
Dipicolylamine coupled rhodamine dyes: new clefts for highly selective naked eye sensing of Cu2+ and CN− ions†
Kumaresh Ghosh*a,
Debojyoti Tarafdara,
Anupam Majumdara,
Constantin G. Daniliucb,
Asmita Samadderc and
Anisur Rahman Khuda-Bukhshc
aDepartment of Chemistry, University of Kalyani, Kalyani-741235, India. E-mail: ghosh_k2003@yahoo.co.in; Fax: +913325828282; Tel: +913325828750
bOrganisch-Chemisches Institut, Universität Münster, Corrensstrasse 40, 48159 Münster, Germany
cDepartment of Zoology, University of Kalyani, Kalyani-741235, India
First published on 4th May 2016
Abstract
The dipicolylamine (DPA) motif which is known as a binder of Zn(II) ions, has been utilized in devising rhodamine labelled compounds 1 and 2. Compound 1 acts as a FRET sensor and shows excellent selectivity for Cu(II) ions over a series of other cations in CH3CN/H2O by exhibiting a colour change (colourless to pink) of the solution. The spectral and colour changes are recovered in the presence of CN− ions and thus, the ensemble 1·Cu2+ in CH3CN/H2O is established as the medium for selective detection of CN− ions. In contrast, the modified compound 2 with the dipicolylamine motif as the principal binding site has been established as the colorimetric sensor of Cu(II) ions and the fluorometric sensor of Hg(II), Zn(II) and Cd(II) ions. Both the compounds 1 and 2 are cell permeable and are successfully employed for the detection of intercellular metal ions through bright field and fluorescence imaging.
Introduction
Copper is an essential transition metal ion that plays an important role in environmental, biological and chemical systems.1 It is toxic to biological systems when the level of Cu2+ ions exceeds the cellular needs. Overloading of this ion can cause oxidative stress and neurological disorders including Alzheimer's, Parkinson's and Wilson's diseases.2 Thus rapid sensing of this ion by simple systems is desirable. In this regard, numbers of fluorescent as well as colorimetric sensors for copper ion have been reported in the past few years.3,4 Of the different chemosensors, rhodamine-labelled sensors draw attention because of visualization of sensing through both color and fluorescence changes.Rhodamine B and its derivatives exhibit excellent photo-physical properties such as high fluorescence quantum yield, large molar extinction coefficient and visible wavelength excitation. It is further mentionable that the rhodamine B and its derivatives in the spirolactam form are non fluorescent (colourless). Upon complexation of metal ions/proton the spirolactam ring is opened and shows strong emission as well as color. Considering this chemistry of rhodamine B, researchers have used this platform in conjunction with different binding sites to construct different metal ion sensors.5
Careful scrutiny of the literature reveals that use of dipicolyl amine (DPA) motif as binding site onto the rhodamine unit is less explored in metal ion sensing.6 Usually this motif binds Zn2+ ions. The wide spread use of DPA in devising receptors of different architectures for Zn2+ ion is worth mentioning. Lippard et al., have reported dipicolylamine coupled some rhodamine derivatives for sensing of Zn2+ ions.6a,b Not only Zn2+ ion but also the complexation of Al3+ and Pb2+ ions using dipicolyl motif in rhodamine platform is known in few cases.7 Inspection of such different reports indicates that the disposition of dipicolylamine motif around the rhodamine part is crucial for tuning the metal ion selectivity. In relation to this, design and synthesis of new dipicolylamine coupled rhodamine derivatives thus draws attention.
In this manuscript, we report a new rhodamine-based structure 1 (Fig. 1) that senses Cu2+ ion through color change involving dipicolyl amine as the binding site. In addition, the ensemble of 1 with Cu2+ ion is observed to detect CN− ion with significant sensitivity. In an effort to understand the role of anthracene in 1 other than FRET, compound 2 was designed and synthesized (Fig. 1). Under identical conditions, compound 2 while selectively senses Cu2+ ions through color change, it fluorometrically detects some multiple ions such as Hg2+, Zn2+ and Cu2+ ions without showing any color change. To explain the role of DPA unit in 1 or 2, the model compound 3 (Fig. 1) was synthesized. Compound 3, under identical conditions, did not show any selectivity in the recognition process.
 | ||
| Fig. 1 Chemdraw structures (left) of 1–3 and XP diagram (30% ellipsoids, right) of 1. Hydrogen atoms were omitted for clarity. | ||
Results and discussion
The synthesis of compound 1 was achieved according to the Scheme 1. Initially, 9-anthraldehyde was transformed into the Schiff base 3 on reaction with 2-picolylamine. Reduction of the Schiff base 3 using NaBH4 afforded the amine 4. The amine was reacted with 2-pivaloylamide-6-bromomethyl pyridine which was obtained from 2-amino-6-methylpyridine by reported procedure8 to obtain the compound 5. Hydrolysis of the amide bond in 5 using KOH in EtOH resulted in amine 6 (Scheme 1a). For compound 2, Scheme 1b was pursued to obtain the precursor amine 10 like amine 6. In this case, reaction of pyridine-2-aldehyde with n-butylamine in dry MeOH gave Schiff base 7 which on in situ reduction with NaBH4 yielded amine 8. The amine was reacted with 2-pivaloylamide-6-bromomethyl pyridine to yield the compound 9. Hydrolysis of the amide bond in 9 using KOH in EtOH resulted in amine 10 (Scheme 1b). For model compound 3, m-nitrobenzaldehyde was converted to the Schiff base 11 which on reduction yielded the amine 12. Benzylation of the amine 12 using benzyl bromide followed by reduction of nitro group furnished the amine 14 in appreciable yield (Scheme 1c). Finally, rhodamine B was converted into its acid chloride 15 with POCl3 in 1,2-dichloroethane and was coupled with the amines 6, 10 and 14 to give the desired compounds 1, 2 and 3, respectively in appreciable yields (Scheme 1d). The structures of 1, 2 and 3 were unambiguously characterized by 1H, 13C NMR and HRMS. For 1, single crystal X-ray analysis further confirmed the structure. | ||
| Scheme 1 (a). (i) 2-Picolylamine, dry MeOH, reflux 6 h; (ii) NaBH4, dry MeOH, reflux 6 h; (iii) 2-pivaloylamide-6-bromomethyl pyridine, dry K2CO3, CH3CN, reflux 8 h; (iv) KOH, EtOH, reflux 6 h; (b). (v) n-Butyl amine, dry MeOH, reflux, 6 h; (vi) NaBH4, dry MeOH, reflux, 6 h; (vii) 2-pivaloylamide-6-bromomethyl pyridine, dry K2CO3, CH3CN, reflux, 8 h; (viii) KOH, EtOH, reflux, 6 h; (c). (ix) n-Butylamine, dry C6H6, reflux, 6 h, (x) NaBH4, dry MeOH, reflux, 6 h; (xi) benzyl bromide, dry K2CO3, CH3CN, reflux 6 h; (xii) SnCl2, EtOH, reflux 3 h; (d). (xiii) POCl3, 1,2-dichloroethane, 8 h; (xiv) 6, Et3N, dry CH2Cl2, 6 h; (xv) 10, Et3N, dry CH2Cl2, 6 h; (xvi) 14, Et3N, dry CH2Cl2, 6 h. | ||
Single crystal of 1 was grown from slow evaporation of acetonitrile solution and was characterized using X-ray crystallography (Fig. 1).9 In the crystal, compound 1 crystallized in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and shows the expected orthogonal arrangement of the xanthene unit towards the lactam ring. The angle between the two planes was found to be 89.8°. The nitrogen atom N5 shows sum of the respective C–N–C angles of ΣN1CCC = 332.6°. In the packing diagram (see ESI, Fig. 1S†) hydrogen bonding interactions involving the carbonyl group C
and shows the expected orthogonal arrangement of the xanthene unit towards the lactam ring. The angle between the two planes was found to be 89.8°. The nitrogen atom N5 shows sum of the respective C–N–C angles of ΣN1CCC = 332.6°. In the packing diagram (see ESI, Fig. 1S†) hydrogen bonding interactions involving the carbonyl group C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the C–H units of the diethylamino moieties were found (C–H⋯O 2.517 Å and 2.761 Å, respectively). Moreover weak π–π-interactions (∼3.48 Å) between two adjacent pyridine groups were detected. No relevant π–π-interactions involving the anthracene moieties were observed.
O and the C–H units of the diethylamino moieties were found (C–H⋯O 2.517 Å and 2.761 Å, respectively). Moreover weak π–π-interactions (∼3.48 Å) between two adjacent pyridine groups were detected. No relevant π–π-interactions involving the anthracene moieties were observed.
The optical responses of compound 1 to various metal cations viz. Hg2+, Cu2+, Cd2+, Fe2+, Mg2+, Co2+, Ni2+, Zn2+, Ag+, Fe3+, Mn2+, Na+, Pd2+, Al3+ and Pb2+ (taken as their perchlorate salts) were investigated in CH3CN–H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) through UV-vis and fluorescence spectroscopic methods. In this context, a high content of water in aqueous measuring solution was desirable, but such an approach was constrained due to the limited solubility of receptor in water. As a reasonable negotiation, CH3CN:H2O (4:1, v/v) was used in the study. It is to note that compound 1 was also found to be soluble in EtOH–H2O (7:3, v/v). However, the UV spectrum of 1 in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) exhibited an intensive band centered at 367 nm which was attributed to the characteristics of anthracene unit. However, addition of Cu2+ ions produced a new band centered at 552 nm which underwent significant change on progression of titration (Fig. 2). On addition of Cu2+ ions, the receptor solution became pink in color.
:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) through UV-vis and fluorescence spectroscopic methods. In this context, a high content of water in aqueous measuring solution was desirable, but such an approach was constrained due to the limited solubility of receptor in water. As a reasonable negotiation, CH3CN:H2O (4:1, v/v) was used in the study. It is to note that compound 1 was also found to be soluble in EtOH–H2O (7:3, v/v). However, the UV spectrum of 1 in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) exhibited an intensive band centered at 367 nm which was attributed to the characteristics of anthracene unit. However, addition of Cu2+ ions produced a new band centered at 552 nm which underwent significant change on progression of titration (Fig. 2). On addition of Cu2+ ions, the receptor solution became pink in color.
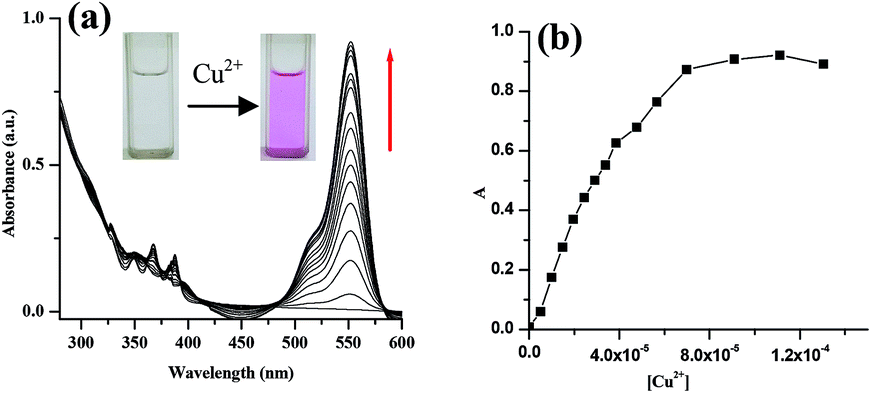 | ||
| Fig. 2 (a) Change in absorbance of 1 (c = 2.5 × 10−5 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) upon gradual addition of 2 equiv. amounts of Cu2+ (c = 1 × 10−3 M), (b) change in absorption intensity with the addition Cu2+ ions. | ||
As reason, the appearance of new peak at 552 nm is ascribed to the binding-induced spirolactam ring opening of rhodamine unit in 1. UV-vis titrations of 1 under similar conditions with other metal ions exhibited negligible perturbation in the spectra showing no peak at 552 nm as well as no color change of the solution (ESI, Fig. 2S†). Thus receptor 1 provides a naked eye detection of Cu2+ ions among the tested metal ions. Fig. 3 highlights the change in absorbance ratio of 1 and Fig. 4 displays the change in color of the receptor solution in presence of the metal ions examined.
 | ||
| Fig. 3 Change in absorption ratio (A − A0/A0) of 1 (c = 2.5 × 10−5 M) at 552 nm upon addition of 2 equiv. amounts of various metal ions in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer). | ||
 | ||
| Fig. 4 Photographs showing the color change of the solution of 1 (c = 2.5 × 10−5 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) in presence of 2 equiv. amounts of metal ions studied. | ||
To understand the interference of other metal ions in the sensing of Cu2+ ion, competition experiment was also carried out by adding Cu2+ ions to the solutions of 1 in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) in presence of other metal ions (Fig. 5). Results in Fig. 5 indicate that the sensing of Cu2+ ion by 1 is hardly affected by the ions considered in the study. The sensor 1 shows 1:1 stoichiometric interaction (Fig. 6a)10 with Cu2+ ion with an association constant11 value of (2.01 ± 0.28) × 104 M−1 [Kd = (4.97 ± 3.57) × 10−5 M] (Fig. 6b). In the sensing of Cu2+ ions, the detection limit12 is determined to be 1.97 × 10−6 M (ESI, Fig. 3S†).
 | ||
| Fig. 5 Competitive selectivity of 1 (c = 2.5 × 10−5 M) towards Cu2+ (c = 1 × 10−3 M) in presence of 2 equiv. of other metal ions in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer). | ||
 | ||
| Fig. 6 (a) UV-vis Job plot of receptor 1 with Cu2+ at 552 nm in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) where [H] = [G] = 2.5 × 10−5 M; (b) binding constant curve from non-linear fitting of UV-vis titration data. | ||
Compound 1 in EtOH–H2O (7:3, v/v, pH = 7.2, 10 mM Tris–HCl buffer) also exhibited similar results as observed in aq. CH3CN (ESI, Fig. 4S and 5S†).
The sensor 1 is a FRET system due to presence of the anthracene at the dipicolyl amine motif. The emission of the intermediate 5 (donor) overlaps with the absorbance of the rhodamine B (acceptor) (Fig. 7a) and thus demonstrates 1 as a FRET system. This FRET-based compound 1 on excitation at the absorbance wavelength 370 nm of anthracene in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) gave structured emission at 416 nm. This emission was due to the anthracene. A broad emission at ∼550 nm in addition to the monomer emission centered at 416 nm is presumed to be either due to intermolecular excimer between the pyridine rings or due to intermolecular exciplex between anthracene and pyridine. However, upon titration with the metal ions except Cu2+ the intensity of broad emission at ∼550 nm was reduced showing no splitting in the region 575 nm to 590 nm due to lactam ring opening of rhodamine part (ESI, Fig. 6S†). Under this condition, the FRET process remained off. In contrast, in presence of Cu2+ ions, the broad peak at 550 nm was split into two peaks of which the peak at ∼580 nm was due to lactam ring opening that activated the FRET process and gave sharp color change (Fig. 7b). However, the intensity of this peak was gradually reduced along with the emission of anthracene at 416 nm on progression of titration and no ratiometric behavior in the emission spectra was observed. This is believed to be due to paramagnetic effect of Cu2+ ion.13 Quenching of fluorescence occurs due to the excitation energy transfer from the ligand to the metal d-orbital and/or ligand to metal charge transfer (LMCT) in aqueous CH3CN.14 This is in accordance with the observations of other researchers.14 The peak at 530 nm arising from splitting was finally abolished on progression of titration. Importantly, the compound 1 is a system where the sensing mechanism is an integration of PET (photoinduced electron transfer) and FRET processes (Scheme 2).15 At pH 7.2, the nitrogen atom of aromatic imino in rhodamine part as well as nitrogen atom of the tripodal centre quench the emission of anthracene to a certain extent. Upon complexation of Cu2+ ion at the DPA moiety involving the lactam part of rhodamine, the FRET process from anthracene to rhodamine moiety occurs. At the same time, the PET process occurring in between the copper complexed site and the excited state of anthracene remains activated that led to a non ratiometric change in the emission spectrum. This was realized from the recording of the emission spectra of 1 at different pHs.
 | ||
| Fig. 7 (a) FRET plot showing the overlapping of emission of donor 5 (λexc = 370 nm) and absorbance of acceptor rhodamine B (c = 2.5 × 10−5 M); (b) change in emission of 1 (c = 2.5 × 10−5 M) upon gradual addition of 10 equiv. amounts of Cu2+ (c = 1 × 10−3 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) (λexc = 370 nm). | ||
 | ||
| Scheme 2 Proposed PET and FRET processes in 1. | ||
In Fig. 7b, the appearance of the peak at 580 nm under the broad emission at ∼550 nm due to Cu2+-induced lactam ring opening was confirmed by recording the fluorescence spectra of 1 in presence of acid where H+-induced lactam ring opening in rhodamine system is usual. In this regard, the fluorescence spectra of 1 were recorded at different pHs. As can be seen from Fig. 8a, the sharp peak at 583 nm in fluorescence with considerable intensity was observed from pH 3 to pH 2. Fig. 8b demonstrates that ratio of intensities at 583 nm and 416 nm (I583/I416) increases with decrease in pH from 4.00 to 2.00. This observation clearly indicates that anthracene acts as FRET donor in receptor 1 and the receptor 1 behaves as an ideal ratiometric sensing platform at strong acidic condition. It is to note that in pH range 4–12, there was no peak at 583 nm for ring opening (ESI, Fig. 7S†). The UV-vis titration spectra with change in pH can be found in the ESI† where the absorbance of 1 at 551 nm at pHs 2 and 3 is in conformity with the opening of lactam ring that resulted in pink color (ESI, Fig. 7S†). Interestingly, a similar pH-response of 1 was observed in EtOH–H2O (7:3, v/v, 10 mM Tris–HCl buffer) (ESI, Fig. 8S†).
 | ||
| Fig. 8 (a) Fluorescence spectra of 1 (c = 2.5 × 10−5 M) in CH3CN–H2O (4:1, v/v, 10 mM Tris–HCl buffer) at different pH values (λexc = 370 nm); (b) change of fluorescence emission ratios (I583/I416) by pH values. | ||
Thus the selective response of 1 toward Cu2+ ion in both UV and fluorescence at pH 7.2 is attributed to the coordination of Cu2+ ion at the dipicolyl moiety involving the lactam part. FTIR spectral analysis of 1 itself and with Cu(ClO4)2 reveals a shifting of carbonyl stretching frequency from 1700 cm−1 to 1650 cm−1 (ESI, Fig. 9S†). This indicates the participation of the amide carbonyl in complexation. In order to realize the participation of the pyridine rings in complexation, we tried to record the 1H NMR of 1 itself and in presence of 1 equiv. amount of Cu(ClO4)2 in CD3CN/D2O (4/1, v/v). Unfortunately, in presence of Cu2+ the sharp signals for different protons of 1 became too broad to interpret. Then we performed the 1H NMR titration of 1 with Cu2+ ion in pure CD3CN solvent. In this solvent system, on progression of titration, signals for the three methylenes (types ‘a’, ‘b’ and ‘c’) around the picolyl motif exhibited downfield shifts of 0.07 ppm upon complexation of equivalent amount of Cu2+ ion. The signals for pyridyl ring protons became broad and underwent small downfield chemical shifts (ESI, Fig. 10S†). Addition of more than 2 equiv. amounts of Cu2+ ions caused broadening of the signals and limits the titration to continue.
This corroborated a weak ground state interaction of 1 with Cu2+ ion at the DPA moiety in the suggested mode shown in Fig. 9a. To be confirmed with the binding role of pyridyl ring nitrogens in 1, compound 3 which lacks of pyridine rings was further explored in binding study. In fluorescence and UV-vis titrations, compound 3 exhibited color change in presence of multiple metal ions (e.g., Cu2+, Hg2+, Fe3+ and Al3+) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) giving no selectivity in the sensing process (ESI, Fig. 11S and 12S†).
 | ||
| Fig. 9 (a) Suggested binding mode of 1 with Cu2+ ion; (b) DFT optimized geometry of the complex 1·Cu2+ in gas phase. | ||
DFT optimization of the copper complex in gas phase (Fig. 9b) reveals that the pyridyl ring nitrogens along with the lactam amide oxygen are intimately involved in coordination of Cu2+ ion. It is important to be pointed out that the DPA unit which is usually the binder of Zn2+ ion, prefers the binding of Cu2+ ion in 1. We believe that this occurs due to the change in metal coordination environment around the dipicolyl motif. To realize this, we considered the intermediate compound 5 which in fluorescence clearly showed the preference for Zn2+ and Cd2+ ions (Fig. 10) like other systems reported in the literature.16
 | ||
| Fig. 10 (a) Change in emission ratio (I − I0/I0) of 5 (c = 2.5 × 10−5 M) at 424 nm upon addition of 2 equiv. amounts of various metal ions in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer), (b) change in emission of 5 (c = 2.5 × 10−5 M) upon gradual addition of 2 equiv. amounts of Zn2+ (c = 1 × 10−3 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) (λexc = 370 nm). | ||
Careful scrutiny of the literature reveals that rhodamine coupled with 2-aminopyridine17a shows responses towards Fe3+, Hg2+ and Pb2+ ions instead of Cu2+. On the other hand, the dipicolylamine motif in 1,8-naphthalimide system as reported by Yoon et al., is observed to bind Cu2+ ion instead of Zn2+ ion.17b In addition, rhodamine-coupled 1,2,3-triazole functionalized 2-amino pyridine exhibits selective response to Zn2+ ions although there was no dipicolyl amine motif in the structure.17c These observations thus validate the fact that the selection of a particular metal ion in a binding site is dependent on the coordination environments that regulate the binding of Cu2+ ions in present example.
In the selective binding of Cu2+ ion, the effect of the counter anions of different copper salts was observed to be negligible. This can be realized from Fig. 13Sa and b†. Furthermore, the reversibility of the binding process between 1 and Cu2+ was established by adding aqueous solution of Na2EDTA to the solution of 1·Cu2+ in both CH3CN/aqueous Tris–HCl buffer (4:1, v/v, pH = 7.2) and EtOH/aqueous Tris–HCl buffer (7:3, v/v, pH = 7.2) solvent systems. In the event, EDTA2− caused demetalation of 1 and regeneration of the spirolactam ring with bleaching of absorption band at ∼552 nm (ESI, Fig. 13Sc and d†).
As practical application, the copper ion ensemble of 1 was explored in studying the interaction of different anions viz. F−, Cl−, Br−, I−, NO3−, CN−, AcO−, ClO4−, HSO4−, HP2O73−, H2PO4− (counter catións: Bu4N+), ATP, ADP, AMP, HPO42−, PO43− and P2O74− (counter catións: Na+). The addition of different anions to the ensemble perturbed the emission to the different extents (Fig. 11a). Among the anions studied, only CN− ion interacted strongly showing a sharp color change (pink to colorless) and recovery of the original absorbance characteristics of 1. This also indicated the reversibility in the binding of Cu2+ ion. In the event, pyrophosphate (P2O74−) responded moderately. The CN−- induced color change from pink to colorless solution associated with complete decrease in absorbance at 552 nm of 1·Cu2+ complex with 1:1 stoichiometric composition suggested that Cu2+ ion was removed from the binding centre through its complexation with CN− ion (Fig. 12a). However, the other tested anions were silent in the process (ESI, Fig. 14S†). Thus, 1·Cu2+ complex can be used as a potential ensemble for selective colorimetric naked eye sensing of CN− over the other tested anions (Fig. 11b).
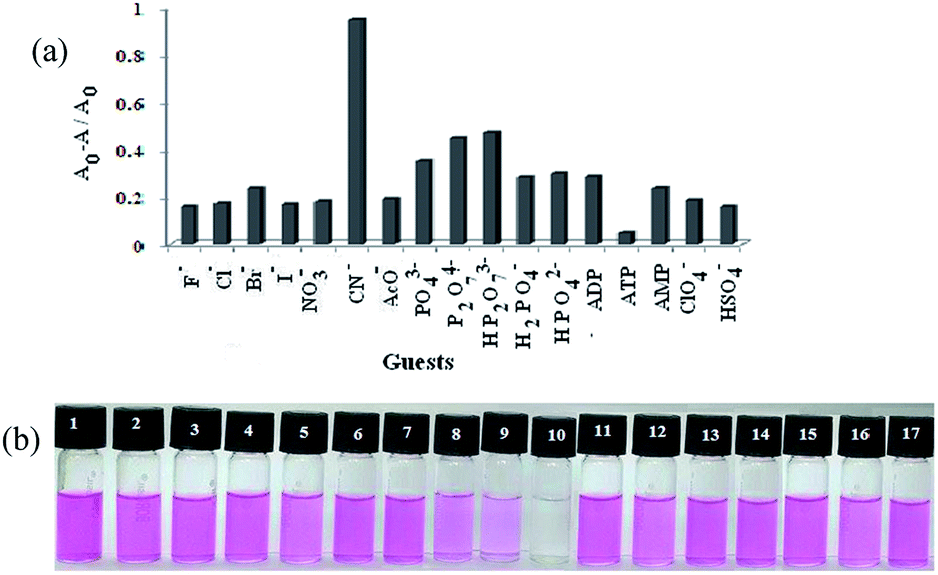 | ||
| Fig. 11 (a) Change in absorbance ratio at 552 nm for ‘1·Cu2+’ ensemble upon addition of 12 equiv. of various anions (c = 1 × 10−3 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer); (b) photographs showing the color change of the solution of 1 (c = 2.5 × 10−5 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) in presence of 12 equiv. of (1) F−, (2) Cl−, (3) Br−, (4) I−, (5) NO3−, (6) AcO−, (7) ClO4−, (8) P2O74−, (9) HP2O73−, (10) CN−, (11) ATP, (12) ADP, (13) AMP, (14) H2PO4−, (15) HPO42−, (16) PO43−, (17) HSO4−. | ||
 | ||
| Fig. 12 (a) Emission spectra for 1·Cu2+ (1:Cu2+ = 1:1 equiv.) ensemble upon gradual addition of 10 equiv. amounts of CN− in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer); (b) change in detection limit of CN− ion for different stoichiometric ensembles of 1·Cu2+. | ||
It is important to be mentioned that the ensembles prepared from the mixing of 1 with more than 1 equiv. amount of Cu2+ responded to CN− ion in a similar way to that of the ensemble 1·Cu2+ having 1:1 equiv. combination. Only there was a slight variation in detection limit of CN− ion and this is due to minor interference of the free Cu2+ ion in the ensemble (Fig. 12b).
Considering the interaction of CN− ion with the copper ensemble of 1, a logic operation was performed. Herein, the sequence dependant “off–on–off” switching of absorption was used to construct a INHIBIT logic gate using Cu2+ and CN− as chemical inputs. In considering the INHIBIT logic gate properties of 1, Cu2+ and CN− ions act as inputs while the absorbance at 552 nm (A552) functions as output (Fig. 13a and b). The output is zero when (i) both the Cu2+ and CN− are absent; (ii) CN− alone is present, or (iii) both Cu2+ and CN− are present, and the gate is OFF. The output is one only when Cu2+ alone is present and the gate is ON. Thus 1 can be used to construct a logic circuit mimicking an INHIBIT gate.
 | ||
| Fig. 13 (a) Logic circuit of receptor 1 with Cu2+ and CN− as chemical inputs and A552 as output of the INHIBIT gate, (b) truth table for the INHIBIT gate of 1 with Cu2+ and CN− as chemical inputs and A552 as output. | ||
In order to understand the role of anthracene (other than FRET process) in the binding event, compound 2 was next explored in the study. The free receptor 2 (c = 3.72 × 10−5 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) exhibited almost negligible absorption peak at 554 nm. However, gradual addition of Cu2+ ion resulted in a significant enhancement of absorption centered at 554 nm and exhibited a sharp color change from colorless to pink (ESI, Fig. 15S†). Such increase in absorbance at 554 nm along with the color change is due to Cu2+-induced spirolactam ring of rhodamine B like the case of 1. The other tested metal ions did not perturb the absorption spectra of 2 and no color change was noticed. The bar plot in Fig. 15S† demonstrates the change in absorption ratio of 2 in presence of 2 equiv. amounts of different metal ions. Thus the binding study clearly reveals that the dipicolylamine motif either in 1 or 2 is sensible to Cu2+ ion.
Like 1, the competitive experiment as shown in Fig. 16Sa,† suggests that 2 is also able to selectively recognize Cu2+ in presence of other metal ions. The 1:1 stoichiometry of the complex was evaluated from the Job plot10 using absorbance data (ESI, Fig. 16Sb†). The association constant11 of 2·Cu2+, derived from nonlinear curve fitting, was calculated to be (4.44 ± 0.14) × 104 M−1 [Kd = (2.23 ± 7.14) × 10−5 M] (ESI, Fig. 17S†). The detection limit12 for 2 towards Cu2+ ion was found to be 8.45 × 10−7 M which is fairly good than the receptor 1 (ESI, Fig. 18S†).
Fluorescence titration experiments were performed to gain an insight into the binding interaction of 2 in the excited state. Sensor 2 (c = 3.72 × 10−5 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer), on excitation at 500 nm, displayed a weak emission at 577 nm. On gradual addition of Cu2+, Hg2+, Zn2+ and Cd2+, significant enhancements in emission of 2 were observed (Fig. 14). Interestingly, in case of addition of Cu2+, an increase in emission centered at 577 nm with concomitant color change of the solution from colorless to pink was observed. This suggested the chelation of Cu2+ ion at the DPA motif involving the amide ion of the spirolactam ring like the mode shown in Fig. 9a. Interestingly, in presence of Hg2+, Zn2+ and Cd2+ ions no color change of the receptor solution was observed. This indicated the intactness of the spirolactam ring during complexation and the increase in emission in such cases is attributed to the complexation of metal ions into the dipicolyl core leading to inhibition of PET process occurring in between the binding site and the excited state of rhodamine. The increment in emission intensity of 2 was significantly higher with the addition of Hg2+ ion (ESI, Fig. 19S†).
 | ||
| Fig. 14 Change in emission of 2 (c = 2.5 × 10−5 M) in CH3CN–H2O (4:1, v/v, pH = 7.2, 10 mM Tris–HCl buffer) upon addition of 10 equiv. of (a) Cu(ClO4)2, (b) Hg(ClO4)2, (c) Cd(ClO4)2, (d) Zn(ClO4)2, [concentration of metal salts were 1 × 10−3 M] (λexc = 500 nm). | ||
However, excitation of 2 in fluorescence at 370 nm instead of 500 nm gave emission at ∼450 nm (presumably for the DPA motif) which underwent small increase in presence of Hg2+, Zn2+ and Cd2+ ions showing no color change (lactam ring intact) in the solution. Under the similar conditions, mere change in emission of 2 at 450 nm with a color change (opening of lactam ring) upon gradual addition of Cu2+ ions was due to paramagnetic effect of Cu2+ ion13 that quenched the emission (ESI, Fig. 20S†). This control experiment highlighted the key role of anthracene in 1 as FRET donor in the selective sensing of Cu2+ ions.
Like 1, receptor 2 was pH sensitive; the color change from colorless to pink was only observed at pHs 2 and 3 (ESI, Fig. 21S†). Receptor 2 was further explored in anion sensing through the use of its copper ensemble like 1. Among the various anions as considered in case of 1, only CN− affected the spectral pattern of the ensemble 2·Cu2+, exhibiting a significant decrease in absorbance (ESI, Fig. 22Sa†). Cyanide ion-induced decrease in absorption of the ensemble 2·Cu2+ resulted in a sharp color change from pink to colorless. Other tested anions perturbed the absorption of the ensemble to the negligible extents (ESI, Fig. 22Sb†). Like 1, the ensembles prepared from mixing of 2 with 1 or more than 1 equiv. amounts of Cu2+ were sensitive to CN− ion and gave a slight variation in detection limit of CN− ion for minor interference of the free Cu2+ ion in the ensembles (ESI, Fig. 23S†).
Thus the experimental findings on structure 2 demonstrate that the replacement of anthracene part in 1 has a marked effect in sensing process. The anthracene part in 1 exerts a steric environment for which the dipicolylamine motif only allows the Cu2+ ion in the interaction process. In contrast, the butyl chain in 2 being less steric allows not only Cu2+ ion but also other ions such as Zn2+, Cd2+ and Hg2+ ions although the binding mechanism is different.
The potentiality of 1 and 2 in biological systems was evaluated for in vitro detection of Cu2+ ions in HepG2 cells. Images of bright field and fluorescence microscopy revealed that 1 and 2 treated cells did not show significantly detectable changes when compared to normal untreated control (Fig. 15a and 16a). But in cell sets of 1 and 2 treated with Cu2+, the cells were found to be red in coloration when observed under bright field (Fig. 15c and 16c). The images also clearly suggests that the receptors are not only permeable through cell membrane (cytoplasm was red in colour) but also receptor effectively cross the nuclear membrane (nucleus were also red in colour). The effect was greater for 2 + Cu2+ than that of 1 + Cu2+. However, no such changes were observed in fluorescent filter (Fig. 15d). On the other hand, cell sets incubated with either 2 + Cd2+, 2 + Zn2+ and 2 + Hg2+ although showed no significant changes in bright field imaging (Fig. 16e, g and i) but a significant increase in fluorescent intensity in red filter was observed in the following order: 2 + Cd2+ < 2 + Zn2+ < 2 + Hg2+ when compared to untreated control cell set (Fig. 16f, h and j).
 | ||
| Fig. 15 Fluorescence and bright field images of HepG2 cells: (a) bright field image of cells treated with receptor 1 (10 μM) for 25 min at 25 °C, (b) fluorescence image of cells treated with 1 (10 μM) for 25 min at 37 °C, (c) bright field image of cells upon treatment with receptor 1 (10 μM) followed by Cu(ClO4)2 (15 μM) for 1 h at 25 °C, (d) fluorescence image of cells upon treatment with 1 (10 μM) followed by Cu(ClO4)2 (15 μM) for 1 h at 37 °C [red filter is used, λex = 480–580 nm]. | ||
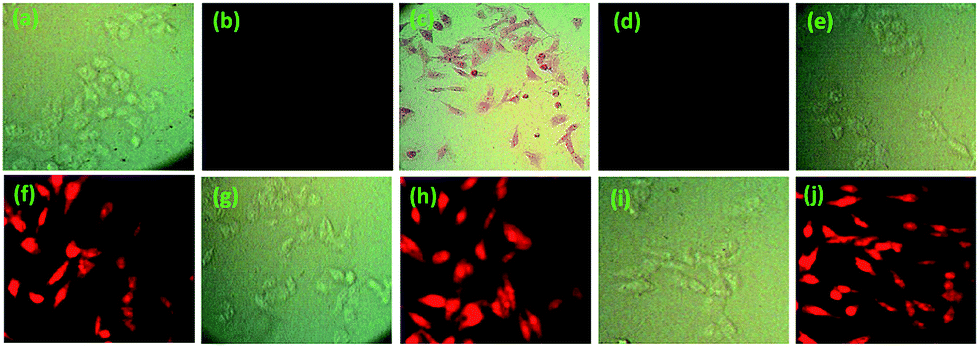 | ||
| Fig. 16 Fluorescence and bright field images of HepG2 cells: (a) bright field image of cells treated with 2 (10 μM) for 25 min at 25 °C, (b) fluorescence image of cells treated with 2 (10 μM) for 25 min at 25 °C, (c) bright field image of cells upon treatment with 2 (10 μM) followed by Cu(ClO4)2 (15 μM) for 1 h at 25 °C, (d) fluorescence image of cells upon treatment with 2 (10 μM) followed by Cu (ClO4)2 (15 μM) for 1 h at 25 °C, (e) bright field image of cells upon treatment with 2 (10 μM) followed by Cd(ClO4)2 (15 μM) for 1 h at 25 °C, (f) fluorescence image of cells upon treatment with 2 (10 μM) followed by Cd(ClO4)2 (15 μM) for 1 h at 25 °C, (g) bright field image of cells upon treatment with receptor 2 (10 μM) followed by Zn(ClO4)2 (15 μM) for 1 h at 25 °C, (h) fluorescence image of cells upon treatment with 2 (10 μM) followed by Zn(ClO4)2 (15 μM) for 1 h at 25 °C, (i) bright field image of cells upon treatment with 2 (10 μM) followed by Hg(ClO4)2 (15 μM) for 1 h at 25 °C, (j) fluorescence image of cells upon treatment with 2 (10 μM) followed by Hg (ClO4)2 (15 μM) for 1 h at 25 °C [red filter is used in fluorescence imaging, λex = 480–580 nm]. | ||
This suggests that both the receptors 1 and 2 are permeable through cellular membrane and can assist in sensing the presence of Cu2+, even in a small concentration, in living tissues without imparting any significant cellular cytotoxicity. Even the receptor 2 in addition, is also able to detect the intercellular metal ions such as Cd2+, Zn2+ and Hg2+ ions effectively by exhibiting marked change in fluorescence.
Results of % cell viability assay suggest that the receptors 1 or 2 have no significant cellular cytotoxicity in HepG2 cells when compared to solvent treated cells (ESI, Fig. 24S†).
Conclusions
In conclusion, a new dipicolylamine coupled rhodamine B molecular sensor 1 has been designed and synthesized. The new sensor 1 displayed strong selectivity for Cu2+ ion over a series of other metal ions examined in CH3CN/aqueous Tris–HCl buffer (4/1, v/v) at pH 7.2 by exhibiting a colour change (from colourless to pink). The sensor 1 has been detected to be insensitive to proton-induced-spirolactam ring opening in the pH range 4 to 12 and thus offers a large pH window for detection of metal ions. The ensemble of 1·Cu2+ has further been successfully used for naked eye detection of CN− ion over a series of other anions examined in the study. Thus the present finding offers a new example of dipicolyl amine (DPA)-based rhodamine molecule for Cu2+ ion in addition to the existing few examples for other metal ions such as Zn2+, Al3+ and Pb2+ in the literature.6,7 The role of DPA motif in 1 for selective binding of Cu2+ ion has been established by considering the model compound 3.On the other hand, replacement of anthracene by butyl chain in receptor 2 shows that the receptor is able to detect Cu2+ colorimetrically and Cd2+, Zn2+ and Hg2+ ions fluorometrically. Like the case of 1, the ensemble 2·Cu2+ is also sensible to CN− ion. Furthermore, the compounds 1 and 2 are cell permeable and can detect the intercellular mentioned metal ions through imaging process.
Experimental
Syntheses
:1, v/v). After completion of the reaction, the solvent was evaporated under reduced pressure. Then, CHCl3–water (2:1, v/v) was added to the residue and the compound was extracted with CHCl3. The organic phase was dried over anhydrous Na2SO4 and evaporated in vacuo. The crude product was purified by column-chromatography using petroleum ether–EtOAc (1:1; v/v) as eluent to afford the amine 4 as a yellow gum (1.1 g, yield: 75.8%); 1H NMR (400 MHz, CDCl3): δ 8.59 (d, 1H, J = 8 Hz), 8.39 (s, 1H), 8.31 (d, 2H, J = 8 Hz), 7.99 (d, 2H, J = 8 Hz), 7.68–7.64 (td, 1H, J1 = 8 Hz, J2 = 4 Hz), 7.53–7.43 (m, 4H), 7.38 (d, 1H, J = 8 Hz), 7.20–7.17 (m, 1H), 4.76 (s, 2H), 4.16 (s, 2H), 2.16 (br s, 1H, NH); FTIR (KBr, cm−1): 3411, 3050, 2856, 1589, 1432.:1, v/v) was added to the residue and the compound was extracted with CHCl3. The combined organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated in vacuo. The crude product was purified through column chromatography using 35% EtOAc in petroleum ether as eluent to give 5 as a yellow solid (0.475 g, yield: 72.5%), mp 160 °C; 1H NMR (400 MHz, CDCl3): δ 8.49 (d, 1H, J = 4 Hz), 8.39 (d, 2H, J = 8 Hz), 8.35 (s, 1H), 8.03 (d, 1H, J = 8 Hz), 7.95 (d, 2H, J = 8 Hz), 7.58–7.55 (m, 2H), 7.48–7.43 (m, 5H), 7.32 (d, 1H, J = 8 Hz), 7.12 (m, 1H), 7.01 (d, 1H, J = 8 Hz), 4.66 (s, 2H), 3.89 (s, 2H), 3.75 (s, 2H), 1.66 (s, 9H); FTIR (KBr, cm−1): 3417, 2959, 2832, 1689, 1449.:1) was added to the residue and extracted with CHCl3. The organic phase was dried over anhydrous Na2SO4 and evaporated in vacuo. The crude product was purified by column-chromatography using petroleum ether/EtOAc, 10:1 (v/v) as eluent to afford the amine 8 as yellow liquid (0.650 g, yield: 84.8%), 1H NMR (400 MHz, CDCl3): δ 8.36 (d, 1H, J = 4.4 Hz), 7.47–7.42 (m, 1H), 7.11 (d, 1H, J = 8 Hz), 6.97 (t, 1H, J = 6.2 Hz), 3.70 (s, 2H), 2.46 (t, 2H, J = 7.2 Hz), 1.92 (brs, 1H), 1.36–1.29 (m, 2H), 1.21–1.12 (m, 2H), 0.71 (t, 3H, J = 7.4 Hz). FTIR (KBr, cm−1): 3389, 2928, 1591, 1570.:1, v/v) was added to the residue and the compound was extracted with CHCl3. The combined organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated in vacuo. The crude product was purified through column chromatography using 25% EtOAc in petroleum ether as eluent to give 9 as yellow gum (0.900 g, yield: 69.5%), 1H NMR (400 MHz, CDCl3): δ 8.44 (d, 1H, J = 4.4 Hz), 8.01 (d, 1H, J = 8 Hz), 7.95 (s, 1H), 7.60–7.55 (m, 2H), 7.45 (d, 1H, J = 8 Hz), 7.19 (d, 1H, J = 8 Hz), 7.06 (t, 1H, J = 8 Hz), 3.72 (s, 2H), 3.61 (s, 2H), 2.44 (t, 2H, J = 8 Hz), 1.46–1.43 (m, 2H), 1.25 (s, 9H), 1.23–1.17 (m, 2H), 0.78 (t, 3H, J = 8 Hz); 13C NMR (100 MHz, CDCl3): δ 177.0, 160.0, 158.4, 150.8, 148.8, 138.6, 136.3, 122.7, 121.8, 118.5, 111.8, 60.4, 60.0, 54.2, 39.7, 29.2, 27.4, 20.4, 13.9; FTIR (KBr, cm−1): 3437, 2958, 1690, 1596.X-ray diffraction
Data sets were collected with a Nonius KappaCCD diffractometer. Programs used: data collection, COLLECT (R. W. W. Hooft, Bruker AXS, 2008, Delft, The Netherlands); data reduction Denzo-SMN;18 absorption correction, Denzo;19 structure solution SHELXS-97;20 structure refinement SHELXL-97 (ref. 21) and graphics, XP (BrukerAXS, 2000). Thermals ellipsoids are shown with 30% probability, R-values are given for observed reflections, and wR2 values are given for all reflections.Computational details
Full geometrical optimizations of all the structures were carried out in the gas phase employing the Becke three-parameter hybrid density functional combined with the Lee–Yang–Parr correlation functional (B3LYP).23–26 The DFT (B3LYP) calculations was performed with the 6-31G(d) basis set27 for carbon, hydrogen, nitrogen and oxygen atoms and for the copper atom SDD basis set28 was used. Frequency calculations were performed at the same level of theory to confirm that each stationary point was a local minimum (with zero imaginary frequency). All DFT calculations were performed with the Gaussian 09 suite of programs.29Material and methods for cell culture
Acknowledgements
We thank DST and UGC, New Delhi, Govt. of India for providing facilities in the university under SAP program. DT thanks CSIR, New Delhi, India for a fellowship.References
- (a) H. Tapiero, D. M. Townsend and K. D. Tew, Biomed. Pharmacother., 2003, 57, 386 CrossRef CAS PubMed; (b) M. C. Linder and M. Hazegh-Azam, Am. J. Clin. Nutr., 1996, 63, 797 Search PubMed; (c) R. Uauy, M. Olivares and M. Gonzalez, Am. J. Clin. Nutr., 1998, 67, 952 Search PubMed; (d) R. A. Lovstad, BioMetals, 2004, 17, 111 CrossRef CAS PubMed.
- (a) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS PubMed; (b) G. L. Millhauser, Acc. Chem. Res., 2004, 37, 79 CrossRef CAS PubMed; (c) G. Multhaup, A. Schlicksupp, L. Hess, D. Beher, T. Ruppert, C. L. Masters and K. Beyreuther, Science, 1996, 271, 1406 CAS; (d) F. Tisato, C. Marzano, M. Porchia, M. Pellei and C. Santini, Med. Res. Rev., 2010, 30, 708 CAS.
- (a) J. W. Lee, H. S. Jung, P. S. Kwon, J. W. Kim, R. A. Bartsch, Y. Kim, S. J. Kim and J. S. Kim, Org. Lett., 2008, 10, 3801 CrossRef CAS PubMed; (b) H. J. Kim, Y. S. Park, S. Yoon and J. S. Kim, Tetrahedron, 2008, 64, 1294 CrossRef CAS; (c) S. Kaur and S. Kumar, Chem. Commun., 2002, 2840 RSC; (d) S. M. Park, M. H. Kim, J.-I. Choe, K. T. No and S. K. Chang, J. Org. Chem., 2007, 72, 3550 CrossRef CAS PubMed; (e) Y. Xiang, A. Tong, P. Jin and Y. Ju, Org. Lett., 2006, 8, 2863 CrossRef CAS PubMed; (f) H. S. Jung, M. Park, D. Y. Han, E. Kim, C. Lee, S. Ham and J. S. Kim, Org. Lett., 2009, 11, 3378 CrossRef CAS PubMed; (g) S. H. Kim, J. S. Kim, S. M. Park and S. K. Chang, Org. Lett., 2006, 8, 371 CrossRef CAS PubMed; (h) X. Qi, E. J. Jun, L. Xu, S. J. Kim, J. S. J. Hong, Y. J. Yoon and J. Yoon, J. Org. Chem., 2006, 71, 2881 CrossRef CAS PubMed; (i) H. S. Jung, P. S. Kwon, J. W. Lee, J. I. Kim, C. S. Hong, J. W. Kim, S. Yan, J. Y. Lee, J. H. Lee, T. Joo and S. J. Kim, J. Am. Chem. Soc., 2009, 131, 2008 CrossRef CAS PubMed; (j) K. Ghosh and T. Sen, Beilstein J. Org. Chem., 2010, 6, 44, DOI:10.3762/bjoc.6.44; (k) T. Gunnlaugsson, J. P. Leonard and N. S. Murray, Org. Lett., 2004, 6, 1557 CrossRef CAS PubMed; (l) K. M. K. Swamy, S. K. Ko, S. K. Kwon, H. N. Lee, C. Mao, J. M. Kim, K. H. Lee, J. Kim, I. Shin and J. Yoon, Chem. Commun., 2008, 5915 RSC; (m) S. Goswami, D. Sen and N. K. Das, Org. Lett., 2010, 12, 856 CrossRef CAS PubMed; (n) Z. Xu, J. Pan, D. R. Spring, J. Cui and J. Yoon, Tetrahedron, 2010, 66, 1678 CrossRef CAS; (o) H. Li, Y. Zhu, B. Shi, W. Qu, Y. Zhang, Q. Lin, H. Yao and T. Wei, Supramol. Chem., 2015, 27, 471 CrossRef CAS; (p) G. C. Yu, Z. B. Zhang, C. Y. Han, M. Xue, Q. Zhou and F. H. Huang, Chem. Commun., 2012, 2958 RSC; (q) X. F. Ji, Y. Yao, J. Y. Li, X. Z. Yan and F. H. Huang, J. Am. Chem. Soc., 2013, 135, 74277 Search PubMed; (r) P. Wang, X. Z. Yan and F. H. Huang, Chem. Commun., 2014, 5017 RSC; (s) Z. Hu, J. Hu, Y. Cui, G. Wang, X. Zhang, K. Uvdal and H. Gao, J. Mater. Chem. B, 2014, 2, 4467 RSC.
- (a) K. Ghosh, T. Sarkar, A. Samadder and A. R. Khuda-Bukhsh, New J. Chem., 2012, 36, 2121 RSC; (b) G. He, X. Zhang, C. He, X. Zhao and C. Duan, Tetrahedron, 2010, 66, 9762 CrossRef CAS; (c) L. Yuan, W. Lin, B. Chen and Y. Xie, Org. Lett., 2012, 14, 432 CrossRef CAS PubMed; (d) M. Kumar, N. Kumar, V. Bhalla, P. R. Sharma and T. Kaur, Org. Lett., 2012, 14, 406 CrossRef CAS PubMed; (e) C. Kaewtong, J. Noiseephum, Y. Uppa, N. Morakot, N. Morakot, B. Wanno, T. Tuntulanic and B. Pulpoka, New J. Chem., 2010, 34, 1104 RSC; (f) D. Maity, D. Karthigeyan, T. K. Kundu and T. Govindaraju, Sens. Actuators, B, 2013, 176, 831 CrossRef CAS; (g) C. Kar, M. D. Adhikari, A. Ramesh and G. Das, Inorg. Chem., 2013, 52, 743 CrossRef CAS PubMed; (h) M. Kaur and D. H. Choi, Sens. Actuators, B, 2014, 190, 542 CrossRef CAS; (i) S. Goswami, D. Sen, A. K. Das, N. K. Das, K. Aich, H. K. Fun, C. K. Quah, A. K. Maity and P. Saha, Sens. Actuators, B, 2013, 183, 518 CrossRef CAS; (j) M. Wang, F. Yan, Y. Zou, L. Chen, N. Yang and X. Zhou, Sens. Actuators, B, 2014, 192, 512 CrossRef CAS; (k) X. Zenga, L. Donga, C. Wua, L. Mua, S. F. Xuea and Z. Taoa, Sens. Actuators, B, 2009, 141, 506 CrossRef; (l) X. Zhang, Y. Shiraishi and T. Hirai, Org. Lett., 2007, 9, 5039 CrossRef CAS PubMed.
- (a) X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910 CrossRef CAS PubMed and references cited therein; (b) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed.
- (a) P. W. Du and S. J. Lippard, Inorg. Chem., 2010, 49, 10753 CrossRef CAS PubMed; (b) E. Tomat and S. J. Lippard, Inorg. Chem., 2010, 49, 9113 CrossRef CAS PubMed; (c) Y. Chen, Y. Bai, Z. Han, W. He and Z. Guo, Chem. Soc. Rev., 2015, 44, 4517 RSC; (d) L. Zhu, Z. Yuan, J. T. Simmons and K. Sreenath, RSC Adv., 2014, 4, 20398 RSC; (e) E. M. Nolan and S. J. Lippard, Acc. Chem. Res., 2009, 42, 193 CrossRef CAS PubMed; (f) L. Zhu, A. H. Younes, Z. Yuan and R. J. Clark, J. Photochem. Photobiol., A, 2015, 311, 1 CrossRef CAS PubMed; (g) J. T. Simmons, Z. Yuan, K. L. Daykin, B. T. Nguyen, R. J. Clark, M. Shatruk and L. Zhau, Supramol. Chem., 2014, 26, 214 CrossRef CAS; (h) Z. Yuan, A. H. Younes, J. R. Allen, M. W. Davidson and L. Zhu, J. Org. Chem., 2015, 80, 5600 CrossRef CAS PubMed; (i) K. Sreenath, Z. Yuan, J. R. Allen, M. W. Davidson and L. Zhu, Chem.–Eur. J., 2015, 21, 867 CrossRef PubMed.
- (a) X. He, N. Zhu and V. W. W. Yam, Organometallics, 2009, 28, 3621 CrossRef CAS; (b) X. Bao, Q. Cao, Y. Xu, Y. Gao, Y. Xu, X. Nie, B. Zhou, T. Pang and J. Zhu, Bioorg. Med. Chem., 2015, 23, 694 CrossRef CAS PubMed; (c) J. Y. Kwon, Y. J. Jang, Y. J. Lee, K. M. Kim, M. S. Seo, W. Nam and J. Yoon, J. Am. Chem. Soc., 2005, 127, 10107 CrossRef CAS PubMed.
- S. Goswami, R. Mukherjee and J. Ray, Org. Lett., 2005, 7, 1283 CrossRef CAS PubMed.
- X-ray crystal structure analysis of 1: formula C55H52N6O2, M = 829.03, pale yellow crystal, 0.12 × 0.10 × 0.08 mm, a = 11.9474(2), b = 12.2845(3), c = 16.6972(5) Å, α = 101.213(1), β = 90.250(1), γ = 98.643(1)°, V = 2375.1(1) Å3, ρcalc = 1.159 g cm−3, μ = 0.071 mm−1, empirical absorption correction (0.991 ≤ T ≤ 0.994), Z = 2, triclinic, space group P (no. 2), λ = 0.71073 Å, T = 223(2) K, ω and φ scans, 21481 reflections collected (±h, ±k, ±l), [(sinθ)/λ] = 0.59 Å−1, 8200 independent (Rint = 0.063) and 4915 observed reflections [I > 2σ(I)], 572 refined parameters, R = 0.086, wR2 = 0.224, max. (min.) residual electron density 0.21 (−0.20) e Å−3, hydrogen atoms were calculated and refined as riding atoms ESI.†.
- P. Job, Ann. Chim., 1928, 9, 113 CAS.
- P. T. Chou, G. R. Wu, C. Y. Wei, C. C. Cheng, C. P. Chang and F. T. Hung, J. Phys. Chem. B, 2000, 104, 7818 CrossRef CAS.
- A. Caballero, R. Martinez, V. Lloveras, I. Ratera, J. Vidal-Gancedo, K. Wurst, A. Tarraga, P. Molina and J. Vaciana, J. Am. Chem. Soc., 2005, 127, 15666 CrossRef CAS PubMed.
- M. H. Lim, B. A. Wong, W. H. Pitcock Jr, D. Mokshagundam, M. H. Baik and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 14364 CrossRef CAS PubMed.
- (a) S. Dalapati, S. Jana, M. A. Alam and N. Guchhait, Sens. Actuators, B, 2011, 60, 1106 CrossRef; (b) A. Rai, N. Kumari, R. Nair, K. Singh and L. Mishra, RSC. Adv., 2015, 5, 14382 RSC; (c) Y. L. Duan, Y. G. Shi, J. H. Chen, X. H. Wu, G. K. Wang, Y. Zhou and J. F. Zhang, Tetrahedron Lett., 2012, 53, 6544 CrossRef CAS; (d) G. Li, F. Taoa, H. Wang, Y. Li and L. Wang, Sens. Actuators, B, 2015, 211, 325 CrossRef CAS.
- S. L. Shen, X. F. Zhang, S. Y. Bai, J. Y. Miao and B. X. Zhao, RSC Adv., 2015, 5, 13341 RSC.
- (a) A. P. De Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385 RSC; (b) A. J. Moro, P. J. Cywinski, S. Korstena and G. J. Mohrac, Chem. Commun., 2010, 1085 RSC; (c) S. Y. Kim and J. I. Hong, Tetrahedron Lett., 2009, 50, 2822 CrossRef CAS; (d) B. Roy, A. S. Rao and K. H. Ahn, Org. Biomol. Chem., 2011, 9, 7774 RSC.
- (a) X. Zhang, Y. Shiraishi and T. Hirai, Tetrahedron Lett., 2007, 48, 5455 CrossRef CAS; (b) K. Sreenath, R. J. Clark and L. Zhu, J. Org. Chem., 2012, 77, 8268 CrossRef CAS PubMed; (c) Z. Xu, J. Pan, D. R. Spring, J. Cui and J. Yoon, Tetrahedron, 2010, 66, 1678 CrossRef CAS.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- Z. Otwinowski, D. Borek, W. Majewski and W. Minor, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 228 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Mossam, J. Immunol. Methods, 1983, 65, 55 CrossRef.
- A. Samadder, S. Das, J. Das and A. R. Khuda-Bukhsh, Colloids Surf., B, 2013, 109, 10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures showing the change in fluorescence and UV-vis titrations of 1 and 2 with various cations, packing diagram, Job plots, binding constant and detection limit plots, MTT assay, spectral data. CCDC 1408733. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra05036k |
| This journal is © The Royal Society of Chemistry 2016 |