
Nitrogen-doped carbon with mesoporous structure as high surface area catalyst support for methanol oxidation reaction†
Li-Mei Zhanga,
Zhen-Bo Wang*a,
Xu-Lei Suia,
Cun-Zhi Liab,
Lei Zhaoa and
Da-Ming Gub
aSchool of Chemical Engineering and Technology, Harbin Institute of Technology, No. 92 West-Da Zhi Street, Harbin, 150001 China. E-mail: wangzhb@hit.edu.cn; Fax: +86-451-86418616; Tel: +86-451-86417853
bSchool of Science, Harbin Institute of Technology, No. 92 West-Da Zhi Street, Harbin, 150001 China
First published on 13th April 2016
Abstract
Mesoporous nitrogen-doped carbon (MNC) with a high surface area has been synthesized via carbonizing polyaniline using silica nanoparticles as template. The more silica nanoparticles, the smaller the micropore surface area is and the larger the mesoporous surface area is. Moreover, with an increase in the amount of silica nanoparticles, the electrocatalytic activity of Pt/MNC catalysts shows a downward trend after an intimal increase, and the Pt/MNC-1/6 (with the weight ratio of aniline monomer to silica nanoparticles of 1/6) catalyst has the highest activity, ascribed to the optimal Pt nanoparticles size, which is closely related to the pore structure of the support. In addition, the electrocatalytic activity and stability of Pt/MNC-1/6 catalyst are significantly superior to that of Pt/nitrogen-doped carbon (Pt/CNx) catalyst. For the same electrocatalytic activity, the Pt loading of Pt/MNC-1/6 catalyst is reduced by 33.3% compared to the Pt/CNx catalyst. The high electrocatalytic activity originates from the introduction of mesoporous structures that can facilitate mass transfer and improve the dispersion of Pt nanoparticles. Furthermore, the Ostwald ripening behavior of Pt nanoparticles is limited in the mesoporous structure of MNC-1/6, which weakens the aggregation effect of Pt nanoparticles during the electrocatalytic processes, thus enhancing the electrocatalytic stability of the catalyst.
1. Introduction
In recent years, direct methanol fuel cells (DMFCs) have attracted enormous attention due to their low-cost, high power density and low operating temperatures, and are envisioned as promising power sources for portable devices and electric vehicles.1–5 To date, carbon supported Pt-based catalysts are generally considered to be the most common electrocatalysts for methanol oxidation reaction (MOR). However, carbon supported Pt-based catalysts suffer from some disadvantages such as the high cost of Pt and being susceptible to corrosion of carbon support, which severely restrict their commercial applications for DMFCs.6,7 Therefore, it is urgent to develop advanced carbon materials, such as carbon nanotubes,8–10 graphene,11–13 and nitrogen-doped carbon,14,15 with large surface area, high chemical stability, and excellent electrical conductivity. Recently, nitrogen-doped carbon derived from conducting polymer (e.g. polyaniline,16,17 polypyrrole,18,19 polyacrylonitrile20) has been considered to be a promising alternative to carbon since N species in the carbon support can lead to high dispersion of fine Pt nanoparticles with the synergistic interaction of Pt and support, resulting in the improved catalytic activity and stability toward MOR.21In view of the electrocatalysts used in fuel cells, high surface areas and large pore volumes of the support materials are required due to the fact that higher surface areas and larger pore volumes allow for better dispersion of Pt nanoparticles and provide an open network around the active catalysts for facile diffusion of fuels and products.22,23 Recently, highly nanoporous carbon (NPC) has attracted considerable attention due to its high surface area, large pore volume and good electrochemical properties.24–26 Generally, NPC can be prepared via several methods, such as nanocasting with hard-templates,27 activation (physical or chemical) of carbon materials,28,29 carbonization of polymeric aerogels,30,31 and so on. Among them, nanocasting with hard-templates is one of the most common and controllable method since the size of hard-template can be used to determine the pore diameter of carbon material. For example, Su et al.32,33 synthesized ordered mesoporous carbon (OMC) using sucrose as carbon precursor, and ordered mesoporous silica SBA-15 as hard-template. Pt nanoparticles on the OMC for oxygen reduction reaction and methanol oxidation reaction were discussed systematically. The results indicate that the mesoporous structure of the carbon support is important for liquid-phase electrochemical reactions. Liang et al.34 prepared a family of mesoporous metal-nitrogen-doped carbon electrocatalysts using silica nanoparticles, ordered mesoporous silica SBA-15, and montmorillonite as templates, respectively. The unprecedented performance of these electrocatalysts for oxygen reduction reaction was ascribed to high surface area, well-defined mesoporous structure, and homogeneous distribution of abundant metal-Nx active sites. However, very few reports on porous nitrogen-doped carbon as Pt-based catalyst support.28 Su et al. reported the preparation and characterization of nitrogen-doped porous carbon nanospheres (PCNs), which were prepared by carbonization and chemical activation of polypyrrole nanospheres. PCNs possessed high BET surface area (up to 1010 m2 g−1) and well-defined microporous structure with a pore size of around 1.1 nm, which had prominent advantages as an alternative Pt-based catalyst support. Introducing microporous structures can enhance surface area of the support, further resulting in the better dispersion of Pt nanoparticles. However, microporous structure would not play a role in the diffusion of fuels and products. In order to provide an open network around the active catalysts for facile diffusion of fuels and products, as well as to further improve the electrocatalytic property of Pt-based catalyst, the development and preparation of mesoporous nitrogen-doped carbon as Pt-based catalyst support are very meaningful.
Our research group35 successfully synthesized mesoporous nitrogen-doped carbon (MNC) material using polyaniline as carbon and nitrogen precursors, and silica nanoparticles as template for achieving mesoporous structure. Subsequently, Pt nanoparticles were dispersed on MNC support through a microwave-assisted polyol process to obtain Pt/MNC catalyst. Effects of different pyrolysis temperatures on the physical and electrochemical performance of catalyst were systematically investigated. The N contents and distribution proportion of different types N of MNC were closely related to the pyrolysis temperature. When to achieve optimal proportion of pyridinic N, pyrrolic N and graphitic N, MNC support reveals not only intense anchoring effect of Pt nanoparticles but also enhanced electric conductivity, further improving electrocatalytic activity and stability of the catalyst. However, the effects of specific surface area and pore structure of support on the electrochemical performance of catalyst have not been thoroughly discussed.
In order to reduce the Pt loading of catalyst, further lowering the catalyst cost, we profoundly discussed the effects of specific surface area and pore structure of support on the physical and electrochemical performance of Pt/MNC catalyst. For comparison, nitrogen-doped carbon (CNx) was also synthesized and characterized in the absence of template and used as Pt catalyst support.
2. Experimental
2.1 Synthesis of MNC support
500 mg aniline monomer was dissolved in 10 mL 1.0 mol L−1 HCl successively and then 40 wt% silica colloid (3.75, 7.50 or 11.25 g) was added. After stirring for 1 h, 2.5 mL 1.0 mol L−1 HCl solution containing 1.23 g ammonium persulfate (APS) was added dropwise with vigorous stirring. The polymerization was conducted in an ice bath for 8 h. After evaporation of water at 70 °C, the obtained polyaniline coated silica nanoparticle composite was then pyrolyzed under flowing argon at 900 °C for 2 h. The silica template was removed by 10 wt% HF etching. According to the feed weight ratio of aniline monomer and silica nanoparticles, the resulting samples were denoted as MNC-1/3, MNC-1/6, and MNC-1/9, respectively. For comparison, nitrogen-doped carbon (CNx) was also prepared under the similar conditions, without using silica nanoparticles as template.2.2 Preparation of Pt/MNC catalyst
Pt nanoparticles were deposited on the MNC support by a microwave-assisted ethylene glycol process. Briefly, 40 mg MNC support was dispersed into 60 mL mixed solution of ethylene glycol and isopropyl alcohol (v/v = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under ultrasonic treatment for 1 h, and then 1.34 mL 0.0383 mol L−1 H2PtCl6–EG solution was added with urgent agitation for 3 h. The pH value of the mixture was then adjusted to 12.0 with an appropriate amount of NaOH–EG solution. After saturated with argon, the mixture was heated using a microwave oven for 55 s. When the mixture cooled down to room temperature, dilute HNO3 was added dropwise to adjust the pH value of the mixture to 2.0–3.0. The mixture was kept stirring for 12 h and then the product was washed repeatedly with ultrapure water (18.2 M cm). The obtained Pt/MNC catalyst was dried for 5 h at 80 °C and then stored in a vacuum vessel. For comparison, Pt/CNx catalyst was also prepared by the same process.
:1) under ultrasonic treatment for 1 h, and then 1.34 mL 0.0383 mol L−1 H2PtCl6–EG solution was added with urgent agitation for 3 h. The pH value of the mixture was then adjusted to 12.0 with an appropriate amount of NaOH–EG solution. After saturated with argon, the mixture was heated using a microwave oven for 55 s. When the mixture cooled down to room temperature, dilute HNO3 was added dropwise to adjust the pH value of the mixture to 2.0–3.0. The mixture was kept stirring for 12 h and then the product was washed repeatedly with ultrapure water (18.2 M cm). The obtained Pt/MNC catalyst was dried for 5 h at 80 °C and then stored in a vacuum vessel. For comparison, Pt/CNx catalyst was also prepared by the same process.
2.3 Characterizations of physical properties
The homemade product was characterized by scanning electron microscope (SEM, Hitachi Ltd. S-4700), Brunauer–Emmett–Teller surface area (BET, QUADRASORB SI), transmission electron microscopy (TEM, TECNAI G2 F30), inductively coupled plasma (ICP, OPTIMA 5300DV), X-ray diffraction (XRD, D/max-RB diffractometer) and X-ray photoelectron spectroscopy (XPS, PHI model 5700).2.4 Electrochemical measurements
The electrochemical performances of Pt/MNC and Pt/CNx catalysts were evaluated at 25 °C in a three-electrode cell connected to a CHI650E workstation. The cell consists of a working electrode, an Hg/Hg2SO4 (0.68 V relative to reversible hydrogen electrode, RHE) reference electrode and a platinum foil counter electrode. All of the potentials are relative to the RHE electrode, unless otherwise noted. The working electrode was prepared as follows: 4 mg catalyst was dispersed in 2 mL mixed solution of ethanol and ultrapure water (v/v = 1:1) and ultrasonicated for 20 min to form a uniform catalyst ink. A total of 5 μL of well-dispersed catalyst ink was pipetted onto the glassy carbon electrode surface and onto which 5 μL of a dilute aqueous Nafion solution (5 wt% solution in a mixture of lower aliphatic alcohols and DuPont water) was added. The prepared electrode was dried at room temperature before electrochemical tests.
The cyclic voltammograms (CV) were recorded within a potential range from 0.05 V to 1.2 V (vs. RHE). The electrochemically active specific surface area (ESA) of catalyst was calculated from the formula ESAPt = QH/(0.21 MPt) where QH is the charge due to the hydrogen adsorption/desorption in the hydrogen region of the CV, 0.21 is the electrical charge associated with monolayer adsorption of hydrogen on Pt, and MPt is the loading of Pt metal on the working electrode.36,37 In order to activate and clean the catalyst surface, the working electrode was treated by continuous cycle at a scan rate of 50 mV s−1 in argon-purged 0.5 mol L−1 H2SO4 solution until a stable response was obtained. Electrochemical impedance spectroscopy (EIS) was obtained at frequencies between 100 kHz and 0.01 Hz with 12 points per decade. The amperometric i–t curve was measured at a constant potential of 0.6 V (vs. RHE) for 3600 s in argon-purged 0.5 mol L−1 H2SO4 + 0.5 mol L−1 CH3OH solutions.
3. Results and discussion
3.1 Physical characterization
SEM is used to investigate the morphology of the catalyst support. As shown in Fig. 1(a), CNx support prepared in the absence of template exhibits irregular blocky structure. However, MNC supports shown in Fig. 1(b–d) present in the form of evenly dispersed spherical-like particles. In addition, it can be seen from the inset of Fig. 1(c) that the surfaces of spherical-like particles appear a typical porous structure in nanoscale. Those results indicate that silica nanoparticles (12 nm in diameter) not only play a role in pore-creating but also promote the formation of spherical-like particles, further to obtain high specific surface area of the support. Moreover, with the increase of the amount of silica nanoparticles, the size of spherical-like particles decreases, probably suggesting that the sizes of spherical-like particles are determined by the amount of silica nanoparticles. When the spherical-like particles are in an optimal size, MNC support can better load and disperse Pt nanoparticles, and then enhance electrocatalytic activity and stability of catalyst. | ||
| Fig. 1 SEM images of CNx (a), MNC-1/3 (b), MNC-1/6 (c) and MNC-1/9 (d). | ||
In order to precisely verify different specific surface areas of supports in the presence or absence of template, N2 adsorption–desorption isotherms and the pore size distribution of CNx and MNC supports are characterized as shown in Fig. 2 and S1.† Clearly, the adsorption isotherm of CNx support as shown in Fig. 2(a) is of type I according to the IUPAC classification, indication of microporous material. While the adsorption–desorption isotherms of MNC supports shown in Fig. 2(b) and S1† exhibit a remarkable hysteresis loop, and the distributions of the pore sizes are centered at about 12 nm, demonstrating MNC support possesses well-defined mesoporous structure. The BET surface areas and pore volumes of MNC-1/3, MNC-1/6 and MNC-1/9 supports are 713 m2 g−1 and 1.0 cm3 g−1, 728 m2 g−1 and 1.3 cm3 g−1, and 714 m2 g−1 and 2.2 cm3 g−1, respectively, which are obviously higher than 278 m2 g−1 and 0.1 cm3 g−1 of the CNx support (Table S1†). Those results manifest that introducing mesoporous structures can yield an increase of support specific surface area and pore volume. This is because, a better dispersion of Pt nanoparticles can be obtained when it is used as support. In addition, the analyses of experimental data indicate that the more silica nanoparticles, the smaller micropore surface area is and the larger mesoporous surface area is. The increase of the mesoporous in MNC is better able to limit the Ostwald ripening behavior of Pt nanoparticles during the electrocatalytic processes, resulting in a better durability for electrocatalytic reactions.
 | ||
| Fig. 2 N2 adsorption–desorption isotherms and the pore size distribution from the BJH method (inset) of CNx (a) and MNC-1/6 (b). | ||
The morphology and nanostructure of Pt/CNx and Pt/MNC catalysts are examined by TEM. As shown in Fig. 3, fine Pt nanoparticles are homogeneously deposited onto CNx and MNC-1/6 supports. However, the mass fractions of Pt in the Pt/CNx and Pt/MNC-1/6 catalysts are significantly different. The mass fraction of Pt in the Pt/CNx catalyst is only 8.29 wt% (ICP result), which is much lower than 21.92 wt% (ICP result) in Pt/MNC-1/6 catalyst. This phenomenon could be ascribed to the high surface area of MNC-1/6 support, which might load more Pt nanoparticles, and better dispersed Pt nanoparticles. With the increase of the amount of silica nanoparticles, the mean sizes of Pt nanoparticles on the MNC-1/3, MNC-1/6, and MNC-1/9 supports gradually become large, around 1.7, 2.0 and 2.5 nm, respectively (Fig. 3(b) and S2†). The possible reason is that the mean sizes of Pt nanoparticles closely rely on the pore structure of support.
 | ||
| Fig. 3 TEM images and Pt size distribution (statistic number 100) of Pt/CNx catalyst (a) and Pt/MNC-1/6 catalyst (b). | ||
The crystalline structures of Pt/CNx and Pt/MNC catalysts are characterized by XRD. Representative diffraction peaks of Pt (111), Pt (200), Pt (220) and Pt (311) are distinctly observed in the XRD patterns (Fig. 4(a)), which means that Pt forms the face-centered cubic (fcc) crystal structure. The broad diffraction peak at 24.9° corresponds to the (002) plane of carbon.
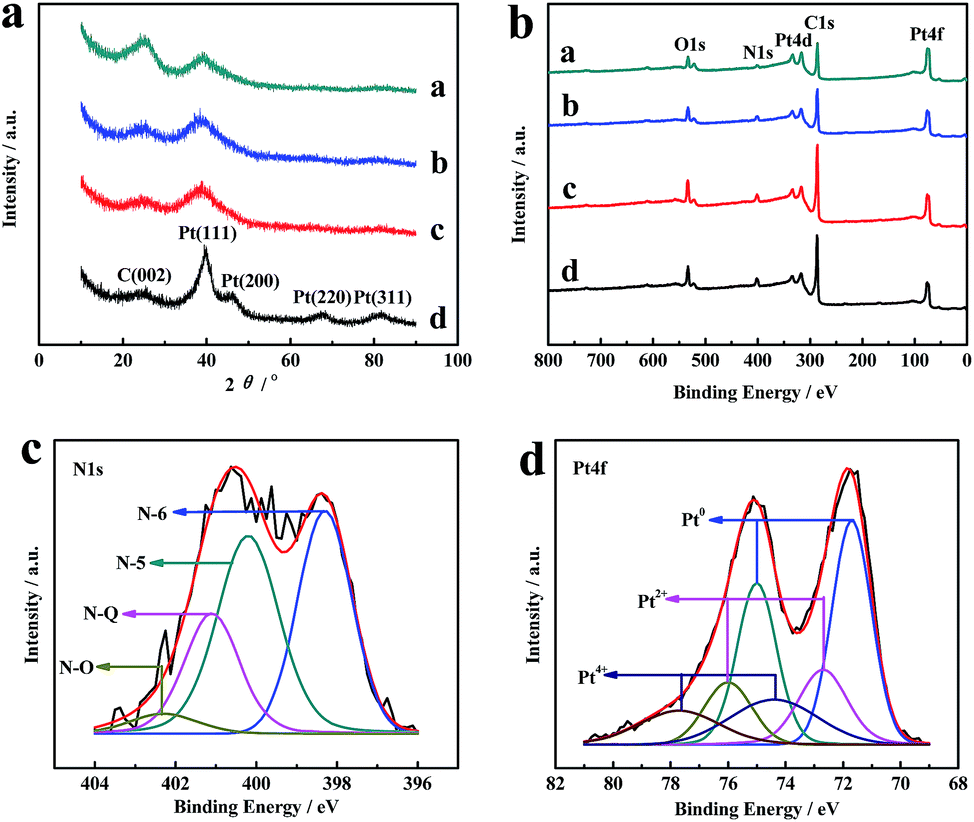 | ||
| Fig. 4 XRD patterns (a), XPS survey spectra (b) of Pt/CNx (a), Pt/MNC-1/3 (b), Pt/MNC-1/6 (c) and Pt/MNC-1/9 (d) catalysts; the high resolution XPS spectra of N 1s (c) and Pt 4f (d) of Pt/MNC-1/6 catalyst. | ||
XPS measurements are performed to probe the surface composition and chemical states of Pt/CNx and Pt/MNC catalysts. The XPS survey spectra are depicted in Fig. 4(b). As expected, C 1s, N 1s, O 1s, Pt 4d and Pt 4f peaks are clearly visible. The N 1s spectrum of Pt/MNC-1/6 catalyst shown in Fig. 4(c) can be deconvoluted into four peaks at 398.3, 400.2, 401.1 and 402.3 eV, which can be assigned to N-6 (pyridinic N), N-5 (pyrrolic N or pyridinic-N in association with phenolic or carbonyl group), N–Q (graphitic N) and N–O (oxidized N).38,39 N-6 and N-5 mainly serve the functions of dispersing and anchoring Pt nanoparticles, while N–Q could improve the electric conductivity of support.40,41
Fig. 4(d) and S3† present the Pt 4f spectra of Pt/CNx and Pt/MNC catalysts, which can be deconvoluted into three pairs of doublets (Table S2†). The most intense doublet with binding energy of 71.3, 71.7, 71.8 or 71.9 eV (Pt 4f7/2) and 74.6, 75.0, 75.1 or 75.2 eV (Pt 4f5/2) is assigned to metallic Pt, the peaks found at around 72.7 and 76.0 eV can be attributed to the Pt2+ chemical state, and the peaks at around 74.4 and 77.7 eV is most likely ascribed to Pt4+ species.42 The integration of peak areas indicates that most Pt species exist as metallic Pt for Pt/CNx and Pt/MNC catalysts. In addition, the binding energies of metallic Pt for Pt/CNx, Pt/MNC-1/3 and Pt/MNC-1/6 catalysts appear a positive shift of 0.6, 0.5 and 0.4 eV, respectively, in comparison with that of Pt/C,43 which is associated with the size effects in metal nanoparticle/cluster.44–46 However, the positive shift of metallic Pt for Pt/MNC-1/9 catalyst has not been observed, which can be attributed to bigger Pt nanoparticles.
3.2 Electrochemical measurement
The cyclic voltammograms of Pt/MNC catalysts in acidic medium (0.5 mol L−1 H2SO4) are illustrated in Fig. 5(a). The electrochemically active specific surface areas (ESAPt) are obtained by the measurements of the hydrogen adsorption–desorption (HAD) integrals. The ESAPt of Pt/MNC-1/3, Pt/MNC-1/6 and Pt/MNC-1/9 catalysts are 83.8, 100.0 and 56.6 m2 g−1, respectively. The Pt/MNC-1/6 catalyst exhibits the biggest electrochemical active area. Moreover, the electrocatalytic activities of the catalysts toward MOR are shown in Fig. 5(b). The peak current densities of Pt/MNC catalysts increase initially and then decrease with the increase of the amount of silica nanoparticles. The methanol oxidation activity are maximized for Pt/MNC-1/6 catalyst with the peak current density of 0.6 A mg−1Pt and decrease successively for Pt/MNC-1/3 and Pt/MNC-1/9 catalysts, with the peak current densities of 0.5 and 0.4 A mg−1Pt, respectively. Pt/MNC-1/6 catalyst exhibits the best electrocatalytic activity toward MOR, which is attributed to optimal Pt nanoparticles size. It is well known that the good distribution and small particle sizes of Pt nanoparticles on the supports are key factors for enhancing their electrocatalytic activity and efficiency.47 However, smaller Pt nanoparticles (<2 nm) inhibit the better dissociative adsorption of methanol, thus is not conducive to the oxidation of methanol.48 In addition, smaller Pt nanoparticles are frequently better incorporated into the micropores of support and thus show blocked surface, which can lead to the decline of methanol electrocatalytic activity.49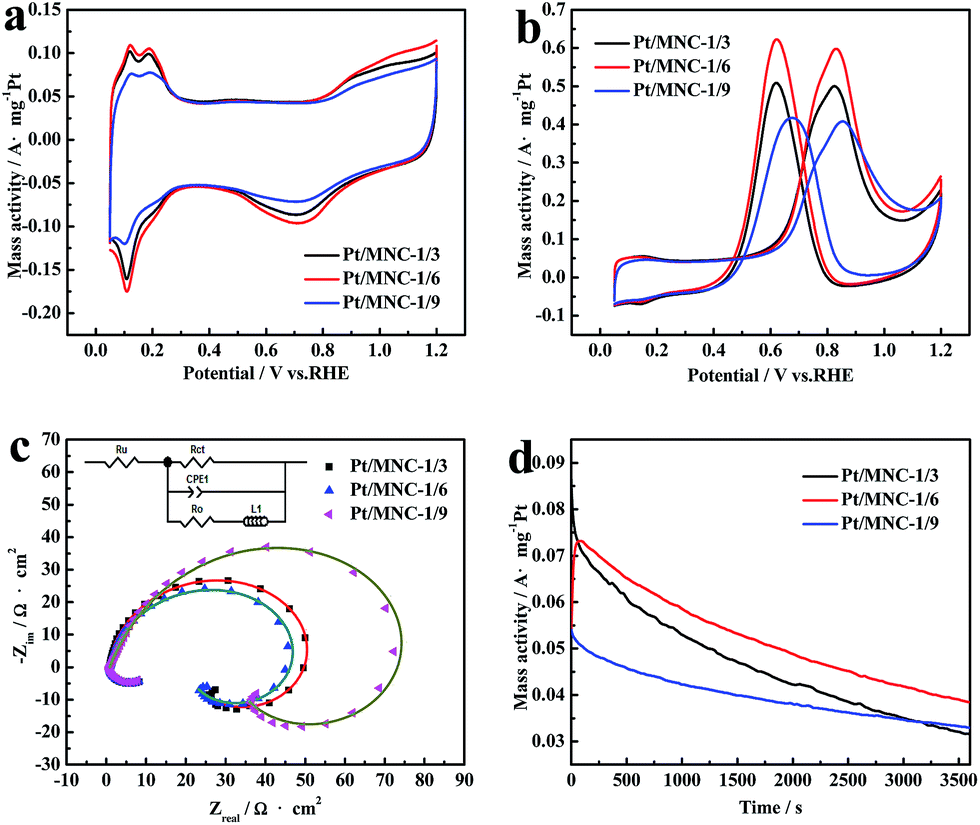 | ||
| Fig. 5 Electrochemical performance of Pt/MNC catalysts: cyclic voltammograms in acidic medium (a) and in methanol acidic medium (b), Nyquist plots (c) and amperometric i–t curves in methanol acidic medium (d). | ||
Electrochemical impedance spectroscopy (EIS) can be used as an effective method for measuring the charge transfer resistance (Rct), which reflects the electrocatalytic activity of the catalyst toward MOR. The Nyquist plots of Pt/MNC catalysts in methanol acidic medium are displayed in Fig. 5(c). In order to quantitatively analyse the impedance behavior, the resistances are analyzed by using EIS with the software of ZsimpWin based on an equivalent electric circuit.50 The Rct of Pt/MNC-1/3, Pt/MNC-1/6 and Pt/MNC-1/9 catalysts are 61.7, 60.3 and 131.0 Ω cm2, respectively. The experimental data reflect the highest electrocatalytic activity for Pt/MNC-1/6 catalyst.
The long-term stability of the catalyst is another important factor influencing their practical application in DMFCs. For comparison, the amperometric i–t curves of Pt/MNC catalysts are measured at a constant potential of 0.6 V (vs. RHE) for 3600 s in methanol acidic medium (Fig. 5(d)). The retention rate (the ratio of the final current density to the maximum current density) of Pt/MNC-1/3, Pt/MNC-1/6 and Pt/MNC-1/9 catalysts after 3600 s are 36.6, 52.5 and 61.0%, respectively, indicating that electrocatalytic stabilities of Pt/MNC catalysts are enhanced gradually with the increasing of the amount of silica nanoparticles. The possible reason is that the confining effect on Pt nanoparticles is gradually enhanced with the increasing of mesoporous.
Considering comprehensively electrocatalytic activity and stability toward MOR, MNC-1/6 is an appropriate support for Pt catalyst.
To verify the effect of introducing mesoporous structures on the electrochemical performance of catalyst, a further comparison is conducted between Pt/MNC-1/6 and Pt/CNx catalysts. Fig. 6(a) shows the cyclic voltammograms in acidic medium. According to the ESAPt formula, the ESAPt of Pt/MNC-1/6 catalyst with 100.0 m2 g−1 is significantly higher than 27.7 m2 g−1 of Pt/CNx catalyst, demonstrating that the electrochemical active area of Pt/MNC-1/6 catalyst is evidently superior to that of Pt/CNx catalyst. In addition, the electrochemical performance of Pt/MNC-1/6 and Pt/CNx catalysts toward MOR are shown in Fig. 6(b–d). From Fig. 6(b), it can be clearly seen that the peak current density of Pt/MNC-1/6 catalyst is 0.6 A mg−1Pt, which is 1.5 times higher than 0.4 A mg−1Pt of Pt/CNx catalyst. Meanwhile, the Rct of Pt/MNC-1/6 catalyst is 60.3 Ω cm2, which is much smaller than 316.5 Ω cm2 of Pt/CNx catalyst (Fig. 6(c)). The results of the cyclic voltammograms and Nyquist plots indicate that methanol electrooxidation activity of Pt/MNC-1/6 catalyst is obviously better than that of Pt/CNx catalyst. Mesoporous introduced to MNC-1/6 support enhance specific surface area and pore volume of the support. High surface area and large pore volume allow for better dispersion of Pt nanoparticles and provide an open network around the active catalysts for facile diffusion of fuels and products, which is consistent with the results reported in the literature.32 Under the same electrocatalytic activity, the Pt loading of Pt/MNC-1/6 catalyst is reduced by 33.3% comparing with Pt/CNx catalyst. The amperometric i–t curves (Fig. 6(d)) can be used for reflecting the electrocatalytic stability toward MOR. As indicated in Fig. 6(d), Pt/MNC-1/6 and Pt/CNx catalysts show a similar trend during the test period. However, the retention rate of Pt/MNC-1/6 catalyst after 3600 s is 52.5%, much higher than 25.7% of Pt/CNx catalyst, indicating that Pt/MNC-1/6 catalyst has better electrocatalytic stability, comparing with Pt/CNx catalyst. The possible reason is N species in the MNC-1/6 and CNx supports are helpful for the enhanced interaction of Pt and support, thus Pt would be anchored tightly on the surface of support. Therefore, the aggregation of Pt nanoparticles on supported catalyst is a big problem for the stability in electrochemical environment.51 The Ostwald ripening behavior of Pt nanoparticles is limited in the mesoporous structure of the MNC-1/6, which weakens the aggregation effect of Pt nanoparticles during the electrocatalytic processes, thus enhancing the electrocatalytic stability of catalyst.
 | ||
| Fig. 6 Electrochemical performance of Pt/CNx and Pt/MNC-1/6 catalysts: cyclic voltammograms in acidic medium (a) and in methanol acidic medium (b), Nyquist plots (c) and amperometric i–t curves in methanol acidic medium (d). | ||
4. Conclusions
In summary, mesoporous nitrogen-doped carbon (MNC) could be prepared via carbonizing polyaniline using silica nanoparticles as template. The physical and electrochemical properties of Pt/MNC catalysts are closely related to the amount of silica nanoparticles. With the increase of the amount of silica nanoparticles, the electrocatalytic activity of Pt/MNC catalysts increase initially and then decrease, while the electrocatalytic stability of Pt/MNC catalysts gradually increase. Considering comprehensively electrocatalytic activity and stability toward MOR, MNC-1/6 can be considered as an appropriate support for Pt catalyst. Moreover, the peak current density of Pt/MNC-1/6 catalyst is 0.6 A mg−1Pt, which is 1.5 times higher than 0.4 A mg−1Pt of Pt/CNx catalyst. The retention rate of Pt/MNC-1/6 catalyst after 3600 s is 52.5%, which is much higher than 25.7% of Pt/CNx catalyst. Under the same electrocatalytic activity, the Pt loading of Pt/MNC-1/6 catalyst is reduced by 33.3% comparing with Pt/CNx catalyst. The excellent electrocatalytic activity arises from introducing mesoporous structures, which enhance surface area and pore volume of the support, further disperses Pt nanoparticles and facilitates the mass transfer. In addition, the Ostwald ripening behavior of Pt nanoparticles is limited in the mesoporous structure of the MNC-1/6, which weakens the aggregation effect of Pt nanoparticles during the electrocatalytic processes, thus enhancing the electrocatalytic stability of catalyst. In view of this, MNC-1/6 is expected to be applied as functional materials in energy conversion and storage and chemical catalysis.Acknowledgements
We acknowledge the National Natural Science Foundation of China (Grant No. 21273058), China postdoctoral science foundation (Grant No. 2012M520731 and 2014T70350), Heilongjiang postdoctoral financial assistance (LBH-Z12089) for their financial support.Notes and references
- J. N. Tiwari, R. N. Tiwari, G. Singh and K. S. Kim, Nano Energy, 2013, 2, 553–578 CrossRef CAS
.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. P. Liu, J. H. Liao, T. H. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 CAS
- H. Shi, Y. F. Shen, F. He, Y. Li, A. R. Liu, S. Q. Liu and Y. J. Zhang, J. Mater. Chem. A, 2014, 2, 15704–15716 CAS
- Z. H. Wen, J. Liu and J. H. Li, Adv. Mater., 2008, 20, 743–747 CrossRef CAS
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 CAS
- H. J. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 CAS
- C. Galeano, J. C. Meier, M. Soorholtz, H. Bongard, C. Baldizzone, K. J. J. Mayrhofer and F. Schüth, ACS Catal., 2014, 4, 3856–3868 CrossRef CAS
- W. M. Zhang, J. Chen, G. F. Swiegers, Z. F. Ma and G. G. Wallace, Nanoscale, 2010, 2, 282–286 RSC
- L. Zhao, Z. B. Wang, X. L. Sui and G. P. Yin, J. Power Sources, 2014, 245, 637–643 CrossRef CAS
- A. B. A. A. Nassr, I. Sinev, M.-M. Pohl, W. Grünert and M. Bron, ACS Catal., 2014, 4, 2449–2462 CrossRef CAS
- Y. Chen, J. Yang, Y. Yang, Z. Y. Peng, J. H. Li, T. Mei, J. Y. Wang, M. Hao, Y. L. Chen, W. L. Xiong, L. Zhang and X. B. Wang, Chem. Commun., 2015, 51, 10490–10493 RSC
- Y. Noh, Y. Kim, S. Lee, E. J. Lim, J. G. Kim, S. M. Choi, M. H. Seo and W. B. Kim, Nanoscale, 2015, 7, 9438–9442 RSC
- M. Z. Li, Y. X. Pan, X. Y. Guo, Y. H. Liang, Y. P. Wu, Y. Wen and H. F. Yang, J. Mater. Chem. A, 2015, 3, 10353–10359 CAS
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed
- W. J. Lee, U. N. Maiti, J. M. Lee, J. Lim, T. H. Han and S. O. Kim, Chem. Commun., 2014, 50, 6818–6830 RSC
- J. Zhang, Q. D. Li, H. Wu, C. Y. Zhang, K. Cheng, H. Zhou, M. Pan and S. C. Mu, J. Mater. Chem. A, 2015, 3, 10851–10857 CAS
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed
- Y. W. Ma, S. J. Jiang, G. Q. Jian, H. S. Tao, L. Yu, X. B. Wang, X. Z. Wang, J. M. Zhu, Z. Hu and Y. Chen, Energy Environ. Sci., 2009, 2, 224–229 CAS
- X. X. Yuan, X. L. Ding, C. Y. Wang and Z. F. Ma, Energy Environ. Sci., 2013, 6, 1105–1124 CAS
- C. A. Cao, X. D. Zhuang, Y. Z. Su, Y. Zhang, F. Zhang, D. Q. Wu and X. L. Feng, Polym. Chem., 2014, 5, 2057–2064 RSC
- Y. Y. Shao, J. H. Sui, G. P. Yin and Y. Z. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed
- S. L. Zhang, L. Chen, S. X. Zhou, D. Y. Zhao and L. M. Wu, Chem. Mater., 2010, 22, 3433–3440 CrossRef CAS
- Y. H. Su, H. L. Jiang, Y. H. Zhu, W. J. Zou, X. L. Yang, J. D. Chen and C. Z. Li, J. Power Sources, 2014, 265, 246–253 CrossRef CAS
- A. G. Kong, X. F. Zhu, Z. Han, Y. Y. Yu, Y. B. Zhang, B. Dong and Y. K. Shan, ACS Catal., 2014, 4, 1793–1800 CrossRef CAS
- S. Y. Gao, K. R. Geng, H. Y. Liu, X. J. Wei, M. Zhang, P. Wang and J. J. Wang, Energy Environ. Sci., 2015, 8, 221–229 CAS
- C. W. Zhang, L. B. Xu, N. N. Shan, T. T. Sun, J. F. Chen and Y. S. Yan, ACS Catal., 2014, 4, 1926–1930 CrossRef CAS
- F. B. Su, Z. Q. Tian, C. K. Poh, Z. Wang, S. H. Lim, Z. L. Liu and J. Y. Lin, Chem. Mater., 2010, 22, 832–839 CrossRef CAS
- B. B. Chang, Y. Z. Guo, Y. C. Li, H. Yin, S. R. Zhang, B. C. Yang and X. P. Dong, J. Mater. Chem. A, 2015, 3, 9565–9577 CAS
- Z. Li and L. Yin, Nanoscale, 2015, 7, 9597–9606 RSC
- J. Li, Q. L. Zhu and Q. Xu, Chem. Commun., 2015, 51, 10827–10830 RSC
- F. B. Su, C. K. Poh, Z. Q. Tian, G. W. Xu, G. Y. Koh, Z. Wang, Z. L. Liu and J. Y. Lin, Energy Fuels, 2010, 24, 3727–3732 CrossRef CAS
- F. B. Su, C. K. Poh, J. H. Zeng, Z. Y. Zhong, Z. L. Liu and J. Y. Lin, J. Power Sources, 2012, 205, 136–144 CrossRef CAS
- H. W. Liang, W. Wei, Z. S. Wu, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed
- L. M. Zhang, Z. B. Wang, J. J. Zhang, X. L. Sui, L. Zhao and D. M. Gu, Carbon, 2015, 93, 1050–1058 CrossRef CAS
- X. L. Sui, Z. B. Wang, C. Z. Li, J. J. Zhang, L. Zhao, D. M. Gu and S. Gu, J. Mater. Chem. A, 2014, 3, 840–846 Search PubMed
- L. Zhao, Z. B. Wang, J. Liu, J. J. Zhang, X. L. Sui, L. M. Zhang and D. M. Gu, J. Power Sources, 2015, 279, 210–217 CrossRef CAS
- J. Zhang, D. P. He, H. Su, X. Chen, M. Pan and S. C. Mu, J. Mater. Chem. A, 2014, 2, 1242–1246 CAS
- H. L. Peng, Z. Y. Mo, S. J. Liao, H. G. Liang, L. J. Yang, F. Luo, H. Y. Song, Y. L. Zhong and B. Q. Zhang, Sci. Rep., 2013, 3, 1765 Search PubMed
- S. C. Roy, A. W. Harding, A. E. Russell and K. M. Thomas, J. Electrochem. Soc., 1997, 144, 2323–2328 CrossRef CAS
- B. Yue, Y. W. Ma, H. S. Tao, L. S. Yu, G. Q. Jian, X. Z. Wang, X. S. Wang, Y. N. Lu and Z. Hu, J. Mater. Chem., 2008, 18, 1747–1750 RSC
- A. S. Aricò, A. K. Shukla, H. Kim, S. Park, M. Min and V. Antonucci, Appl. Surf. Sci., 2001, 172, 33–40 CrossRef
- Z. Z. Jiang, Z. B. Wang, D. M. Gu and E. S. Smotkin, Chem. Commun., 2010, 46, 6998–7000 RSC
- D. P. He, Y. L. Jiang, H. F. Lv, M. Pan and S. C. Mu, Appl. Catal., B, 2013, 132–133, 379–388 CrossRef CAS
- P. Kannan, T. Maiyalagan, N. G. Sahoo and M. Opallo, J. Mater. Chem. B, 2013, 1, 4655–4666 RSC
- T. T. P. Cheung, Surf. Sci., 1984, 140, 151–164 CrossRef CAS
- Z. B. Wang, G. P. Yin and P. F. Shi, J. Electrochem. Soc., 2005, 152, A2406–A2412 CrossRef CAS
- K. Bergamaski, A. L. N. Pinheiro, E. Teixeira-Neto and F. C. Nart, J. Phys. Chem. B, 2006, 110, 19271–19279 CrossRef CAS PubMed
- L. Gan, H. D. Du, B. H. Li and F. Y. Kang, New Carbon Mater., 2010, 25, 53–59 CrossRef CAS
- Y. H. Lin, X. L. Cui, C. H. Yen and C. M. Wai, Langmuir, 2005, 21, 11474–11479 CrossRef CAS PubMed
- B. Y. Xia, Y. Yan, X. Wang and X. W. Lou, Mater. Horiz., 2014, 1, 379–399 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06104d |
| This journal is © The Royal Society of Chemistry 2016 |