
Facile O-glycosylation of glycals using Glu-Fe3O4-SO3H, a magnetic solid acid catalyst†
Raju S. Thombal and
Vrushali H. Jadhav*
Department of Organic Chemistry, National Chemical Laboratory (CSIR-NCL), Pune-411008, India. E-mail: vh.jadhav@ncl.res.in; Tel: +91 20 25902275
First published on 16th March 2016
Abstract
A new glucose derived magnetic solid acid catalyst (Glu-Fe3O4-SO3H) was synthesized in a convenient and ecofriendly manner and well characterized using FTIR, PXRD, EDAX, SEM, and XPS which showed the presence of Fe3O4 embedded on the surface of the catalyst along with –SO3H, –OH and –COOH functional groups. This new heterogeneous catalyst was studied for synthesis of 2-deoxy galactosides/glucosides with good yields and selectivity. Moreover, the catalyst can be easily separated from the reaction with an external magnetic force and reused for a minimum of four times without any significant decrease in the yields of the products after every recycle.
1. Introduction
2-Deoxy-O-glycosides are found widely in number of natural products.1,2 The 2-deoxy-O-glycoside component in any natural product plays an important role as it is responsible for the bioactivity of a drug. For chemical synthesis of these natural products large quantities of 2-deoxy-O-glycosides are required. Hence, attention has been extensively paid for synthesis of these glycosides. Till date, number of homogeneous catalyst have been reported for the synthesis of 2-deoxy-O-glycosides by direct addition of alcohols to glycals using protic acids or Lewis acids.3–8 They suffer from drawback of corrosion, contamination and needs neutralization after the reaction and hence the workup is tedious. Such catalysts are difficult to separate from the reaction and they cannot be reused. Also one main disadvantage in these direct glycosylations is the possibility of the Ferrier reaction. Heterogeneous catalysts have advantages over homogeneous catalyst that they are inexpensive, more efficient, stable, easily separable and reusable. Hence there is a need to develop mild and efficient catalyst for glycosylation reactions to overcome the above mentioned problems. Balmond et al.9 in 2012, reported selective synthesis of 2-deoxy-O-galactoside using thiourea as a catalyst. Das et al.10 in 2015, reported selective synthesis of 2-deoxy-O-glycosides using electron deficient pyridinium salts. Our continuous interest in this field prompted us to synthesize glucose derived magnetic solid acid catalyst.11 Herein, we report the synthesis of new Glu-Fe3O4-SO3H catalyst and showed its application for tetrahydropyran protection of alcohols and glycosylation of protected glycals in the synthesis of 2-deoxy-O-glycosides (Fig. 1).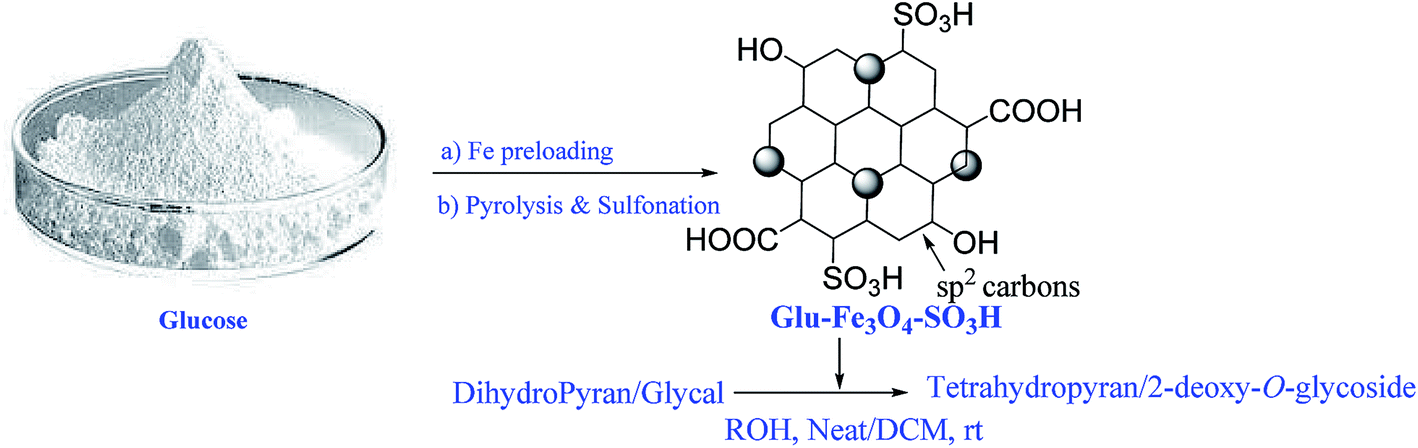 | ||
| Fig. 1 Schematic representation for synthesis of Glu-Fe3O4-SO3H catalyst and its application for THP protection of alcohol and 2-deoxy-O-glycoside synthesis. | ||
2. Experimental
2.1 Catalyst preparation
5.0 g of glucose and 276 mL of FeCl3 solution with a concentration of 100 mmol l−1 were mixed in a flask and stirred for 5 h at rt. Then, the water in the mixture was evaporated, and the solid residue was dried at 353 K overnight. Fe preloaded glucose was then stirred with p-TSA (5 eq.) at 140 °C for 24 h under argon atmosphere. The mixture was then cooled to rt, and then slowly added to a beaker containing 500 mL deionised water. The suspension mixture was then filtered off and washed repeatedly with deionised water to remove all the sulfate ions (SO42−). The solid was further washed with absolute ethanol to remove the water, and then dried at 353 K overnight to get a black magnetic material (Glu-Fe3O4-SO3H).2.2 General procedure for THP protection of alcohols using Glu-Fe3O4-SO3H under neat conditions
Dihydropyran (0.30 mmol), alcohol (0.9 mmol) and catalyst Glu-Fe3O4-SO3H (1 wt%) were stirred together in a screw-capped vial under argon atmosphere. The mixture was stirred neat at rt for 2–5 min. The reaction was monitored by TLC analysis. After completion of the reaction, DCM was added and the catalyst was separated from the reaction mixture by applying external magnetic field. DCM layer was decanted and then evaporated to obtain the product.2.3 General procedure for glycosylation of protected glycals using Glu-Fe3O4-SO3H
Glycal (1.2 mmol), alcohol (1.4 mmol) and catalyst Glu-Fe3O4-SO3H (5 wt%) were stirred together in a screw-capped vial under argon atmosphere neat or in DCM (DCM was used when the glycal and alcohol both are solids). The mixture was stirred for required temperatures and required time. The reaction was monitored by TLC. After completion of the reaction the catalyst was separated from the reaction mixture by applying external magnetic field (in neat conditions, DCM was added after completion of the reaction). DCM layer was then decanted and evaporated to obtain the product. The anomeric ratio's were determined from the 1H NMR of the products.3. Results and discussion
3.1 Characterization of the catalyst
Due to our continuous interest in this field, we have synthesized a magnetic carbonaceous solid acid catalyst Glu-Fe3O4-SO3H. For preparation of this catalyst, we choose readily available D-glucose as a carbon precursor. Fe was preloaded on glucose using FeCl3 and p-TSA was used as a sulphonating agent to create active acidic sites. Initially Fe was preloaded on glucose using FeCl3. The free –OH groups in glucose easily coordinated with adsorbed Fe(III) ions and then evaporation of solvent and drying gave black Fe(III) based complex. This complex was then pyrolysed and sulfonated simultaneously using p-TSA at 140 °C under nitrogen. The Fe preloaded on glucose was partly hydrolysed to FeO(OH) during drying. FeO(OH) was further reduced to Fe3O4 by reducing components like H2, CO2 and CO which are formed during carbonation process.12 Pyrolysis and sulfonation in situ leads to formation of a polycylic aromatic structure embedded with active Fe3O4, –SO3H, –COOH and –OH sites.The catalyst thus obtained was characterized with FT-IR (Spectrum 400), PXRD (Panalytical X'Pert Pro), Elemental analysis from EDAX (Nova Nano SEM 450), SEM (Quanta™ Scanning Electron Microscope), XPS (Prevac Ambient Pressure Photo Electron Spectroscopy) and BET surface area (Quantachrome ASiQwin). The acid densities were measured by acid base titration.13 FT-IR spectrum (Fig. 2a) showed characteristic peaks at 1012 cm−1 and 1040 cm−1 which are attributed to O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO stretching vibrations in –SO3H groups and peak at 1127 cm−1 for SO3H stretching. This indicates that the sulfonic acid groups have been successively incorporated on the catalyst surface. Peaks at 1645 cm−1 attributed for CC stretching vibrations in aromatic carbons and peak at 1670 cm−1 attributed for CO stretching. Peak at 1714 cm−1 attributed to presence of CO stretching vibration of –COOH group. Bands at 2925 cm−1 attributed for C–H stretching band. Bands due to O–H stretching were observed at 3410 cm−1. The PXRD (Fig. 2b) showed a weak but broad peak of 2θ at 15–30°, indicating formation of amorphous carbon having aromatic carbon sheets oriented in a random fashion. The sharp peaks seen indicates the formation of Fe3O4 crystallite in the pyrolysis process. The energy dispersive X-ray analysis (Fig. 2c) confirms that the catalyst surfaces are composed mainly of C, O, Fe and S. Composition of S and Fe is found to be 7.4% and 7.7% respectively from EDAX. The elemental analysis showed composition of C to be 50%, H to be 4% and S to be 9%. SEM (Fig. 2d) image shows formation of porous nature of the catalyst. The surface composition of Glu-Fe3O4-SO3H was analysed by XPS (Fig. 2e). The C 1s spectrum includes six peaks with different binding energy values. The peaks could be assigned to the carbon atoms in the forms of C–S (283.5 eV), C–C (284 and 284.5 eV), C–O (285 eV), CO (285.5 eV), OC–O (286 eV). The S 2p spectrum showed three different peaks that can be assigned to S–C (186.5), S–O (169 eV) and SO (169.5 eV). O 1s spectrum shows peaks in the range 530–535 eV indicating the presence of Fe–O and C–O–Fe groups suggesting the linkage of Fe3O4 with porous carbon. BET surface area, pore size and pore volume was calculated using the standard Brunauer–Emmett–Teller (BET) equation and was found to be 3.38 m2 g−1, 9.53 Å and 6.07 m3 g−1 respectively. The total acid density and the sulphonic acid density of Glu-Fe3O4-SO3H based on acid base titration was found to be 2.87 mmol g−1 and 1.46 mmol g−1 respectively.
SO stretching vibrations in –SO3H groups and peak at 1127 cm−1 for SO3H stretching. This indicates that the sulfonic acid groups have been successively incorporated on the catalyst surface. Peaks at 1645 cm−1 attributed for CC stretching vibrations in aromatic carbons and peak at 1670 cm−1 attributed for CO stretching. Peak at 1714 cm−1 attributed to presence of CO stretching vibration of –COOH group. Bands at 2925 cm−1 attributed for C–H stretching band. Bands due to O–H stretching were observed at 3410 cm−1. The PXRD (Fig. 2b) showed a weak but broad peak of 2θ at 15–30°, indicating formation of amorphous carbon having aromatic carbon sheets oriented in a random fashion. The sharp peaks seen indicates the formation of Fe3O4 crystallite in the pyrolysis process. The energy dispersive X-ray analysis (Fig. 2c) confirms that the catalyst surfaces are composed mainly of C, O, Fe and S. Composition of S and Fe is found to be 7.4% and 7.7% respectively from EDAX. The elemental analysis showed composition of C to be 50%, H to be 4% and S to be 9%. SEM (Fig. 2d) image shows formation of porous nature of the catalyst. The surface composition of Glu-Fe3O4-SO3H was analysed by XPS (Fig. 2e). The C 1s spectrum includes six peaks with different binding energy values. The peaks could be assigned to the carbon atoms in the forms of C–S (283.5 eV), C–C (284 and 284.5 eV), C–O (285 eV), CO (285.5 eV), OC–O (286 eV). The S 2p spectrum showed three different peaks that can be assigned to S–C (186.5), S–O (169 eV) and SO (169.5 eV). O 1s spectrum shows peaks in the range 530–535 eV indicating the presence of Fe–O and C–O–Fe groups suggesting the linkage of Fe3O4 with porous carbon. BET surface area, pore size and pore volume was calculated using the standard Brunauer–Emmett–Teller (BET) equation and was found to be 3.38 m2 g−1, 9.53 Å and 6.07 m3 g−1 respectively. The total acid density and the sulphonic acid density of Glu-Fe3O4-SO3H based on acid base titration was found to be 2.87 mmol g−1 and 1.46 mmol g−1 respectively.
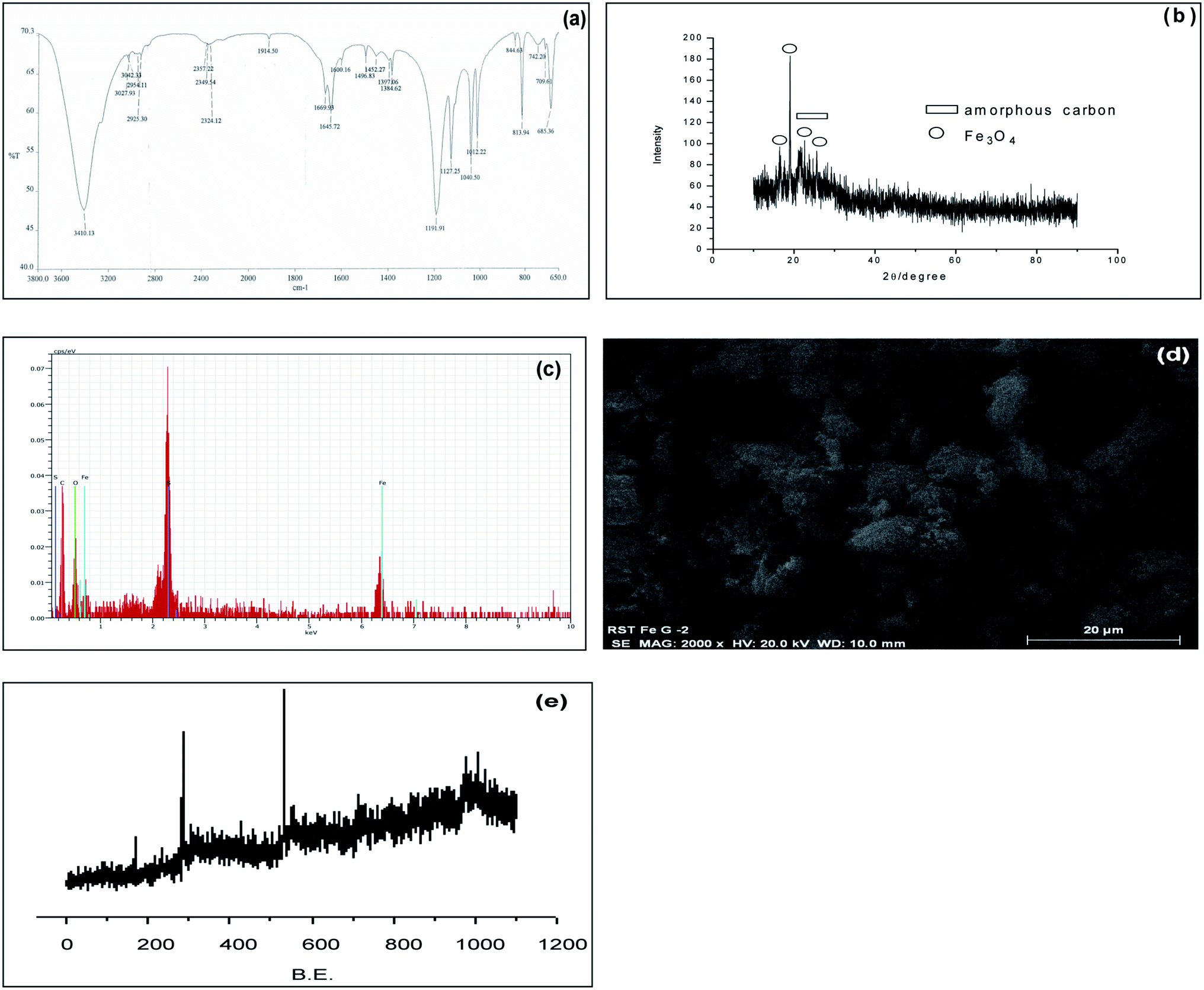 | ||
| Fig. 2 (a) FT-IR of Glu-Fe3O4-SO3H. (b) PXRD of Glu-Fe3O4-SO3H. (c) EDAX of Glu-Fe3O4-SO3H. (d) SEM image of Glu-Fe3O4-SO3H. (e) XPS of Glu-Fe3O4-SO3H. | ||
3.2 Effect of catalyst on tetrahydropyran protection of alcohols
Initially we focused our attention on the tetrahydropyranyl (THP) protection of alcohols. As a first set of experiments, we studied the THP protection of various primary, secondary and tertiary alcohols like cyclohexanol 2a, isopropanol 2b, benzyl alcohol 2c and t-butanol 2d with dihydropyran 1 (Table 1).
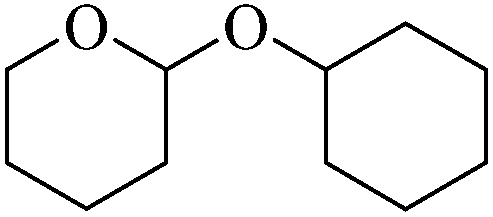
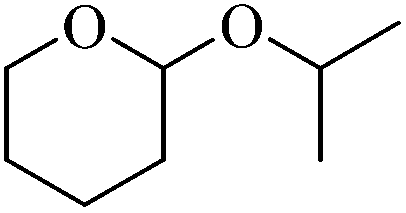
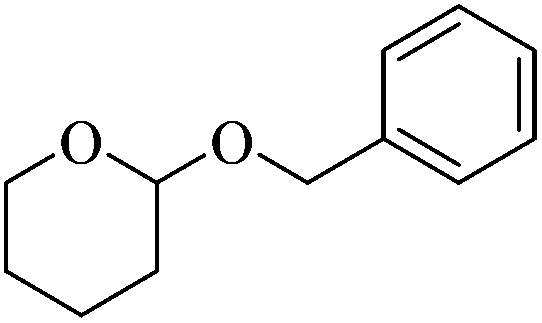
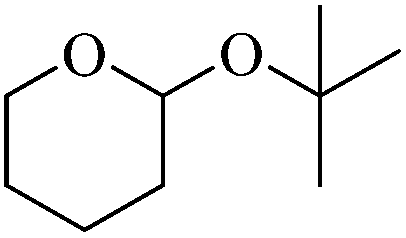
To our delight, the Glu-Fe3O4-SO3H catalyst efficiently catalyzed this reaction under neat reaction conditions at rt within 2–5 minutes to obtain THP-protected products 3a–3d using 1 wt% loading of catalyst to give full conversion and high yields. The reaction when performed without using Glu-Fe3O4-SO3H catalyst, no formation of THP protected product was observed.
3.3 Effect of catalyst on glycosylation of protected glycals
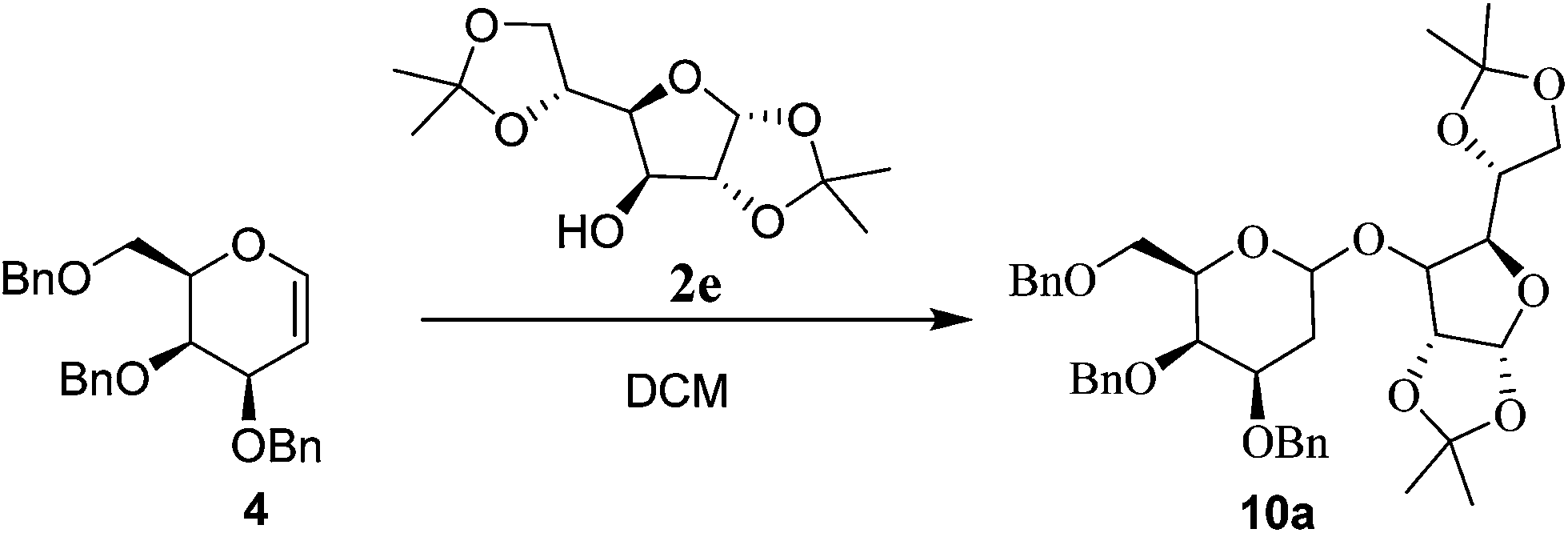
After successful establishment of the THP protection of various alcohols, we turned our attention towards the more challenging glycosylation reaction of different glycals using various acceptors as alcohols. For this study the required glycal components 3,4,6-tri-O-benzyl, 3,4,6-tri-O-silyl and 3,4,6-tri-O-acetyl galactals (4–6) as well as 3,4,6-tri-O-benzyl, 3,4,6-tri-O-silyl and 3,4,6-tri-O-acetyl glucals (7–9) were synthesized by known procedure.10 Glycosyl acceptors chosen for the study were bisacetonide-D-glucose 2e, (−) menthol 2f, cholesterol 2g, octanol 2h, propargyl alcohol 2i and methanol 2j.Initially, we optimized the reaction conditions for glycosylation of 3,4,6-tri-O-benzyl-D-galactal10 4 using bisacetonide-D-glucose 2e using Glu-Fe3O4-SO3H catalyst (Table 2). Since both reagents are solid, DCM was used as a solvent for glycosylation reaction. The reaction was studied at various temperatures such as −20 °C, 0 °C, rt and 40 °C using 10 wt% of catalyst loading. It was observed that the reaction did not proceed at all at −20 °C in DCM even after 24 h. At 0 °C, in DCM after 12 h, the reaction did not proceed to completion thus giving 45% yield of the glycoside 10a in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α:β anomeric ratio. At rt in DCM, glycosylation was found to be complete giving 94% yield of the glycoside 10a in 83:17 α:β anomeric ratio. When the glycosylation was carried out at 40 °C in DCM, only 32% of the glycosylated product 10a was obtained along with the Ferrier rearranged product. Hence the optimum temperature for the reaction was found to be room temperature. The catalyst loading was further studied and it was found out that glycosylation of 4 with 5 wt% catalyst in DCM at rt gave quantitative yield of the glycoside with 83:17 α:β selectivity. When the catalyst loading was further decreased to 1 wt% at rt, it was observed that the reaction was incomplete. Hence 5 wt% catalyst loading was optimized for further glycosylation reactions that were carried out. It was also observed that the isopropylidine groups were stable under these catalytic conditions.
:1 α:β anomeric ratio. At rt in DCM, glycosylation was found to be complete giving 94% yield of the glycoside 10a in 83:17 α:β anomeric ratio. When the glycosylation was carried out at 40 °C in DCM, only 32% of the glycosylated product 10a was obtained along with the Ferrier rearranged product. Hence the optimum temperature for the reaction was found to be room temperature. The catalyst loading was further studied and it was found out that glycosylation of 4 with 5 wt% catalyst in DCM at rt gave quantitative yield of the glycoside with 83:17 α:β selectivity. When the catalyst loading was further decreased to 1 wt% at rt, it was observed that the reaction was incomplete. Hence 5 wt% catalyst loading was optimized for further glycosylation reactions that were carried out. It was also observed that the isopropylidine groups were stable under these catalytic conditions.
Additions of various alcohols 2e–2j on different glycals 4–9 using Glu-Fe3O4-SO3H catalyst are reported in Table 3. It was observed that addition of alcohols 2f, 2g and 2h with 3,4,6-tri-O-benzyl-D-galactal 4, afforded exclusively the α-galactoside with excellent yields and selectivity (Table 3, entries 1–3). Addition of alcohol 2i gave formation of galactoside in α:β 71:29 ratio (Table 3, entry 4). When methanol 2j was used, selectively α-galactoside was formed in good yield (Table 3, entry 5). Both primary and secondary alcohols reacted smoothly to provide the glycoside product. 3,4,6-Tri-O-silyl-D-galactal 5 with alcohol 2e afforded the glycoside in 91:9 α:β ratio with excellent yield (Table 3, entry 6). 3,4,6-Tri-O-acetyl-D-galactal 6 did not undergo addition of alcohol 2e even on prolonged time (Table 3, entry 7). The deactivating effect of acetyl protection in glycosylation has been reported earlier.14
|
||||||
|---|---|---|---|---|---|---|
| Entry | Glycal | Alcohol | Time (h) | Product | Yielda (%) | α:β ratiob |
| a Isolated yields.b Ratio's determined from 1H NMR spectroscopy.c 3 equiv. of alcohols were used.d α:β ratio of 2-deoxy-O-glycoside.e Ratio of the Ferrier product and 2-deoxy-O-glycoside.f α:β ratio of the Ferrier product. |
||||||
| 1 | 4 | 2f | 8 | 10b | 82 | α |
| 2 | 4 | 2g | 6 | 10c | 89 | α |
| 3c | 4 | 2h | 4 | 10d | 91 | α |
| 4c | 4 | 2i | 3 | 10e | 96 | 71:29 |
| 5c | 4 | 2j | 3 | 10f | 94 | α |
| 6 | 5 | 2e | 3 | 10g | 88 | 91:9 |
| 7 | 6 | 2e | 5 | — | No reaction | — |
| 8 | 7 | 2e | 3 | 10h | 84 | 77:23 |
| 9 | 7 | 2f | 6 | 10i | 80 | 83:17 |
| 10 | 7 | 2g | 6 | 10j | 83 | 77:23 |
| 11c | 7 | 2h | 4 | 10k | 80 | 91:9,d 50:50,e 90:10f |
| 12c | 7 | 2i | 3 | 10l | 93 | 90:10,d 50:50,e 83:27f |
| 13c | 7 | 2j | 3.5 | 10m | 94 | 69:31,d 38:62,e 91:9 |
| 14 | 8 | 2e | 3 | 10n | 81 | 91:9 |
| 15 | 9 | 2e | 6 | — | No reaction | — |
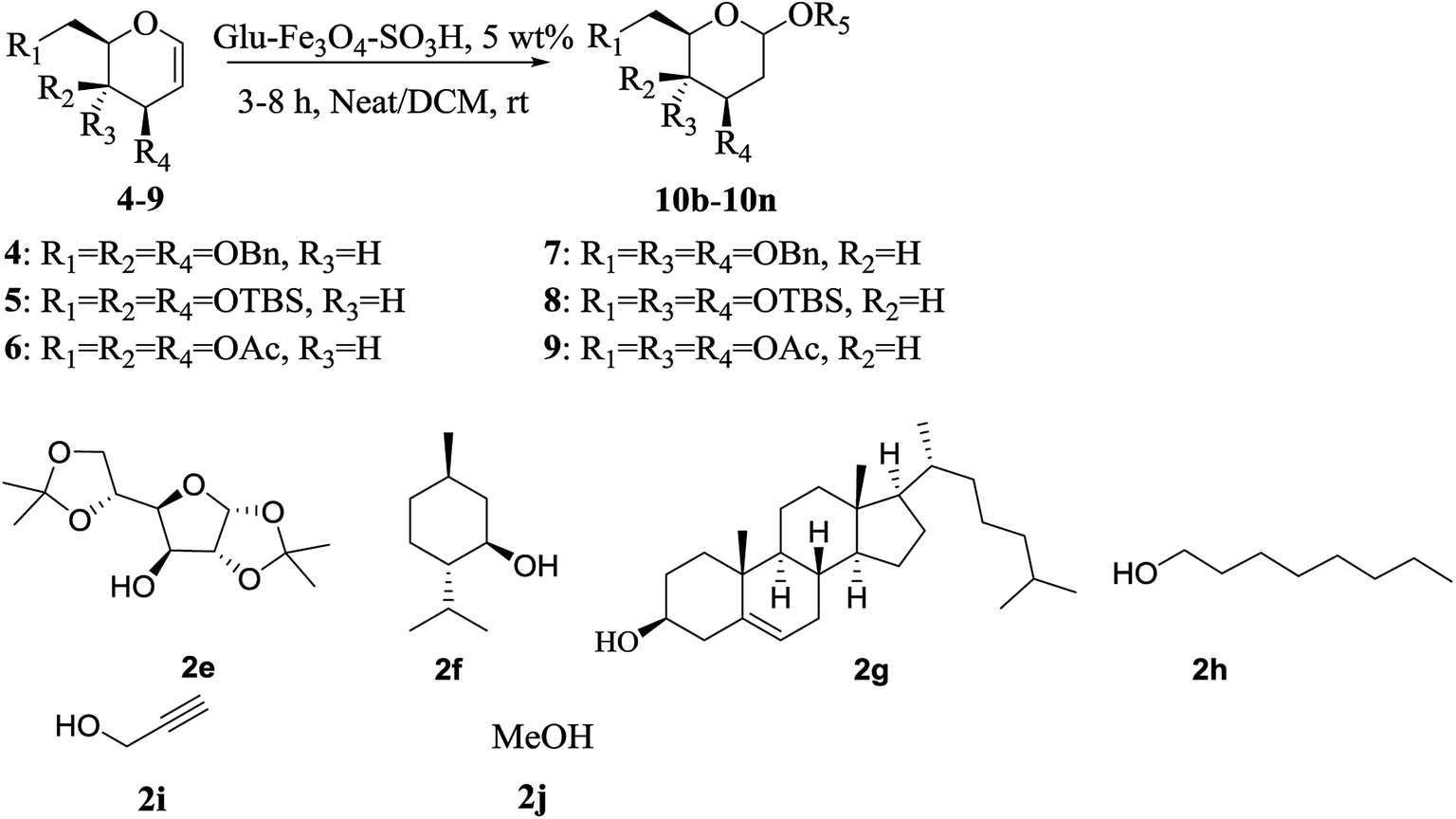
We next turned our attention to glucals 7–9 for glycosylation. 3,4,6-Tri-O-benzyl-D-glucal 7, reacted smoothly with alcohol 2e, 2f and 2g giving α:β glycoside in 77:23, 83:17 and 77:23 ratio respectively (Table 3, entries 8–10). In case of addition of alcohol 2h to 3,4,6-tri-O-benzyl-D-glucal 7,2-deoxy-O-glycoside and Ferrier product were formed in a ratio of 50:50, of which 2-deoxy-O-glycoside was formed in a α:β ratio of 91:9, whereas Ferrier product was formed in a α:β ratio of 90:10 (Table 3, entry 11). Similarly in case of addition of alcohols 2i and 2j to 3,4,6-tri-O-benzyl-D-glucal 7 gave mixture of 2-deoxy-O-glycoside and Ferrier products (Table 3, entries 12 and 13). 3,4,6-Tri-O-sily-D-glucal 8 on addition of alcohol 2e gave 91:9 α:β mixture of the glucoside (Table 3, entry 14). 3,4,6-Tri-O-acetyl-D-glucal 9 did not show formation of any product (Table 3, entry 15) due to deactivating effect of acetyl group.
Comparison of some known catalyst vs. Glu-Fe3O4-SO3H has been shown in Table 4 for addition of alcohols to 3,4,6-tri-O-benzyl-D-galactal 4. Addition of cholesterol to D-galactal 4 in presence of trimethylsilylnitrate gave 81:19 α:β ratio of 2-deoxy-O-glycoside (Table 4, entry 1). Whereas addition of methanol to D-galactal 4 in presence of p-TSA/[bmim]BF4 and CAN gave 4:1 and 32:1 α:β ratio of 2-deoxy-O-glycoside respectively (Table 4, entries 2 and 3). Heterogeneous catalyst such as Y zeolites and SiO2·H2SO4 gave no formation of 2-deoxy-O-glycoside instead gave only Ferrier rearranged product formation (Table 4, entries 4 and 5). Whereas, Glu-Fe3O4-SO3H gave addition of methanol to D-galactal 4 selectively forming α-2-deoxy-O-glycoside which indicates good efficiency of the catalyst for selective formation of 2-deoxy-O-glycoside (Table 4, entry 6).
| Entry | Catalyst | Alcohol | Yield (%) | α:β ratio of 2-deoxy-O-glycoside |
|---|---|---|---|---|
| 1 | TMSONO2 | Cholesterol | 68 | 81:19 (ref. 15) |
| 2 | pTSA/[bmim]BF4 | MeOH | 91 | 4:1 (ref. 16) |
| 3 | Ceric ammonium nitrate | MeOH | 78 | 32:1 (ref. 17) |
| 4 | Y zeolites | BnOH | 80 | Only Ferrier product (ref. 18) |
| 5 | SiO2·H2SO4 | MeOH | 81 | Only Ferrier product (ref. 19) |
| 6 | Glu-Fe3O4-SO3H | MeOH | 94 | Only α |
It was observed that addition reaction of alcohols to galactals displayed high α-stereoselectivity, which is consistent with the anomeric effect. While, addition reaction of alcohols to glucals generally gave a low α:β ratio along with the Ferrier rearranged products in some cases. This phenomenon can be explained by the steric hindrance of the bulky benzyloxy/silyl group at C-4 position of galactals which prevents the attack of acceptors from the top face of the sugar ring, thus promoting the formation of major α-anomer.
3.4 Reusability of the catalyst
Reusability and isolation of the catalysts are important factors for any practical application. It is very convenient to recover Glu-Fe3O4-SO3H catalyst in the end of the reaction by separating it using external magnetic field (Fig. 3). | ||
| Fig. 3 Separation of catalyst Glu-Fe3O4-SO3H using external magnetic field. | ||
The solid catalyst thus separated could be readily reused for the next runs without any prior activation. It was observed that reuse of Glu-Fe3O4-SO3H catalyst gave no significant decrease in yield of products in case of THP protection as well as glycosylation of glycals after four recycles (Table 5).
4. Conclusion
In conclusion, a new porous magnetic carbonaceous solid acid catalyst Glu-Fe3O4-SO3H was efficiently prepared and was found to show excellent catalytic activity for THP protection of alcohols and synthesis of 2-deoxy-O-glycoside under mild reaction conditions giving high yields and selectivity. The catalyst could also be recovered by external magnetic force and reused several times without any loss of activity. This catalytic system is environmentally green and can be used as an ideal method for glycosylation reactions.Acknowledgements
V. H. J. thanks DST, New Delhi for the INSPIRE Faculty Award (IFA 12, CH-44) and Fast Track Grant (CS-041/2013). V. H. J. also thanks Director, NCL for providing all the infra-structural facilities.References
- X. He, G. Agnihotri and H. W. Liu, Chem. Rev., 2000, 100, 4615–4662 CrossRef CAS PubMed.
- A. Kirsching, A. F. W. Rohr and J. Rohr, Bioorganic Chemistry: Deoxysugars,Polyketides and Related Classes, Polyketides and Related Classes: Synthesis; Biosynthesis: Enzymes, Springer, Berlin, 1997, pp. 1–84 Search PubMed.
- J. S. Yadav, B. V. S. Reddy, K. B. Reddy and M. Satyanarayana, Tetrahedron Lett., 2002, 43, 7009–7012 CrossRef CAS.
- K. Toshima, H. Nagai, Y. Ushiski and S. Matsumura, Synlett, 1998, 1007–1009 CrossRef CAS.
- K. Pachamuthu and Y. D. Vankar, J. Org. Chem., 2001, 66, 7511–7513 CrossRef CAS PubMed.
- X. K. Cui, M. Zhong, X. B. Meng and Z. J. Li, Carbohydr. Res., 2012, 358, 19–22 CrossRef CAS PubMed.
- T. Kimura, D. Takahashi and K. Toshima, J. Org. Chem., 2015, 80, 9552–9562 CrossRef CAS PubMed.
- C. Masson, J. Soto and M. Bessodes, Synlett, 2000, 1281–1282 CAS.
- E. I. Balmond, D. M. Coe, M. C. Galan and E. M. McGarrigle, Angew. Chem., Int. Ed., 2012, 51, 9152–9155 CrossRef CAS PubMed.
- S. Das, D. Pekel, J. M. Neudcrfl and A. Berkessel, Angew. Chem., Int. Ed., 2015, 54, 12479–12483 CrossRef CAS PubMed.
- R. S. Thombal, A. R. Jadhav and V. H. Jadhav, RSC Adv., 2015, 5, 12981–12986 RSC.
- W. J. Liu, K. Tian, H. Jiang and H. Q. Yu, Sci. Rep., 2013, 3, 2419–2425 Search PubMed.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS PubMed.
- E. I. Balmond, D. Benito-Alifonso, D. M. Coe, R. W. Alder, E. M. McGarrigle and M. C. Galan, Angew. Chem., Int. Ed., 2014, 53, 8190–8194 CrossRef CAS PubMed.
- B. Gopal Reddy and Y. D. Vankar, ARKIVOC, 2004, viii, 12–19 Search PubMed.
- G. Diaz, A. Ponzinibbio and R. D. Bravo, Top. Catal., 2012, 55, 644–648 CrossRef CAS.
- K. Pachamuthu and Y. D. Vankar, J. Org. Chem., 2001, 66, 7511–7513 CrossRef CAS PubMed.
- P. Levecque, D. W. Gammon, P. Jacobs, D. De Vos and B. Sels, Green Chem., 2010, 12, 828–835 RSC.
- J. Zhang, B. Zhang, J. Zhou, H. Chen, J. Li, G. Yang, Z. Wang and J. Tang, J. Carbohydr. Chem., 2013, 32, 380–391 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra03305a |
| This journal is © The Royal Society of Chemistry 2016 |