
Nanocrystalline alumina-supported ceria–praseodymia solid solutions: structural characteristics and catalytic CO oxidation†
Damma Devaiahab,
Gode Thrimurthulua,
Panagiotis G. Smirniotisb and
Benjaram M. Reddy*a
aInorganic and Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology, Uppal Road, Hyderabad-500 007, India. E-mail: bmreddy@iict.res.in; Fax: +91 40 2716 0921; Tel: +91 40 27193510
bChemical Engineering Program, Biomedical, Chemical, and Environmental Engineering, University of Cincinnati, Cincinnati, OH 45221-0012, USA
First published on 20th April 2016
Abstract
In this work, alumina supported ceria–praseodymia (CP/A) samples were synthesized by a deposition coprecipitation method. The structural, textural, and redox properties of the prepared samples were characterized at different calcination temperatures from 773 to 1073 K and their catalytic activity was assessed in the CO oxidation reaction. In order to determine the promoting effect of the alumina support in the sample, the physicochemical and catalytic properties of CP/A were compared with unsupported ceria–praseodymia (CP) solid solutions. The X-ray diffraction results indicated the formation of ceria–praseodymia solid solutions over the alumina support. The nanocrystalline nature of the samples was confirmed by transmission electron microscopy. The CP/A sample showed an extremely high surface area which remained reasonably high even after calcination at 1073 K. The combined analyses revealed that the CP/A sample had more oxygen vacancies than CP. The H2-temperature programmed reduction results suggested that the active oxygens were significantly improved in CP/A over CP. The characterization results also highlighted the excellent thermal stability of CP/A. The CO oxidation profiles signified that the catalytic activity of CP/A calcined at 773 K was remarkably enhanced in comparison to that of CP. The fine dispersion of ceria–praseodymia solid solutions over the alumina support in the process of deposition coprecipitation and the synergistic effect between ceria–praseodymia and the support, which resulted in very high surface areas, oxygen vacancy concentrations, and active oxygen species, are believed to be responsible for the superior activity of the CP/A sample.
1. Introduction
Air pollution has become a vital issue around the world; carbon monoxide (CO) emitted from the transport, energy production, agriculture, chemical, and steel industries is particularly harmful to human and animal life. The inhalation of CO even at ppm levels causes serious health problems, including respiratory infections, shortness of breath, dizziness, cardiovascular events, and death.1–3 In addition, CO distributes easily in air and poses severe risks to our environment due to the formation of ground-level ozone and the enhancement of greenhouse gases by the stabilization of CH4 in the atmosphere.4 As a result, the mitigation of CO emission is currently mandatory in order to improve the quality of human life and of the environment. To date, various technologies have been investigated to achieve CO elimination; among these, the catalytic oxidation of CO is still the most effective method.5–7 Therefore, global concerns regarding harmful emissions such as CO have spurred the development of suitable catalyst materials.As is well-known, nanostructured ceria (CeO2) has proved to be highly efficient for CO oxidation and many other important catalytic reactions based on its distinctive oxygen vacancies and redox properties.8–10 In general, it is considered to be an essential catalyst support in environmental catalysis.11,12 Thus, the development of CeO2-based catalysts is strongly desirable not only for the reduction of pollutants, but also for other industrial applications. Tremendous efforts have been made to improve the catalytic performance of ceria by doping it with other metal cations.13–16
Especially, doped ceria with multivalent cations is attractive because of its enhanced catalytic properties.17,18 Among the various multivalent elements, praseodymium (Pr) with 3+/4+ states is expected to be the most appropriate for dissolution into the ceria matrix to form a Ce–Pr oxide solid solution, due to its analogous fluorite structural nature (Pr6O11) and because its ionic radius is close to that of Ce4+ ions.18–21 In addition, the presence of Pr in CeO2 is found to be propitious to enhance the formation of oxygen vacancies, resulting in excellent oxygen release ability (redox properties) due to its valence changeability from 3+ to 4+ or from 4+ to 3+. These admirable properties can strengthen CO adsorption on the surfaces and thus significantly activate the CO oxidation reaction from active centers around the vacancies in the Ce–Pr oxide solid solution.21–23 However, noticeable deactivation due to aggregation of these Ce–Pr oxide catalysts is inevitable during the reaction.21
Support materials with high surface areas have proved to be highly beneficial for improving the durability of catalysts, since the high specific surface area of the supports could contribute to the fine dispersion of the active components to avoid sintering.24,25 Among the various supports, alumina is considered to be outstanding, especially for CO oxidation, due to its ideal cost-efficiency, high and thermally stable surface area, relative inertness towards steam, and adhesive properties.26,27 In particular, γ-Al2O3 support has been extensively used to improve the thermal stability of ceria-based materials for three-way catalysis.13,28 Monte et al.29 demonstrated a strong correlation between the γ-Al2O3 support and the oxygen storage capacity (OSC) and thermal stability of supported nanostructured CemZr1−mO2 mixed oxides. Our group also previously reported superior OSC and CO oxidation activity over γ-Al2O3 supported CeO2-based oxides in comparison with unsupported oxides.30,31 Morikawa et al.32 found that the introduction of Al2O3 into Ce1−xZrxO2 primary particles can improve durability at high temperatures and the oxygen release rate of the materials. From these studies, it is clear that the presence of the Al2O3 support is very advantageous for overcoming the potential sintering of ceria-based catalysts during the reaction.
Accordingly, the present investigation aimed at preparing γ-alumina supported ceria–praseodymia (CP/A) solid solutions by a deposition coprecipitation method. The structural, textural, and redox properties of the prepared samples were determined by a combination of several characterization techniques, such as X-ray diffraction (XRD), inductively coupled plasma-optical emission spectroscopy (ICP-OES), transmission electron microscopy (TEM), Raman spectroscopy (RS), ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS), H2-temperature programmed reduction (H2-TPR), BET surface area (BET SA), and X-ray photoelectron spectroscopy (XPS). The obtained results allowed us to study the effect of alumina addition as a support on the physicochemical properties of ceria–praseodymia; this was correlated with the catalytic activity of CP/A and was evaluated in the CO oxidation reaction.
2. Experimental
2.1 Catalyst preparation
The optimized alumina supported ceria–praseodymia solid solutions (CP/A, Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CeO2:Pr6O11 = 10:8:2 mole ratio based on oxides) were synthesized via a deposition coprecipitation method using (NH4)2Ce(NO3)6 (Loba Chemie, GR grade), Pr(NO3)3·6H2O (Aldrich, AR grade), γ-Al2O3 (Harshaw, specific surface area 127 m2 g−1) as precursors. In a typical procedure, first, the required amounts of cerium and praseodymium precursors were dissolved in deionized water. The supporting oxide (γ-Al2O3) was dispersed separately in deionized water and subsequently mixed with the above Ce–Pr precursor solution under stirring. The mixture was further diluted with deionized water and maintained under stirring conditions for another 1 h to give a clear solution. Then, the aqueous NH3 solution was slowly added dropwise to the mixed solution under vigorous stirring until the pH reached ∼8.5. The obtained precipitates were filtered and washed with deionized water to eliminate impurities. In order to remove water and any residual precursors from the precipitation step, the resulting precipitate was oven dried at 393 K for 12 h. Finally, the obtained catalyst was calcined at 773 K for 5 h in air atmosphere and denoted as CP/A 773. To test the thermal stability of the catalyst, portions of the 773 K calcined sample were further treated at 873, 973, and 1073 K for 5 h in air atmosphere and were designated as CP/A 873, CP/A 973, and CP/A 1073, respectively.
:CeO2:Pr6O11 = 10:8:2 mole ratio based on oxides) were synthesized via a deposition coprecipitation method using (NH4)2Ce(NO3)6 (Loba Chemie, GR grade), Pr(NO3)3·6H2O (Aldrich, AR grade), γ-Al2O3 (Harshaw, specific surface area 127 m2 g−1) as precursors. In a typical procedure, first, the required amounts of cerium and praseodymium precursors were dissolved in deionized water. The supporting oxide (γ-Al2O3) was dispersed separately in deionized water and subsequently mixed with the above Ce–Pr precursor solution under stirring. The mixture was further diluted with deionized water and maintained under stirring conditions for another 1 h to give a clear solution. Then, the aqueous NH3 solution was slowly added dropwise to the mixed solution under vigorous stirring until the pH reached ∼8.5. The obtained precipitates were filtered and washed with deionized water to eliminate impurities. In order to remove water and any residual precursors from the precipitation step, the resulting precipitate was oven dried at 393 K for 12 h. Finally, the obtained catalyst was calcined at 773 K for 5 h in air atmosphere and denoted as CP/A 773. To test the thermal stability of the catalyst, portions of the 773 K calcined sample were further treated at 873, 973, and 1073 K for 5 h in air atmosphere and were designated as CP/A 873, CP/A 973, and CP/A 1073, respectively.
2.2 Catalyst characterization
The X-ray diffraction measurements were performed on a Rigaku Multiflex diffractometer equipped with a nickel-filtered Cu-Kα (1.5418 Å) radiation source and a Scintillation counter detector. The diffraction patterns were recorded over a 2θ range of 10 to 80° with a step size of 0.02° using a counting time of 1 s per point. The XRD phases present in the samples were identified with the help of the Powder Diffraction File from the International Centre for Diffraction Data (PDF-ICDD). The mean crystallite size (D) was measured by applying the Scherrer equation. The lattice parameter was calculated by a standard cubic indexation method with the intensity of the most prominent (111) peak using the relation of a = d(h2 + k2 + l2)1/2, where ‘a’ is the lattice parameter and ‘d’ is the interplanar spacing calculated from the Bragg equation.The chemical analysis of the prepared samples was performed by inductively coupled plasma-optical emission spectroscopy (Thermo Jarrel Ash model IRIS Intrepid II XDL, USA) to confirm the respective concentrations of the elements in the system. For ICP analysis, approximately 50 mg of the sample was dissolved in a solution of 25 mL aqua regia and 475 mL distilled water. Then 10 mL of the above solution was diluted to 250 mL.
Transmission electron microscopy studies were carried out on a JEM-2100 (JEOL) instrument equipped with a slow-scan CCD camera at an accelerating voltage of 200 kV. Samples for TEM analysis were prepared by crushing the materials in an agate mortar and dispersing them ultrasonically in ethyl alcohol. Afterward, a drop of the dilute suspension was placed on a perforated-carbon-coated copper grid and allowed to dry by evaporation at ambient temperature.
The Raman spectra were obtained at room temperature using a LabRam HR800UV Raman spectrometer (Horiba Jobin-Yvon) fitted with a confocal microscope and a liquid-nitrogen cooled charge-coupled device (CCD) detector. The samples were excited either with the emission line at 325 nm from a He–Cd laser (Melles Griot Laser) or with the emission line at 632 nm from an Ar+ ion laser (Spectra Physics) which was focused on the sample under the microscope, with the diameter of the analyzed spot being ∼1 μm. The acquisition time was adjusted according to the intensity of the Raman scattering. The wavenumber values obtained from the spectra were precise to within 2 cm−1. The UV-vis DRS measurements were performed using a GBSCintra 10e UV-vis NIR spectrophotometer with an integration sphere diffuse reflectance attachment. BaSO4 was used as the reference, and the spectra were recorded in the range of 200 to 800 nm.
The reducibility of the catalysts was studied by H2-TPR analysis using a thermal conductivity detector with a gas chromatograph (Shimadzu). Prior to the reduction, approximately 30 mg of the sample was loaded in an isothermal zone of the reactor, pre-treated in a helium gas flow at 473 K and then cooled to room temperature. Then, the sample was heated at a rate of 10 K min−1 from ambient temperature to 1100 K in a 20 mL min−1 flow of 5% H2 in Ar. The hydrogen consumption during the reduction process was estimated by passing the effluent gas through a molecular sieve trap to remove the produced water; it was analyzed by gas chromatography using a thermal conductivity detector. The BET surface areas were determined by N2 physisorption at liquid N2 temperature on a Micromeritics Gemini 2360 instrument using a thermal conductivity detector (TCD). Prior to analysis, the samples were degassed at 393 K for 2 h to remove the surface adsorbed residual moisture.
The XPS measurements were performed on a Shimadzu ESCA 3400 spectrometer using Mg-Kα (1253.6 eV) radiation as the excitation source at room temperature. The samples were maintained in a vacuum, typically on the order of less than 10−8 Pa, to avoid a large amount of noise in the spectra from contaminants. The obtained binding energies were corrected by referencing the spectra to the carbon (C 1s) peak at 284.6 eV.
2.3 Catalytic activity studies
The catalytic performance of the samples for CO oxidation was evaluated at atmospheric pressure in the temperature range of 300 to 800 K. About 0.3 g catalyst samples (1 mm mesh size) were supported between glass wool plugs and flanked by inert porcelain beads at the centre of a specially designed quartz reactor. The catalysts were activated at 573 K for 1 h prior to the reaction studies. The reactant gases (1% CO, 5% O2, balanced with 94% N2) passed through the reactor at a space velocity of 30000 h−1. The CO and CO2 in the outlet gas were separated using a “Porapack Q” packed column and were finally detected separately by online GC-TCD.
3. Results and discussion
3.1 X-ray diffraction
The powder XRD patterns of CP/A calcined at 773, 873, 973, and 1073 K are shown in Fig. 1. For comparison purposes, we have also included the XRD patterns of unsupported ceria–praseodymia calcined at 773 (CP 773) and 1073 K (CP 1073), CeO2 calcined at 773 K, Pr6O11 (Aldrich Chemicals), and γ-Al2O3 (Harshaw) samples in Fig. 1. XRD peaks obtained for CP/A and CP samples evidenced the presence of characteristic (111), (200), (220), (311), (222), (400), (331) and (420) crystal faces only, corresponding to the cubic fluorite-structure of CeO2. Additionally, as can be seen from the inset of Fig. 1, the (111) reflection was slightly shifted to lower 2θ positions for the CP/A and CP samples when compared to pure CeO2. Surprisingly, no diffraction peaks related to γ-Al2O3 were observed over the entire CP/A patterns, indicating the X-ray amorphous state of Al2O3. These results clearly suggest the formation of ceria–praseodymia solid solutions by the incorporation of praseodymium ions into the ceria lattice in the CP/A and CP samples. Also, no transition of Al2O3 from the γ- to α-phase and no peaks related to Pr6O11, CeAlO3, and PrAlO3 phases were observed for any of the CP/A samples, even after calcination at a higher temperature of 1073 K. The suppression of the formation of CeAlO3 and PrAlO3 phases and the phase transition of Al2O3 at 1073 K indicate the good thermal stability of the CP/A sample, which is due to the synergistic stabilization effect between ceria–praseodymia and the Al2O3 support. | ||
| Fig. 1 Powder X-ray diffraction patterns of ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at different temperatures along with pure ceria (CeO2) calcined at 773 K, praseodymia (Pr6O11), and alumina (Al2O3) (inset: expanded view of selected region). | ||
The lattice parameter (a) of the CP/A and CP 773 samples (see Table 1) is slightly larger than that of pure CeO2. The ionic radius of Pr4+ (0.096 nm) is slightly smaller than the Ce4+ radius (0.097 nm); therefore, if Ce4+ is replaced by Pr4+, no significant change in the lattice parameter is expected. The presence of Pr3+ ions in the ceria lattice will cause a lattice expansion because of the larger Pr3+ ionic radius (0.113 nm). The lattice expansion is also ascribed to the increased presence of oxygen vacancies conjugated with Ce3+ ions in the samples. Therefore, it can be concluded that the lattice expansion in the CP/A and CP 773 samples is due to the substitution of the parent Ce4+ cations by larger Pr3+ ions and the increased content of Ce3+ ions in the samples, which is consistent with the (111) peak shift to lower 2θ angles.21,33,34 Moreover, it is interesting to note that the lattice parameter of CP/A 773 is higher than that of the CP 773 sample, signifying the presence of more 3+ (Ce3+ and Pr3+) cations in the sample. Although the lattice parameter of the CP/A sample decreased with increasing calcination temperature, CP/A 1073 has a greater “a” value than CP 1073 and pure CeO2, demonstrating that a comparatively high number of 3+ ions are present in CP/A at 773 K and that they are relatively stable up to 1073 K. Therefore, it can be deduced that the alumina support enhances the number and thermal stability of 3+ ions in the CP/A sample, which is attributed to the synergistic interaction between ceria–praseodymia and the alumina support. These 3+ cations can play a crucial role in catalytic reactions by creating oxygen vacancies. On the other hand, the lattice parameter of the CP 1073 sample is equal to the pure ceria value, which may be due to the inferior interaction between CeO2 and Pr6O11 in CP at a higher calcination temperature.
| Sample | SA (m2 g−1) | Da (nm) | aa (Å) | [Ce3+]/[Ce3+ + Ce4+]b | O 1s binding energyc (eV) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OL | OH | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Calculated from XRD analysis.b The surface relative molar ratio of [Ce3+]/[Ce3+ + Ce4+] calculated from the Ce 3d XPS spectra.c The values represent the O 1s binding energy obtained from the O 1s XPS spectra. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CeO2 | 41 | 7.7 | 5.415 | 0.25 | 530.3 | 532.4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CP 773 | 72 | 7.1 | 5.430 | 0.43 | 530.6 | 533.0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CP 1073 | 31 | 16.3 | 5.415 | 0.32 | 530.7 | 532.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CP/A 773 | 156 | 5.4 | 5.437 | 0.47 | 530.6 | 533.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CP/A 873 | 138 | 5.7 | 5.429 | 0.45 | 530.6 | 533.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CP/A 973 | 120 | 6.6 | 5.425 | 0.43 | 530.5 | 533.0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CP/A 1073 | 107 | 8.1 | 5.420 | 0.38 | 530.5 | 532.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The diffraction peaks of the CP/A samples are broader than that of CP, indicating the formation of smaller CeO2 crystallites in the CP/A sample. The crystallite size was calculated from the peak broadening of the (111) diffraction peak using the Debye–Scherrer equation. The average crystallite sizes of CP/A 773 and CP 773 were estimated to be 5.4 and 7.1 nm, respectively. The lower crystallite size of CP/A 773 was likely to be caused by the good dispersion of ceria–praseodymia solid solution over the support. With increasing calcination temperature from 773 to 1073 K, the crystallite size of CP was increased by 2.3 times, while it is enhanced by only 1.5 times in the case of the CP/A sample. This result again clearly indicated that the Al2O3 support serves as the thermal diffusion barrier and effectively inhibits the growth of ceria crystallites in CP/A during calcination, resulting in the lower CeO2 crystallite size for CP/A 1073 compared to CP 1073. These results are in line with the BET surface area values shown in Table 1. The molar ratios of Ce, Pr, and Al were determined by elemental analysis and were found to be very close to the nominal values for all CP/A samples (Table S1, ESI†), indicating the complete precipitation of Ce and Pr cations over the support during the preparation process.
3.2 Transmission electron microscopy
The TEM and HREM images of the CP/A and CP samples are shown in Fig. 2 and 3, respectively; these demonstrate the particle morphology, size, and crystal structures. The obtained TEM images primarily exhibited agglomerated microstructures with irregular shapes and well-defined octahedral crystal facets at both the 773 and 1073 K calcination temperatures. These shapes (encircled areas of Fig. 2) are clearly visible for the high temperature calcined samples, while for the low temperature calcined samples, this visibility is very poor. This observation indicates that the calcination temperature significantly affects the morphology of the samples. Interestingly, the octahedral facets can be clearly observed from the HREM micrographs (enlarged views of Fig. 3) at both calcination temperatures. However, the Al2O3 particles cannot be distinguished from ceria even in the HREM images, most likely due to their low atomic weights and poor contrast.35 As can be seen from the HREM images, the particles of the samples are composed of small crystallites. In addition, these crystals are randomly oriented towards each other with a relatively narrow size range; the average crystallite sizes of CP/A 773 and CP/A 1073 are about 5 and 8 nm, whereas they are about 7 and 16 nm for the CP 773 and CP 1073 samples, respectively. From these results, it can be noted that the average crystallite size of the CP/A samples is lower than that of CP at both calcination temperatures. Meanwhile, the growth of the crystallites with calcination temperature is larger for CP (i.e., from ∼7 to 16 nm) than for the CP/A samples (i.e., from ∼5 to 8 nm only). These consequences may be due to the fine dispersion of nanosized ceria–praseodymia solid solution over the alumina support and the synergistic interaction between the solid solution and support, which can inhibit the growth of ceria crystallite, thereby enhancing the thermal stability of the CP/A samples. These results are also in good agreement with the conclusions drawn from the XRD measurements. Based on the interplanar distance (d) of 0.31 nm shown in the HREM images, the octahedral facets of the samples preferentially expose the (111) crystal plane, which can be indexed to the cubic fluorite phase of CeO2.36 Moreover, the overlapping regions of the crystallites are responsible for the improved catalytic activity. | ||
| Fig. 2 TEM images of ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at 773 K and 1073 K (inset: encircled views of selected areas). | ||
 | ||
| Fig. 3 HREM images of ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at 773 K and 1073 K (inset: enlarged views of selected areas). | ||
3.3 Raman spectroscopy
Raman spectroscopy is a very useful technique for the direct characterization (without probing molecules) of catalysts; it can provide additional structural information regarding the phase, the oxygen environment around the host cations and the formation of oxygen vacancies. Literature reports have demonstrated that the Raman spectral features are quite different when UV and visible region excitation laser lines are used, due to the different detection depths of UV and visible excitation lasers. Therefore, in the present investigation, Raman spectra of the samples were acquired in the UV region (325 nm) as well as the visible region (632 nm). Generally, for solids, Raman scattering comes from both the surface and the bulk of the samples. However, the signal from the bulk is attenuated when the sample strongly absorbs the excitation laser and scattering light. Hence, more Raman scattering comes from the surface region than the bulk of the sample when the sample absorbs the excitation laser and scattering light. Accordingly, UV Raman spectroscopy is more sensitive to the surface region than the bulk of the sample due to the strong absorption at 325 nm, while visible Raman spectroscopy gives information from both the bulk and the surface of the sample because there is a weak absorption at 632 nm.37,38The UV and visible Raman spectra of the samples, presented in Fig. 4, show the presence of two characteristic peaks which arise solely from the cubic CeO2 fluorite phase. The most prominent feature in the spectra is the F2g mode characteristic of a cubic fluorite crystal structure, which is positioned around 466.2 and 465.5 cm−1 in the UV and visible Raman spectra of pure CeO2, respectively. Interestingly, the F2g mode in the CP/A and CP samples experiences a red shift and broadening (in both the UV and visible Raman spectra) in comparison with pure ceria. This is due to the lattice expansion with the increased concentration of 3+ (Pr3+ and Ce3+) ions in the samples. Therefore, the shift and broadening of the F2g mode in the samples confirms that Pr has been incorporated into the ceria lattice, forming ceria–praseodymia solid solutions. In addition, the broader F2g peaks in CP/A and CP suggest a lower CeO2 crystallite size in the samples compared to pure ceria. These results are in agreement with the XRD data. In addition to the F2g peak, the other feature at a higher wave number (∼568 to 600 cm−1) is usually ascribed to oxygen vacancies (OV) in the samples. According to the literature, this mode originates from the existence of both intrinsic and extrinsic oxygen vacancies in the samples.19 The intrinsic oxygen vacancies are created in the samples due to the presence of Ce3+ ions, whereas the extrinsic oxygen vacancies are introduced into the ceria in order to maintain charge neutrality when Ce4+ ions are replaced by Pr3+ ions. For every two substituted Pr3+ ions, one O2− leaves the crystal lattice.
 | ||
| Fig. 4 (A) UV (B) visible Raman spectra of ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at different temperatures along with pure ceria (CeO2) calcined at 773 K. | ||
For all the samples, the UV Raman spectral features are quite different from those in the visible Raman spectra in terms of the relative band intensities, which results from the absorption properties of the samples. The F2g band is weak in the UV Raman spectrum due to the strong absorption of the samples at 325 nm, while it is strong in the visible Raman spectra owing to the weak absorption at 632 nm. Considering the characteristic band of the oxygen vacancy (OV), it is strong in the UV Raman spectra but weak in the visible Raman spectra. Evidently, UV Raman spectroscopy is more sensitive to oxygen vacancies present in the samples than visible Raman spectroscopy. This is due to the resonance Raman effect,38,39 since the samples strongly absorb in the UV region. The ratio of the integrated peak area of the oxygen vacancies (AOV) to that of the main peak (AF2g), defined as AOV/AF2g, is used here to characterize the relative amount of oxygen vacancies among the samples.37,38,40 Fig. 5 illustrates the AOV/AF2g values of the samples in the UV and visible Raman spectra. As can be observed in this figure, the AOV/AF2g value is much higher in the UV region than that in the visible region in all samples, which is due to the fact that the vacancies are enriched on the surface of the samples.38,41 In both the spectra, the AOV/AF2g value follows the sequence: CP/A 773 > CP 773 > CP/A 873 > CP/A 973 > CP/A 1073 > CP 1073 > CeO2, suggesting that CP/A and CP samples have higher oxygen vacancies than pure ceria. This could be due to the enhanced 3+ ions in the samples by the synergistic interaction between ceria and praseodymia. Interestingly, CP/A 773 possesses a greater number of oxygen vacancies in comparison to CP 773. It is clear that the 3+ ions were further augmented (as evidenced from the XRD results); therefore, there are larger oxygen vacancies in CP/A 773 than in CP 773, which is mainly attributed to the increased synergistic effect between CeO2 and Pr6O11 over the Al2O3 support. More oxygen vacancies may result in high mobility of activated oxygens, which are crucial to the catalytic performances of the samples. Moreover, it is found that the AOV/AF2g ratio decreases obviously in both the UV and visible Raman spectra for the CP/A and CP samples as the calcination temperature increases from 773 to 1073 K. This result implies that the relative concentration of oxygen vacancies decreases in the samples with increasing calcination temperature. Especially, the decreasing trend of the oxygen vacancies is more pronounced in the visible Raman spectra than in the UV Raman spectra during the calcination process, indicating that the vacancies in the surface region are more stable than in the bulk of the samples. However, the CP/A sample showed more vacancies than the CP sample at 1073 K in both the UV and visible Raman spectra. This observation confirms the enhanced thermal stability of the oxygen vacancies in CP/A, which is again due to the synergistic interaction between ceria–praseodymia and the alumina support.
 | ||
| Fig. 5 Relative quantity of oxygen vacancies (AOV/AF2g ratio) for ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at different temperatures along with pure ceria (CeO2) calcined at 773 K in UV and visible Raman spectra. | ||
3.4 UV-visible diffuse reflectance spectroscopy
To investigate the surface coordination and oxidation states of the metal ions, UV-visible diffuse reflectance spectroscopy was used. Fig. 6 shows the deconvoluted UV-vis DR spectra of the samples. In the spectra of all the samples, the bands centered at ∼252, ∼285, and ∼340 nm are related to the O2− → Ce3+ and O2− → Ce4+ charge transfers (CT) and the interband transitions (IBT), respectively.21 Apart from these three bands, one more strong absorption band at ∼520 nm is observed for the CP/A and CP samples. The presence of this band is associated with Pr3+ ion transitions in the samples.38 Hence, these findings confirm the presence of Ce3+, Ce4+, and Pr3+ ions in the CP/A and CP samples. Moreover, the absorption edge of the samples follows the order: CP/A 773 < CP 773 < CP/A 1073 < CP 1073 < CeO2. The lowest absorption edge of CP/A 773 could be ascribed to the fact that it has the highest amount of oxygen vacancies among the samples,42 in accordance with the Raman results. Additionally, the UV-vis DRS studies clearly indicate the formation of ceria–praseodymia solid solutions in the CP/A and CP samples by the absence of phases other than CeO2, in agreement with the XRD and Raman measurements.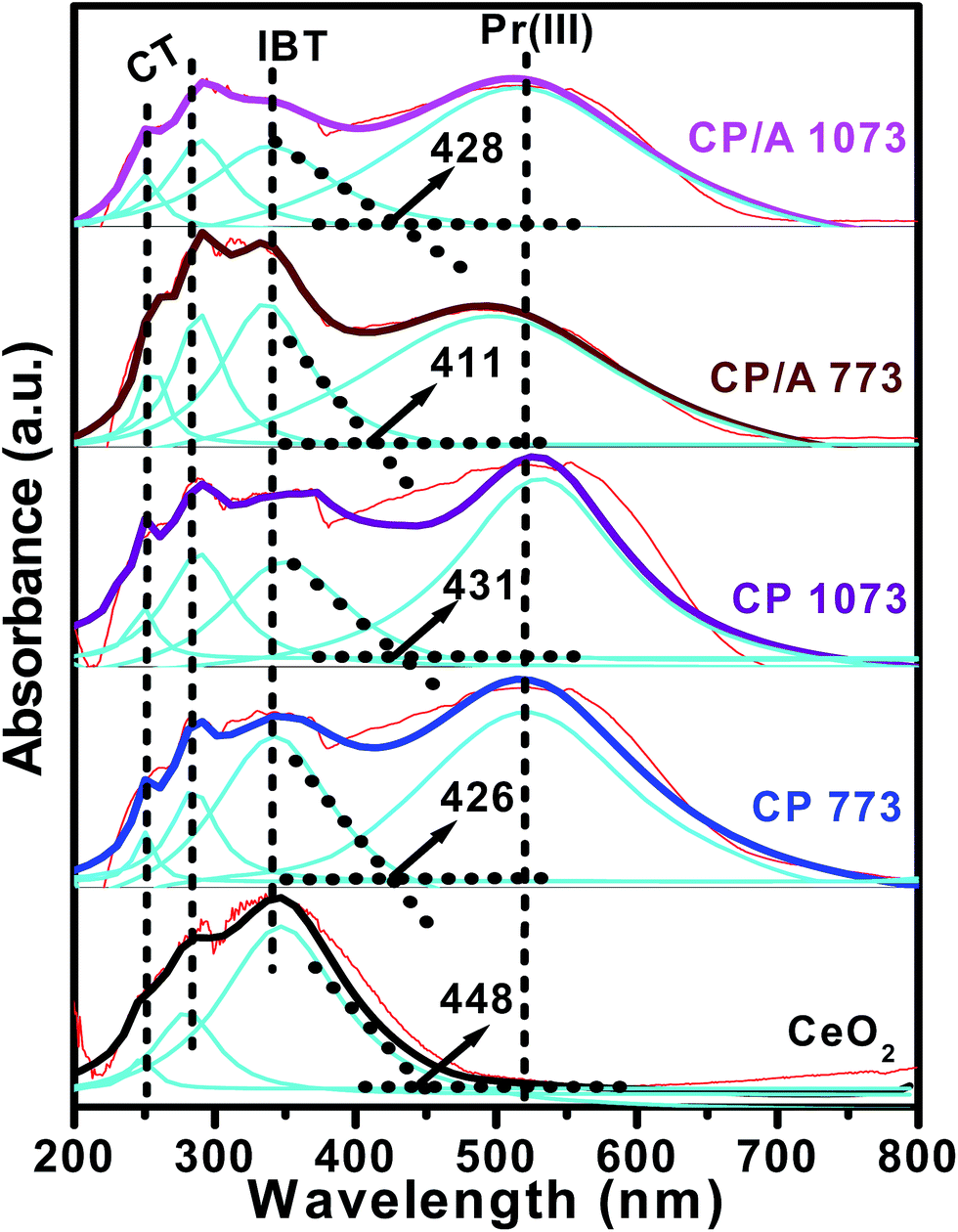 | ||
| Fig. 6 UV-vis DR spectra of ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at 773 and 1073 K along with pure ceria (CeO2) calcined at 773 K. | ||
3.5 H2-temperature programmed reduction
The reducibility of the samples is an important factor influencing their catalytic performance; this was probed using H2-TPR experiments. The TPR profiles of the samples are depicted in Fig. 7. Two characteristic reduction peaks can be discerned at 755 and 1005 K for pure CeO2. The low-temperature peak is assigned to a reduction of the surface oxygen species of ceria, which is generally considered as the active oxygen in the oxidation catalysis. The high-temperature peak is due to the reduction of the bulk oxygen in ceria, which has an insignificant contribution to oxidation reactions.43 Like pure CeO2, both the CP/A and CP samples show the surface and bulk reduction peaks. However, in the TPR profiles of the CP/A and CP samples, the two peaks were shifted to lower temperatures compared with bare CeO2. This result implies that the synergistic interaction between ceria and praseodymia with the formation of ceria–praseodymia solid solutions (as evidenced from XRD, Raman, and UV-vis DRS results) can promote the reduction of CeO2 at lower temperatures. The conceivable valence change of both Ce and Pr from 4+ to 3+ or from 3+ to 4+, since Pr4+ is reduced more easily than Ce4+, might be the cause for the synergistic interaction in the samples. Moreover, as the calcination temperature increased to 1073 K, the surface and bulk reduction of CP/A and CP shifted to higher temperatures, which may be due to the decrease in the synergetic interaction between the ceria and praseodymia with increasing calcination temperature. | ||
| Fig. 7 H2-TPR profiles of ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at 773 and 1073 K along with pure ceria (CeO2) calcined at 773 K. | ||
An important observation in the TPR patterns is that the surface reduction temperature of CP/A is significantly lower than that of the CP sample. As the support is hardly reducible in the explored temperature range, the H2 consumption in the CP/A samples is mainly attributed to the reduction of Ce4+ and Pr4+ ions. Hence, the reduced surface reduction temperature of CP/A should be attributed to the increased synergistic interaction between the Ce4+/Ce3+ and Pr4+/Pr3+ redox couples on the surface due to the high dispersion of ceria–praseodymia solid solutions over the alumina support, which is corroborated by the surface area data. Notably, the greatly decreased surface reduction temperature of CP/A 773 indicates that it has a higher number of active oxygen species compared with the other samples. This enhancement of the active oxygen species in the CP/A 773 sample is strongly related to its highest number of oxygen vacancies (confirmed from the Raman and UV-visible DR spectral results) among the samples, which could play a crucial role in catalytic oxidation reactions. Thus, it can be expected that the CP/A 773 sample will have high activity in oxidation catalysis. On the other hand, the CP/A sample has a higher bulk reduction temperature than the CP sample. The strong interaction between ceria–praseodymia and the alumina support in the CP/A sample could be responsible for this result.
3.6 BET surface area
The BET surface areas of the CP/A, CP, and CeO2 samples are compiled in Table 1. As seen from Table 1, the surface areas of the CP/A 773 and CP 773 samples are remarkably higher than that of pure CeO2, which illustrates that the formation of ceria–praseodymia solid solution in the CP/A and CP samples leads to an increase in their surface area. Meanwhile, it is worth noting that the CP/A 773 sample shows a much larger surface area (almost two times) than the CP 773 sample. This result implies that the surface area of CP/A 773 was enormously increased by the presence of Al2O3 support in the sample, which could be caused by the fine dispersion of ceria–praseodymia over the support and the synergistic interaction of ceria–praseodymia and alumina. It is obvious that a larger surface area leads to an increase in catalytic activity, due to sufficient contact with the reactant molecules by the presence of more adsorption sites. In addition, the surface area is found to decrease with increasing calcination temperature from 773 to 1073 K for the CP/A and CP samples, which is due to the sintering of the samples at higher temperature. The percentage of the decline in surface area from 773 to 1073 K for the CP/A sample is 31.4%, while it is 56.9% for the CP sample. This result demonstrates that the CP/A sample exhibited a reasonably high surface area, even at 1073 K. Therefore, it can be concluded that the Al2O3 support provides good thermal stability for the CP/A sample, possibly due to the synergistic interaction between ceria–praseodymia and the alumina support.443.7 X-ray photoelectron spectroscopy
XPS is an effective technique to investigate the surface chemical compositions, metal oxidation states, nature of oxygen, and adsorbed species of a solid sample. The Ce 3d photoelectron peaks of the samples are shown in Fig. 8(A). The Ce 3d spectrum is very complex in nature, owing to the dual oxidation states of Ce and the mixing of Ce 4f levels and O 2p states during the primary photoemission process. Consequently, this complexity is accompanied by the splitting of the spectral peaks into five spin–orbit doublets (or ten peaks), which are designated as v0–u0, v–u, v′–u′, v′′–u′′, and v′′′–u′′′. The characteristic lines u (u0, u, u′, u′′, and u′′′) and v (v0, v, v′, v′′, and v′′′) originate from the two sets of spin–orbital multiplets, corresponding to the 3d3/2 and 3d5/2 contributions, respectively. Among these, three doublets, v–u, v′′–u′′, and v′′′–u′′′, represent the 3d104f0 state related to Ce4+ ions, and the two other doublets, v0–u0 and v′–u′, represent the 3d104f1 state of Ce3+ ions.45,46 From Fig. 8(A), it can be observed that the CP/A, CP, and CeO2 samples exhibit contributions from all five doublets, indicating the coexistence of Ce3+ and Ce4+ species on the surface of the samples. Further, the surface Ce3+ ions are closely associated with oxygen vacancies because they create these oxygen vacancies in order to maintain electrostatic equilibrium in the samples. Namely, the high relative amount of surface Ce3+ ions indicated the high concentration of surface oxygen vacancies, which could enhance the catalytic oxidation activity of the samples.45,47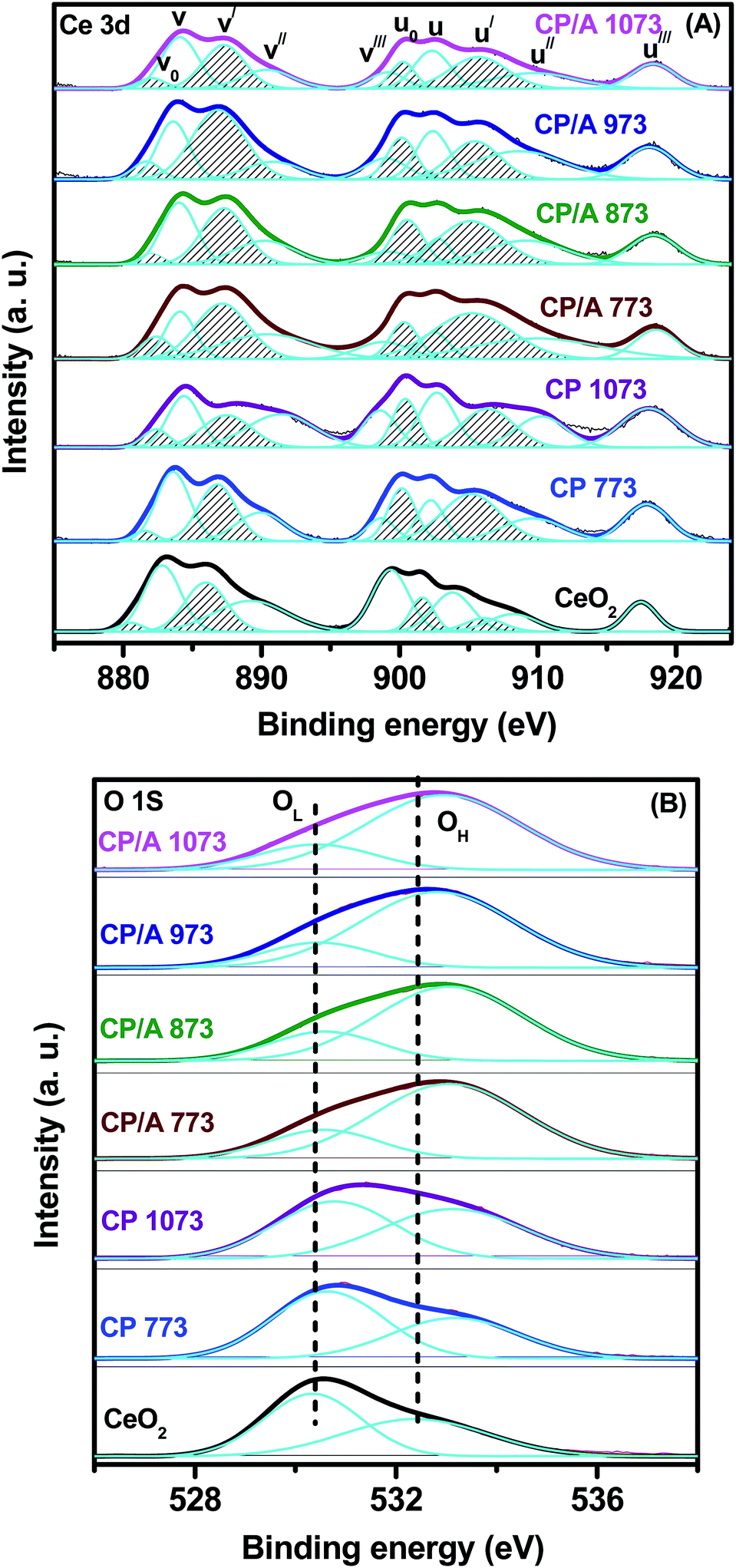 | ||
| Fig. 8 XPS spectra of (A) Ce 3d, (B) O 1s for ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at different temperatures, along with pure ceria (CeO2) calcined at 773 K. | ||
The relative amount of Ce3+ ions in the samples can be estimated by the [Ce3+]/[Ce3+ + Ce4+] ratio, and the obtained values are shown in Table 1. It can be clearly seen that the relative concentration of Ce3+ ions for the samples increased in the order CeO2 < CP 1073 < CP/A 1073 < CP/A 973 < CP/A 873 < CP 773 < CP/A 773. That is to say, the CP/A and CP samples have a higher relative amount of Ce3+ ions than CeO2. This observation demonstrates that the formation of ceria–praseodymia solid solution by the doping of Pr ions into the CeO2 lattice could facilitate the generation of more surface oxygen vacancies in the samples. Notably, the CP/A 773 sample is found to have the highest amount of Ce3+ ions on the surface; therefore, it has the most oxygen vacancies among the samples. This result suggests that not only the substitution of the CeO2 lattice by Pr ions but also the alumina support could further increase the quantity of the oxygen vacancies in the CP/A 773 sample due to its synergistic effect with ceria–praseodymia, which can change the electronic structure of the Ce ions and thus alter the coordination between the Ce and oxygen ligands.48,49 Additionally, with increasing calcination temperature, the content of Ce3+ ions, and therefore the oxygen vacancies, decreased in both the CP/A and CP samples. However, CP/A 1073 had a higher concentration of Ce3+ ions compared to CP 1073. Hence, it could be concluded that the thermal stability of CP/A is obviously improved by the presence of the Al2O3 support. These observations are in good agreement with our previous Raman, UV-vis DRS, and H2-TPR results.
The XPS spectra of the CP/A, CP, and CeO2 samples in the O 1s region are displayed in Fig. 8(B), and the corresponding binding energy values are demonstrated in Table 1. The O 1s spectra for all samples are asymmetric due to the non-equivalence of the surface oxygen ions, which causes the spectra to split into two peaks. This result clearly illustrates that the two kinds of oxygen species are present on the surface of all the samples. The peak labeled with OL at a lower binding energy (530.3 to 530.7 eV) is characteristic of the lattice oxygen (O2−) for metallic oxides in the samples, while the other sub-band labeled with OH at a high binding energy (532.4 to 533.2 eV) is attributed to the surface chemisorbed oxygen, such as O22− or O− attributed to oxide defects, surface oxygen ions with low coordination or surface hydroxyl and/or carbonate impurities.50,51 Meanwhile, it can be noted from Table 1 that the binding energies (OH peak) of CP/A and CP are slightly shifted to higher values compared with CeO2. The increase in the binding energy of the OH peak in the CP/A and CP samples is ascribed to their greater surface oxygen vacancy concentration compared to pure CeO2.52,53
Fig. 9(A) illustrates the Pr 3d core level XPS spectra of the CP/A and CP samples. Usually, the Pr 3d spectrum is characterized by two sets of 3d3/2 and 3d5/2 spin–orbit multiplets. However, the peaks corresponding to the 3d3/2 sublevel are very complex in nature due to the multiplet effect, whereas the 3d5/2 sublevel exhibits only two peaks, which are related to the possible 3+ and 4+ oxidation states of Pr.54,55 As a result, we studied only the 3d5/2 region of the Pr 3d spectrum to determine the oxidation state of Pr in the samples. The Pr 3d5/2 spectra of the samples show two prominent peaks at ∼930.7 and ∼935.3 eV, which correspond to Pr3+ and Pr4+ ions, respectively.23,55 This clearly indicates that the Pr ions display both 3+ and 4+ oxidation states in both the CP/A and CP samples at the surface region. Moreover, the intensity of the Pr4+ ion peak is greater than that of Pr3+, indicating the greater concentration of Pr4+ ions on the surface of all samples. However, the figure clearly demonstrates that the calcination temperature has no significant effect on the intensity of the Pr 3d5/2 spectrum in both the CP/A and CP samples. It is interesting to note that the binding energies of the CP/A samples are higher than that of CP, which is probably due to the interaction between ceria–praseodymia and the alumina support in the samples.
 | ||
| Fig. 9 XPS spectra of (A) Pr 3d5/2 for ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP). (B) Al 2p for ceria–praseodymia/alumina (CP/A) samples calcined at different temperatures. | ||
The Al 2p XPS spectra of the CP/A samples are shown in Fig. 9(B). The Al 2p peaks for all samples are observed to be centered at ∼76 eV, which is slightly higher than the values previously reported for Al2O3.56,57 This shift could be ascribed to the strong interaction between ceria–praseodymia and the alumina support, which is also supported by the XPS spectra of Ce 3d and Pr 3d5/2 as discussed earlier.58 In addition, the binding energy values of Al 2p do not vary with calcination temperature, suggesting the presence of one type of aluminium oxide (γ-alumina) with an oxidation state of 3+ at all calcination temperatures.
3.8 CO oxidation activity
In the blank experiment (only quartz particles were loaded in the reactor), we did not notice a significant conversion of CO below 1000 K. This result reveals that no homogeneous reactions took place below 1000 K. The catalytic performances of the CP/A samples for CO oxidation as a function of temperature are shown in Fig. 10 and for comparison; the activity results for CP and CeO2 samples are also reported. The catalytic activities of the samples were evaluated using the temperatures (T50 and T100) required for CO conversions of 50 and 100%, respectively. It can be clearly observed from Fig. 10 that the T50 of the samples follows the sequence: CP/A 773 > CP 773 > CP/A 1073 > CP 1073 > CeO2. In addition, it is found that nearly 100% conversion is observed over the CP/A 773, CP 773, and CP/A 1073 samples within 780 K; however, the CP 1073 and CeO2 samples exhibit only 92% and 88% conversion, respectively. As well, the T100 values of CP/A 773, CP 773, and CP/A 1073 are ∼704, ∼769, and ∼780 K, respectively. Interestingly, at the T100 of CP/A 773, the CP 773, CP/A 1073, CP 1073, and CeO2 samples showed only ∼73%, ∼58%, ∼47%, and ∼32% conversion, respectively. These results obviously demonstrated that the CP/A samples have better activity than the CP and pure CeO2 samples. Particularly, the CP/A 773 sample exhibited the highest activity among the samples. In order to determine the importance of the Al2O3 support in increasing the activity of the catalyst, the CO oxidation profile of the CP/A 773 sample was compared with that of the CP 773 sample. It is worth noting that the T50 and T100 of CP/A 773 were much lower, by 78 and 65 K, respectively, than those of the CP 773 sample. Evidently, this result suggests that the γ-Al2O3 support was beneficial for enhancing the catalytic activity of the CP/A 773 sample for CO oxidation. Additionally, the catalytic activity decreased in both the CP/A and CP samples with increasing calcination temperature from 773 to 1073 K. However, the T50 of the CP/A 1073 sample was ∼20 K lower than that of the CP 1073. This result reflects the promoting effect of the Al2O3 support in improving the thermal stability of the CP/A sample. It is clear that CP/A showed reasonably good thermal stability compared to CP, which could be attributed to the strong interaction between ceria–praseodymia and Al2O3 by the fine dispersion of ceria–praseodymia over the support. | ||
| Fig. 10 Catalytic activity of ceria–praseodymia/alumina (CP/A) and ceria–praseodymia (CP) samples calcined at 773 and 1073 K along with pure ceria (CeO2) calcined at 773 K for CO oxidation. | ||
The difference in the CO oxidation activity of the samples is better understood from the characterization results. Usually, the surface area of the catalyst plays an important role in the oxidation of CO; a larger surface area can expose more active sites for the oxidation reaction, which is advantageous to increase the catalytic activity.42 Obviously, the CP/A 773 sample with the highest surface area of 156 m2 g−1 demonstrated the best activity among the samples. However, the CP 1073 sample shows higher catalytic activity by 25 K, even though its surface area is smaller than that of pure CeO2. This finding indicates that the surface area does not solely determine the catalytic performance, but that additional factors such as oxygen vacancies and active oxygen species may play a vital role in the activity.15,59 Interestingly, the order of the AOV/AF2g (Raman results) values, absorption edges (UV-vis DRS), and relative quantities of Ce3+ ions (XPS spectra) in the samples exactly coincided with the order of the CO oxidation activity. This reflects that the oxygen vacancies play a major role in the CO oxidation reaction. Generally, the CO oxidation occurs by CO + [M − O]* → CO2 + M, where [M − O]* is an active oxygen species. The active oxygen site is notably favoured by the presence of oxygen vacancies that improve the surface dynamics.60 According to the H2-TPR results, the CP/A 773 sample had more active oxygen species than the other samples. Hence, it should be concluded that the CP/A 773 sample with highest number of oxygen vacancies, which in turn increases the number of active oxygen species, showed the most remarkable activity for CO oxidation among the investigated samples. Overall, the combined results illustrate that the surface area, oxygen vacancies, and active oxygen species are the key factors that influence the catalytic performance of the present samples.
Further, the catalytic activity of the present CP/A 773 sample was also compared with our earlier reports of Al2O3 supported samples. Surprisingly, CP/A 773 shows a lower T50 than Ce0.8Tb0.2O2/Al2O3 (CT/A),31 Ce0.8Hf0.2O2/Al2O3 (CH/A),31 Ce0.5Zr0.5O2/Al2O3 (CZ/A),31 and Ce0.8La0.2O2/Al2O3 (CL/A)48 samples calcined at 773 K. In particular, at the T50 of CP/A 773, the CT/A, CH/A, CL/A, and CZ/A samples exhibit only 31%, 27%, 23%, and 20% conversions, respectively (Table 2). Evidently, CP/A 773 showed the best performance for CO oxidation among the various alumina supported ceria-based mixed oxides.
To examine the stability of the CP/A 773 catalyst in the CO oxidation reaction, cycling tests were performed over the sample. Fig. S1 (ESI†) illustrates the T50 values over the CP/A 773 sample for five successive runs. It can be clearly observed that the T50 of the sample remained almost constant up to five cycles. This result indicates the good stability of the CP/A 773 sample for long-term CO oxidation reactions. Moreover, a UV Raman spectrum was also acquired for the CP/A 773 sample after the cycling test; this is shown in Fig. S2 (ESI†). Quite impressively, no significant loss of oxygen vacancies was observed over the spent CP/A 773 catalyst. This finding provides further evidence for the stability of the CP/A 773 sample. Therefore, it can be assumed that the present CP/A 773 sample is the most promising catalyst for CO oxidation and may be envisioned for use in various other catalyzed oxidation reactions; thus, its ultimate industrial applications are also promising.
4. Conclusions
In conclusion, ceria–praseodymia/alumina samples have been successfully prepared via a simple deposition coprecipitation method and calcined at different temperatures from 773 K to 1073 K. The calcined samples were characterized by means of different techniques and their catalytic activities were evaluated for CO oxidation. The ceria–praseodymia was found to be finely dispersed on the alumina support in the form of a nanocrystalline solid solution. The CP/A showed very high surface area, which remained reasonably good even after calcination at 1073 K. CP/A had remarkable oxygen vacancies in comparison to the unsupported CP, as confirmed by Raman, UV-vis DRS, and XPS techniques. The active oxygen species were higher in the CP/A sample than in CP. The CP/A sample also exhibited excellent thermal stability. These results obviously revealed the promoting effect of alumina as a support in the CP/A samples. The synergistic interaction between ceria–praseodymia and the alumina support was most likely the reason for the excellent properties of the CP/A samples. The CP/A samples exhibited superior catalytic performance for CO oxidation compared with the CP and CeO2 samples. The results collectively point to a strong correlation between the surface area, oxygen vacancies, and active oxygen species in the investigated samples and their catalytic performance. Especially, the oxygen vacancies in the samples were found to have a dramatic effect on the CO oxidation activities. Therefore, the superior activity of CP/A 773 among the samples should be attributed to its high surface area, large quantity of oxygen vacancies, and high fraction of active oxygen species. In addition, this sample showed good catalytic stability during the CO oxidation process. Finally, it is believed that the Al2O3 support has great significance in the design of highly active and stable CeO2-based catalysts in real-world applications.Acknowledgements
We gratefully acknowledge Department of Science and Technology (DST), New Delhi for financial support of this work under SERB Scheme (SB/S1/PC-106/2012). D. D. thanks the CSIR, New Delhi for providing senior research fellowship.References
- C. M. Somers, B. E. McCarry, F. Malek and J. S. Quinn, Science, 2004, 304, 1008 CrossRef CAS PubMed.
- A. Ernst and J. D. Zibrak, N. Engl. J. Med., 1998, 339, 1603–1608 CrossRef CAS PubMed.
- J. Xu, Y.-Q. Deng, X.-M. Zhang, Y. Luo, W. Mao, X.-J. Yang, L. Ouyang, P. Tian and Y.-F. Han, ACS Catal., 2014, 4, 4106–4115 CrossRef CAS.
- A. Biabani-Ravandi, M. Rezaei and Z. Fattah, Chem. Eng. J., 2013, 219, 124–130 CrossRef CAS.
- J. Lee and J. H. Chang, J. Solid State Chem., 2012, 188, 100–104 CrossRef CAS.
- H.-J. Freund, G. Meijer, M. Scheffler, R. Schlögl and M. Wolf, Angew. Chem., Int. Ed., 2011, 50, 10064–10094 CrossRef CAS PubMed.
- P. Venkataswamy, K. N. Rao, D. Jampaiah and B. M. Reddy, Appl. Catal., B, 2015, 162, 122–132 CrossRef CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CAS.
- R. Si and M. Flytzani-Stephanopoulos, Angew. Chem., 2008, 120, 2926–2929 CrossRef.
- J. Ke, J.-W. Xiao, W. Zhu, H. Liu, R. Si, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 15191–15200 CrossRef CAS PubMed.
- A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50, 353–367 CrossRef CAS.
- S. Sun, X. Zhao, H. Lu, Z. Zhang, J. Wei and Y. Yang, CrystEngComm, 2013, 15, 1370–1376 RSC.
- B. M. Reddy, P. Bharali, P. Saikia, A. Khan, S. Loridant, M. Muhler and W. Grünert, J. Phys. Chem. C, 2007, 111, 1878–1881 CAS.
- B. M. Reddy, L. Katta and G. Thrimurthulu, Chem. Mater., 2010, 22, 467–475 CrossRef CAS.
- D. Devaiah, T. Tsuzuki, C. U. Aniz and B. M. Reddy, Catal. Lett., 2015, 145, 1206–1216 CrossRef CAS.
- G. K. Reddy, K. Gunasekara, P. Boolchand and P. G. Smirniotis, J. Phys. Chem. C, 2011, 115, 920–930 CAS.
- M. Balaguer, C. Solís and J. M. Serra, J. Phys. Chem. C, 2012, 116, 7975–7982 CAS.
- C. Tiseanu, V. Parvulescu, D. Avram, B. Cojocaru, N. Apostol, A. V. Vela-Gonzalez and M. Sanchez-Dominguez, Phys. Chem. Chem. Phys., 2014, 16, 5793–5802 RSC.
- K. Ahn, D. S. Yoo, D. H. Prasad, H.-W. Lee, Y.-C. Chung and J.-H. Lee, Chem. Mater., 2012, 24, 4261–4267 CrossRef CAS.
- J. Mikulová, S. Rossignol, J. Barbier Jr, D. Duprez and C. Kappenstein, Catal. Today, 2007, 124, 185–190 CrossRef.
- B. M. Reddy, G. Thrimurthulu, L. Katta, Y. Yamada and S.-E. Park, J. Phys. Chem. C, 2009, 113, 15882–15890 CAS.
- G. Xiao, S. Li, H. Li and L. Chen, Microporous Mesoporous Mater., 2009, 120, 426–431 CrossRef CAS.
- Z.-Y. Pu, J.-Q. Lu, M.-F. Luo and Y.-L. Xie, J. Phys. Chem. C, 2007, 111, 18695–18702 CAS.
- M. Moser, C. Mondelli, T. Schmidt, F. Girgsdies, M. E. Schuster, R. Farra, L. Szentmiklósi, D. Teschner and J. Pérez-Ramírez, Appl. Catal., B, 2013, 132–133, 123–131 CrossRef CAS.
- R. Zhang, P. Li, N. Liu, W. Yang, X. Wang, R. Cui and B. Chen, Catal. Today, 2013, 216, 169–177 CrossRef CAS.
- B. L. Moroz, P. A. Pyrjaev, V. I. Zaikovskii and V. I. Bukhtiyarov, Catal. Today, 2009, 144, 292–305 CrossRef CAS.
- P. Ammendola, P. S. Barbato, L. Lisi, G. Ruoppolo and G. Russo, Surf. Sci., 2011, 605, 1812–1817 CrossRef CAS.
- Q. Dong, S. Yin, C. Guo and T. Sato, Catal. Sci. Technol., 2012, 2, 2521–2524 CAS.
- R. D. Monte, P. Fornasiero, J. Kašpar, M. Graziani, J. M. Gatica, S. Bernal and A. Gómez-Herrero, Chem. Commun., 2000, 2167–2168 RSC.
- B. M. Reddy, P. Saikia, P. Bharali, S.-E. Park, M. Muhler and W. Grünert, J. Phys. Chem. C, 2009, 113, 2452–2462 CAS.
- P. Bharali, P. Saikia and B. M. Reddy, Catal. Sci. Technol., 2012, 2, 931–933 CAS.
- A. Morikawa, T. Suzuki, T. Kanazawa, K. Kikuta, A. Suda and H. Shinjo, Appl. Catal., B, 2008, 78, 210–221 CrossRef CAS.
- N. Paunović, Z. Dohčević-Mitrović, R. Scurtu, S. Aškrabić, M. Prekajski, B. Matović and Z. V. Popović, Nanoscale, 2012, 4, 5469–5476 RSC.
- N. Guillén-Hurtado, A. García-García and A. Bueno-López, Appl. Catal., B, 2015, 174–175, 60–66 CrossRef.
- Y. Shen, S. Zhu, T. Qiu and S. Shen, Catal. Commun., 2009, 11, 20–23 CrossRef CAS.
- Y. Tang, H. Qiao, H. Wang and P. Tao, J. Mater. Chem. A, 2013, 1, 12512–12518 CAS.
- M.-F. Luo, Z.-L. Yan, L.-Y. Jin and M. He, J. Phys. Chem. B, 2006, 110, 13068–13071 CrossRef CAS PubMed.
- L. Li, F. Chen, J.-Q. Lu and M.-F. Luo, J. Phys. Chem. A, 2011, 115, 7972–7977 CrossRef CAS PubMed.
- Z. Wu, M. Li, J. Howe, H. M. Meyer III and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- Q. Shen, G. Lu, C. Du, Y. Guo, Y. Wang, Y. Guo and X. Gong, Chem. Eng. J., 2013, 218, 164–172 CrossRef CAS.
- B. M. Reddy and A. Khan, Langmuir, 2003, 19, 3025–3030 CrossRef CAS.
- D. Devaiah, T. Tsuzuki, T. Boningari, P. G. Smirniotis and B. M. Reddy, RSC Adv., 2015, 5, 30275–30285 RSC.
- G. Chen, Q. Xu, Y. Wang, G. Song and W. Fan, J. Mater. Chem. A, 2015, 3, 7022–7028 CAS.
- F. Lin, X. Wua, S. Liu, D. Weng and Y. Huang, Chem. Eng. J., 2013, 226, 105–112 CrossRef CAS.
- J. He, G. K. Reddy, S. W. Thiel, P. G. Smirniotis and N. G. Pinto, J. Phys. Chem. C, 2011, 115, 24300–24309 CAS.
- N. Tsud, R. G. Acres, M. Iakhnenko, D. Mazur, K. C. Prince and V. Matolín, J. Phys. Chem. B, 2013, 117, 9182–9193 CrossRef CAS PubMed.
- Z. Qu, F. Yu, X. Zhang, Y. Wang and J. Gao, Chem. Eng. J., 2013, 229, 522–532 CrossRef CAS.
- L. Katta, G. Thrimurthulu, B. M. Reddy, M. Muhler and W. Grünert, Catal. Sci. Technol., 2011, 1, 1645–1652 CAS.
- P. J. Schmitz, R. K. Usmen, C. R. Peters, G. W. Graham and R. W. McCabe, Appl. Surf. Sci., 1993, 72, 181–187 CrossRef CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Catal. Today, 2012, 184, 160–165 CrossRef CAS.
- D. Mrabet, A. Abassi, R. Cherizol and T.-O. Do, Appl. Catal., A, 2012, 447–448, 60–66 CrossRef CAS.
- Z. Song, W. Liu, H. Nishiguchi, A. Takami, K. Nagaoka and Y. Takita, Appl. Catal., A, 2007, 329, 86–92 CrossRef CAS.
- A. K. Venugopal, A. T. Venugopalan, P. Kaliyappan and T. Raja, Green Chem., 2013, 15, 3259–3267 RSC.
- H. Borchert, Y. V. Frolova, V. V. Kaichev, I. P. Prosvirin, G. M. Alikina, A. I. Lukashevich, V. I. Zaikovskii, E. M. Moroz, S. N. Trukhan, V. P. Ivanov, E. A. Paukshtis, V. I. Bukhtiyarov and V. A. Sadykov, J. Phys. Chem. B, 2005, 109, 5728–5738 CrossRef CAS PubMed.
- D. H. Prasad, S. Y. Park, H.-I. Ji, H.-R. Kim, J.-W. Son, B.-K. Kim, H.-W. Lee and J.-H. Lee, J. Phys. Chem. C, 2012, 116, 3467–3476 CAS.
- C. D. Wagner, W. M. Riggs, I. E. Davis and J. F. Moulder, in Handbook of X-ray Photoelectron Spectroscopy, ed. G. E. Muilenberg, Perkin-Elmer Corporation, Minnesota, 1978 Search PubMed.
- P. Dufresne, E. Payen, J. Grlmblot and J. P. Bonnelle, J. Phys. Chem., 1981, 85, 2344–2351 CrossRef CAS.
- K. V. R. Chary, C. P. Kumar, D. Naresh, T. Bhaskar and Y. Sakata, J. Mol. Catal. A: Chem., 2006, 243, 149–157 CrossRef CAS.
- D. Devaiah, D. Jampaiah, P. Saikia and B. M. Reddy, J. Ind. Eng. Chem., 2014, 20, 444–453 CrossRef CAS.
- F. Romero-Sarria, J. J. Plata, O. H. Laguna, A. M. Márquez, M. A. Centeno, J. Fdez Sanz and J. A. Odriozol, RSC Adv., 2014, 4, 13145–13152 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical composition, cycling test of CP/A 773 for CO oxidation, and UV Raman spectra of spent CP/A 773 sample. See DOI: 10.1039/c6ra06679h |
| This journal is © The Royal Society of Chemistry 2016 |