
One-pot synthesis of 2,5-diformylfuran from fructose using a magnetic bi-functional catalyst†
Neha Mittal,
Grace M. Nisola,
Lenny B. Malihan,
Jeong Gil Seo,
Hern Kim,
Seong-Poong Lee and
Wook-Jin Chung*
Energy and Environment Fusion Technology Center (E2FTC), Department of Energy Science and Technology (DEST), Myongji University, Myongji-ro 116, Namdong, Cheoingu, Yongin, Gyeonggi-Do 449-728, Republic of Korea. E-mail: wjc0828@gmail.com; Fax: +82-31-337-2902; Tel: +82-31-336-8471
First published on 2nd March 2016
Abstract
A magnetic bi-functional WO3HO-VO(salten)-SiO2@Fe3O4 nanocatalyst was prepared to directly synthesize 2,5-diformylfuran (2,5-DFF) from fructose. The chlorosilylated SiO2@Fe3O4 (Cl-SiO2@Fe3O4) nanoparticles served as the platform for the two functionalities. Tungstic acid was generated via protonation of sodium tungstate, which was directly attached on the platform via nucleophilic –Cl displacement. Meanwhile, oxovanadium was complexed with a salten ligand which was functionalized on the Cl-SiO2@Fe3O4. Characterization results confirmed the successful preparation of the WO3HO-VO(salten)-SiO2@Fe3O4 nanocatalyst. Under the optimal one-pot system, tungstic acid-mediated fructose dehydration afforded 82% 5-hydroxymethylfurfural (5-HMF) in 1 h. Upon co-oxidant H2O2 addition, in situ 5-HMF oxidation by the activated oxoperoxovanadium species produced 71% of 2,5-DFF after 15 h under ambient air. The stability of 5-HMF formation was found critical to 2,5-DFF production. Aside from the catalytic efficiency and process simplicity, the WO3HO-VO(salten)-SiO2@Fe3O4 nanocatalyst was readily retrieved magnetically and re-used multiple times with marginal losses in its activity.
1. Introduction
Production of biomass-based platform chemicals typically involves a multi-step conversion of the substrate through sequential transformations of bridging molecules. A common practice is to carry out reactions independently wherein each step requires a suitable down-stream process and a unique catalyst, which functions in a narrow set of reaction conditions and solvent system. This rigorous procedure ensures that the desired product quality and yield are achieved.1,2Particular interest is currently focused on 2,5-diformylfuran (2,5-DFF), a specialty chemical with a wide array of uses as a precursor of fungicides, pharmaceuticals, ligands of various poly-Schiff bases, cross-linking agents, and polymers.3 The 2,5-DFF can be produced via a two-step reaction using biomass-derived carbohydrates as substrates (Scheme 1). Initial step would require dehydration of C6-carbohydrates like fructose into 5-hydroxymethylfurfural (5-HMF), a valuable key intermediate molecule.1,4,5 In the second step, the isolated 5-HMF can be selectively oxidized to afford 2,5-DFF.6,7
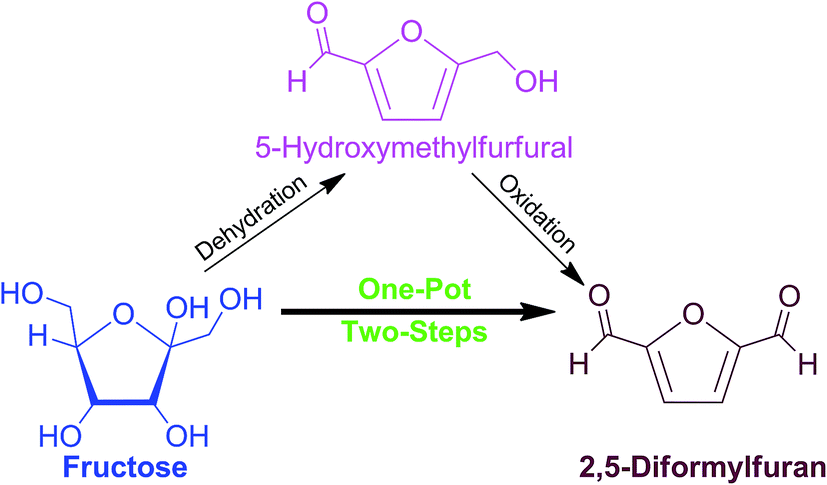 | ||
| Scheme 1 One-pot, two-step conversion of fructose to 2,5-diformylfuran. | ||
Fructose dehydration into 5-HMF has been commonly performed with several types of acid catalysts1,4 either as homogeneous inorganic mineral acids, metal chlorides or heterogeneous5 ion-exchange resins, supported heteropolyacids, zeolites, sulphonic acid functionalized polymer and carbon nanotubes, graphite oxide, mesoporous silica and magnetic silica supported acid catalysts. Likewise, various reaction systems which employed homogenous catalysts6 like Pb(OAc)4-pyridine, pyridinium chlorochromate, K2Cr2O7–DMSO, Co/Mn acetates, 2,2,6,6-tetramethylpiperidine-1-oxide, V2O5 and Cu(NO3)2/VOSO4 and heterogeneous catalysts7 like vanadylphosphate, porous manganese oxide, V2O5-zeolite, silver-manganese oxide, Mn3O4/Fe3O4, TEMPO-SBA-15, Ru-PVP–CNTs and Ru-covalent triazine frameworks were reported for the selective 5-HMF oxidation to 2,5-DFF.
Although adequate yields have been achieved from each independent reaction, the use of 5-HMF as an isolated precursor is impractical due to its difficult separation from high-boiling point reaction solvents and un-stable nature.4c Thus, an alternative approach is to directly produce 2,5-DFF from fructose. Tandem reactions, which include dehydration and oxidation in a one-pot system could significantly simplify the whole process, reduce the production cost, and render the production route environmentally benign.
Few studies have reported one-pot 2,5-DFF synthesis from fructose using a duo of catalysts such as (1) Amberlyst-15 combined with Ru/HT, (2) AlCl3·6H2O/NaBr with supported vanadium oxide, (3) protonated graphitic carbon nitride (g-C3N4) with vanadium doped g-C3N4, (4) magnetic sulphonic acid with magnetic hydroxyapatite, and (5) Amberlyst-15 with polyaniline-VO(acac)2.7c,8–11 Recently, more advanced catalysts with bi-functional properties such as (1) Pd-V@MIL-101 (MOFs),12 (2) protonated and vanadium doped g-C3N4,9 and (3) heteropolyacids containing molybdenum (H3PMo12O40),13a combined with cesium (Cs3HPMo11VO40)13b have also been reported; the combination of two catalysts on a single material platform further simplified the one-pot 2,5-DFF system.
But so far, acceptable 2,5-DFF yields and facile one-pot reaction systems are yet to be achieved. The lack of reaction “switch” that activates the required catalyst for the subsequent reaction suggests unregulated production of intermediates like 5-HMF, which could limit 2,5-DFF production.9 Also, the use of high boiling point solvents like DMSO and DMF requires energy-intensive and complex product isolation techniques.8,12,13b While some bi-functional nanocatalysts were recycled via filtration or centrifugation, these post-reaction steps add cost to the entire production.9,13b With these current limitations, a robust recyclable catalyst with dual functionality that can be easily retrieved via a simple technique, must be developed. Furthermore, the use of nanoparticles (NPs) as support could provide a pseudo-homogeneous character wherein significant activity can be achieved despite the heterogeneity of the catalyst system. In this regard, magnetic iron oxide NPs (Fe3O4) are attractive catalyst supports due to their low cost, easy synthesis and functionalization, high surface area, and facile separation by an external magnetic force.7g,14–19
Thus herein, a bi-functional catalyst with magnetic property was developed for direct synthesis of 2,5-DFF from fructose. The bi-functional catalyst was anchored with (1) tungstic acid for fructose dehydration and (2) oxovanadium complex for 5-HMF oxidation. Both active sites were easily attached covalently on the magnetic NPs (Scheme 2). Tungstic acid was selected due to its reportedly high catalytic activity.20,21 Meanwhile, the oxovanadium complex is a dormant moiety which can be activated upon addition of H2O2.22 This reaction ‘switch’ mechanism could mimic the sequential dehydration and oxidation of conventional two-step 2,5-DFF synthesis in a one-pot system.
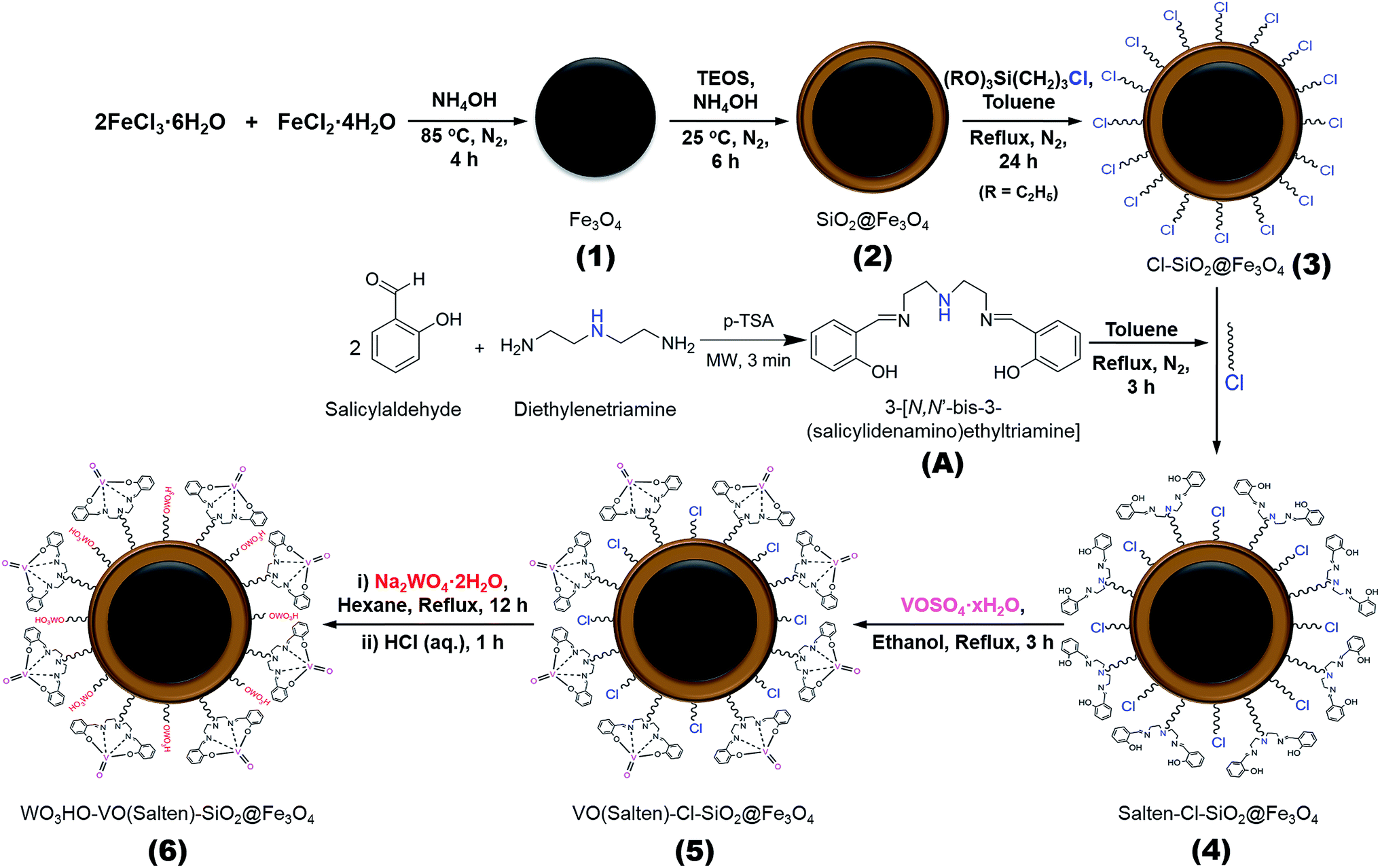 | ||
| Scheme 2 Synthesis of bi-functional WO3HO-VO(salten)-SiO2@Fe3O4 (6) nanocatalyst. | ||
2. Experimental
2.1. Materials
Iron(II) chloride tetrahydrate (>99% FeCl2·4H2O), iron(III) chloride hexahydrate (>99% FeCl3·6H2O), tetraethylorthosilicate (98% TEOS), (3-chloropropyl)-triethoxysilane (95% CPTES), salicylaldehyde (99%), diethylenetriamine (>98%), sodium tungstate dihydrate (>99% Na2WO4·2H2O), and 5-hydroxymethylfurfural (98% 5-HMF) were purchased from Acros Organics. Vanadium(IV) oxide sulfate hydrate (97% VOSO4·xH2O), p-toluenesulfonic acid monohydrate (98.5% p-TSA), D-(−)fructose (99%), and dry toluene (99.8%) were obtained from Sigma-Aldrich (USA). 2,5-diformylfuran (>98% 2,5-DFF) was supplied by Tokyo Chemicals (Japan). Hydrochloric acid (35–37% HCl) was bought from Showa Chemicals whereas other reagents and solvents including ammonia hydroxide (25–28% NH3·H2O), hydrogen peroxide (30% H2O2), sodium sulfate (99%), anhydrous ethanol (99.9% EtOH), n-hexane (95%), ethyl acetate (99.8% EtOAc), and isopropanol (99.5%) were purchased from Dae Jung Chemicals (South Korea). All chemicals were of reagent grade or higher, and were directly used without further purification.2.2. Bi-functional catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio) was continuously stirred (400 rpm) under N2 blanket at 85 °C. Ammonia water (28% NH3·H2O) was then added dropwise until pH = 9. After 4 h, the black precipitates were collected and washed with deionized (DI) water (pH = 7) and EtOH. The NPs were vacuum-dried for 12 h to obtain Fe3O4 (1) NPs.
:1 mole ratio) was continuously stirred (400 rpm) under N2 blanket at 85 °C. Ammonia water (28% NH3·H2O) was then added dropwise until pH = 9. After 4 h, the black precipitates were collected and washed with deionized (DI) water (pH = 7) and EtOH. The NPs were vacuum-dried for 12 h to obtain Fe3O4 (1) NPs.Product 1 was acid treated in 0.1 M HCl (2 g 100 mL−1) for 10 min and then washed with DI water before coating with SiO2. The NPs were dispersed in (5 + 1 v/v) EtOH/H2O solution, followed by pH adjustment to 10 using NH3·H2O (28%). The SiO2 precursor (2 mL TEOS) was fractionally added and reacted for 6 h under N2 blanket at room temperature (RT). The collected dark brown NPs were washed thrice with EtOH and finally vacuum-dried to obtain SiO2@Fe3O4 (2).
2.3. Preparation of mono-functional catalysts
2.4. Catalyst characterization
Wide-angle powder X-ray diffraction (XRD) patterns were recorded on PANalytical X'pert-Pro diffractometer, operated at a 40 kV and 40 mA. The FT-IR spectra of the samples were scanned in Varian spectrophotometer using standard KBr disc method between wavenumbers 400 and 4000 cm−1 at RT. Thermogravimetric analysis (TGA) was carried out in SCINCO TGA (N-1000) from 35 °C to 1000 °C at 10 °C min−1 ramping rate, in N2 atmosphere (50 mL min−1). Nitrogen adsorption–desorption isotherms were obtained using BELSORP Mini-II (Japan) surface area analyzer at 77 K. The samples were outgassed in vacuo at 100 °C for 24 h prior to the adsorption experiments. The samples were inspected under Field Emission Transmission Electron Microscope (FE-TEM TECNAI G2 F30). The magnetic strength of NPs was measured on a vibrating sample magnetometer (VSM) at 25 °C from −15000 to 15000 Oe. Vanadium content was quantified by inductively coupled plasma mass spectrometer (ICP-MS 7500, Agilent, USA). The tungstic acid sites in nanocatalyst (6) was measured using acid–base titration. The catalyst was treated with standardized aqueous NaOH solution. The remaining excess (un-neutralized) NaOH was used to titrate an oxalic acid standard using phenolphthalein as the indicator to determine the concentration of remaining NaOH.21
2.5. 2,5-DFF production from fructose
Known amounts of fructose substrate, magnetic nanocatalyst, H2O2 (if added), and isopropanol solvent were added in a 100 mL round bottom flask fitted with a mechanical stirrer and water cooled condenser. The temperature was kept constant in an oil bath and the reaction mixture was mechanically stirred to facilitate the one-pot reaction. At certain time intervals, aliquots of the reaction mixture were collected and the nanocatalyst was magnetically separated. The liquid sample was dried under reduced pressure, dissolved in DI water and then analyzed in high pressure liquid chromatography (HPLC).2.6. Product analysis and quantification
Samples were pre-filtered through a syringe filter (0.2 μm Nylon). The Waters HPLC was equipped with Waters 2414 refractive index (RI) detector maintained at 40 °C and Biorad Aminex HPX-87H ion-exclusion column (300 × 7.8 mm) kept at 60 °C. A 5 mM H2SO4 mobile phase was delivered at a flow rate of 0.6 mL min−1. The concentrations of 2,5-DFF (i.e. [DFF]) were quantified against calibration curves of standard samples. The amount of produced 2,5-DFF (WDFF) and corresponding reaction yields were calculated according to eqn (1) and (2) where V is the sample volume, MW is the molecular weight of fructose (180.16 g mol−1) or 2,5-DFF (124.09 g mol−1) while WRM, WAL, and Wfructose are the masses of the reaction mixture, aliquot for analysis, and fructose, respectively.
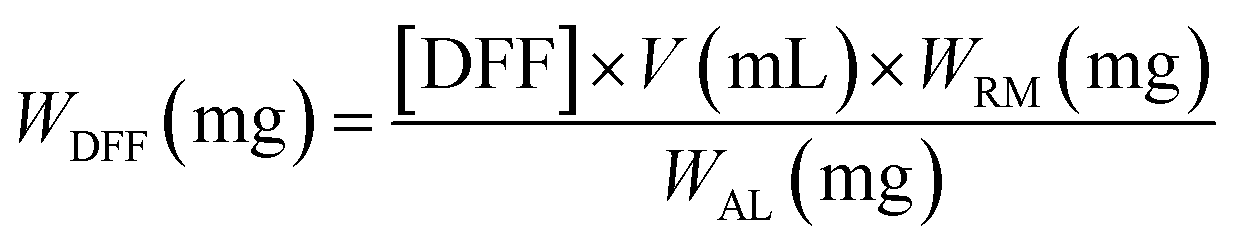 | (1) |
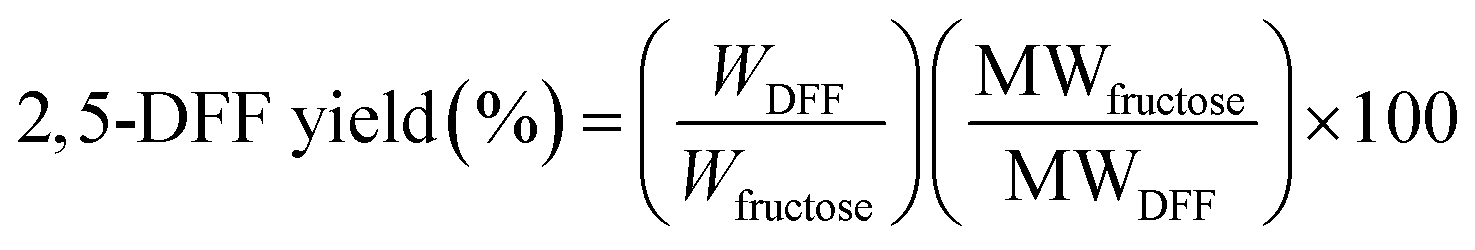 | (2) |
2.7. Isolation and purification of 2,5-DFF
After catalyst removal, the vacuum-dried reaction sample (under reduced pressure) was washed with hexane and re-crystallized in ethyl acetate to obtain the pale white crystalline 2,5-DFF product (0.1464 g, isolated yield = 60%). Identification of the product 2,5-DFF was confirmed by 1H NMR on a Varian 400 MHz NMR spectrometer in deuterated chloroform (CDCl3) as shown in Fig. S2.† 1H NMR (CDCl3): δ 7.30 (s, 2H), 9.81 (s, 2H) ppm.2.8. Recycling of catalyst (6)
After reaction, the bi-functional magnetic nanocatalyst was magnetically retrieved from the mixture. The used catalyst was washed with isopropanol to remove traces of reaction debris and then dried at RT before re-use as detailed in Section 2.5.3. Results and discussion
3.1. Synthesis and characterization of bi-functional catalyst
The XRD patterns (Fig. 1) verified the crystallinity of bare Fe3O4 which exhibited six well-resolved diffraction peaks at 2θ = 30.6°, 36.0°, 43.5°, 54.1°, 57.7° and 63.2°. These peaks correspond to (2 2 0), (3 1 1), (4 0 0), (4 2 2), (5 1 1) and (4 4 0) reflection planes, which is consistent with a cubic inverse spinel structure as reported in literature (JCPDS 19-0629).25 Meanwhile, the identical XRD pattern of SiO2@Fe3O4 with bare Fe3O4 and absence of characteristic peaks for SiO2 indicate the amorphous nature of the SiO2 coating layer.26 Likewise, attachments of salten ligand and WO3HO moieties on SiO2@Fe3O4 neither altered the textural property of Fe3O4 (Fig. 1 WO3HO-VO(salten)-SiO2@Fe3O4), which confirms the structural preservation of the bulk NP support after multi-functionalization steps.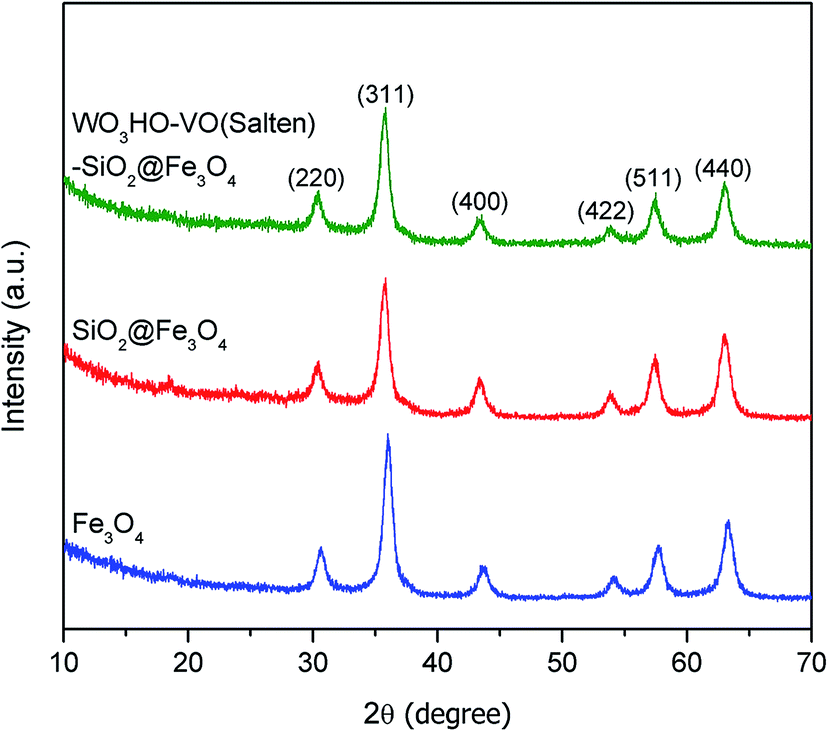 | ||
| Fig. 1 Powder XRD patterns of Fe3O4, SiO2@Fe3O4 NPs and WO3HO-VO(salten)-SiO2@Fe3O4 catalyst. | ||
While the SiO2 shell was not indicated in the XRD results, its presence was confirmed via TEM (Fig. 2). Bare Fe3O4 NPs (Fig. 2a) were spherical with average nanosize diameter (Dave) of 9 ± 0.9 nm (Fig. 2b). Its SAED pattern (inset: Fig. 2a), which corroborated the XRD result (Fig. 1), further ascertained the mono-crystalline nature of Fe3O4. The SiO2@Fe3O4 apparently had similar morphology (Fig. 2c) with the bare Fe3O4 but close inspection (inset: Fig. 2c) revealed the SiO2 layer with an average thickness of 1.36 ± 0.6 nm, ranging from 0.82–1.92 nm; this is consistent with the increased Dave of SiO2@Fe3O4 to 11.8 ± 0.8 nm (Fig. 2b). Statistical comparison of the average diameters of the two samples via unpaired t-test reveal their significant difference (p < 0.0001), which further affirms the presence of SiO2 layer in SiO2@Fe3O4. Additional confirmatory test via EDS confirmed the presence of SiO2 layer on SiO2@Fe3O4 (Fig. 2d).
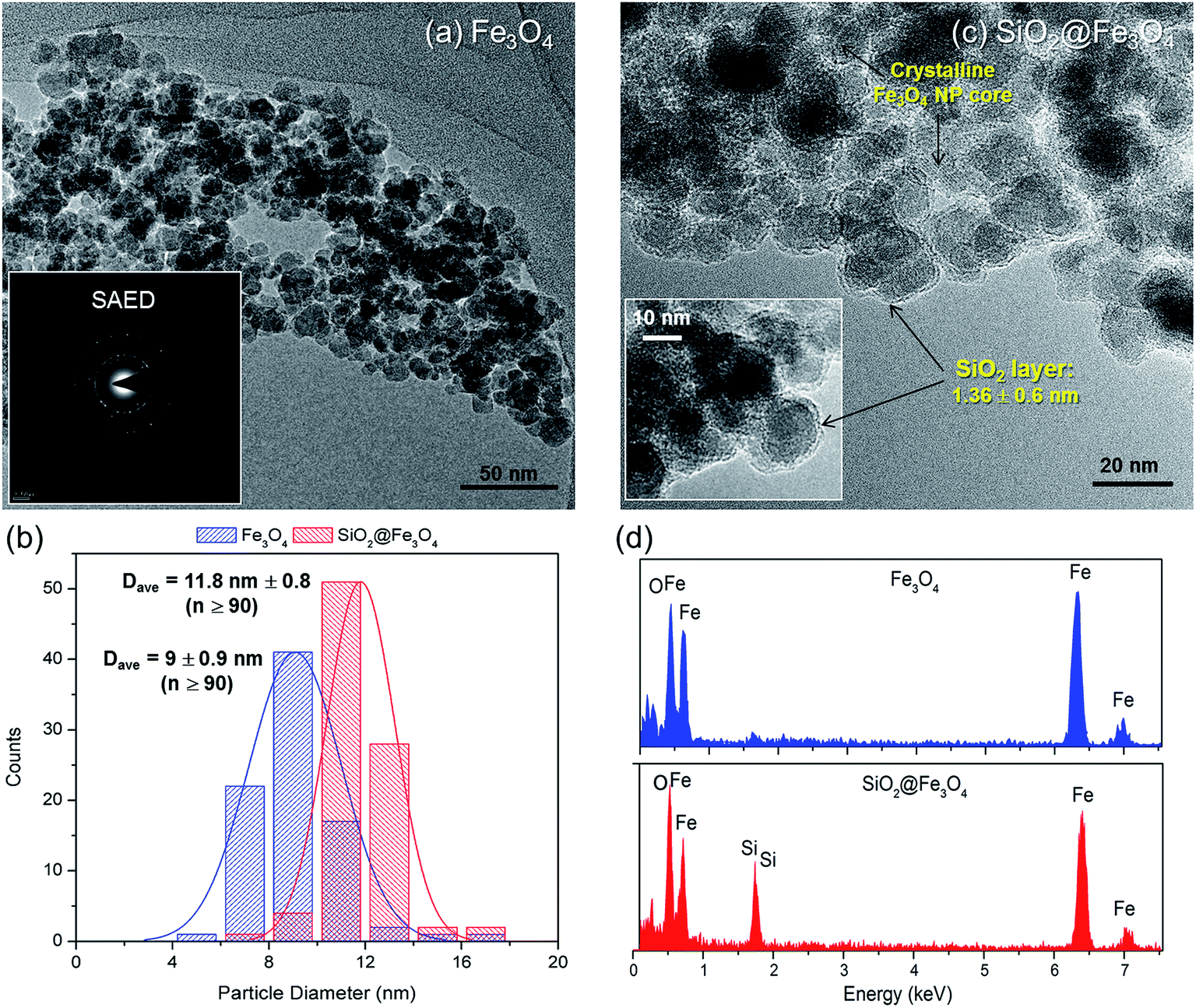 | ||
| Fig. 2 TEM images of (a) Fe3O4, (c) SiO2@Fe3O4 NPs, and their (b) size distribution histograms and (d) EDAX analysis. The insets show (a) SAED pattern of Fe3O4 NPs and (c) HRTEM image of silica layer over the Fe3O4 core. | ||
Though the nanocatalyst WO3HO-VO(salten)-SiO2@Fe3O4 appeared similar with SiO2@Fe3O4 (Fig. S1a†), it had the largest Dave (12.4 nm) among the samples (Fig. S1b†). Based on the measured Dave from TEM, the surface area (SA = πr2) ratio of spherical bare Fe3O4 and nanocatalyst was estimated to be around 1.90. On the other hand, the BET SA of WO3HO-VO(salten)-SiO2@Fe3O4 decreased to 50.9 m2 g−1 (vs. 100.2 m2 g−1 of bare Fe3O4) from which the SA ratio was 1.96. The very close SA ratios from TEM and BET SA results strongly suggest growth in size of the nanocatalyst due to functionalization.
The thermal stability and presence of organic moieties on WO3HO-VO(salten)-SiO2@Fe3O4 catalyst were examined by comparing the TGA curves of the NPs (Fig. 3). Initial weight loss (1.4–2.0%) in all samples below 200 °C is attributable to the evaporation of physically adsorbed (or crystalline) water.14 The bare Fe3O4 experienced only 7.2% of total weight loss while that of SiO2@Fe3O4 had similar value (data not shown). On the other hand, SiO2@Fe3O4 functionalized with CPTES organic linker (3) had a total weight loss of 9.8% while that of the nanocatalyst WO3HO-VO(salten)-SiO2@Fe3O4 was 12.3%. The higher weight loss in both samples indicates the presence of organic functional groups (i.e. CPTES linker and salten ligand), which were thermally decomposed from 200 to 800 °C. Thus, TGA results suggest the successful preparation of WO3HO-VO(salten)-SiO2@Fe3O4. Moreover, the minimal weight loss in the nanocatalyst below 200 °C indicates its thermal stability and feasible application at its typical operating condition of 100 °C.14
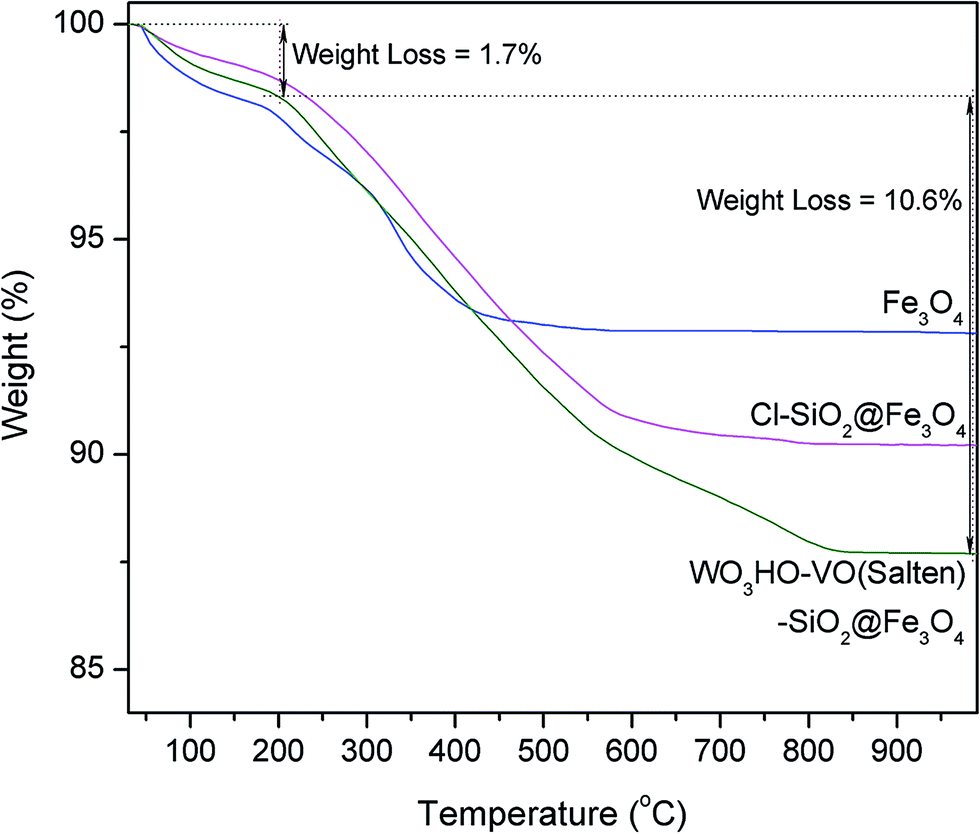 | ||
| Fig. 3 TGA curves of Fe3O4, Cl-SiO2@Fe3O4 NPs and WO3HO-VO(salten)-SiO2@Fe3O4 catalyst. | ||
The FTIR results provided a more definite indication of the functional groups present on the modified Fe3O4 NPs (Fig. 4). The characteristic peak of bare Fe3O4 (Fig. 4a) at 578 cm−1 was due to the Fe–O stretching.27 The silica coating layer in SiO2@Fe3O4 sample was confirmed by the Si–OH stretching at 3425 cm−1 and Si–O–Si bonds (antisymmetric) at 1080 cm−1 (Fig. 4b).7h Peak appearances at 2978 and 2916 cm−1 in Cl-SiO2@Fe3O4, due to asymmetric and symmetric C–H stretching of propyl chain of the linker, indicate successful silylation of SiO2@Fe3O4 with CPTES (Fig. 4c). Peaks at 1489 cm−1 and 1458 cm−1 were due to the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the ligand in salten-Cl-SiO2@Fe3O4 (Fig. 4d). Likewise, the vibrational signal at 1627 cm−1 was due to the CN stretching of the salten ligand.28 However, the complexation of vanadyl sulfate with salten-Cl-SiO2@Fe3O4 (5) showed weak intensities at 980 cm−1 (for VO) and 555 cm−1 (for V–N) which might have been masked by strong Si–O–Si and Fe–O bands (Fig. 4e).29 The final nanocatalyst product exhibited sharp signals for WO bond at 956 cm−1 and W–O bond at 902 cm−1,21,30 suggesting the successful synthesis of the bi-functional WO3HO-VO(salten)-SiO2@Fe3O4 nanocatalyst.
C stretching of the ligand in salten-Cl-SiO2@Fe3O4 (Fig. 4d). Likewise, the vibrational signal at 1627 cm−1 was due to the CN stretching of the salten ligand.28 However, the complexation of vanadyl sulfate with salten-Cl-SiO2@Fe3O4 (5) showed weak intensities at 980 cm−1 (for VO) and 555 cm−1 (for V–N) which might have been masked by strong Si–O–Si and Fe–O bands (Fig. 4e).29 The final nanocatalyst product exhibited sharp signals for WO bond at 956 cm−1 and W–O bond at 902 cm−1,21,30 suggesting the successful synthesis of the bi-functional WO3HO-VO(salten)-SiO2@Fe3O4 nanocatalyst.
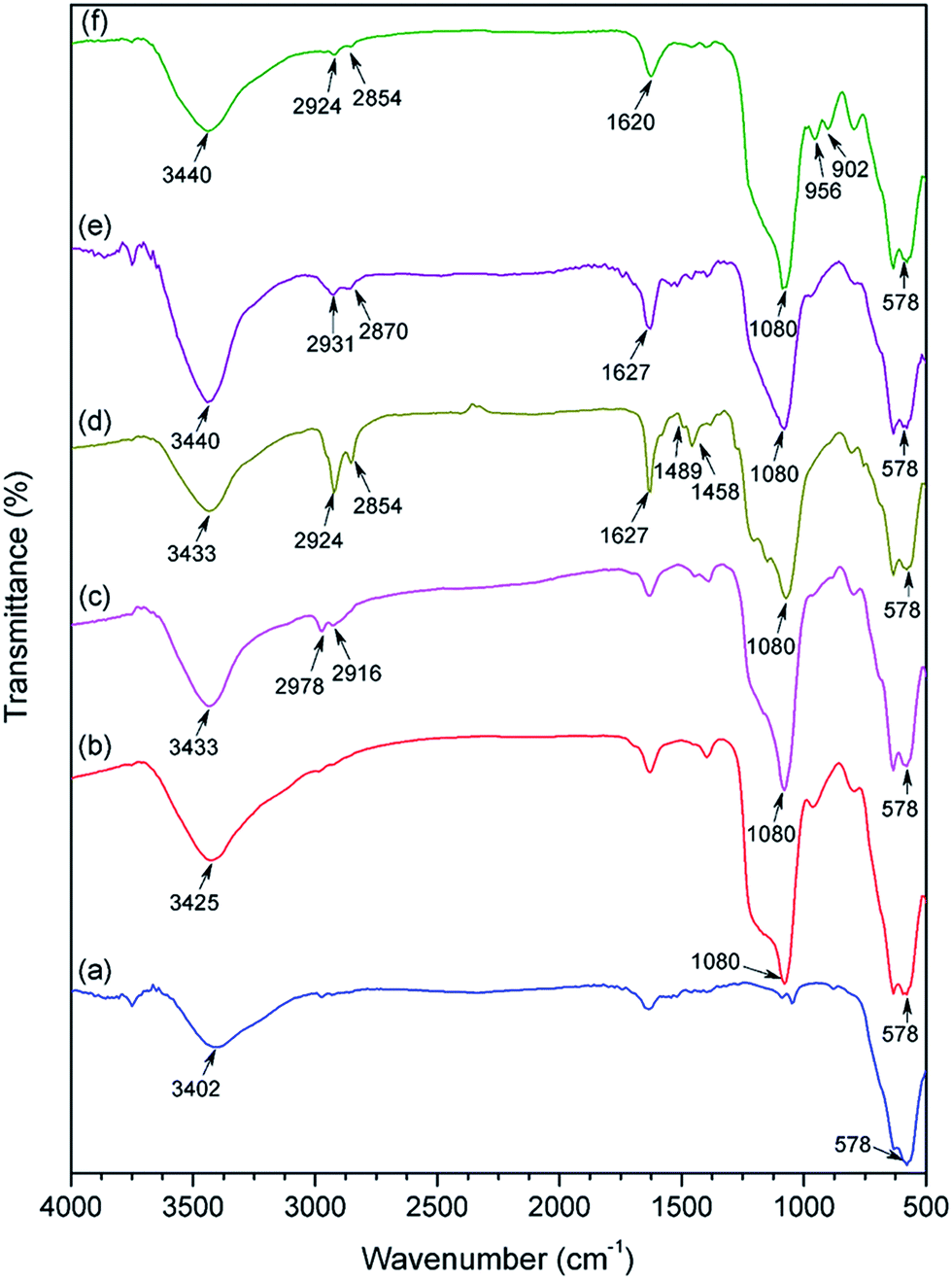 | ||
| Fig. 4 FT-IR spectra of (a) Fe3O4, (b) SiO2@Fe3O4, (c) Cl-SiO2@Fe3O4, (d) salten-Cl-SiO2@Fe3O4, (e) VO(salten)-Cl-SiO2@Fe3O4 and (f) WO3HO-VO(salten)-SiO2@Fe3O4 catalyst. | ||
Overall characterization results confirm the successful preparation of WO3HO-VO(salten)-SiO2@Fe3O4. The spherical bi-functional nanocatalyst was found to contain 0.4 mmol g−1 of VO(salten) complex from the V content of ICP-MS results and 0.25 mmol g−1 tungstic acidic sites (WO3OH-) based on acid–base titration. No material deterioration is anticipated if it is used at elevated temperature (∼100 °C).
While the preparation of WO3HO-VO(salten)-SiO2@Fe3O4 was found successful, its utility as a heterogeneous bi-functional nanocatalyst was further assessed in terms of its recovery from the reaction mixture. Primarily, its magnetic character must remain sufficiently strong to facilitate its convenient magnetic separation from the reaction mixture. Magnetization curves (Fig. 5) of bare Fe3O4 and WO3HO-VO(salten)-SiO2@Fe3O4 exhibited no hysteresis loop, suggesting the superparamagnetic nature of both NPs.14 Compared with the bare Fe3O4 (67.2 emu g−1), the reduced magnetization saturation (Ms) value of the nanocatalyst (41.6 emu g−1) was due to its increased mass and size after SiO2 coating and functionalization. Nonetheless, the Ms value of the nanocatalyst was still reasonably strong for its easy and efficient magnetic separation from the reaction mixture (inset: Fig. 5).
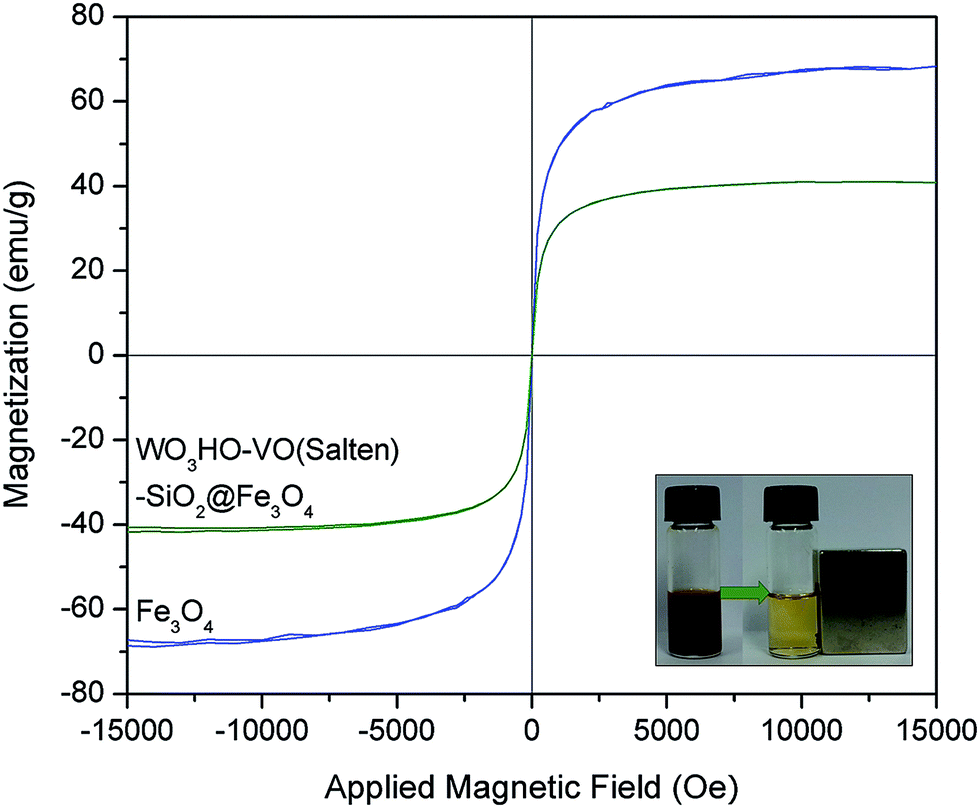 | ||
| Fig. 5 Magnetization curves of Fe3O4 NPs and WO3HO-VO(salten)-SiO2@Fe3O4 catalyst. The inset shows the magnetic behavior of the catalyst after reaction. | ||
3.2. Catalytic performance of mono-functional catalysts (7) and (8)
To elucidate the performance of the bi-functional catalyst, the role of each catalytic component was decoupled by synthesizing the mono-functionalized nanocatalysts which were solely anchored with either tungstic acid (WO3HO-SiO2@Fe3O4) or VO(salten) complex [VO(salten)-SiO2@Fe3O4] (Schemes S1 and S2†). Acid–base titration result revealed 0.64 mmol g−1 of tungstic acid in WO3HO-SiO2@Fe3O4 (7) while ICP-MS analysis of V estimated 0.70 mmol g−1 of VO(salten) complex in VO(salten)-SiO2@Fe3O4 (8).As a Brønsted acid catalyst, WO3HO-SiO2@Fe3O4 supplies the proton that activates the anomeric –OH site in fructose which initiates the triple dehydration reaction.20,31–33 It is believed that the released protons are captured back by the tungstic acid sites, which closes the catalytic cycle for fructose dehydration.20,32 Time profile (Fig. 6a) reveals a gradual decline in fructose concentration concomitant with the steady increase in 5-HMF until a maximum yield of 86.3% and 90% fructose conversion was achieved after 60 min. No rehydration products4c were detected even after 120 min when the 5-HMF yield eventually declined to 27.6%. However, the decrease in 5-HMF yield after 60 min could be due to the onset of polymeric humins formation, a degradation product of 5-HMF.4a,34 Polymeric humins are typically formed in a highly acidic environment (i.e. surplus acid supply from WO3HO-SiO2@Fe3O4) and prolonged reaction time at an elevated temperature. This finding suggests that regulation of acid catalyst loading is critical to minimize the extent of 5-HMF degradation.
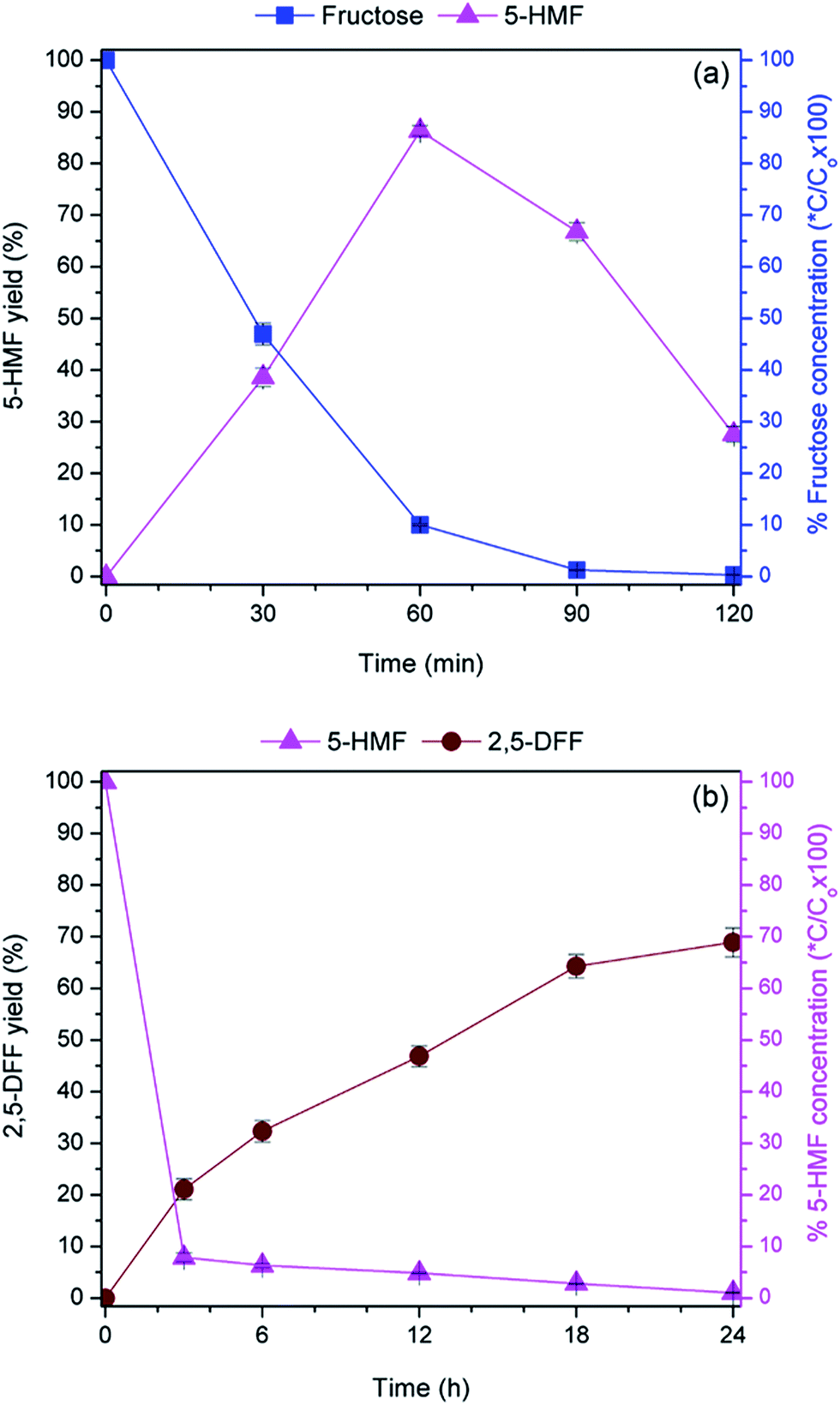 | ||
| Fig. 6 (a) Dehydration of fructose using WO3HO-SiO2@Fe3O4 catalyst. Reaction conditions: fructose (1.0 mmol), catalyst (50 mg, 0.032 mmol tungstic acid), isopropanol (solvent), 80 °C and, (b) oxidation of 5-HMF using VO(salten)-SiO2@Fe3O4 catalyst. Reaction conditions: 5-HMF (1.0 mmol), catalyst [50 mg, 0.035 mmol VO(salten)], H2O2 (1.0 mmol), isopropanol (solvent), RT. *C/Co = final concentration/initial concentration. | ||
Oxidation of 5-HMF is facilitated by the intermediate oxoperoxovanadium species which are conveniently generated via activation of the VO(salten) complex in the presence of H2O2.36 Time profile (Fig. 6b) shows the gradual formation of 2,5-DFF while the consumption of 5-HMF was rapid and complete, while over-oxidation products remained undetected. Production of 2,5-DFF eventually stalled after 24 h at which a maximum yield of 68.9% was achieved. The non-concurrent rates of product generation and substrate consumption could be due to the limiting effects of intermediate formation, which consequently hampered the final step of 2,5-DFF formation. Since the reaction was performed at RT, oxidation temperature increase could be needed to accelerate either the generation of oxoperoxovanadium species or the oxidation of 5-HMF by oxoperoxovanadium complex. Nonetheless, the absence of over-oxidation products along with the decent 2,5 DFF yield makes the VO(salten)-SiO2@Fe3O4 a suitable and selective 5-HMF oxidation catalyst.
Overall results from monocatalyst activities demonstrate the individual function and effectiveness of each component of the bi-functional catalyst. To figure out the synergy between the tungstic acid and VO(salten) complex, one-pot reactions using the bi-functional nanocatalyst WO3HO-VO(salten)-SiO2@Fe3O4 was carried out and tuned to maximize the 2,5-DFF yield from fructose at the shortest period possible.
3.3. One-pot synthesis of 2,5-DFF from fructose
Initial one-pot 2,5-DFF synthesis from 1.0 mmol fructose (Fig. 7a) was performed using 100 mg of the bi-functional nanocatalyst (0.025 mmol tungstic acid and 0.04 mmol of VO(salten) complex). Fructose dehydration was initiated at 80 °C wherein a maximum of 82.2% 5-HMF yield and 94.2% fructose conversion were achieved after 1 h. This reaction behavior coincided with that of the acidic monocatalyst (WO3HO-SiO2@Fe3O4). The slightly lower 5-HMF yield can be ascribed to the lower level of tungstic acid supply from the bi-functional nanocatalyst (0.025 mmol vs. 0.032 mmol in Section 3.2.1.). | ||
| Fig. 7 Reaction profiles of one-pot conversion of fructose to 2,5-DFF using WO3HO-VO(salten)-SiO2@Fe3O4 bi-functional catalyst at different temperatures (oxidation step). Reaction conditions: fructose (1.0 mmol), catalyst [100 mg, 0.04 mmol VO(salten) and 0.025 mmol tungstic acid], H2O2 (1.0 mmol, when added), isopropanol (solvent), 80 °C (for initial 1 h). *C/Co = final concentration/initial concentration. | ||
After 1 h of fructose dehydration, the reaction mixture was cooled to RT (25 °C) and then injected with H2O2 to initiate 5-HMF oxidation into 2,5-DFF. The 5-HMF gradually declined and reached a minimum value of 4.8% after 24 h. Coupled with 5-HMF consumption is the gradual production of 2,5-DFF which reached 58.9% yield after 24 h oxidation (i.e. 25 h overall reaction). Albeit the supply of VO(salten) complex was higher in the current experiment (0.04 mmol vs. 0.035 mmol in Section 3.2.2.), the 2,5-DFF yield was lower than that from the monocatalyst VO(salten)-SiO2@Fe3O4. Apart from humins formation due to 5-HMF degradation, the accessibility of VO(salten) complex in WO3HO-VO(salten)-SiO2@Fe3O4 to the 5-HMF or H2O2 might have been reduced due to the simultaneous presence of tungstic acid sites. Moreover, the lower yield of 5-HMF which was the substrate of 2,5-DFF might have also contributed to its lower production. Nonetheless, the findings from this reaction exhibited the potential production of 2,5-DFF from fructose via-one pot reaction using the developed bi-functional WO3HO-VO(salten)-SiO2@Fe3O4.
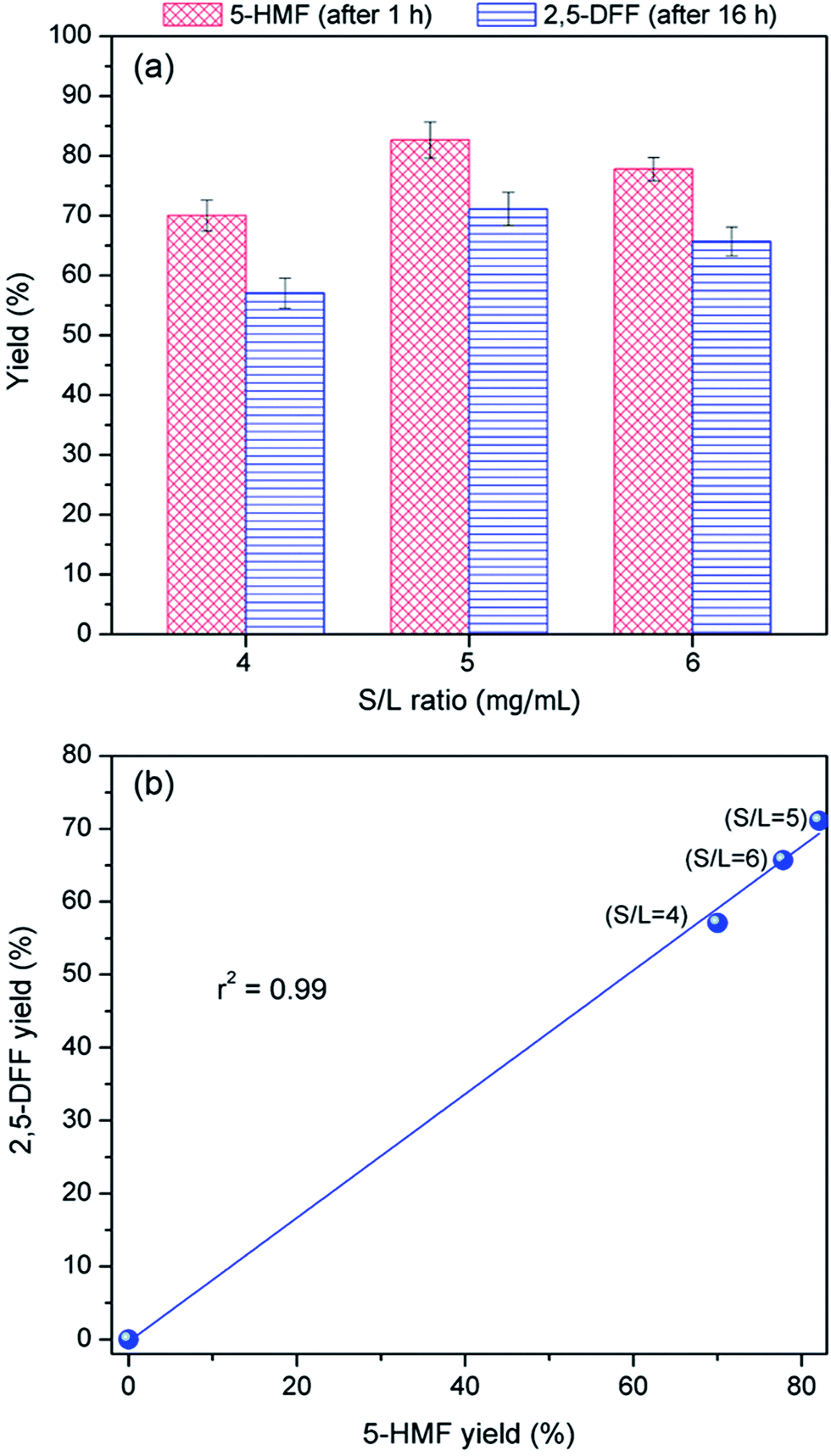 | ||
| Fig. 8 (a) Effect of S/L ratio (catalyst dosage/reaction volume) on one-pot conversion of fructose to 2,5-DFF using WO3HO-VO(salten)-SiO2@Fe3O4 catalyst and (b) plot of dependence of 2,5-DFF yield on 5-HMF produced in the reaction with respect to S/L ratio. Reaction conditions: fructose (1.0 mmol), catalyst [0.4 mmol g−1 VO(salten) and 0.25 mmol g−1 tungstic acid], H2O2 (1.0 mmol, added after 1 h), isopropanol (solvent), 80 °C (for initial 1 h) and 60 °C (for subsequent 15 h). | ||
To elucidate the effect of nanocatalyst dosage, 5-HMF yield was plotted against 2,5-DFF yield (Fig. 8b). The strong correlation between the values (r2 = 0.99) indicates that 2,5-DFF production was mainly dependent on the availability of 5-HMF. This supports the earlier finding (Section 3.3.) that 2,5 DFF yield at fixed 5-HMF was not dependent on the amount of available VO(salten) complex. Thus, this suggests that it is the availability of tungstic acid sites (i.e. controlled via bi-functional catalyst dosage) that influenced 5-HMF production, which in turn affected the 2,5-DFF yield. At low S/L ratio = 4, insufficient tungstic acid sites led to low 5-HMF production whereas a harsher acidic environment at S/L ratio = 6 hampered the 5-HMF yield perhaps due to the observed humins formation. The lower 5-HMF yields in both S/L ratios consequently resulted in sub-optimal 2,5-DFF yields. The highest 5-HMF yield and 2,5-DFF production at S/L = 5 suggests that this catalyst dosage provided the most suitable tungstic acid level for the reaction (Fig. 8b).
 | ||
| Fig. 9 Effect of H2O2 dosage on one-pot conversion of fructose to 2,5-DFF using WO3HO-VO(salten)-SiO2@Fe3O4 catalyst. Reaction conditions: fructose (1.0 mmol), catalyst [100 mg, 0.04 mmol VO(salten) and 0.025 mmol tungstic acid], H2O2 (added after 1 h), isopropanol (solvent), 80 °C (for initial 1 h) and 60 °C (for subsequent 15 h). | ||
Results from one-pot synthesis of 2,5-DFF from fructose using WO3HO-VO(salten)-SiO2@Fe3O4 as a bi-functional nanocatalyst reveal that temperature control in each reaction step, nanocatalyst dosage via S/L ratio, and H2O2 addition were critical in achieving the maximum 5-HMF yield (82.2%) and 2,5-DFF production (71.1%).
3.4. Recycling of WO3HO-VO(salten)-SiO2@Fe3O4 catalyst
Recycling and isolation of any catalyst are essential characteristics for its application at industrial scale. The nanocatalyst WO3HO-VO(salten)-SiO2@Fe3O4 was readily retrieved due to its magnetic core support. After each reaction, it was magnetically separated from the mixture, washed with isopropanol, and then dried for further use. The performance of the bi-functional nanocatalyst was reliable even after repeated use (Fig. 10). After five cycles of reaction, the decline in 2,5-DFF yield was very minor (≅5%) relative to the first cycle. Furthermore, the FTIR of used nanocatalyst (Fig. S3†) showed consistent signature peaks with that of fresh WO3HO-VO(salten)-SiO2@Fe3O4 (Fig. 4f). This suggests resilience of the nanocatalyst to deterioration despite its exposure to different chemical conditions during the reaction. The consistent performance of WO3HO-VO(salten)-SiO2@Fe3O4 after several uses demonstrates its potential as an economically viable route for one-pot synthesis of 2,5-DFF from biomass-based carbohydrates like fructose. | ||
| Fig. 10 Recyclability of WO3HO-VO(salten)-SiO2@Fe3O4 catalyst. Reaction conditions: fructose (1.0 mmol), catalyst [100 mg, 0.04 mmol VO(salten) and 0.025 mmol tungstic acid], H2O2 (1.0 mmol, added after 1 h), isopropanol (solvent), 80 °C (for initial 1 h) and 60 °C (for subsequent 15 h). | ||
4. Conclusions
The bi-functional magnetic nanocatalyst WO3HO-VO(salten)-SiO2@Fe3O4 which contained two types of active sites was successfully prepared through a simple functionalization procedure. The nanocatalyst had a stable structure which can withstand the needed reaction temperature. Its dual catalytic performance is attributable to a reaction “switch” mechanism, which afforded sequential conversions. The catalyst effectively produced 2,5-DFF from fructose in a one-pot system with reasonable yields under mild reaction conditions. The nanocatalyst was readily separated from the reaction mixture by an external magnet and it performed consistently for several cycles of reaction without any form of deterioration. These findings strongly indicate the industrial potential of WO3HO-VO(salten)-SiO2@Fe3O4 nanocatalyst for direct 2,5-DFF production from fructose. Overall findings can provide insights for the development of one-pot systems for the syntheses of biomass-derived specialty chemicals.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2009-0093816) and by the Ministry of Science, ICT & Future Planning (2015R1C1A2A01054605). The authors would like to thank Korea Basic Science Institute (KBSI), Seoul for the use of TEM and Korea Institute of Science and Technology (KIST), Seoul for the use of VSM.References
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- Z. Yuan, Z. Zhang, J. Zheng and J. Lin, Fuel, 2015, 150, 236 CrossRef CAS.
- (a) K. T. Hopkins, W. D. Wilson, B. C. Bender, D. R. McCurdy, J. E. Hall, R. R. Tidwell, A. Kumar, M. Bajic and D. W. Boykin, J. Med. Chem., 1998, 41, 3872 CrossRef CAS PubMed; (b) M. D. Poeta, W. A. Schell, C. C. Dykstra, S. Jones, R. R. Tidwell, A. Czarny, M. Bajic, M. Bajic, A. Kumar, D. Boykin and J. R. Perfect, Antimicrob. Agents Chemother., 1998, 42, 2495 Search PubMed; (c) D. T. Richter and T. D. Lash, Tetrahedron Lett., 1999, 40, 6735 CrossRef CAS; (d) A. Gandini and M. N. Belgacem, Prog. Polym. Sci., 1997, 22, 1203 CrossRef CAS.
- (a) N. Mittal, G. M. Nisola and W.-J. Chung, Tetrahedron Lett., 2012, 53, 3149 CrossRef CAS; (b) J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC; (c) Y. Qiao, N. Theyssen and Z. Hou, Recyclable Catal., 2015, 2, 36 Search PubMed.
- (a) Y.-M. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59 CrossRef CAS PubMed; (b) Y. Li, H. Liu, C.-H. Song, X.-M. Gu, H.-M. Li, W.-S. Zhu, S. Yin and C.-R. Han, Bioresour. Technol., 2013, 133, 347 CrossRef CAS PubMed; (c) B. Liu, Z.-Z. Gou, A.-Q. Liu and Z.-H. Zhang, J. Ind. Eng. Chem., 2015, 21, 338 CrossRef CAS; (d) V. V. Ordomsky, J. C. Schouten, J. van der Schaaf and T. A. Nijhuis, Chem. Eng. J., 2012, 207–208, 218 CrossRef CAS; (e) G.-X. Nie, X.-L. Tong, Y.-Y. Zhang and S. Xue, Catal. Lett., 2014, 144, 1759 CrossRef CAS; (f) X.-C. Guo, Q. Cao, Y.-J. Jiang, J. Guan, X.-Y. Wang and X.-D. Mu, Carbohydr. Res., 2012, 351, 35 CrossRef CAS PubMed; (g) X.-C. Zhang, M. Wang, Y.-H. Wang, C.-F. Zhang, Z. Zhang, F. Wang and J. Xu, Chin. J. Catal., 2014, 35, 703 CrossRef CAS.
- (a) J. W. Reijendam, G. J. Heeres and M. J. Janssen, Tetrahedron Lett., 1970, 26, 1291 CrossRef; (b) L. Cottier, G. Descotes, J. Lewkowski and R. Skowronski, Pol. J. Chem., 1994, 68, 693 CAS; (c) L. Cottier, G. Descotes, J. Lewkowski and R. Skowronski, Org. Prep. Proced. Int., 1995, 27, 564 CrossRef CAS; (d) W. Partenheimer and V. V. Grushin, Adv. Synth. Catal., 2001, 343, 102 CrossRef CAS; (e) G. A. Halliday, R. J. Young and V. V. Grushin, Org. Lett., 2003, 5, 2003 CrossRef CAS PubMed; (f) J. P. Ma, Z. T. Du, J. Xu, Q. H. Chu and Y. Pang, ChemSusChem, 2011, 4, 51 CrossRef CAS PubMed; (g) N. Mittal, G. M. Nisola, L. B. Malihan, J. G. Seo, S.-P. Lee and W.-J. Chung, Korean J. Chem. Eng., 2014, 31, 1362 CrossRef CAS.
- (a) C. Carlini, P. Patrono, A. M. R. Galletti, G. Sbrana and V. Zima, Appl. Catal., A, 2005, 289, 197 CrossRef CAS; (b) O. C. Navarro, A. C. Canos and S. I. Chornet, Top. Catal., 2009, 52, 304 CrossRef CAS; (c) A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562 CrossRef CAS; (d) Z. Z. Yang, J. Deng, T. Pan, Q. X. Guo and Y. Fu, Green Chem., 2012, 14, 2986 RSC; (e) I. Sadaba, Y. Y. Gorbanev, S. Kegnaes, S. S. R. Putluru, R. W. Berg and A. Riisager, ChemCatChem, 2013, 5, 284 CrossRef CAS; (f) G. D. Yadav and R. V. Sharma, Appl. Catal., B, 2014, 147, 293 CrossRef CAS; (g) B. Liu, Z. Zhang, K. Lv, K. Deng and H. Duan, Appl. Catal., A, 2014, 472, 64 CrossRef CAS; (h) N. Mittal, G. M. Nisola, J. G. Seo, S.-P. Lee and W.-J. Chung, J. Mol. Catal. A: Chem., 2015, 404, 106 CrossRef; (i) J. Chen, J. Zhong, Y. Guo and L. Chen, RSC Adv., 2015, 5, 5933 RSC; (j) J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672 CrossRef CAS PubMed.
- S. Zhang, W. Li, X. Zeng, Y. Sun and L. Lin, BioResources, 2014, 9, 4568 Search PubMed.
- J. Z. Chen, Y. Y. Guo, J. Y. Guo, L. S. Song and L. M. Chen, ChemCatChem, 2014, 6, 3174 CrossRef CAS.
- Z. Zhang, Z. Yuan, D. Tang, Y. Ren, K. Lv and B. Liu, ChemSusChem, 2014, 7, 3496 CrossRef CAS PubMed.
- F. Xu and Z. Zhang, ChemCatChem, 2015, 7, 1470 CrossRef CAS.
- F. Tao, Y. Cui, P. Yang and Y. Gong, Russ. J. Phys. Chem. A, 2014, 88, 1091 CrossRef CAS.
- (a) Y. Liu, L. Zhu, J. Tang, M. Liu, R. Cheng and C. Hu, ChemSusChem, 2014, 7, 3541 CrossRef CAS PubMed; (b) R. Liu, J. Chen, L. Chen, Y. Guo and J. Zhong, ChemPlusChem, 2014, 79, 1448 CrossRef CAS.
- S. Otokesh, E. Kolvari, A. Amoozadeh and N. Koukabi, RSC Adv., 2015, 5, 53749 RSC.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036 CrossRef CAS PubMed.
- Y. Zhang and C. G. Xia, Appl. Catal., A, 2009, 366, 141 CrossRef CAS.
- S. Shylesh, A. Wagener, A. Seifert, S. Ernst and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 184 CrossRef CAS PubMed.
- J. Lee, Y. Lee, J. K. Youn, H. B. Na, T. Yu, H. Kim, S. M. Lee, Y. M. Koo, J. H. Kwak, H. G. Park, H. N. Chang, M. Hwang, J. G. Park, J. Kim and T. Hyeon, Small, 2008, 4, 143 CrossRef CAS PubMed.
- G. Chouhan, D. S. Wang and H. Alper, Chem. Commun., 2007, 4809 RSC.
- B. Karami, V. Ghashghaee and S. Khodabakhshi, Catal. Commun., 2012, 20, 71 CrossRef CAS.
- S. K. Kundu, J. Mondal and A. Bhaumik, Dalton Trans., 2013, 42, 10515 RSC.
- (a) A. L. Cánepa, C. M. Chanquía, V. M. Vaschetti, G. A. Eimer and S. G. Casuscelli, J. Mol. Catal. A: Chem., 2015, 404–405, 65 CrossRef; (b) J. Pisk, J. –C. Daran, R. Poli and D. Agustin, J. Mol. Catal. A: Chem., 2015, 403, 52 CrossRef CAS.
- A. Ying, S. Liu, Y. Ni, F. Qiu, S. Xu and W. Tang, Catal.: Sci. Technol., 2014, 4, 2115 CAS.
- S. Das, V. K. Das, L. Saikia and A. J. Thakur, Green Chem. Lett. Rev., 2012, 5(3), 457 CrossRef CAS.
- F. C. Zheng, Q. W. Chen, L. Hu, N. Yana and X. K. Kong, Dalton Trans., 2014, 43, 1220 RSC.
- J. Choi, J. C. Kim, Y. B. Lee, I. S. Kim, Y. K. Park and N. H. Hur, Chem. Commun., 2007, 1644 RSC.
- M. Yamaura, R. L. Camilo, L. C. Sampaio, M. A. Macedo, M. Nakamura and H. E. Toma, J. Magn. Magn. Mater., 2004, 279, 210 CrossRef CAS.
- P. Sharma, A. Lazar and A. P. Singh, Appl. Catal., A, 2012, 439–440, 101 CrossRef CAS.
- J. R. Zamian and E. R. Dockal, Transition Met. Chem., 1996, 21, 370 CrossRef CAS.
- S. Wang, Z. Zhang, B. Liu and J. Li, Catal.: Sci. Technol., 2013, 3, 2104 CAS.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Catal., 2012, 295, 122 CrossRef CAS.
- M. Farahi, B. Karami, S. Alipour and L. T. Moghadam, Acta Chim. Slov., 2014, 61, 94 CAS.
- J. Zhang, A. Das, R. S. Assary, L. A. Curtiss and E. Weitza, Appl. Catal., B, 2016, 181, 874 CrossRef CAS.
- D. Ray, N. Mittal and W.-J. Chung, Carbohydr. Res., 2011, 346, 2145 CrossRef CAS PubMed.
- S. Wang, Z. Zhang, B. Liu and J. Li, Ind. Eng. Chem. Res., 2014, 53, 5820 CrossRef CAS.
- A. M. E. Hendaway, A. M. Fayed and M. R. Mostafa, Transition Met. Chem., 2011, 36, 351 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01549b |
| This journal is © The Royal Society of Chemistry 2016 |