
Biomimetic matrix mediated room temperature synthesis and characterization of nano-hydroxyapatite towards targeted drug delivery†
Lubna Sheikhabc,
Sucheta Tripathyab and
Suprabha Nayar*ac
aAcademy of Scientific and Innovative Research (AcSIR), New Delhi, India. E-mail: suparabhanayar@gmail.com
bCSIR-Indian Institute of Chemical Biology, Kolkata, India
cCSIR-National Metallurgical Laboratory, Jmashedpur-831007, India
First published on 20th June 2016
Abstract
The nucleation and growth of hydroxyapatite is closely associated with the extracellular matrix environment. Bovine serum albumin, collagen and polyvinyl alcohol were used to mimic the extracellular matrix. An attempt to understand the role of these matrices on the synthesis and function of hydroxyapatite has been made. XRD, FT-IR, XPS, TG-DTA, SEM and TEM confirmed the formation of hydroxyapatite synthesized by the biomimetic route. Further, the role of the organic matrix in controlling the nucleation and growth of hydroxyapatite particles at the nano level is understood by in-depth analysis of the XPS spectra. The in vitro release of the anti-cancer drug methotrexate in aqueous solution was studied, and the in vitro release profile was assayed by elution in phosphate buffered saline with pH 7.4 and pH 5 at 37 °C. The percentage of loading and release profiles of the drug were evaluated. The results show that the use of the matrix increased the drug release efficiency from 44.5% to 66% at pH 7.4 and 78% to 98.92% at pH 5. These results suggest that the synthesized hydroxyapatite can be used as a pH responsive vehicle for delivering drugs. Further, the release profile was predicted by the Higuchi and Peppas models. The results suggest that the release mechanism is governed by Fickian diffusion for the initial 8 h followed by anomalous transport for longer times. The cytocompatibility of the materials was evaluated by in vitro cytotoxicity tests. Both MTT and live/dead assay observations indicated that the material had no adverse impact on cell proliferation. The results imply that these composites are bioactive with good cytocompatibility. Although all the matrices showed good results, the one with polyvinyl alcohol exhibited higher biocompatibility and drug release efficiency. A plausible explanation is proposed for the enhanced drug delivery efficiency of these materials.
1. Introduction
An ever-increasing number of people suffer from osteoporosis, and bone diseases lead to a large requirement for bone building agents.1 Though osteoporosis has been studied for a number of years, no effective prevention method exists to date. It has been speculated that current orthopaedic implants often fail since they do not sufficiently mimic the nanometer surface roughness that bone cells naturally are accustomed to in the body.2–4 It is the hope that new materials will repair bone damage quickly and effectively. Calcium phosphate-based compounds serve as promising materials in this direction for both delivering drugs and increasing bone mass.5–7 Calcium phosphate-based biomaterials have been used in medicine for more than two decades, among which hydroxyapatite (Ca10(PO4)6(OH)2, HA), the most stable calcium phosphate salt with chemical and structural similarity to the mineral phase of bone and teeth,8–10 is a compound of great interest, be it in the field of agriculture, pharmacy, waste management or as a biocompatible material.11–13 It has attracted the attention of many researchers in recent years because of its versatile nature and exotic properties such as potential for bone substitution, osteointegrativity and osteoconductivity, stability towards bio-resorption, drug carrying capability and biocompatibility.14–19Being such a useful candidate and an extensively researched material, many methods have been proposed by researchers worldwide for its synthesis. Previously explored methods of HA synthesis include plasma spraying,20 hydrothermal synthesis,21,22 freeze drying,23,24 sol–gel25,26 phase transformation,27 mechanochemical synthesis,28 chemical precipitation,29 and precipitation in simulated body fluid (SBF).30,31 All of these methods produce products with varying levels of purity, size, crystallinity, and yield.32 Further, most of these methods involve elevated temperatures and/or extreme pH, which results in larger crystal growth and an increase in coarseness of the microstructure, limiting its biological application.33,34 Furthermore, precipitation in simulated body fluid has such a low yield and long reaction time that it would not be practically feasible for large scale production.
Therefore, development of a simple, one-pot, cost-effective and scalable process for the synthesis of HA at ambient reaction conditions is needed, and this problem is solved by an interesting approach termed “biomimetics”, which underlines the present study.35–37 We are not the first to report biomimetic synthesis of HA. In fact Wang et al. used this process to produce a composite by deposition of HA onto a collagen substrate and reported a Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P molar ratio of 1.58.38 Similarly there are many reports on collagen mediated mineralization, for example Mendes et al. where a temperature of 80 °C for approximately 40 min was required for the synthesis.39 Choi reported a room temperature process for the synthesis of HA using organic solvents such as tetrahydrofuran, which formed particles with diameters between 1 and 10 mm.1 Similarly, the synthesis of biomimetic Ca-hydroxyapatite powders at 37 °C in synthetic body fluids was reported by A. Cuneyt Tas, where the synthesized precursors were found to reach phase purity after 6 h of calcination in an air atmosphere at 900 °C.40 This suggests the need of post heat treatment/calcination for achieving an optimum level of crystallinity and phase purity, however the supramolecular matrix that is used as a template for this type of synthesis degrades at such high temperatures, precluding the preparation of in situ HA/biodegradable protein or polymer composites.
:P molar ratio of 1.58.38 Similarly there are many reports on collagen mediated mineralization, for example Mendes et al. where a temperature of 80 °C for approximately 40 min was required for the synthesis.39 Choi reported a room temperature process for the synthesis of HA using organic solvents such as tetrahydrofuran, which formed particles with diameters between 1 and 10 mm.1 Similarly, the synthesis of biomimetic Ca-hydroxyapatite powders at 37 °C in synthetic body fluids was reported by A. Cuneyt Tas, where the synthesized precursors were found to reach phase purity after 6 h of calcination in an air atmosphere at 900 °C.40 This suggests the need of post heat treatment/calcination for achieving an optimum level of crystallinity and phase purity, however the supramolecular matrix that is used as a template for this type of synthesis degrades at such high temperatures, precluding the preparation of in situ HA/biodegradable protein or polymer composites.
We have attempted to overcome the above mentioned challenges with a simple process without any post heat treatment, which is possible due to the involvement of biological systems as they offset thermodynamic limitations by establishing kinetic control on nucleation and growth of the crystals, leading to the synthesis of phase pure and crystalline HA nanoparticles.41 It is worth mentioning that though there are some reports on the use of matrix mediated synthesis of HA using alginate and chitosan as the matrix, but there was not much emphasis on the role of the matrix in the synthesis leading to the enhanced properties of HA.42,43 In view of this, we for the first time have used three different matrices and tried to understand the role of the matrix on both the synthesis and functional properties of the synthesized nanoparticles. The matrices used in the present investigation are polyvinyl alcohol (P), bovine serum albumin (B) and collagen (C), and the last one without a matrix, was treated as a control. The samples were named as P-HA, B-HA, C-HA and bare-HA respectively. The intention behind using these matrices stems from the following reasons: (1) gelation of the solution under optimum conditions and formation of a non-covalent spatial network inspired us to select P as one of the matrices, (2) B was used because of its nano size cavities in its native form which helps in attracting cations to a particular site,35,44 and (3) C was chosen to mimic HA crystallization occurring in bone naturally.45
The use of matrices for bio-inspired synthesis lies in its (1) faster rate of precipitation, (2) feasibility of fabricating particles in the nanoscale regime at room temperature with a lesser degree of agglomeration under mild reaction conditions, (3) directing the formation of hierarchically assembled inorganic structures, (4) reduction of energy input and avoidance of harmful solvents, which makes the synthesis inherently green, (5) elegant control of the size, shape and crystal structure of the product, and (6) the potential to produce materials with highly specific or multiple functions.46
The formation of HA by the adapted protocol is confirmed by the X-ray diffraction (XRD) and Fourier transform-infrared (FT-IR) techniques. Detailed studies of the interaction of the matrix with HA, chemical composition, Ca:P ratio, oxidation state and chemical linkage of various elements are confirmed by X-photoelectron spectroscopy (XPS). The thermal stability of the synthesized HA particles is determined using thermogravimetric and differential thermogravimetric (TG-DTA) tests. The microstructural investigation was performed by Scanning Electron (FEG-source) Microscopy (SEM) and Transmission Electron Microscopy (TEM). Altogether the above techniques proved the biomimetic approach as a powerful method for the synthesis of monodisperse nanoparticles with hierarchical structures produced by self-assembly, self-construction and self-organization of the basic building components.
Further, the large surface to volume ratio and unique ability to adsorb different chemical species of HA nanoparticles on their surfaces has inspired us to explore its application as an anti-cancer drug delivery vehicle.47 Drug loading and release studies were performed with an anti-cancer drug, methotrexate, in aqueous solution at neutral and pH 5. pH 5 was chosen deliberately to mimic the acidic environment of tumours and inflammatory tissues. The results of the drug loading/release revealed that the use of the matrix enhances the efficiency of HA as a drug delivery vehicle. It was also observed that the efficiency at acidic pH was greater than the neutral pH. The combined results show that these HAs can also be used as a pH responsive drug delivery agent.48,75 Based on the results, it was found that PVA is the best matrix for synthesizing nano-HA for application in drug delivery. Further, to understand the release mechanism, the experimental data were fitted using the Higuchi model, and the diffusion coefficient of the each sample was calculated. The fitting model reveals that initially (up to 8 h) the release is governed by Fickian transport, while for longer times (8 h ≤ t ≤ 144 h) it is the anomalous transport that dominates the release profile. A plausible explanation for the enhanced drug delivery efficiency was proposed, taking into account the particle size, surface area and diffusion coefficients of the HA nanoparticles. This type of drug carrier can be used in a specific field for the pharmacological treatment of musculoskeletal disorders, such as osteoporosis, osteoarthritis and osteosarcoma.
2. Materials and methods
The experimental portion is divided into two parts. The first part is related to the synthesis of HA and the second part is related to the drug study.2.1 HA synthesis
Collagen (C), PVA (95% hydrolysed, avg. MW = 95000) and BSA were purchased from Sigma Aldrich and Acros Organics. Liquor ammonia (30% v/v) was procured from Merck. Calcium nitrate tetrahydrate (Ca(NO3)2·4H2O) and diammonium hydrogen phosphate ((NH4)2HPO4) were purchased from Himedia and Merck. All the chemicals were of analytical grade and were used as received. Double distilled water was used throughout the experiment. HA was synthesized as per our patented process. Briefly 0.4 M alkaline solution of calcium nitrate (pH-9.5) was added to different organic matrices viz. 0.5% of PVA, BSA, and collagen. These solutions were equilibrated for a day followed by the addition of 0.156 M diammonium hydrogen phosphate and ammonia solution, and then the precipitates were aged for 7 days, and washed thoroughly to bring down the pH to neutral. The obtained precipitate was HA. The experimental details for HA synthesis are given in Table 1. The precipitation of HA can be described by eqn (1) and/or (2):| 10Ca2+ + 6HPO4− + 2OH− → Ca10(PO4)6(OH)2 + 6H+ | (1) |
| 10Ca2+ + 6H2PO4− + 2OH− → Ca10(PO4)6(OH)2 + 12H+ | (2) |
| Matrix | Weight% of matrix | Concentration of Ca(NO3)2·4H2O | Concentration of (NH4)2HPO4 |
|---|---|---|---|
| BSA | 0.5 | 0.4 M | 0.156 M |
| Collagen | 0.5 | 0.4 M | 0.156 M |
| PVA | 0.5 | 0.4 M | 0.156 M |
| Without matrix (bare) | NA | 0.4 M | 0.156 M |
The obtained precipitate was oven dried at 60 °C, crushed and the resulting powders were used for further characterization.
Phase identification of the powder sample was confirmed by room temperature XRD in a D8 DISCOVER BRUKER Diffractometer, with a Cu-Kα (λ = 1.5418 Å) source, radiation generated at 40 kV and 40 mA within a scanning range of 20–60 (2θ) at a step size of 0.02 per step, and a scanning rate of 1 s per step. The functional groups of the synthesized samples were identified by FTIR (NICOLET-759) using the KBr-disc method from 400–4000 cm−1. Structural investigations were carried out on analytical TEM, JEOL JEM 2010 instrument using a 200 kV electron source. The samples for TEM were prepared by dispersing different HA powders in ethanol by ultrasonication in a low power ultrasonic cleaning bath (150 W, 40 kHz) for 30 min. A single drop of the dispersion was dried on carbon-coated copper grids (∼300 mesh) and then viewed under the microscope. The surface morphology was studied by FESEM on an FEI Nova Nano SEM 430 instrument operated at 15 kV. TG-DTA was performed using the Shimadzu DTG-TA-50 and DTA 50 analyser, over a 25–1000 °C temperature range, in an air environment, with Al2O3 as the reference. XPS was performed in an ESCA system M/s SPECS with a Mg twin (1253.6 eV) and Al monochromator (1486.6 eV) source. The vacuum of the chamber was better than 10−9 Torr while recording the spectra. The spectra were deconvoluted by using CASA XPS software. Prior to curve fitting, the background was subtracted from each spectrum by the Shirley method. The binding energy scale was calibrated for electrostatic charging with respect to the standard C1s peak at 284.6 eV.
Biocompatibility tests were performed by seeding cells at densities of 1 × 105 cells per well in a 96 well plate and allowing them to attach for 10 h in a CO2 incubator maintained at 37 °C. Different concentrations of each sample were weighed (0.05, 0.1, 0.5 and 1 mg ml−1), sterilized with 70% ethyl alcohol (v/v), followed by UV treatment. Powders were suspended in DMEM medium and then were filter sterilized (himedia 0.2–0.222 μm). The extract was replaced for the media in the well plate and the constructs were incubated for 1, 3, 5 and 7 days at 37 °C and 5% CO2 in a humidified atmosphere to examine the growth of cells in the presence of our samples. The numbers of living cells were determined using the MTT assay. The culture media was replaced and washed with PBS to remove the non-adhered cells. 100 μl per well of MTT solution prepared in DMEM was added to the wells, and the cells were cultured for an additional 4 h. The medium was then removed, and 200 μl of dimethyl sulfoxide (DMSO) was added to each well. The optical density (OD) was measured at a wavelength of 595 nm using a micro plate reader (Model Perkin-Elmer 2030 explorer). The experiments were done in triplicate and the best concentration (1 mg ml−1) for cell growth is given.
Similarly the drug binding and release studies were conducted using different HA samples, namely bare-HA, B-HA, C-HA and P-HA. Methotrexate tablets (Folitrax®-2.5) were obtained from Ipca Laboratories, India. UV-VIS was performed by a VARIAN-50 BIO spectrophotometer. The main objective besides checking the in vitro drug release kinetics that concern the non-in vivo arena are the physicochemical characterization of the systems, system optimization such as the concentration used, the amount of drug desired, criteria for quality assurance etc.
2.2 The drug load and release protocol
The drug release was evaluated using the following equation:
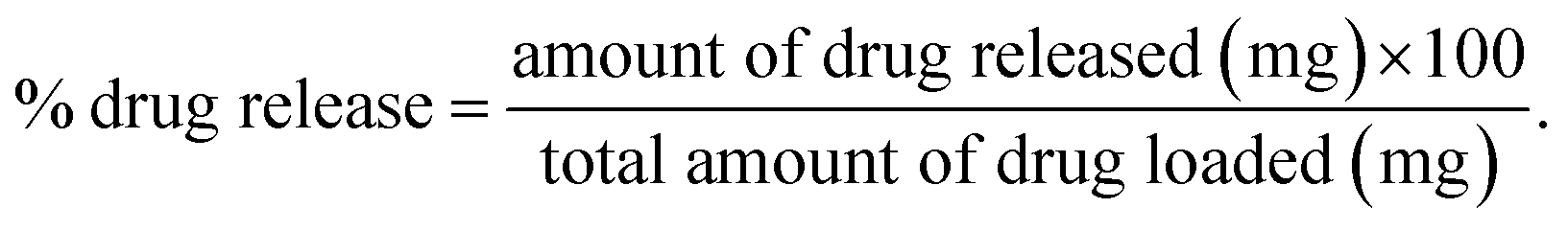
(1) Perfect sink conditions were maintained throughout the experiment.
(2) The pellets are square in shape.
(3) The diffusion coefficient of the drug is constant.
(4) The drug is uniformly distributed in the tablet before exposure to the release medium.
(5) There is no swelling of the pellets in the medium.
By using Higuchi’s mathematical expression, the derived expression of the fraction of drug released at time t, specifically for the given sample geometry, is
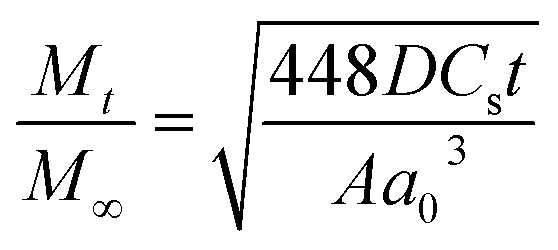 | (3) |
3. Results and discussion
3.1 XRD analysis
The X-ray diffraction (XRD) technique is widely used for the identification of the crystal structure and its phases. The calcium phosphate-based material forms a wide range of compounds with different phases. In order to confirm the formation of the HA phase, the synthesized powders were characterized by XRD. The XRD characterization of HA is well established in the literature. Fig. 1 represents the XRD data of the synthesized HA with and without the matrices. The XRD patterns reveal the formation of HA, and it matches well with the standard JCPDS card no. 09-0432. Three high-intensity peaks located at 2θ = 31.7°, 32.2° and 32.9° with Cu Kα radiation are difficult to be exactly recognized from their diffraction patterns, and appeared as a single peak with shoulders at 32.2° and 32.9°. An underlying broadening in the peaks reflects the nanometer range of the particle size. Interaction of the matrices is revealed from the slight shift in the peaks, symbolising a matrix mediated synthesis otherwise absent in bare-HA.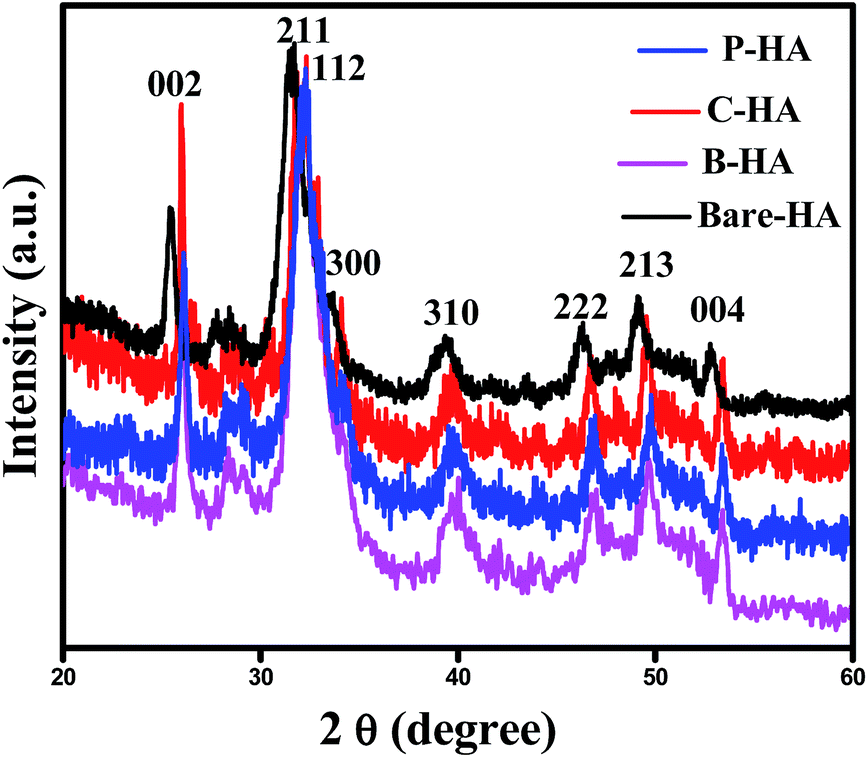 | ||
| Fig. 1 XRD data of the HA synthesized with and without the matrix. | ||
Further, the slight shift in the peak positions of HA with the matrix towards higher 2θ values may be attributed to the shrinkage in the lattice structure leading to the decrease in the ‘d’ value, which indirectly affects the crystallite size. To further cross check this we have calculated the crystallite size, using the Scherrer formula
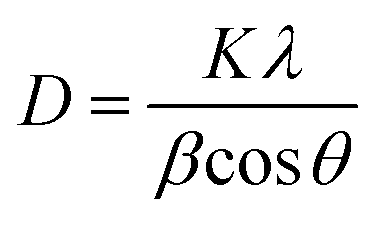
The values of the crystallite size for the (211), (112) and (300) reflections were calculated as 32 nm, 11.80 nm, 14.09 nm and 8.52 nm, and the average crystallite size was 37 nm, 18.29 nm, 19.89 nm, and 14.85 nm for the bare-HA, B-HA, C-HA and P-HA respectively. The beauty of biomimetics is further confirmed by different matrices dictating the size of the particle, and as the bare-HA was without any matrix, it showed a huge deviation. The crystallite size calculation indicated that the use of selective matrices can yield HA with different crystallite sizes. It is worth mentioning that different methods have been employed by different researchers to reduce the particle size of HA to increase the functional properties of HA. Indeed Cai et al. have reported that the smaller the particle size, the greater is the cell viability and proliferation and hence the more bioactive is the compound.49 The XRD results of the HA synthesized by our process revealed that the crystallite size of the HA particles are different with different matrices, indicating that the matrix plays a major role in the growth of HA crystals. In addition it is also clear that our process is capable of controlling the particle size of HA in the nano regime.
3.2 FT-IR analysis
To identify the different functional groups present in the synthesized HA samples we have characterized the samples by FT-IR spectroscopy. Fig. 2 shows the FT-IR spectra of bare HA compared to the HA synthesized with different matrices. The FT-IR spectra show the characteristic hydroxyl and phosphate peaks in all the samples. In the case of bare-HA, the peaks are not well developed. This could be because the reaction temperature was ambient, and for the HA peak to develop, it either needed high temperature or the addition of additives to bring down the reaction kinetics. The absorptivity in FT-IR is an absolute measure of infrared absorbance intensity for a specific molecule at a specific wavenumber. For pure samples, the concentration is at its maximum, and the peak intensities are true representations of the values at different vibrations. However, in a composite system, two peaks may have different intensities because there are molecules present in different concentrations.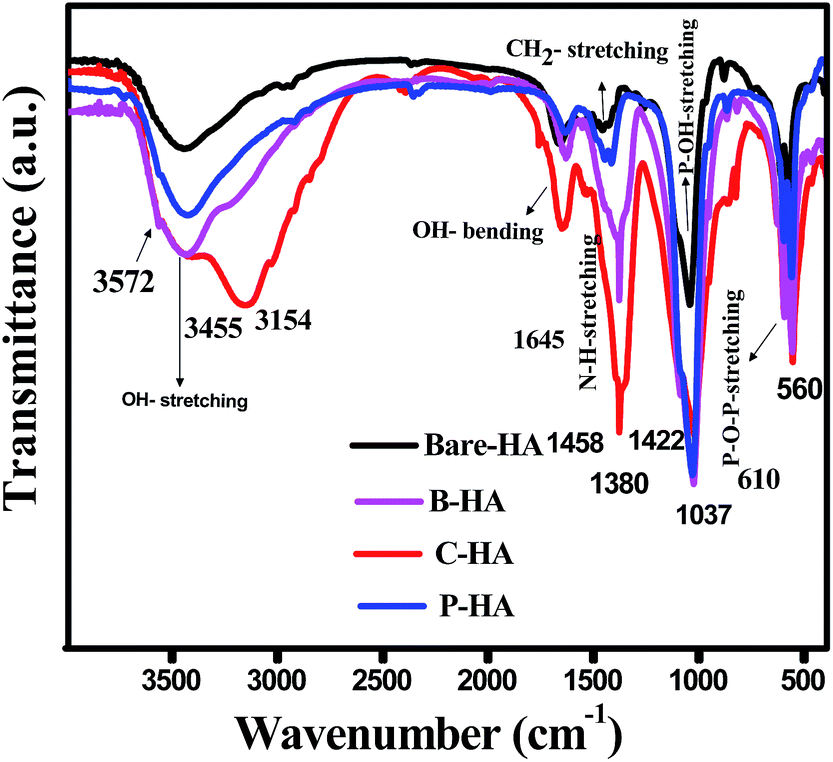 | ||
| Fig. 2 FT-IR spectra of the bare-HA as compared to the HA synthesized using matrices. | ||
In addition, in any sample where hydrogen bonding occurs, the number and strength of intermolecular interactions varies greatly within the sample, causing the bands in these samples to be particularly broad, as seen in the cases of B-HA, C-HA and P-HA.
Further, the major bonds associated with PO43− (1037, 961 and 606 cm−1) and lattice OH at 3572 cm−1 appeared in all four samples.50 The peaks at 1037 cm−1 are associated with the stretching modes of the P–O bonds of HA, and the doublets at 606 cm−1 and 560 cm−1 are due to the bending modes of P–O bonds in the phosphate group, further confirming the presence of phosphate in the system.51 Three well-defined strong peaks at 1380 cm−1, 1645 cm−1 and 3154 cm−1 are assigned to C–N, C–O and stretching of the N–H bond respectively, and are seen only in C-HA and B-HA due to the presence of proteins/amino acids.52 Presence of the CO32− group is revealed by a weak peak at 870 cm−1 in the case of bare-HA. To further cross-check our FT-IR results we have characterized our samples by the XPS technique, which will conclude the chemical linkage of different species present in the sample, and most importantly the interaction of the matrix with HA, which was not visible by FT-IR.
3.3 XPS analysis
XPS is a very powerful surface technique which gives information about the chemical composition, chemical state, chemical linkage and atomic ratio in a sample.Many researchers have tried to characterize HA by XPS.53–58 However, there are no systematic and organized reports in the literature pertaining to the characterization of HA by XPS. In this report we have systematically characterized HAs synthesized with and without matrices, and an attempt has been made to find out the composition, Ca:P ratio, chemical state of Ca, P and O, and the chemical linkage so that a detailed characterisation of HA by this powerful technique is available in one paper. In addition, an attempt has also been made to understand the role of matrices on the synthesis of HA through XPS. Fig. 3 represents the survey scan of bare-HA and HA with matrices. The survey scan is a quick scan that gives immediate information about the type of elements present in the sample. The survey scan of our sample shows the presence of Ca, P, O and C elements.
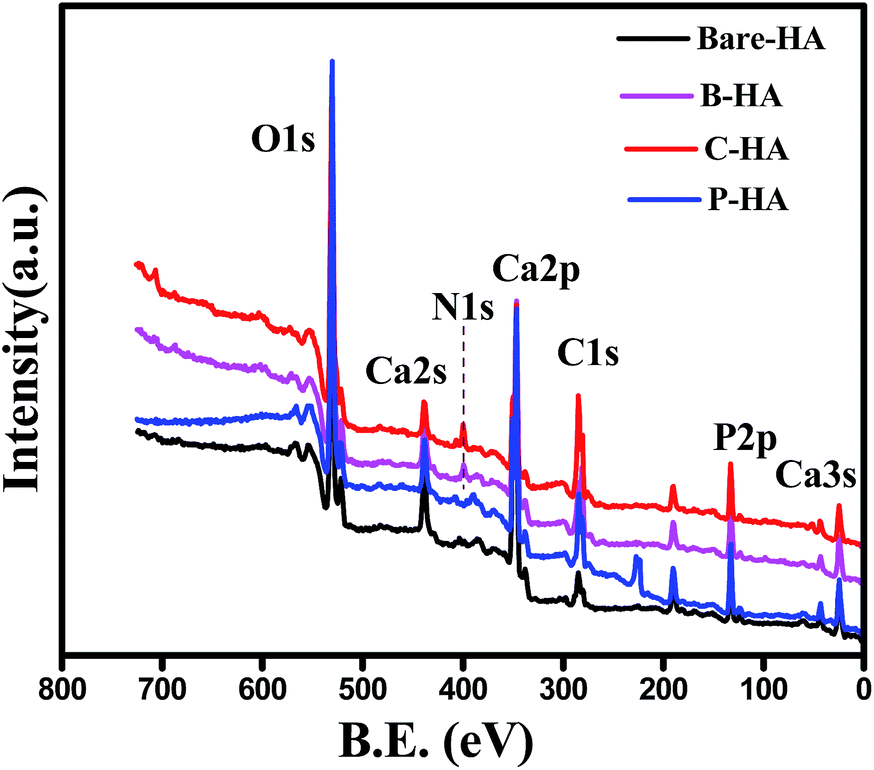 | ||
| Fig. 3 Survey scans of bare-HA and HA with matrices. | ||
The Ca, P and O is the composition of the HA, whereas the appearance of the C peak is due to the use of polymer matrices while synthesizing HA, and in the case of bare-HA it may be due to carbon contamination from the environment. The appearance of the N1s peak in B-HA and C-HA is due to the presence of nitrogen in the polymer matrix.
In order to draw more insight into the role of the matrices in the synthesis of HA, slow scans for individual elements were performed and the peaks were analysed using CASA XPS software. The Ca:P ratio was also calculated using the CASA XPS software. It is well reported in the literature that for stoichiometric HA the Ca:P ratio is 1.67 and its value differs if HA is non-stoichiometric.59 From our XPS data it was observed that in the case of P-HA the ratio is 1.67, and the value is 1.63, 1.65 and 1.70 for B-HA, C-HA and bare-HA respectively. It is noted that the Ca:P ratio when the matrices are used is less than or equal to 1.67, whereas in the case of the bare-HA the ratio increased beyond 1.67. The increase in the Ca:P ratio in the case of bare-HA indirectly predicts the presence of some other compound along with HA. To verify this we have analysed the deconvoluted Ca2p peak.
Fig. 4(a) shows the deconvoluted Ca2p peak of the HA samples synthesized with and without matrices. In all the HA samples the Ca2p peak appeared as a typical doublet corresponding to Ca2p3/2 and Ca2p1/2 as a result of spin orbit coupling. The peak position and peak energy separation (ΔE) are the most important parameters for determining the chemical state of an element. The most common observation in all the Ca2p peaks is the peak energy separation value (energy difference between the Ca2p1/2 and Ca2p3/2) that is 3.5 eV, which is a match for Ca in the +2 oxidation state.60 Therefore, from the peak energy separation value we can safely conclude that “Ca” in all our synthesized HA is in the +2 oxidation state, which is the chemical state of “Ca” in HA.61 To know about the chemical environment around Ca2+ and its linkage with other elements, we have analysed the fitted Ca2p peak of the bare-HA and compared the results with those synthesized using matrices. An immediate observation is that both the Ca2p3/2 and Ca2p1/2 peaks fit two peaks in bare-HA, whereas it fits only one peak in the case of the other HAs. The fitting of two peaks predicts that the “Ca” in bare-HA is attached to two different types of functionalities. In fact, similar observations were noted by Rossi et al. when they have deposited HA films using the plasma deposition method.62 In their case they attributed the extra peak towards a lower binding energy to Ca–CO3 bond. In our case, the main peak of Ca2p3/2 for bare-HA appeared at 346.73 eV and a small peak fits towards a lower binding energy at 344.74 eV. We attribute the peak at 346.73 eV to Ca–Ca, Ca–OH and Ca–O bonds in the HA crystal structure,63 while the peak at 344.74 eV is due to Ca–O bonds from carbonates that may have formed due to the entrapment of air from the environment during the synthesis.62 The striking observation is that, in the case of B-HA, C-HA and P-HA, it fits only one peak at 346.62 eV, 346.64 eV and 346.94 eV respectively, which indicates the absence of carbonate in these samples. We attribute the appearance of these peaks to Ca–Ca, Ca–OH and Ca–O bonds in the HA crystal. A little shift in the binding energy positions of the Ca2p peak is due to the interaction of the organic matrix with HA, which is discussed in detail in the later part of the manuscript. Analysis of the Ca2p peak reveals that carbonate is formed only in the case of bare-HA, which explains the reason for the increased Ca:P ratio and the appearance of a C1s peak in bare-HA. It can be inferred from the analysis of the Ca2p peak of the synthesized HA that the use of the matrix prevents formation of unwanted byproducts related to calcium. It is also observed from the literature that the binding energy position of the Ca2p peak varies from author to author; therefore we have repeated the XPS scan of all the samples at least five times, and we noted the reproducibility of the results each time. Our experimental results match with Abe et al., where they have tried to establish an acid etching procedure to alter the Ca:P ratio of nanostructured HA.63 In our case, the X-ray take-up angle is 90° and our XPS results are in good agreement with the values reported by Abe et al. Next, we present the deconvoluted P2p data to understand the chemical environment around P and its linkage to other elements.
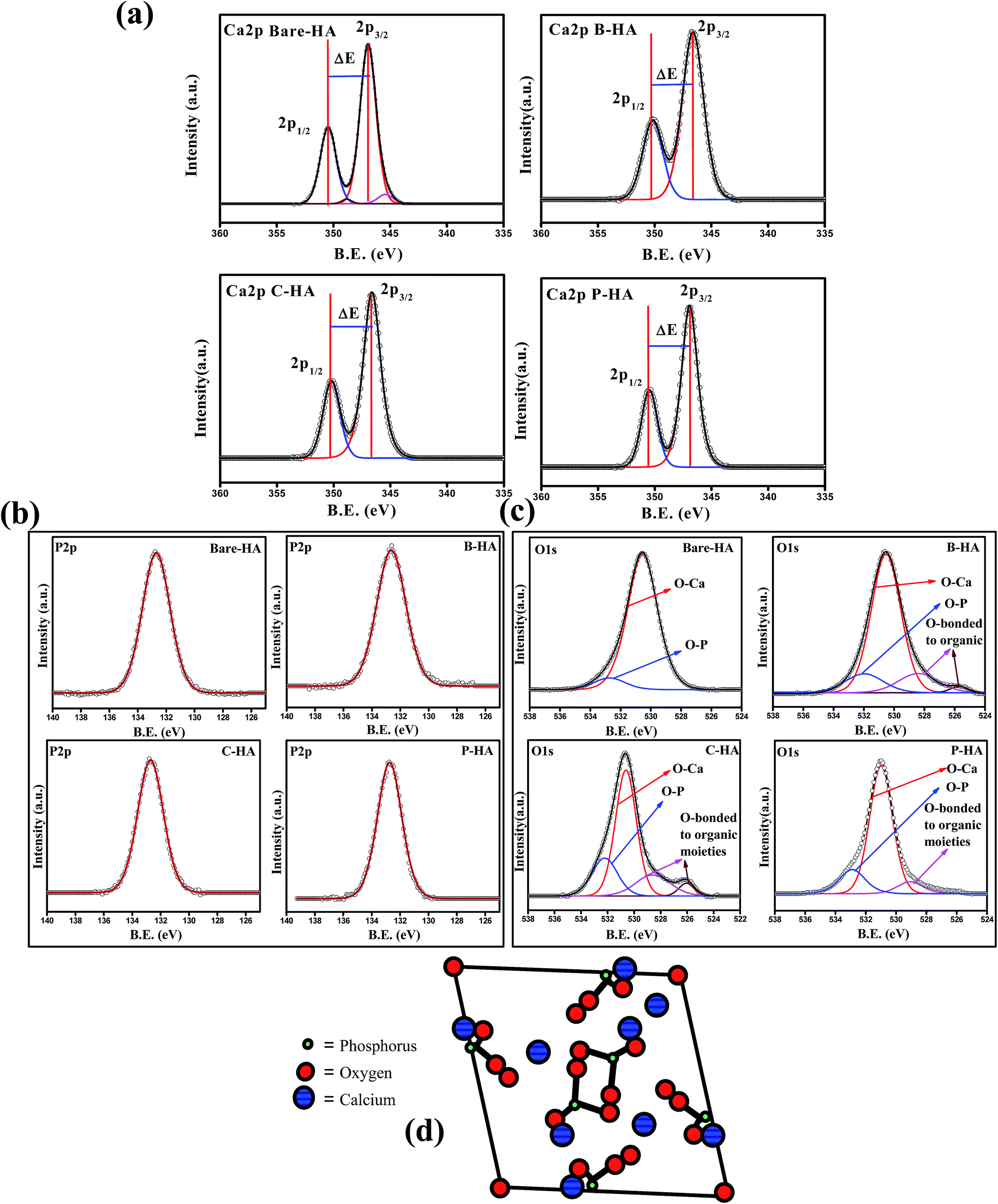 | ||
| Fig. 4 (a)–(c) Ca2p, P2p and O1s peaks of bare-, B-, C- & P-HA, respectively. (d) Model showing the arrangement of P in the HA crystal structure. | ||
Fig. 4(b) shows the P2p peaks of bare, B-, C-, and P-HA. The deconvolution of all the P2p peaks fits only one peak, indicating that the chemical environment is similar around P, i.e. P is bonded to the same functional groups in all cases otherwise it would have fitted in more than one peak as for “Ca” in bare-HA. The phosphorus 2p peak in our case appears at 132.76, 132.60, 132.8 and 132.66 eV for bare-HA, B-HA, C-HA and P-HA respectively. The peak position is matching with the phosphate group.58 Therefore, we assign the P2p peak to P–O bonds in the PO43− of HA. The fitting of one peak in P2p is also supported by the fact that “P” in the crystal structure of HA is bonded to four O atoms in a tetrahedron.64–66 The position of P in the HA crystal is given in Fig. 4(d) for a better understanding. Therefore, the chemical environment of P is similar in the HA crystal structure and hence fitted with one peak. Thus from the XPS analysis of the P2p peak it is confirmed that no other byproducts related to P are formed in our case unlike for Rossi et al.,62 where P2O5 was formed in addition to P–O bonds in HA. Finally, to verify our results we present the O1s spectra which will provide useful information about the presence of Ca–O and P–O bonds.
Fig. 4(c) presents the O1s deconvoluted spectra of the HA samples. The O1s of bare-HA fits two peaks at 530.5 and 532.3 eV. The peak at 532.3 eV is attributed to the P–O bond in HA, while the peak at 530.5 eV is due to the contribution from OI, OII, and OIII bound to CaI and CaII in the hydroxyapatite structure, and it also has contribution from carbonate groups as these two groups cannot be distinguished by routine peak fitting. However, the presence of carbonate in bare-HA is confirmed from the Ca2p peak, as explained earlier. Similar peaks are observed in the O1s spectra of HA synthesized using matrices. In addition there are two more peaks fitted in the lower binding energy side of O1s in the case of B-HA and C-HA, while only one extra peak towards a lower binding energy is observed for P-HA. We attribute these peaks to the contribution from the used organic matrices.
In order to understand the interaction of the organic matrix with HA, we have analysed the C1s peak of the B, C and P samples with the corresponding C1s peaks of HA with matrices (i.e. B-HA, C-HA and P-HA). Fig. 5 represents the C1s peak of the used matrix and corresponding HA with matrix. First we, present the C1s data of the B, C and P. The deconvoluted C1s spectra of B and C fit three peaks, while the spectrum fits two peaks in the case of P. The peak at 284.6 eV is due to the aliphatic carbon skeleton of these compounds. The fitting of peaks at 285.5 and 288 eV in the case of B and C is attributed to the contribution from the HN–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HR group and O–NH group respectively.67 The peak fitted at 286.4 eV for P is attributed to the methine α to the hydroxyl group.68 Next, we present the C1s data of the HA synthesized with the matrix. It can be clearly seen that all the C1s spectral features of HA with matrices are quite different from the corresponding polymer/protein matrix where two distinct peaks with a shoulder in the second peak appeared towards the higher binding energy side. Such type of splitting in the C1s spectrum is not observed in the case of B, C and P. The splitting of the peak indicates that HA is interacting with the matrix, otherwise the spectral features would be similar to the pure organic matrices B, C or P.
HR group and O–NH group respectively.67 The peak fitted at 286.4 eV for P is attributed to the methine α to the hydroxyl group.68 Next, we present the C1s data of the HA synthesized with the matrix. It can be clearly seen that all the C1s spectral features of HA with matrices are quite different from the corresponding polymer/protein matrix where two distinct peaks with a shoulder in the second peak appeared towards the higher binding energy side. Such type of splitting in the C1s spectrum is not observed in the case of B, C and P. The splitting of the peak indicates that HA is interacting with the matrix, otherwise the spectral features would be similar to the pure organic matrices B, C or P.
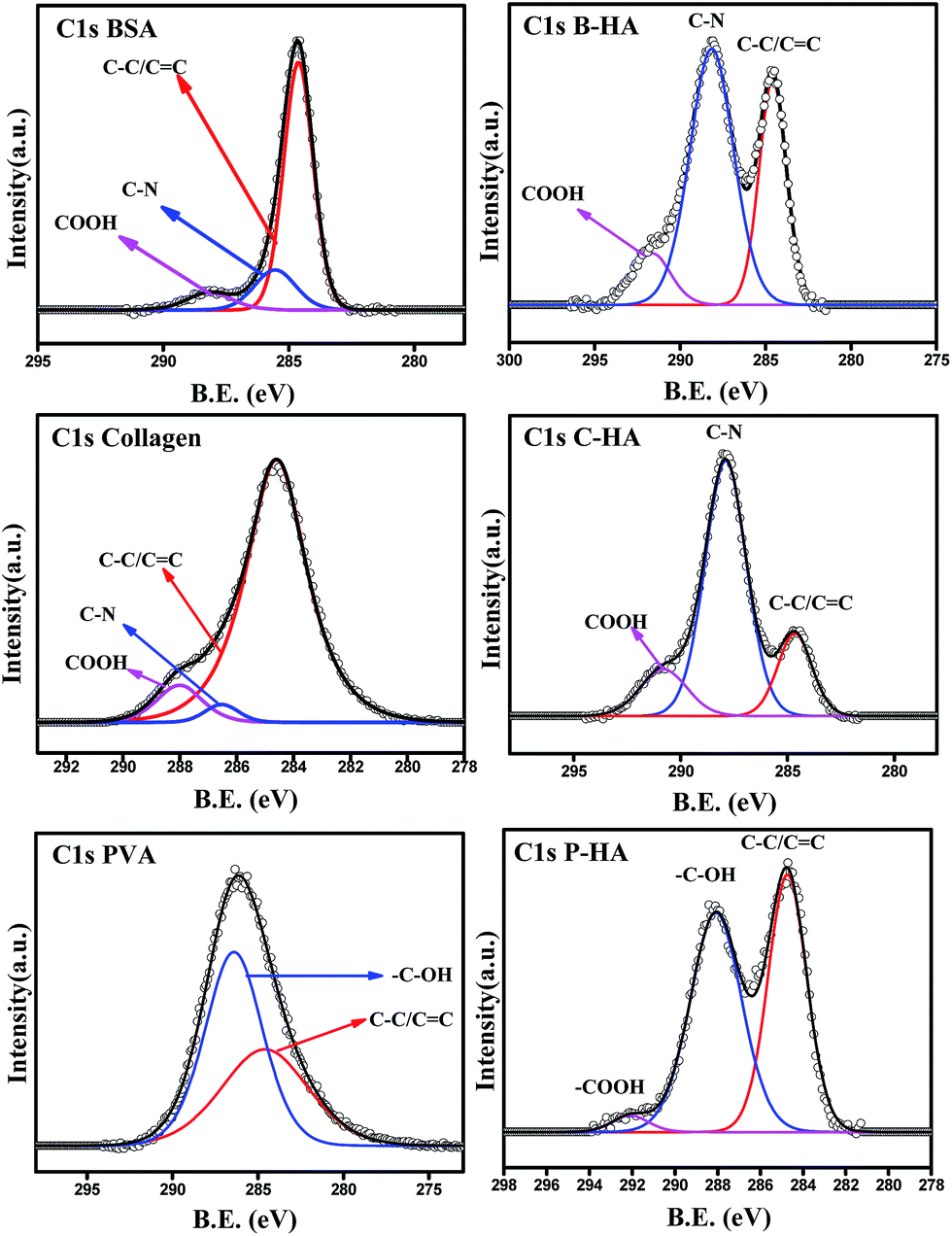 | ||
| Fig. 5 C1s spectra of the matrices & respective HAs. | ||
The deconvolution of these peaks reveals that the peak position due to the aliphatic carbon skeleton is unaltered and appears at 284.6 eV in all cases, while a large shift in the peak position of the other functional groups towards the higher binding energy side is observed. The peak at 285.5 eV for BSA and collagen is shifted to 288 eV and 287.8 eV, while the peak at 288 eV is shifted to 291.67 eV and 290.7 eV for B-HA and C-HA respectively. In the case of PVA the peak at 286.4 is shifted to 288 eV with an extra peak fitted at 292 eV. The shift in the peak position towards the higher binding energy side will occur only when some electron withdrawing entities are interacting with the corresponding functional groups. Therefore, the shift in the binding energy of these functional groups indicates there is an electron withdrawing group interacting with them. However, the XPS analysis of the HA samples synthesized with the matrix ruled out the formation of any kind of byproduct, pointing towards the fact that HA is interacting with these groups. Indeed, while synthesizing HA with the matrix, the calcium precursor is added to the matrix first and then the solution is allowed to equilibrate for 24 h, so it may be that the electropositive Ca2+ ions are attracted towards electronegative N atoms in the case of B and C, and towards O atoms in the case of the P matrix due to electrostatic attraction, and on the addition of phosphate ions to these solutions, the nucleation of HA begins from those sites where Ca2+ ions are attached. As Ca2+ is electropositive it withdraws electrons from the N and O atoms, which leads to the electron deficiency in the C atoms to which these N and O atoms are attached, leading to the shift in the corresponding peaks to the higher binding energy side. Therefore, it is clear from the results that the use of the matrix in the synthesis of HA makes a number of preferential sites available for the controlled nucleation and growth of HA crystals.
The combined results of XRD, FT-IR and XPS revealed that the use of matrices in the synthesis of HA prevents the formation of unwanted byproducts and yields nano-crystalline HA by providing selective nucleation sites for controlled growth of the HA crystals. Based on the Ca:P ratio and crystallite size it is predicted that among the different matrices the PVA yielded the best result in terms of stoichiometry and crystallite size.
One of the well-known properties of HA is its thermal stability, and in order to check this we performed TG-DTA analysis of the synthesized samples.
3.4 TG-DTA analysis
Fig. 6 shows the TG-DTA plots of the different HAs analysed in an air atmosphere. The spectra of all the four samples show a weight loss in three steps. The first step of weight loss is between 50 °C and 100 °C due to weakly entrapped water in the sample and dehydration of the precipitating complex. Indeed, Liang et al. reported similarly slow weight loss in this temperature range in the case of HA–alginate.42 The second weight loss step between 200 °C and 400 °C may be due to the decomposition of the organic components, and the third step between 400 °C and 800 °C may be attributed to the loss of lattice water.69,70 No rapid weight loss was observed, suggesting no unreacted calcium. The total weight loss percentage was approximately 44%, 33%, 21%, and 18% for C-HA, B-HA, P-HA and bare-HA respectively. The hygroscopic nature of collagen could be the reason for the increased weight loss in C-HA. The DTA curve shows a sharp exothermic peak near 230 °C, which shows the thermal decomposition of P in P-HA, and the exothermic peaks at 200 °C and 400 °C show the degradation of proteins or the elimination of organic components in C-HA and B-HA respectively. The weight loss for the bare-HA in this temperature region is due to the decarboxylation of carbonates formed as a byproduct along with HA, otherwise pure HA is stable in this range.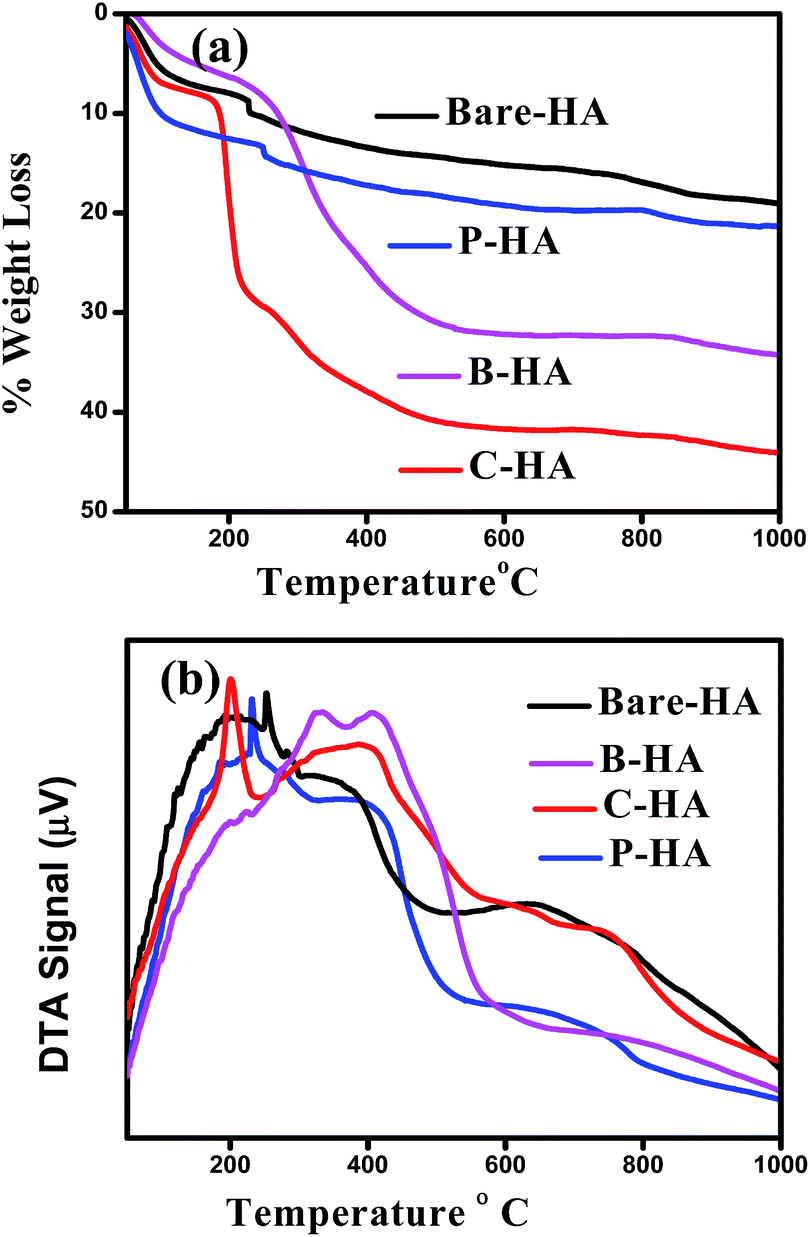 | ||
| Fig. 6 (a) TGA and (b) DTA plots of bare-HA and HA with different matrices. | ||
In order to check the morphology and size of the different HAs we performed microstructural investigations by Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM).
3.5 SEM analysis
Fig. 7 represents a wide perspective of different HAs over a micron range (20 microns) depicting their uniformity. All the four samples show different and unique morphologies. The particles are self-assembled in different patternt. B-HA shows small particles of almost similar shapes and sizes distributed homogeneously. In C-HA the morphology looks like a flower arranged in a hierarchical order.71 Similarly P-HA shows a unidirectional arrangement of particles which may be due to the influence of PVA arranged in a tubular pattern on the growth of particles in a particular direction. The best thing we noticed was the irregular shape and structure of bare-HA particles, suggesting the importance of matrices in dictating the shape, size and self-assembly of the nanoparticles. The inset shows the microstructure of the different matrices. Globular cavities typical of BSA are seen in matrix B, while matrices C and P reveal tubular arrangements typical of the helical structure of C, and tubular polymeric chain arrangement seen in P respectively. The elemental analysis was also performed to identify the different elements present in the system along with its approximate Ca:P ratio. The atomic Ca:P ratio was found to be 1.79, 1.648, 1.639, and 1.679 for bare-HA, B-HA, C-HA and P-HA respectively, which are very close to the results obtained from XPS. Altogether, the SEM results revealed that the HA synthesized with the matrix exhibited unique morphology and homogeneity unlike the bare HA.
 | ||
| Fig. 7 Morphology of the synthesized HAs with and without the matrix under SEM. | ||
In order to have a closer view of the synthesized nanoparticles, TEM was performed.
3.6 TEM analysis
Fig. 8 shows the TEM micrographs of HAs with their corresponding selected area diffraction pattern (SAED) patterns. The TEM micrographs show a typical acicular needle-like morphology for those synthesized with matrices, and a platelet-like morphology for bare-HA. It was observed that almost all the particles were of similar shape and size except bare-HA, where a change in the shape and size of the particles was noticed. In fact, the bare-HA particles were no longer rod-shaped but became platelet-like. The absence of the matrix in bare-HA led to uncontrolled nucleation and growth, leading to the precipitation of irregular particles. From the micrographs it is clear that the HA crystals are embedded in the matrix in the case of B-HA, C-HA and P-HA, representing matrix mediated synthesis, while individual particles are seen in the case of bare-HA due to the absence of the matrix. The average particle distribution was found to be 6 nm, 7.2 nm, 7.8 nm and 30 nm in P-HA, B-HA, C-HA and bare-HA respectively. Precipitation of nano-sized HA particles could be explained based on a matrix surface-induced high nucleation rate followed by a sluggish growth rate, whereas the absence of the matrix led to the random and uncontrolled nucleation and growth of bare-HA particles. Further, the SAED patterns in all the cases show a polycrystalline ring pattern depicting the smaller size of the synthesized particles. The C-HA SAED pattern reveals a typical HA crystal structure with an apparent (002) plane. It is known that in human bone the HA crystals grow in parallel with the collagenous fibres along the c-axis of the (002) plane.72 The collagen matrix in the case of C-HA also play a role in aligning the growth of HA crystals along the c-axis, resulting in an elongated shape. This was also seen in XRD where the 002 peak of C-HA is more pronounced compared to the others. Combining the results of XRD and TEM in association with XPS, it can be concluded that the matrices help the controlled nucleation and growth of HA particles. | ||
| Fig. 8 TEM micrographs of HAs with and without the matrix. The inset to each image is the corresponding SAED pattern. | ||
Since the synthesized HA is mainly for biomedical applications, its biocompatibility test is a must and in order to do so we report its compatibility test using the MTT assay.
3.7 Biocompatibility test
The MTT assay was performed on human mesenchymal cells (hMSCs) in order to check the cell viability as a test of biocompatibility. The results of the MTT assay for MSCs incubated in different extracts for 1, 3, 5 and 7 days are shown in Fig. 9. None of the tested extracts showed significant cytotoxicity for the cells. There was rather an increase in the cell growth compared to the one without HA nanoparticles (control), but P-HA showed the best performance with a geometric increase in the absorbance which is directly related to the number of viable cells. The reason for this enhanced cell activity is probably because, for optimal functioning and support, cells in our body rely on their interaction with an extracellular matrix (ECM), which is a complex network of substrates with intricate features that are nanoscale in dimension. The ECM stimulates and interacts with cell physiology on a nanoscale level which is present in the form of hydroxyapatite nanoparticles here. Live/dead staining was performed with fluorescein diacetate (FDA) and propidium iodide (PI). FDA is taken up by live cells, which convert the non-fluorescent FDA into the green fluorescent metabolite fluorescein. In contrast, the nuclei staining dye PI cannot pass through a viable cell membrane. It reaches the nucleus by passing through disordered areas of dead cell membranes, and intercalates with the DNA double helix of the cell. Almost all cells were live in the presence of different types of HA, further confirming their compatibility and rendering our material nontoxic.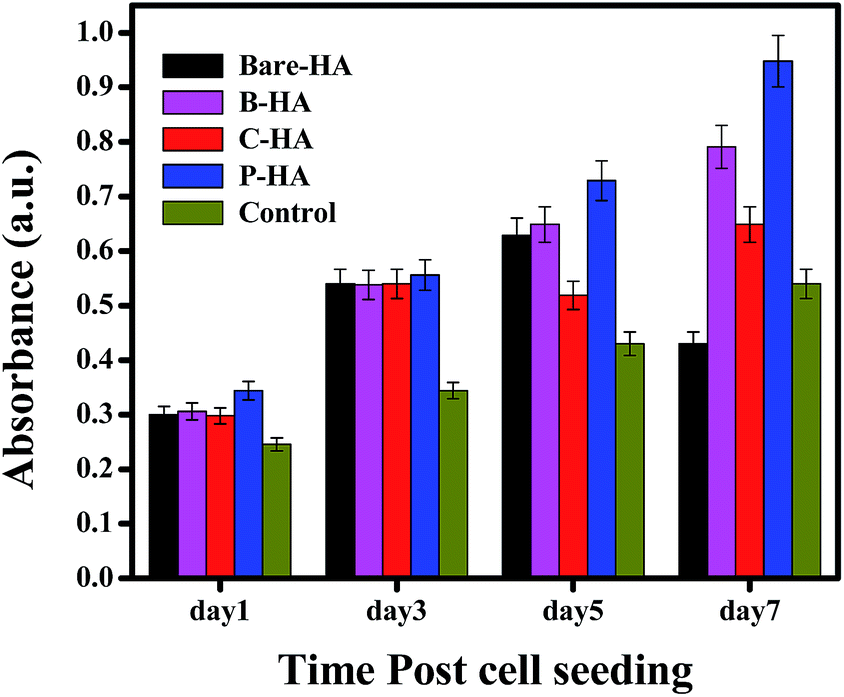 | ||
| Fig. 9 MTT assay of bare-HA, B-HA, C-HA and P-HA. | ||
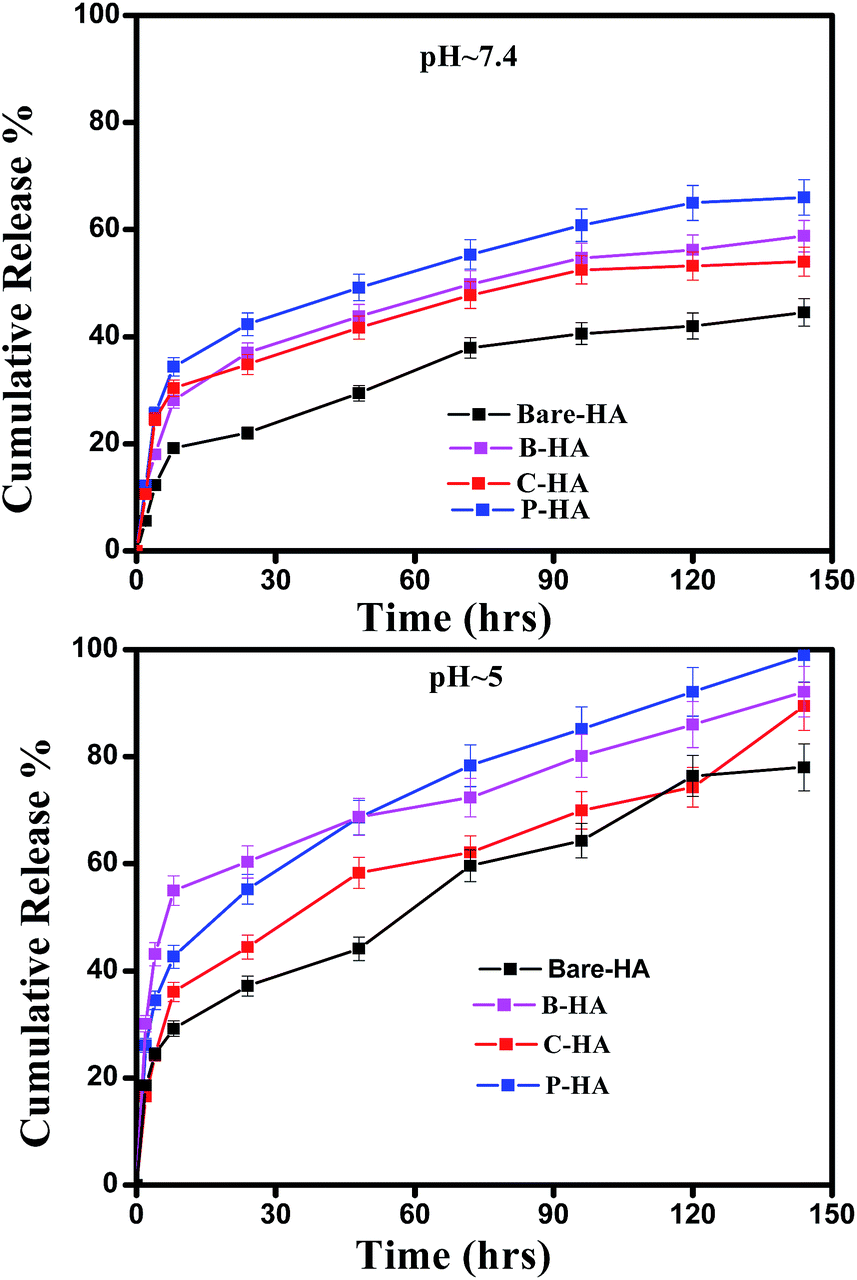 | ||
| Fig. 10 Drug release profile at pH 7.4 & 5 along with calibration curve. | ||
Further, to understand the mechanism of the in vitro release kinetics, we have fitted the experimental results using the Higuchi model and calculated the diffusion coefficients of each HA-tablet, which would yield more information about the observed release profile in our case.
where the first term in the right hand side is the Fickian contribution and the second term is the case-II contribution. Consequently, Brophy et al. have demonstrated that the fraction of drug release of any shape of matrix will follow the general equation of the following type:83
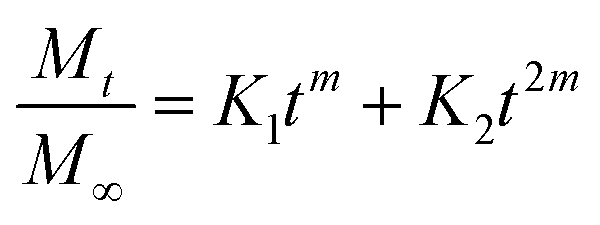
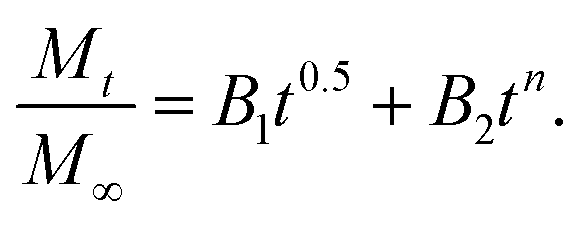
By considering the models of Peppas and Borphy et al., and after careful observation of our in vitro release profile, we have fitted our experimental results with the following equation:
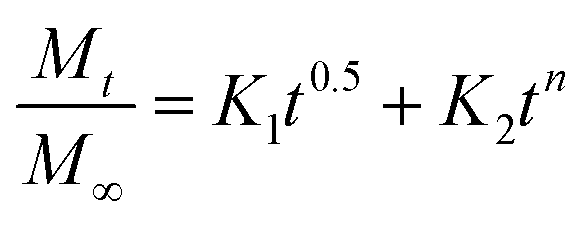
The reason behind using two terms in the equation is due to the fact that the data fitted well by considering the first part of the equation for an initial 8 h, while the second part of equation takes care of the experimental data for times greater than 8 h. The diffusion coefficients of the tablets in the release medium are calculated by plotting the release profile as a function of the square root of time up to 8 h. The linear fitting of this graph gives the value of the slope, which is the value of K1 (448DCs/Aa03)1/2. Using the slope value the diffusion coefficients were calculated by using the formula D = (slope)2/(448Cs/Aa03)1/2. Plots of the release versus the square root of time with linear fitting are shown in Fig. S1 and S2 in the ESI.†
The calculated values of the diffusion coefficients for both pH 7 and pH 5 are given in Tables 2 and 3 respectively.
| Sample | Diffusion coefficient (D) (m2 s−1) × 10−8 | n | K2 × 10−5 |
|---|---|---|---|
| Bare-HA | 1.425 | 0.77 | −1.312 |
| B-HA | 3.9 | 0.827 | −1.512 |
| C-HA | 2.96 | 0.91 | −0.376 |
| P-HA | 4.82 | 0.83 | −1.517 |
| Sample | Diffusion coefficient (D) (m2 s−1) × 10−8 | n | K2 × 10−5 |
|---|---|---|---|
| Bare-HA | 3.4 | 0.74 | −2.907 |
| B-HA | 12.1 | 0.80 | −3.708 |
| C-HA | 4.96 | 0.77 | −2.633 |
| P-HA | 7.314 | 0.83 | −1.063 |
Next, we present the fitted in vitro release profile of our samples at pH 7.4 (Fig. 11). The fitting results show that two mechanisms govern the release profile in our case. For the initial hours, i.e. up to 8 h, it is the Fickian transport that predominates the release. These plots clearly indicate that up to 8 h the release profile follows the equation:
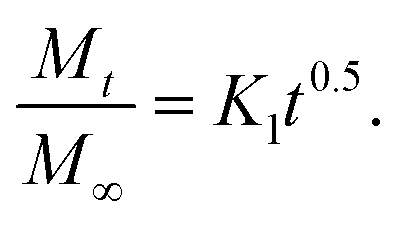
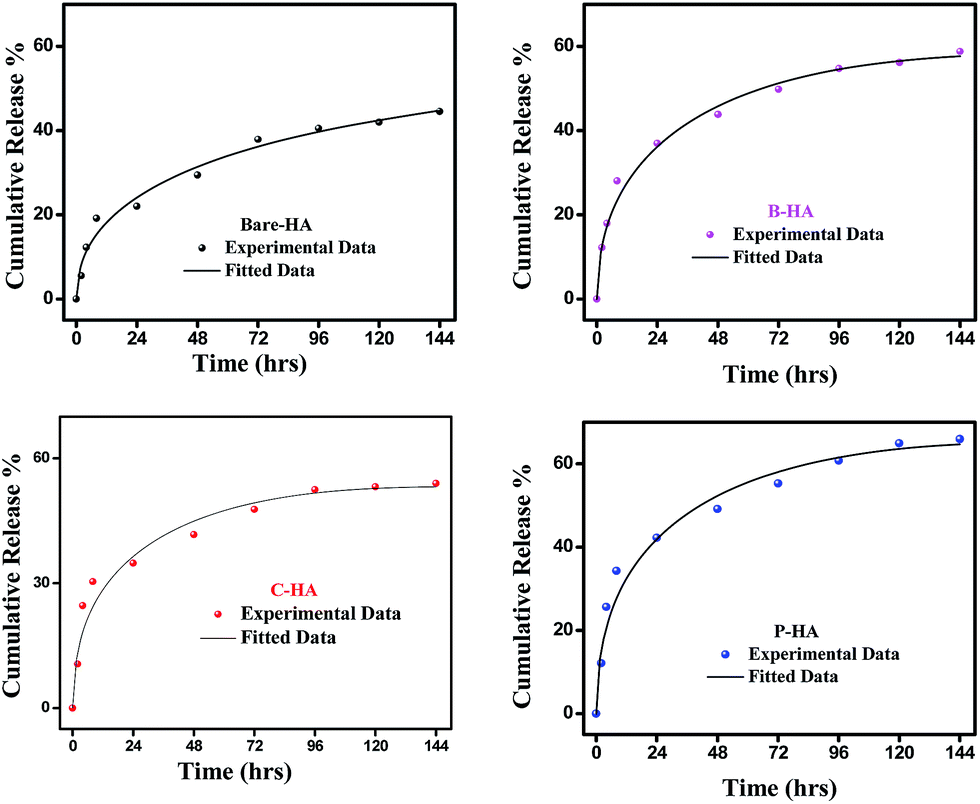 | ||
| Fig. 11 Fitted drug release profiles of the synthesized HA tablets at pH 7.4. | ||
Therefore, we attribute the initial release to be diffusion controlled in our case. This again proves why there is a burst release initially. The plot of the release profile data with Fickian fitting up to 8 h is shown in Fig. S4 in the ESI.† Further, the diffusion coefficient values of each tablet follow the order: P-HA > B-HA > C-HA > bare-HA, which is the drug release order of the HA tablets in the initial period. After 8 h the release profile deviates from the Fickian transport mechanism and follows the equation:
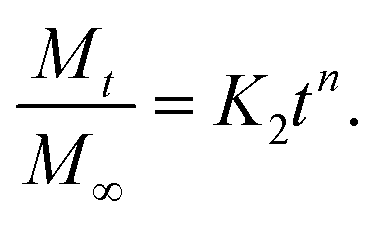
The plot of the release profile with the above equation is also shown in Fig. S4 in the ESI† for a longer period of time, i.e. up to 144 h. We have calculated the K2 and n values from the fitting data and an attempt has been made to describe the release mechanism for a longer period of time. Table 2 represents the K2 and n values of each HA-tablet obtained from the fitting of the experimental data for the second term of eqn (3). It can be seen from the table that the coefficient of n is greater than 0.5 and less than 1, i.e. the n values lie between 0.5 < t < 1. These values lies between the Fickian transport and case-II mechanism and hence are attributed to anomalous transport.83 Therefore, we propose that Fickian transport dominates the release profile in the initial period, i.e. up to 8 h, while for longer times the anomalous transport takes over the Fickian mechanism. Further, the careful observation of the K2 values indicates that it follows the order P-HA < B-HA < bare-HA < C-HA, and the n value varies as C-HA > P-HA > B-HA > bare-HA. The K2 and n values support our drug release order that varies as P-HA > B-HA > C-HA > bare-HA. It can be seen that a small deviation is observed in the case of C-HA in the order of K2 and n when compared with the release order. However, the highest value of n nullifies the lower value of K2 as compared to bare-HA. Indeed, Peppas et al. have shown that with the decrease in the K2 value the Fickian fraction of the drug release becomes larger, and this is also related to the particle size distribution, i.e. the smaller the particle size the larger the release rate.80 Our TEM results show that P-HA has the smallest particle size and hence the highest release rate, while bare-HA has the largest particle size leading to the lowest release rate.
Next we present the fitted data of the in vitro release profile at pH 5 (Fig. 12). The different parameters and the diffusion coefficient of the different HAs are listed in Table 3. The trend is the same as for pH 7.4 for longer times. However, an anomaly is observed in the initial time of release. For the initial time the drug release is highest for B-HA and follows the order B-HA > P-HA > C-HA > bare-HA, as given in Table 3. However, after 40 h again the release rate of P-HA is the highest. This may be because over longer periods, diffusion is no longer controlling the release mechanism and anomalous transport takes over the Fickian transport. The fitting trend of pH 5 was similar to that of pH 7.4, i.e. the Fickian transport controls the release mechanism for the initial 8 h and the anomalous transport takes over beyond 8 h. The separate plots for the Fickian transport and anomalous transport are shown in Fig. S3 in the ESI.† The fitting parameters for pH 5 are given in Table 3.
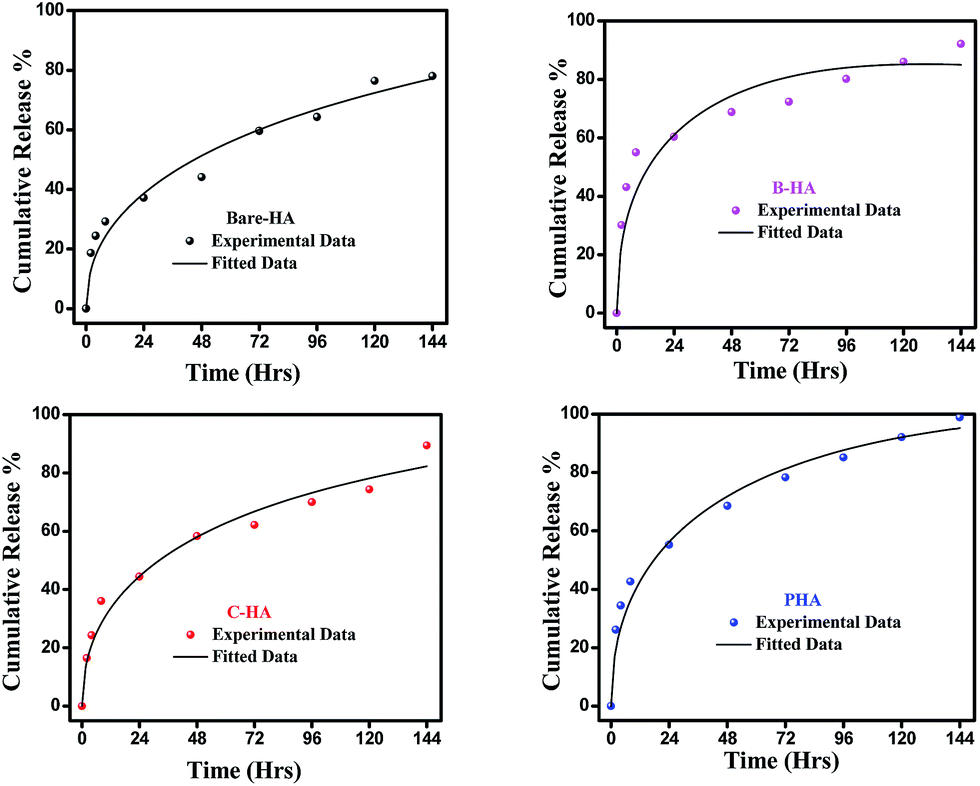 | ||
| Fig. 12 Fitted drug release profile of the synthesized HA tablets at pH 5. | ||
The release pattern showed that, in all the cases, there is an initial burst release followed by prolonged release. The initial rapid release is attributed to the initial high difference in the drug concentration gradient between the scaffolds and the release medium. This is further supported by the fitting data, i.e. initially the release mechanism is governed by diffusion, i.e. Fickian transport. This burst release is advantageous for some applications such as wound treatment (where a burst release followed by diminishing need for the drug is required), pulsatile release and targeted drug delivery where triggered burst release and rapid action of the drug is needed.74
These experiments reveal that these nano-HAs also have a good pH-controlled release function and are promising for applications as pH responsive drug nanocarriers that can respond to physiopathological pH signals to control the drug release process. The drug loading and release study revealed that the matrix-mediated synthesis of HA enhances the drug delivery efficiency. Among the three matrices, the P matrix showed the best release efficiency in both neutral and acidic environments.
Altogether these results suggest that the drug loading and release capability is governed by factors like particle size, surface area and surface properties, which therefore explains the superior performance of P-HA.
4. Conclusion
In summary, this work highlights a bio-inspired approach of materials synthesis where interfacial molecular interactions at the organic–inorganic interface assist the site specific nucleation and growth of HA nanoparticles, ideal for future clinical applications. Further, the use of an organic matrix governs the morphology, function and self-assembled architecture of these nanoparticles. Among the three matrices used, P yielded the best results in terms of the particle size, stoichiometry and drug loading capacity. In addition we have shown that the use of matrices led to the increase of the drug releasing efficiency from 44.5% to 66% at pH 7.4, while at pH 5 the drug release capacity increased from 78% to 98.92% and the biocompatibility test stood the test of time. Further, the fitting of the in vitro release profile shows that Fickian transport governs the release mechanism initially for all HA-tablets, while for longer times the release is governed by anomalous transport. This study throws light on the possible use of templating agents for HA synthesis in order to achieve the desired HA with monodisperse size and uniform shape. This study will definitely open up the scope of using selective matrices for HA synthesis according to the desired properties. Additionally, a study as to why the particle size follows the order of bare-HA > C-HA > B-HA > P-HA is underway and will be reported soon.Acknowledgements
The authors would like to thank the CSIR-Ministry of Science and Technology, OLP-230 and ESC-103 for their financial support. Lubna Sheikh acknowledges DST for the INSPIRE Fellowship. Thanks to Dr Ranjan K. Sahu, Y. N. Singhababu for help in the XPS study and Shivendra Sinha for his help with the drug data analysis. The authors acknowledge Prof. Krishna Pramanick, NIT Rourkela, for the cell study facilities.References
- D. Choi, K. G. Marra and P. N. Kumta, Chemical synthesis of hydroxyapatite/poly(ε-caprolactone) composites, Mater. Res. Bull., 2004, 39, 417–432 CrossRef CAS.
- G. Balasundaram, M. Sato and T. J. Webster, Using hydroxyapatite nanoparticles and decreased crystallinity to promote osteoblast adhesion similar to functionalizing with RGD, Biomaterials, 2006, 27, 2798–2805 CrossRef CAS PubMed.
- T. J. Webster, Nanophase ceramics: the future orthopedic and dental implant material, Adv. Chem. Eng., 2001, 27, 125–166 Search PubMed.
- F. S. Kaplan, W. C. Hayes, T. M. Keaveny, A. Boskey, T. A. Einhorn and J. P. Iannotti, Biomaterials, in Orthopedic basic science, ed. S. P. Simon, American Academy of Orthopedic Surgeons, Columbus, OH, 1994, pp. 460–478 Search PubMed.
- M. Nelson, G. Balasundaram and T. J. Webster, Increased osteoblast adhesion on nanoparticulate crystalline hydroxyapatite functionalized with KRSR, Int. J. Nanomed., 2006, 1(3), 339–349 CAS.
- M. E. Nimni, Polypeptide growth factors: targeted delivery systems, Biomaterials, 1997, 18, 1201–1225 CrossRef CAS PubMed.
- M. Otsuka, Y. Matsuda, Y. Suwa, J. L. Fox and W. I. Higuchi, A novel skeletal drug-delivery system using self-setting calcium-phosphate cement. 4. Effects of the mixing solution volume on the drug-release rate of heterogeneous aspirin-loaded cement, J. Pharm. Sci., 1994, 83, 259–263 CrossRef CAS PubMed.
- S. V. Dorozhkin, Calcium orthophosphates, Biomatter, 2011, 1(2), 121–164 CrossRef PubMed.
- A. Singh, Hydroxyapatite, a biomaterial: its chemical synthesis, characterization and study of biocompatibility prepared from shell of garden snail, Helix aspersa, Bull. Mater. Sci., 2012, 35(6), 1031–1038 CrossRef CAS.
- Z. Wang, Z. Xu, W. Zhaoa and N. Sahai, A potential mechanism for amino acid-controlled crystal growth of hydroxyapatite, J. Mater. Chem. B, 2015, 3, 9157–9167 RSC.
- J. S. Al-Sanabani, A. A. Madfa and F. A. Al-Sanabani, Application of Calcium Phosphate Materials in Dentistry, Int. J. Biomater., 2013, 2013, 876132, DOI:10.1155/2013/876132.
- N. Gupta, A. K. Kushwaha and M. C. Chattopadhyaya, Adsorption of cobalt(II) from aqueous solution onto hydroxyapatite/zeolite composite, Adv. Mater. Lett., 2011, 2(4), 309–312 CrossRef CAS.
- K. Yamashita, K. Kitagaki and T. Umegaki, Thermal instability and proton conductivity of ceramic hydroxyapatite at high temperatures, J. Am. Ceram. Soc., 1995, 78(5), 1191–1197 CrossRef CAS.
- K. T. Mahan and M. J. Carey, Hydroxyapatite as a bone substitute, J. Am. Podiatric Med. Assoc., 1999, 89(8), 392–397 CrossRef CAS PubMed.
- T. Albrektsson and C. Johansson, Osteoinduction, osteoconduction and osseointegration, Eur. Spine J., 2001, 10, S96–S101 CrossRef PubMed.
- P. He, S. Sahoo, K. S. Ng, K. Chen, S. L. Toh and J. C. H. Goh, Enhanced osteoinductivity and osteoconductivity through hydroxyapatite coating of silk-based tissue-engineered ligament scaffold, J. Biomed. Mater. Res., Part A, 2013, 101A(2), 555–566 CrossRef CAS PubMed.
- V. P. Orlovskii, V. S. Komlev and S. M. Barinov, Hydroxyapatite and Hydroxyapatite-Based Ceramics, Inorg. Mater., 2002, 38(10), 973–984 CrossRef CAS.
- T. L. Moore, A. S. Schreurs, R. A. Morrison, E. K. Jelen, J. Loo, R. K. Globus and F. Alexis, Polymer-Coated Hydroxyapatite Nanoparticles for the Delivery of Statins, J. Nanomed. Nanotechnol., 2014, 5(5), 237 Search PubMed.
- J. Chen, K. Nan, S. Yin, Y. Wang, T. Wu and Q. Zhang, Characterization and biocompatibility of nanohybrid scaffold prepared via in situ crystallization of hydroxyapatite in chitosan matrix, Colloids Surf., B, 2010, 81, 640–647 CrossRef CAS PubMed.
- E. O. Lopez, A. Mello, H. Senda, L. T. Costa, A. L. Rossi, R. O. Ospina, F. F. Borghi, J. G. S. Filho and A. M. Rossi, Growth of Crystalline Hydroxyapatite Thin Films at Room Temperature by Tuning the Energy of the RF-Magnetron Sputtering Plasma, ACS Appl. Mater. Interfaces, 2013, 5, 9435–9445 Search PubMed.
- M. Kikuchi, A. Yamazaki, R. Otsuka, M. Akao and H. Aoki, Crystal structure of hydroxyapatite synthesized by hydrothermal method, J. Solid State Chem., 1994, 113, 373–378 CrossRef CAS.
- P. Hui, S. L. Meena, G. Singh, R. D. Agarawal and S. Prakash, Synthesis of Hydroxyapatite Bio-Ceramic Powder by Hydrothermal Method, J. Miner. Mater. Charact. Eng., 2010, 9(8), 683–692 Search PubMed.
- H. W. Kim, J. C. Knowles and H. E. Kim, Hydroxyapatite and gelatin composite foams processed via novel freeze-drying and crosslinking for use as temporary hard tissue scaffolds, J. Biomed. Mater. Res., Part A, 2005, 72A(2), 136–145 CrossRef CAS PubMed.
- H. Lu, Z. Qu and Y. Zhou, Preparation and mechanical properties of dense polycrystalline hydroxyapatite through freeze-drying, J. Mater. Sci.: Mater. Med., 1998, 9(10), 583–587 CrossRef CAS PubMed.
- K. Agrawal, G. Singh, D. Puri and S. Prakash, Synthesis and Characterization of Hydroxyapatite Powder by Sol–Gel Method for Biomedical Application, J. Miner. Mater. Charact. Eng., 2011, 10(8), 727–734 Search PubMed.
- D. O. Costa, S. J. Dixon and A. S. Rizkalla, One- and Three-Dimensional Growth of Hydroxyapatite Nanowires during Sol–Gel-Hydrothermal Synthesis, ACS Appl. Mater. Interfaces, 2012, 4, 1490–1499 Search PubMed.
- S. F. Ou, S. Y. Chiou and K. L. Ou, Phase transformation on hydroxyapatite decomposition, Ceram. Int., 2013, 39(4), 3809–3816 CrossRef CAS.
- A. S. F. Alqap, S. Adzila, I. Sopyan, M. Hamdi and S. Ramesh, Thermal Analysis on Hydroxyapatite Synthesis through Mechanochemical Method, BIOMED 2011, IFMBE Proceedings, 2011, vol. 35, pp. 108–111 Search PubMed.
- E. Hossein, S. H. Mehran and T. Mohammadreza, Synthesis and Characterization of Hydroxyapatite Nanocrystals via Chemical Precipitation Technique, Iran. J. Pharm. Sci., 2008, 4(2), 127–134 Search PubMed.
- L. J. Zhanga, X. S. Fenga, H. G. Liua, D. J. Qiana, L. Zhanga, X. L. Yub and F. Z. Cui, Hydroxyapatite/collagen composite materials formation in simulated body fluid environment, Mater. Lett., 2004, 58, 719–722 CrossRef.
- P. N. Chavan, M. M. Bahir, R. U. Mene, M. P. Mahabole and R. S. Khairnar, Study of nanobiomaterial hydroxyapatite in simulated body fluid: formation and growth of apatite, Mater. Sci. Eng., B, 2010, 168, 224–230 CrossRef CAS.
- K. Yamashita, H. Owada, H. Nakagawa, T. Umegaki and T. Kanazawa, Trivalent-Cation-Substituted Calcium Oxyhydroxyapatite, J. Am. Ceram. Soc., 1986, 69(8), 590–594 CrossRef CAS.
- R. Riman, Biomimetic Hydroxyapatite Synthesis, Christina Sever Patent No: US 8,287,914 B2, Oct. 16, 2012.
- H. Elhendawi, R. M. Felfel, M. Bothaina Abd El-Hady and F. M. Reicha, Effect of Synthesis Temperature on the Crystallization and Growth of In Situ Prepared Nanohydroxyapatite in Chitosan Matrix, ISRN Biomater., 2014, 897468 Search PubMed , 8 pages.
- A. Sinha, S. Nayar, A. Agrawal, D. Bhattacharyya and P. Ramachandrarao, Synthesis of nanosized and microporous precipitated hydroxyapatite in synthetic polymers and biopolymers, J. Am. Ceram. Soc., 2003, 86, 357–359 CrossRef CAS.
- S. Nayar, M. K. Sinha, D. Basu and A. Sinha, Synthesis and sintering of biomimetic hydroxyapatite nanoparticles for biomedical applications, J. Mater. Sci.: Mater. Med., 2006, 17, 1063–1068 CrossRef CAS PubMed.
- S. Bhattacharya, L. Sheikh, V. Tiwari, M. Ghosh, J. N. Patel, A. B. Patel and S. Nayar, Protein-polymer functionalized aqueous ferrofluids showing high T2 relaxivity, J. Biomed. Nanotechnol., 2014, 10(5), 811–819 CrossRef CAS PubMed.
- Y. Wang, C. Yang, X. Chen and N. Zhao, Biomimetic formation of hydroxyapatite/collagen matrix composite, Adv. Eng. Mater., 2006, 8, 97–100 CrossRef CAS.
- L. C. Mendes, G. L. Ribeiro and R. C. Marques, In Situ Hydroxyapatite Synthesis: Influence of Collagen on its Structural and Morphological Characteristic, Mater. Sci. Appl., 2012, 3, 580–586 Search PubMed.
- A. Cuneyt Tas, Synthesis of biomimetic Ca-hydroxyapatite powders at 37 °C in synthetic body fluids, Biomaterials, 2000, 21, 1429–1438 CrossRef.
- S. Mann, Biomimetic materials chemistry, VCH publishers, Inc., New York, 1996 Search PubMed.
- Y. H. Liang, C. H. Liu, S. H. Liao, Y. Y. Lin, H. W. Tang, S. Y. Liu, I. R. Lai and K. C.-W. Wu, Cosynthesis of Cargo-Loaded Hydroxyapatite/Alginate Core–Shell Nanoparticles (HAP@Alg) as pH-Responsive Nanovehicles by a Pregel Method, ACS Appl. Mater. Interfaces, 2012, 4, 6720–6727 Search PubMed.
- L. Chen, J. Hu, J. RaN, X. Shen and H. Tong, A novel nanocomposite for bone tissue engineering based on chitosan-silk sericin/hydroxyapatite: biomimetic synthesis and its cytocompatibility, RSC Adv., 2015, 5, 56410–56422 RSC.
- S. Nayar, A. Sinha, S. K. Das, S. Das and P. Ramachandrarao, In situ synthesis of nanosized cadmium sulphide using bovine serum albumin, J. Mater. Sci. Lett., 2001, 20, 2099–2100 CrossRef CAS.
- T. V. Thamaraiselvi, K. Prabakaran and S. Raeswari, Synthesis of Hydroxyapatite that Mimic Bone Minerology, Trends. Biomater. Artif. Organs., 2006, 19(2), 81–83 Search PubMed.
- X. Liu and S. K. Mallapragada, Bioinspired synthesis of organic/inorganic nanocomposite materials mediated by biomolecules, On Biomimetics, In Tech publishers, Inc., 2011 Search PubMed.
- V. Uskoković and D. P. Uskoković, Nanosized hydroxyapatite and other calcium phosphates: chemistry of formation and application as drug and gene delivery agents, J. Biomed. Mater. Res., Part B, 2011, 96B(1), 152–191 CrossRef PubMed.
- H. W. Kim, J. C. Knowles and H. E. Kim, Hydroxyapatite/poly(ε-caprolactone) composite coatings on hydroxyapatite porous bone scaffold for drug delivery, Biomaterials, 2004, 25(7–8), 1279–1287 CrossRef CAS PubMed.
- Y. Cai, Y. Liu, W. Yan, Q. Hu, J. Tao, M. Zhang, Z. Shic and R. Tang, Role of hydroxyapatite nanoparticle size in bone cell proliferation, J. Mater. Chem., 2007, 17, 3780–3787 RSC.
- S. Koutsopoulos, On of hydroxyapatite crystals: a review study on the analytical methods, J. Biomed. Mater. Res., 2002, 4, 62 Search PubMed.
- L. Berzina-Cimdina and N. Borodajenko, Research of Calcium Phosphates Using Fourier Transform Infrared Spectroscopy, Infrared Spectroscopy – Materials Science, Engineering and Technology, ed. T. Theophile, Publisher InTech, published online, 25 April 2012, published in print edition April 2012, ISBN 978-953-51-0537-4 Hard cover, 510 pages Search PubMed.
- L. Sheikh, N. Mahto and S. Nayar, In Situ Synthesis of Hydroxyapatite Nanocomposites Using Iron Oxide Nanofluids at Ambient Conditions, J. Mater. Sci.: Mater. Med., 2015, 26, 5393 CrossRef PubMed.
- Y. W. Gu, K. A. Khor and P. Cheang, Bone-like apatite layer formation on hydroxyapatite prepared by spark plasma sintering (SPS), Biomaterials, 2004, 25, 4127–4134 CrossRef CAS PubMed.
- S. Kaciulis, G. Mattogno, L. Pandolfi, M. Cavalli, G. Gnappi and A. Montenero, XPS study of apatite-based coatings prepared by sol–gel technique, Appl. Surf. Sci., 1999, 151, 1–5 CrossRef CAS.
- M. A. Stranick and M. J. Root, Influence of strontium on monofluorophosphate uptake by hydroxyapatite XPS characterization of the hydroxyapatite surface, Surf. Interface Anal., 2002, 34, 45–49 CrossRef.
- M. P. Casaletto, S. Kaciulis, G. Mattogno, A. Mezzi, L. Ambrosio and F. Branda, XPS characterization of biocompatible hydroxyapatite-polymer coatings, Surf. Interface Anal., 2002, 34, 45–49 CrossRef CAS.
- K. McLeod, S. Kumar, R. S. C. Smart, N. Dutta, N. H. Voelcker, G. I. Anderson and R. Sekel, XPS and bioactivity study of the bisphosphonate pamidronate adsorbed onto plasma sprayed hydroxyapatite coatings, Appl. Surf. Sci., 2006, 253, 2644–2651 CrossRef CAS.
- M. C. Changa and J. Tanaka, XPS study for the microstructure development of hydroxyapatite–collagen nanocomposites cross-linked using glutaraldehyde, Biomaterials, 2002, 23, 3879–3885 CrossRef.
- G. Vazquez, C. P. Barba and N. Munguia, Stoichiometric hydroxyapatite obtained by precipitation and sol gel processes, Rev. Mex. Fis., 2005, 51(3), 284–293 Search PubMed.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Eden Prairie Minnesota, 1st edn, 1979 Search PubMed.
- S. V. Dorozhkin, Calcium orthophosphates – occurrence, properties, biomineralization, pathological calcification and biomimetic applications, Biomatter, 2011, 1(2), 121–164 CrossRef PubMed.
- E. O. Lopez, A. Mello, H. Senda, L. T. Costa, A. L. Rossi, R. O. Ospina, F. F. Borghi, J. G. S. Filho and A. M. Rossi, Growth of Crystalline Hydroxyapatite Thin Films at Room Temperature by Tuning the Energy of the RF-Magnetron Sputtering Plasma, ACS Appl. Mater. Interfaces, 2013, 5, 9435–9445 Search PubMed.
- Y. Abe, Y. Okazaki, K. Hiasa, K. Yasuda, K. Nogami, W. Mizumachi and I. Hirata, Bioactive Surface Modification of Hydroxyapatite, BioMed Res. Int., 2013, 626452, DOI:10.1155/2013/626452.
- T. I. Ivanova, O. V. Frank-Kamenetskaya, A. B. Koltsov and V. L. Ugolkov, Crystal Structure of Calcium-Deficient Carbonated Hydroxyapatite. Thermal Decomposition, J. Solid State Chem., 2001, 160, 340–349 CrossRef CAS.
- M. Kikuchi, A. Yamazaki and R. Otsuka, Crystal Structure of Sr-Substituted Hydroxyapatite synthesized by Hydrothermal Method, J. Solid State Chem., 1994, 113, 373–378 CrossRef CAS.
- K. Sato, Y. Suetsugu, J. Tanaka, S. Ina and H. Monma, The Surface Structure of Hydroxyapatite Single Crystal and the Accumulation of Arachidic Acid, J. Colloid Interface Sci., 2000, 224, 23–27 CrossRef CAS PubMed.
- G. Iucci, G. Polzonetti, G. Infante and L. Rossi, XPS and FT-IR spectroscopy study of albumin adsorption on the surface of a p-conjugated polymer film, Surf. Interface Anal., 2004, 36, 724–728 CrossRef CAS.
- K. M. Shakesheff, C. Evora, I. Soriano and R. Langer, The Adsorption of Poly(vinyl alcohol) to Biodegradable Microparticles Studied by X-Ray Photoelectron Spectroscopy (XPS), J. Colloid Interface Sci., 1997, 185, 538–547 CrossRef CAS PubMed.
- T. I. Ivanova, O. V. Frank-Kamenetskaya, A. B. Koltsov and V. L. Ugolkov, Crystal Structure of Calcium-Deficient Carbonated Hydroxyapatite. Thermal Decomposition, J. Solid State Chem., 2001, 160, 340–349 CrossRef CAS.
- A. S. F. Alqap, S. Adzila, I. Sopyan, M. Hamdi and S. Ramesh, Thermal Analysis on Hydroxyapatite Synthesis through Mechanochemical Method, Biomed, IFMBE Proceedings, 2011, vol. 35, pp. 108–111 Search PubMed.
- L. C. Mendes, G. L. Ribeiro and R. C. Marques, In Situ Hydroxyapatite Synthesis: Influence of Collagen on its Structural and Morphological Characteristic, Mater. Sci. Appl., 2012, 3, 580–586 Search PubMed.
- M. C. Chang, C. C. Ko and W. H. Douglas, Preparation of hydroxyapatite–gelatin nanocomposite, Biomaterials, 2003, 24, 2853–2862 CrossRef CAS PubMed.
- M. Betsiou, C. Sikalidis and A. Papageorgiou, Adsorption of Oxaliplatin by Hydroxyapatite, Bioautomation, 2007, 8, 138–145 Search PubMed.
- Z. N. Al-Sokanee, A. A. H. Toabi, M. J. Al-assadi and E. A. S. Alassadi, The Drug Release Study of Ceftriaxone from Porous Hydroxyapatite Scaffolds, AAPS PharmSciTech, 2009, 10, 772, DOI:10.1208/s12249-009-9265-7.
- L. Palanikumar, S. Ramasamy, G. Hariharan and C. Balachandran, Influence of particle size of nano zinc oxide on the controlled delivery of amoxicillin, Appl. Nanosci., 2013, 3, 441–451 CrossRef CAS.
- N. F. Mohammad, R. Othman and F. Y. Yeoh, Nanoporous hydroxyapatite preparation methods for drug delivery applications, Rev. Adv. Mater. Sci., 2014, 38, 138–214 Search PubMed.
- X. Zhao, Y. Zhu, F. Chen, B. Lu and J. Wu, Nanosheet-assembled hierarchical nanostructures of hydroxyapatite: surfactant-free microwave hydrothermal rapid synthesis, protein/DNA adsorption and pH-controlled release, CrystEngComm, 2013, 15, 206–212 RSC.
- T. Higuchi, Mechanism of Sustained-Action Medication. Theoretical Analysis of Rate of Release of Solid Drugs Dispersed in Solid Matrices, J. Pharm. Sci., 1963, 52(12), 1145–1149 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, A simple equation for description of solute release. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- N. A. Peppas and J. J. Sahlin, A simple equation for the description of solute release. III. Coupling of diffusion and relaxation, Int. J. Pharm., 1989, 57, 169–172 CrossRef CAS.
- A. Cosijns, C. Vervaet, J. Luyten, S. Mullen, F. Siepman, L. Van Hoorebek, B. Masschaele, V. Cnuddan and J. P. Remon, Porous hydroxyapatite tablets as carriers for low-dosed drugs, Eur. J. Pharm. Biopharm., 2007, 67(2), 498–550 CrossRef CAS PubMed.
- M. Öner, E. Yetiz, E. Ay and U. Uysal, Ibuprofen release from porous hydroxyapatite tablets, Ceram. Int., 2011, 37, 2117–2125 CrossRef.
- M. R. Brophy and P. B. Deasy, Application of the Higuchi model for drug release from dispersed matrices to particles of general shape, Int. J. Pharm., 1981, 31, 41 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra06759j |
| This journal is © The Royal Society of Chemistry 2016 |