q. Xu, ChemCatChem, 2015, 7, 526–531 CrossRef CAS.
- N. Cao, L. Yang, H. M. Dai, T. Liu, J. Su, X. J. Wu, W. Luo and G. Z. Cheng, Inorg. Chem., 2014, 53, 10122–10128 CrossRef CAS PubMed.
- P. P. Zhao, N. Cao, W. Luo and G. Z. Cheng, J. Mater. Chem. A, 2015, 3, 12468–12475 CAS.
- N. Cao, L. Yang, C. Du, J. Su, W. Luo and G. Z. Cheng, J. Mater. Chem. A, 2014, 2, 14344–14347 CAS.
- N. Cao, J. Su, W. Luo and G. Z. Cheng, Int. J. Hydrogen Energy, 2014, 39, 9726–9734 CrossRef CAS.
- Y. S. Du, J. Su, W. Luo and G. Z. Cheng, ACS Appl. Mater. Interfaces, 2015, 7, 1031–1034 CAS.
- F. Z. Song, Q. L. Zhu and Q. Xu, J. Mater. Chem. A, 2015, 3, 23090–23094 CAS.
- L. Wen, X. Q. Du, J. Su, W. Luo, P. Cai and G. Z. Cheng, Dalton Trans., 2015, 44, 6212–6218 RSC.
- S. K. Singh and Q. Xu, Inorg. Chem., 2010, 49(13), 6148–6152 CrossRef CAS PubMed.
- D. Bhattacharjee, K. Mandal and S. Dasgupta, J. Power Sources, 2015, 287, 96–99 CrossRef CAS.
- W. Gao, C. M. Li, H. Chen, M. Wu, S. He, M. Wei, D. G. Evansa and X. Duan, Green Chem., 2014, 16, 1560–1568 RSC.
- P. P. Zhao, N. Cao, J. Su, W. Luo and G. Z. Cheng, ACS Sustainable Chem. Eng., 2015, 3, 1086–1093 CrossRef CAS.
- Y. T. Gong, M. M. Li, H. R. Li and Y. Wang, Green Chem., 2015, 17, 715–736 RSC.
- J. J. Zhu, P. Xiao, H. L. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CAS.
- S. M. Yin, J. Y. Han, T. H. Zhou and R. Xu, Catal. Sci. Technol., 2015, 5, 5048–5061 CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- T. Yuan, H. F. Gong, K. Kailasam, Y. X. Zhao, A. Thomas and J. J. Zhu, J. Catal., 2015, 326, 38–42 CrossRef CAS.
- S. K. Singh, Z. H. Lu and Q. Xu, Eur. J. Inorg. Chem., 2011, 14, 2232–2237 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01335j |
| This journal is © The Royal Society of Chemistry 2016 |