
Fabrication of C3N4 ultrathin flakes by mechanical grind method with enhanced photocatalysis and photoelectrochemical performance†
Yuyu Bua,
Zhiwei Chenb,
Tian Xiea,
Weibing Lib and
Jin-Ping Ao*a
aInstitute of Technology and Science, Tokushima University, 2-1 Minami-Josanjima, Tokushima 770-8506, Japan. E-mail: jpao@ee.tokushima-u.ac.jp; Fax: +81 88 656 7442; Tel: +81 88 656 7442
bSchool of Environment and Safety Engineering, Qingdao University of Science and Technology, 53 Zhengzhou Road, Qingdao, 266042, China
First published on 4th May 2016
Abstract
In this paper, we prepared a large-sized multilayer C3N4 ultrathin flake photocatalyst with an approximately 90% yield rate by a facile wet mechanical grinding method. The blue shift phenomenon of the UV-Vis diffuse reflectance spectrum and photoluminescence spectrum indicated that a quantum effect was formed with the thickness of g-C3N4 (g-CN) when decreased to ultrathin flakes. The photocatalysis water reduction for hydrogen evolution performance of ultrathin flake C3N4 (UF-CN) was approximate 3.2 times higher than that of g-CN. In addition, the photoelectrochemical (PEC) property of UF-CN was increased more dramatically than that of g-CN. There are two possible reasons for photocatalysis and PEC performances of UF-CN promotion. First, the valence band (VB) potential of UF-CN was 1.59 eV, which was 0.25 eV more positive than that of g-CN, thus the oxidation capacity of UF-CN photogenerated holes would be stronger than the bulk counterpart. Secondly, the electrons transfer capacity on the horizontal plane of UF-CN was increased with the layered structure of the g-CN exfoliated to an ultrathin structure, thus prolonging the life time of the photogenerated electrons.
1. Introduction
Conversion of solar energy into chemical energy using a photocatalysis technique is a good method to help resolve the energy crises and lower environment pollution.1,2 To date, numerous photocatalysts have been explored to improve photocatalytic performance. Although researchers have made many efforts in this area, performance of the photocatalysts is limited by their weak photo quantum effects and poor physical chemistry stability.3–5Recently, an organic layered high polymer, so-called graphic like carbon nitride, has been reported as an excellent photocatalyst with its abundant composition elements, visible light responsive capacity, and high stability in harsh working environments.6–11 However, the photocatalytic property of this material is limited by its weak photo quantum conversion effect and poor specific surface area, due to its layered structure.12–14
So, as described above, it is urgent to improve the photo quantum conversion effect and specific surface area of g-C3N4. Recently, some literature references reported that the layered structure of g-C3N4 could be exfoliated to multilayered C3N4 nanosheets that increase the photo quantum effect and specific surface areas. Because of the relatively weak van der Waals's force interactions between the layers, g-C3N4 could be exfoliated by several methods. Zhang et al.15 prepared two-dimensional C3N4 nanosheets by ultrasonic exfoliation of bulk g-C3N4 in water, and found that the photoluminescence (PL) intensity of the ultrathin C3N4 could be increased dramatically. Yang et al.16 improved the ultrasonic exfoliation process of bulk g-C3N4 in a solution of IPA, NMP, ethanol, or acetone, and found that the ultrathin C3N4 nanosheets with large sizes possessed better photocatalysis performance than bulk g-C3N4. Niu et al.17 prepared graphene-like carbon nitride nanosheets by a facial post annealing method, and found that the as prepared nanosheet C3N4 showed more and stronger photocatalytic activity than its bulk counterpart. Wang et al.18 prepared atomic single layered C3N4 nanosteels by combining the processes of post annealing and ultrasonic exfoliation; the as prepared C3N4 nanosteels showed a considerable photocatalytic performance for ammonia degradation. Although the ultrathin C3N4 nanosheets could be prepared by above methods, the actual yield rate of the target product was only about 10%.15–20 In addition, the organic promoters used in an exfoliation process would increase pollution of the environment. So, it is urgent to optimize the process of ultrathin C3N4 nanosheet preparation by increasing the productivity rate of it using a green synthesis route.
Recently, some literature references reported that ultrathin graphene could be prepared through a mechanical grinding graphic method. Zhao et al.21 developed a feasible way to produce graphene from graphite flakes using ball grind exfoliation. Their results found that the electrical conductivity of the as prepared graphene powder with a thickness of approximately 0.8–1.8 nm was 1.2 × 103 S m−1 at room temperature. Jeon et al.22 employed the ball grind method to prepare edge-selectively functionalized graphene nanoplatelets with large-scale production, and the as prepared graphene showed a high efficiency for an oxygen reduction reaction. Inspired by these results, g-C3N4 with weak interlayer van der Waals interactions may be exfoliated by this method. So, in this paper, we employed the wet mechanical grind method to prepare ultrathin C3N4 nanoflakes, expecting to enhance the production rate and photocatalytic property of this material.
2. Experimental section
Preparation of large-scaled ultrathin C3N4 nanoflakes
All reagents used in this study were of analytical grade and purchased from Aladdin Industrial Corporation, China, without any further purification. For bulk g-C3N4 synthesis, 10 g dicyanodiamide was annealed at 550 °C with a ramp rate of 10 °C min−1 in air for 3 h to form a yellow powder. Then 5 g of as prepared g-C3N4, 30 mL water, and 250 g corundum balls with diameters of 0.5 cm were added into a Teflon container with a 100 mL volume. Subsequently, the mixture was ground using a speed rate of 350 rpm for 17 h. The resultant homogenate was filtered with a silk net having a pore diameter of 100 μm, and centrifuged at 7000 rpm; the sediment was washed with alcohol and water for three times and dried at 60 °C overnight to give a light yellow powder with a yield of nearly 90%.Characterization
X-ray diffraction (XRD, D/MAX-2500/PC; Rigaku Co., Tokyo, Japan) was used to identify the crystalline structures of the series samples. The micro-morphology of as prepared g-CN and UF-CN was observed using a scanning electron microscope (SEM, JSM-6700F; JEOL, Tokyo, Japan). The ultrathin structure of the materials was observed using a field emission transmission electron microscope (FE-HRTEM, Tecnai G2 F20, FEI Company, USA), and atomic force microscope (AFM, Agilent, USA). The elementary composition and bonding information of the synthesized materials were analyzed using X-ray photoelectron spectroscopy (XPS, Axis Ultra, Kratos Analytical Ltd., England). A UV-visible diffuse reflectance spectrophotometer (UV-Vis DRS, U-41000, HITACHI, Tokyo, Japan) was used to test the optical absorption properties of the samples. The photoluminescence of the as prepared materials was characterized using a fluorescence spectrometer (PL, Fluoro Max-4, HORIBA Jobin Yvon, France). Raman spectra were recorded by a Raman spectrometer (Horiba JY HR-800; Japan) with an excitation wavelength of 785 nm.Photocatalytic water splitting and dye degradation testing
For testing of photocatalytic water splitting, 0.05 g photocatalyst was added to a 100 mL mixture containing 10 mL methanol and 90 mL water. Before the test, the photo-reaction system was placed under vacuum until the pressure gage was stable. An externally illuminating 150 W Xe lamp (PLS-SXE300, Beijing Changtuo Co., Ltd., China) was used as the light source, and the visible light was achieved using a 420 nm cut-off filter to remove ultraviolet light; the photo density control was set at 250 mW cm−2. The testing temperature was maintained at 5 °C by a condensate water system. In the dye degradation testing process, 0.1 g photocatalyst was added to 100 mL rhodamine B (RhB) with a concentration of 10 mg L−1. Before illumination, the mixture was stirred for 30 m in the dark. The light source was kept the same with the photocatalytic water splitting test. The temperature of the dye liquid was maintained at 25 °C using circulating water. The changes of the light absorptivity of RhB with photocatalysis time going was measured using a UV-visible absorption spectrophotometer (U-41000, HITACHI, Tokyo, Japan).Fabrication of photoelectrodes and the photoelectrochemical performance characterization
We prepared the photoelectrodes by an electrophoresis method. In brief, 20 mg photocatalyst was dispersed in 50 mL water, and 5 mg I2 was added to the solution with string for 30 m. Subsequently, two FTO conductive glasses (1 cm × 2 cm) separated by a distance of 1 cm were immersed in the mixture, then 20 V of bias was applied between them for 3 m using a potentiostat. The as prepared thin film on the cathode was annealed at 300 °C for 1 h. A three electrode system was employed to measure the PEC performance. The photoelectrodes, a large piece of platinum, and Ag/AgCl (saturated KCl) were used as the working, counter, and reference electrodes, respectively. All measurements were performed in 0.1 M Na2SO4 electrolyte by a Par4000 electrochemical system (Par4000, Princeton Ltd., USA). The photoinduced current densities with time (I–t) curves were measured at a bias potential of 1 V (vs. Ag/AgCl). The electrochemical impedance spectroscopy (EIS) was carried out at a bias potential of 0 V and the frequency range was from 105 to 10−1 Hz. The Mott–Schottky plots were measured in the dark using a potential range of −1.0 V to 1.0 V with a frequency of 10 Hz.3. Results and discussion
Fig. 1 presents the SEM images of g-CN and UF-CN. The surface SEM morphology of g-CN is shown in Fig. 1A; we can observe that the particle size of g-CN is larger than 5 μm with a typical layer-structure. However, after mechanical grinding in water, the surface morphology of g-CN was changed significantly, and the result is shown in Fig. 1B. It is interesting to note that the layer-structure present on g-CN was replaced by ultrathin nanoflakes with a planar structure, indicating that the layer-structure of g-CN could be exfoliated by the facile mechanical grinding method. | ||
| Fig. 1 SEM images of (A) g-CN and (B) UF-CN. | ||
Fig. S1† shows the SEM element mapping result of the UF-CN. From this result, the N, C, and O elements can be found on the UF-CN powder. The extra oxygen element was formed during the thermal condensation process of the melem. However, the dots density of oxygen (Fig. S1D†) is far less than that of N (Fig. S1B†) and C (Fig. S1C†), indicating that the content of the oxygen element on the UF-CN is very low.
To develop a better understanding of the thickness change of the g-CN after mechanical grinding, we employed the TEM technique to observe the as prepared materials, and the results are shown in Fig. 2. Fig. 2A and B show the low and high resolution TEM images of g-CN respectively. From Fig. 2A, we can find large sized particles with a low light transmittance, indicating a great thickness of g-CN. Fig. 2B presents the high resolution TEM of the edge region of g-CN and it is clearly shows a layered stacking. Fig. 2C and D present the low and high resolution images of UF-CN. Compared with Fig. 2A, the light transmittance degree of UF-CN in Fig. 2C increased significantly; meanwhile, the size of the flakes decreased to approximate 500 nm. The high resolution TEM image of UF-CN is shown in Fig. 2D and from this result we can see that an ultrathin CN flake is stacked less than three layers, presenting a well-defined two dimension quantum structure.16
 | ||
| Fig. 2 TEM results of (A) low resolution and (B) high resolution of g-CN; (C) low resolution and (D) high resolution of UF-CN. | ||
For further research on the layered structure of UF-CN, the AFM technique was carried out to observe it, and the results are shown in Fig. 3A and B. The ultrathin 2-D structure of UF-CN can be observed clearly. However, this large-sized 2-D nanoflake does not show an absolute monolayer, it is formed with 3–6 layers overlapped, and also with some nano asperities distributed on its surface. This phenomenon may be formed during the grinding process because high energy was exerted on the g-CN during this process. Fig. 3C shows the thickness change of the UF-CN corresponding to the marked area in Fig. 3B. From this picture, we can see that the thickness changed between different layers; in addition, the thickness of the CN monolayer is near 2 nanometers. The XRD results of g-CN and UF-CN are presented in Fig. 3B. The XRD peak at 27.5° corresponds to interlayer stacking of the g-CN. A strongly diffuse peak at 27.5° on the g-CN curve can be found, indicating that the g-CN presented an obvious interlayer stacking structure. However, for the UF-CN material, compared with g-CN, the diffuse peak density at 27.5° decreased dramatically, indicating the layered structure of g-CN was exfoliated to a multilayer ultrathin structure.23
 | ||
| Fig. 3 (A) and (B) AFM image of UF-CN; (C) thickness change of the area marked in (B); and (D) XRD results of g-CN and UF-CN. | ||
Fig. 4 presents the XPS results of g-CN and UF-CN. Fig. 4A shows the survey spectrums of g-CN and UF-CN. Except for C, N and O element peaks, no other element peak was observed, indicating that the element composition of the UF-CN did not change after mechanical grinding. Fig. 4B shows the high-resolution C 1s spectra, the peaks at 286.1 eV and 288.2 eV on both materials corresponding to the C–C and C–N–C bonds, respectively.24,25 For the material of bulk g-CN, the peak area of the C–C bond is larger than that of C–N–C bond. However, for UF-CN, this situation reversed, the peak area of the C–C bond is less than that of the C–N–C bond; this situation probably resulted in the C–C bond present between the interlayer when it was dissociated during the exfoliation process. The high-resolution spectra of N 1s is presented in Fig. 4C; the peak has been deconvoluted into three peaks, including 398.8 eV, 399.7 eV, and 401 eV, corresponding to the bonds of C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, N–[C]3, and C–NHx, respectively.24,25 No obvious differences of the N 1s can be observed with these two materials. Fig. 4D shows the O 1s high-resolution XPS results of g-CN and UF-CN, indicating that some oxidation groups were formed during the polymerization process.
C, N–[C]3, and C–NHx, respectively.24,25 No obvious differences of the N 1s can be observed with these two materials. Fig. 4D shows the O 1s high-resolution XPS results of g-CN and UF-CN, indicating that some oxidation groups were formed during the polymerization process.
 | ||
| Fig. 4 XPS results of g-CN and UF-CN (A) survey; (B) high resolution of C 1s; (C) N 1s; and (D) O 1s. | ||
Fig. 2S† displays the dispersive capacity of g-CN and UF-CN in water. In this test, 0.02 g g-CN and UF-CN powders were ultrasonically dispersed in 20 mL water for 30 m, respectively. After that, the mixtures rested for 3 days for follow-up observing. Fig. 2SA† shows the photography of g-CN and it can be seen that the powder settled to the bottom of the water after 3 days of resting. Compared to the bulk g-CN, at the same condition, the UF-CN kept a good dispersion in water. This phenomenon contributed to the ultrathin structure of CN to improve the dispersion capacity of it.
To develop a better understanding of the relative characteristics between photocatalysis performance and ultrathin exfoliation of g-CN and UF-CN, we employed photocatalytic water splitting for hydrogen evolution and photocatalytic RhB degradation techniques, and the relevant results are shown in Fig. 5. Fig. 5A shows the photocatalytic water splitting for hydrogen evolution performances of g-CN and UF-CN photocatalysts. For g-CN, the photocatalytic hydrogen evolution yield in 4 hours reached 190 μmol g−1, however, for UF-CN, the corresponding value increased to 608 μmol g−1 in the 1st round, which is approximately 3.2 times larger than that of the former. We also compared the photocatalytic hydrogen evolution performance of this UF-CN with other similar ultrathin C3N4 made with different preparation methods, and the results are listed in Table 1. From this table, we can observe that UF-CN shows the better photocatalytic hydrogen evolution performance. The photocatalysis stability of UF-CN was tested further; the hydrogen yields could be reproduced in 4 cycles, indicating a stable photocatalysis performance of UF-CN.
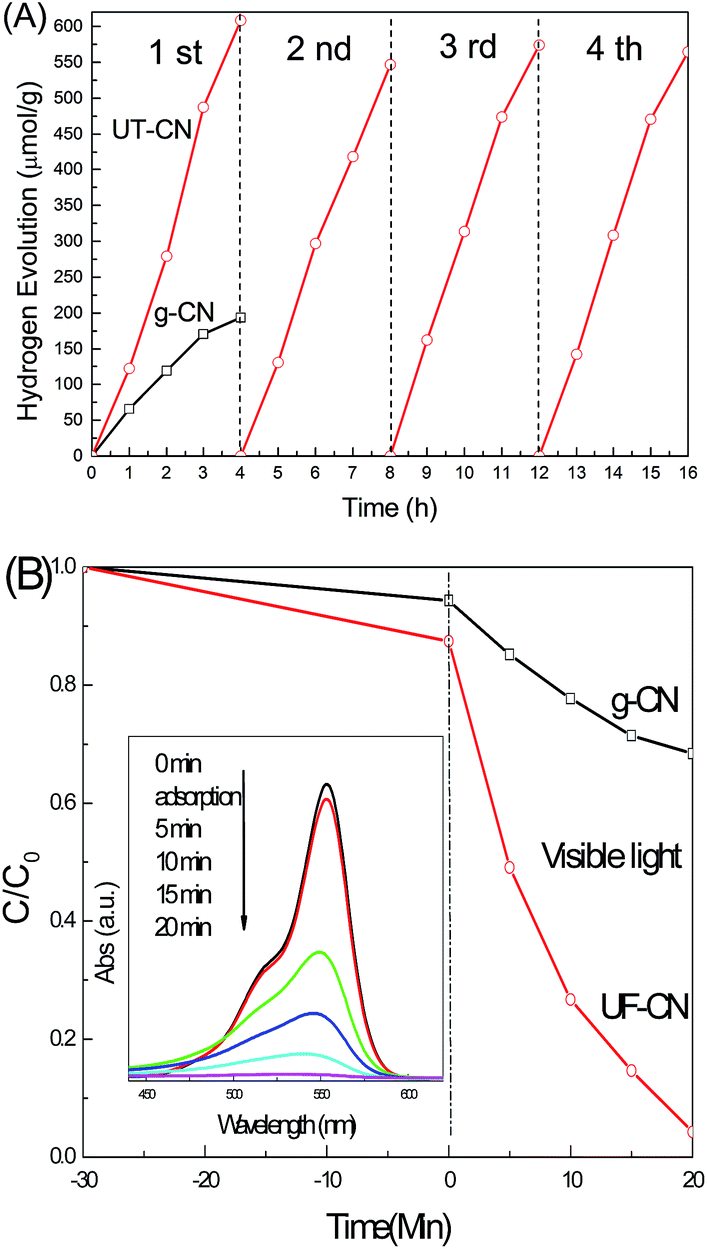 | ||
| Fig. 5 (A) Photocatalysis water splitting performances of g-CN and UF-CN under visible light (λ > 420 nm) in methanol solution with a volume 10%, and 1 wt% Pt added before illumination; (B) photocatalysis RhB degradation under visible light (λ > 420 nm) illumination (inset is the RhB absorption spectrum intensity changes during UF-CN photocatalytic degradation). | ||
| Method | Xe light intensity (W) | Sacrificial agent | Hydrogen evolution rate (μmol h−1) | Ref. |
|---|---|---|---|---|
| Sonication | 300 W (420 nm) | Triethanolamine | 93 | 16 |
| Thermal oxidation | 300 W (400 nm) | Triethanolamine | 160 | 17 |
| Liquid grinding (disorder g-C3N4) | 300 W (420 nm) | Triethanolamine | 80 | 29 |
| Mechanical grind | 150 W (420 nm) | Methanol | 150 | This study |
Fig. 5B shows the result of photocatalytic RhB degradation of g-CN and UF-CN under visible light. Before visible light illumination, the photocatalyst-dye mixture was stirred in the dark 30 m for adsorption equilibrium. From the corresponding curves in Fig. 5B, we can see that the RhB adsorption ability of UF-CN is slightly higher than that of g-CN; this phenomenon may be contributed to by the comprehensible larger specific surface area of ultrathin CN. After visible light illumination for the photocatalysis of g-CN, only 35% RhB was degraded in 20 m. However, for UF-CN, the RhB was degraded completely in 20 m, which was dramatically improved over g-CN. Corresponding absorption spectrum curves of RhB with illumination times are shown as an inset in Fig. 5B; the absorption peak intensity of RhB decreased from 0 to 20 m gradually, and disappeared completely at 20 m, indicating that the RhB did not merely decolor, but mineralized during the photocatalysis process.26 The cyclic RhB degradation property of UF-CN is present in Fig. S3†; from this result we can see that this photocatalyst shows a high stability of RhB degradation after 5 test cycles. It is meaningful that the UF-CN is also a stable photocatalyst for dye degradation.
Energy band structure is an important parameter for photocatalysis performance of semiconductor photocatalysts, because the oxide and reduction capacities of the photogenerated electrons and holes are mainly decided by it. So, we employed UV-Vis DRS, PL, and Mott–Schottky plot techniques to characterize the energy band structures of g-CN and UF-CN, and the results are shown in Fig. 6. Fig. 6A presents the typical UV-Vis DRS results of g-CN and UF-CN; the absorption threshold of g-CN is near 470 nm, corresponding to a bandgap width of 2.64 eV. Meanwhile, the absorption threshold of UF-CN presents at 445 nm with a bandgap width 2.78 eV. From this result, a 0.14 eV blue shift could be found after the layered structure of g-CN transferred into the ultrathin 2-D structure. Fig. 6B shows the PL results of the g-CN and UF-CN; the emission peak of UT-CN shifted to the blue direction compared to its bulk counterpart. The blue shift of the UV-Vis DRS and PL results should have contributed to the thickness decrease of the layered g-CN that resulted in a quantum confinement effect. Some analogous results have been reported by Zhang et al.15 when they prepared ultrathin C3N4 nanosheets for bioimaging application. As with width changing of the bandgap of CN, the energy band potential would be changed simultaneously. As we know, one serious limiting factor for the photocatalysis performance of g-C3N4 is the deficient potential of the valence band (VB), which value is less than 1.4 eV. So, the photocatalysis performance of g-C3N4 would be decreased to its weak oxidation capacity of the photogenerated holes. To further explore the change situation of the energy band of UF-CN, the Mott–Schottky plots method was employed to research the relation between capacitance of the space charge region and the applied potential. The description formula is as follows:
| 1/C2 = 2(eεε0ND)−1 × (E − Efb − κT/e) | (1) |
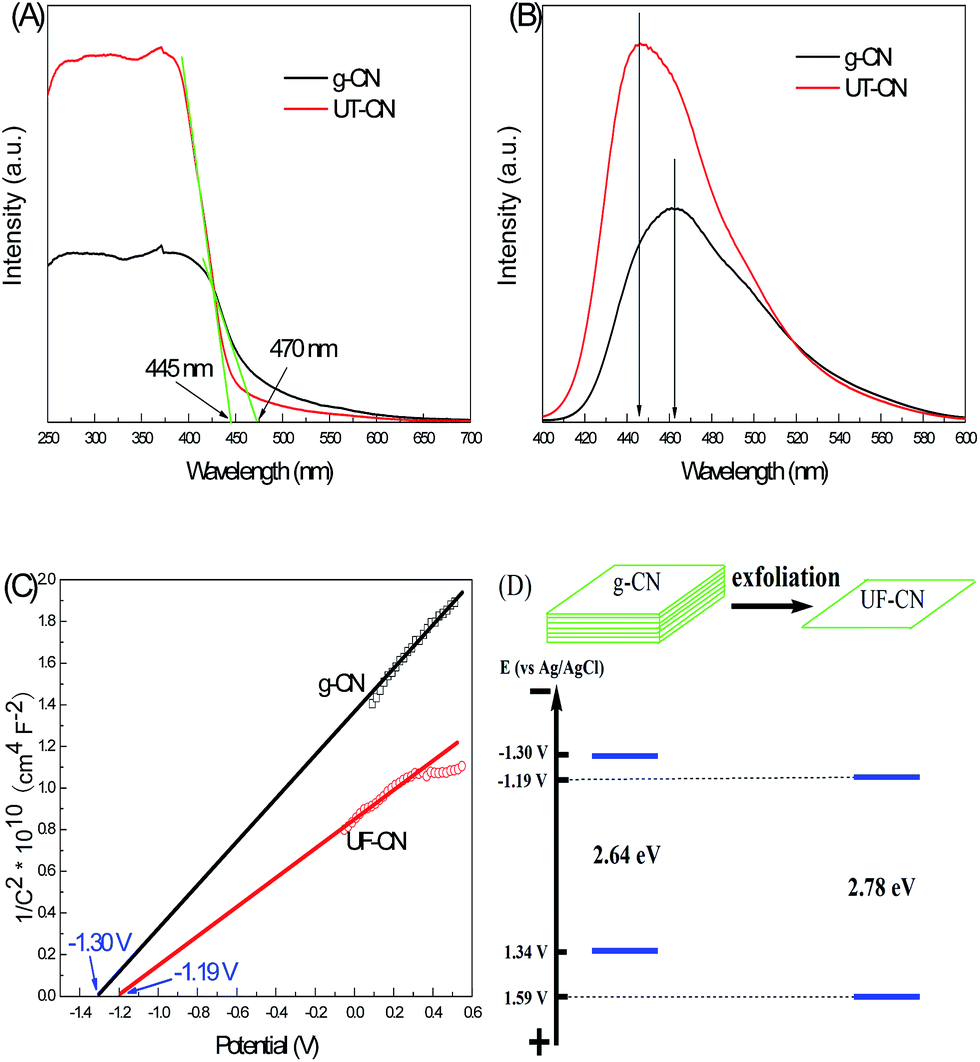 | ||
| Fig. 6 (A) UV-Vis diffuse reflection of g-CN and UF-CN; (B) photoluminescence of g-CN and UF-CN; (C) Mott–Schottky plots of g-CN and UF-CN; and (D) schematic of energy band change with g-CN transfer to UF-CN. | ||
Fig. 7A shows the photoinduced I–t curves of g-CN and UF-CN photoelectrodes. A photoinduced I–t curve is an indirect technique to reflect the photo quantum effect of a semiconductor photoelectrode. As the results presented in Fig. 6C show, it is clear that the photoinduced current of the UF-CN electrode is much larger compared to the bulk counterpart one, indicating a higher photo quantum effect of UF-CN. The electron migration capacity in a semiconductor thin film can be characterized by the EIS method.28 With the electrons migration capacity increasing, the life time of the photoinduced electrons can be prolonged and that improves the photocatalysis performance of the photocatalyst. The EIS results of g-CN and UF-CN are presented in Fig. 7B. The resistance arc radius of UF-CN is much smaller than that of g-CN, indicating that the electron mobility of UF-CN is larger than the latter's. This phenomenon means that the electron transfer capacity on the horizontal plane of g-CN may be influenced by the interlayer van der Waals force, so the electron transfer capacity of UF-CN was increased with the layered structure of the g-CN exfoliated to an ultrathin structure by the wet mechanical grind method.
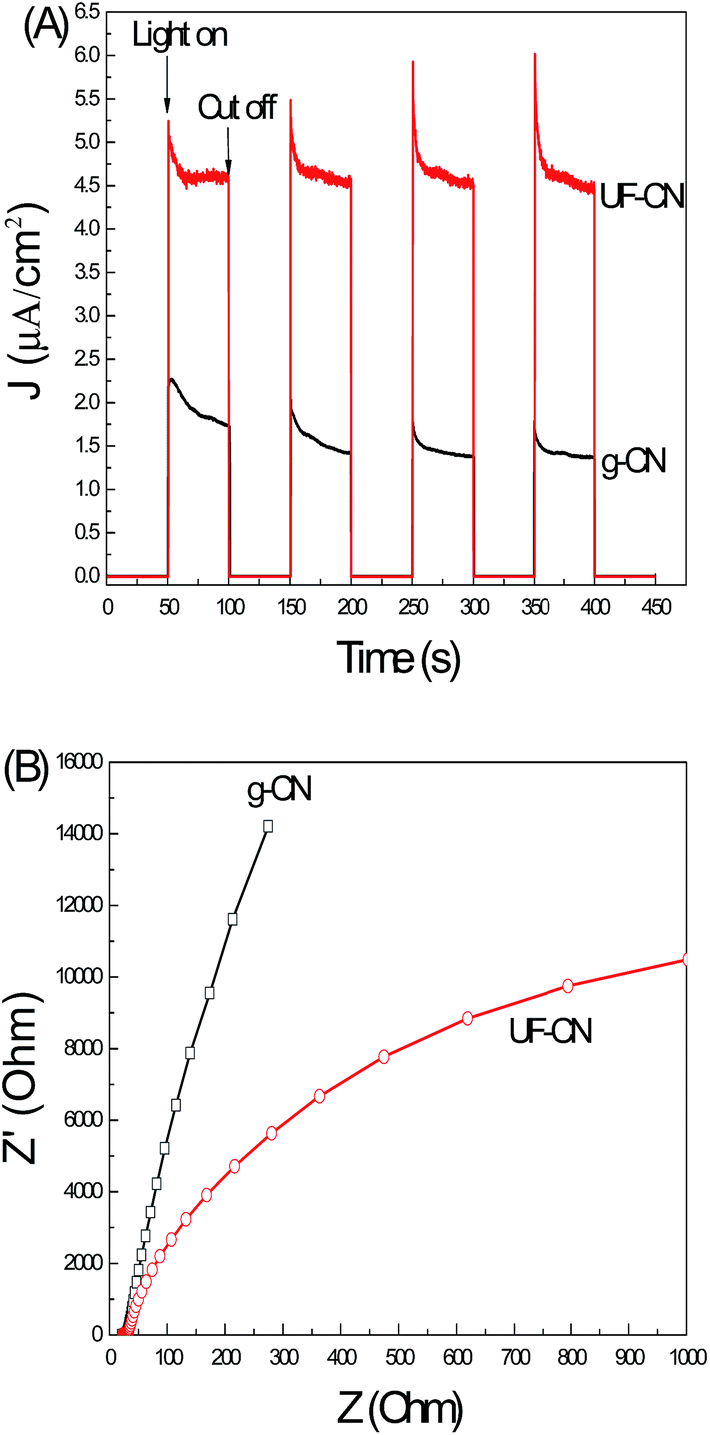 | ||
| Fig. 7 (A) Photoinduced current–time curves and (B) EIS results of g-CN and UF-CN thin film electrodes. | ||
Fig. 8 shows the main mechanism of the photocatalysis property improvement with the g-C3N4 transfer to ultrathin C3N4. As shown in Fig. 8, the VB potential of g-C3N4 located at 1.34 V, which resulted in a weak oxidation energy of the photogenerated holes. Thus, if the holes can't react with the surroundings quickly, they will combine with the photogenerated electrons that decrease the photo quantum of this photocatalyst. With the g-CN transfer form a layered structure to an ultrathin structure, these band structures were changed by the quantum size effect. The bandgap width of g-CN is 2.64 eV, which is 0.14 eV narrower than UF-CN's, and means that the visible light absorption range of g-CN is larger than the latter. However, the photoelectrochemical and photocatalysis performances of the UF-CN are higher than that of g-CN, so this phenomenon contributed to the VB potential positive move of UF-CN, and that increased the oxidation capacity of the photogenerated holes.
 | ||
| Fig. 8 Mechanism of the photocatalysis performance improvement with the g-C3N4 transfer to ultrathin C3N4. | ||
4. Conclusion
In this study, we prepared an ultrathin C3N4 by a simple wet mechanical grind method with a 90% yield rate. The as prepared UF-CN showed a 3.2 times higher photocatalytic water reduction for hydrogen evolution performance than that of g-CN. Meanwhile, the photocatalytic RhB degradation property of UF-CN was improved dramatically compared to the bulk counterpart, and the RhB solution could be degraded in 20 m under visible light illumination. The reasons for increased photocatalysis performance were investigated by UV-Vis, DRS, PL, Mott–Schottky plots, and EIS techniques. The results indicated that the oxidation capacity of photogenerated holes and electron mobility on the horizontal plane were increased after exfoliating the g-CN into UF-CN, thus improving the photocatalysis performance of UF-CN.Acknowledgements
This work was financially supported by the National Basic Research Program of China (No. 41506093).Notes and references
- M. R. Hoffmann, T. M. Scot, W. Choi and D. W. Bahnenmann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- L. Jing, W. Zhou, G. Tian and H. Fu, Chem. Soc. Rev., 2013, 42, 9509 RSC.
- Y. Bu, Z. Chen and W. Li, Dalton Trans., 2013, 42, 16272 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniecc and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 CAS.
- Y. Bu and Z. Chen, Electrochim. Acta, 2014, 144, 42 CrossRef CAS.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397 CrossRef CAS PubMed.
- X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen, Y. Hou, X. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294 CrossRef CAS PubMed.
- C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Funct. Mater., 2012, 22, 1518 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18 CrossRef CAS PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. Cheng, Adv. Funct. Mater., 2012, 22, 4763 CrossRef CAS.
- H. Wang, Y. Su, H. Zhao, H. Yu, S. Chen, Y. Zhang and X. Quan, Environ. Sci. Technol., 2014, 48, 11984 CrossRef CAS PubMed.
- J. Tian, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2013, 85, 5595 CrossRef CAS PubMed.
- X. L. Zhang, C. Zheng, S. S. Guo, J. Li, H. H. Yang and G. Chen, Anal. Chem., 2014, 86, 3426 CrossRef CAS PubMed.
- W. Zhao, M. Fang, F. Wu, H. Wu, L. Wang and G. Chen, Chemistry, 2010, 20, 5817 CAS.
- Y. I. Jeon, H. J. Choi, S. M. June, J. M. Seo, M. J. Kim, L. Dai and J. B. Baek, J. Am. Chem. Soc., 2013, 135, 1386 CrossRef PubMed.
- J. Tian, Q. Liu, C. Ge, Z. Xing, A. M. Asiri, A. O. Al-Youbicd and X. P. Sun, Nanoscale, 2013, 5, 8921 RSC.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017 RSC.
- L. Zhang, D. Jing, X. She, H. Liu, D. Yang, Y. Lu, J. Li, Z. Zheng and L. Guo, J. Mater. Chem. A, 2014, 2, 2071 CAS.
- H. Fu, S. Zhang, T. Xu, Y. Zhu and J. Chen, Environ. Sci. Technol., 2008, 42, 2085 CrossRef CAS PubMed.
- Y. Y. Bu, Z. Y. Chen and W. B. Li, Appl. Catal., B, 2014, 144, 622 CrossRef CAS.
- X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117, 9952 CAS.
- X. L. Wang, W. Q. Fang, S. Yang, P. Liu, H. Zhao and H. G. Yang, RSC Adv., 2014, 4, 10676 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra05524a |
| This journal is © The Royal Society of Chemistry 2016 |