
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–charge transfer property relationship in self-assembled discotic liquid-crystalline donor–acceptor dyad and triad thin films
Kwang Jin
Lee†
a,
Jae Heun
Woo†
ab,
Yiming
Xiao
c,
Eunsun
Kim
a,
Leszek Mateusz
Mazur
cd,
David
Kreher
c,
André-Jean
Attias
 c,
Katarzyna
Matczyszyn
d,
Marek
Samoc
d,
Benoît
Heinrich
e,
Stéphane
Méry
e,
Frédéric
Fages
f,
Loic
Mager
e,
Anthony
D'Aléo
f,
Jeong Weon
Wu
a,
Fabrice
Mathevet
*c,
Pascal
André
*ag and
Jean-Charles
Ribierre‡
*a
c,
Katarzyna
Matczyszyn
d,
Marek
Samoc
d,
Benoît
Heinrich
e,
Stéphane
Méry
e,
Frédéric
Fages
f,
Loic
Mager
e,
Anthony
D'Aléo
f,
Jeong Weon
Wu
a,
Fabrice
Mathevet
*c,
Pascal
André
*ag and
Jean-Charles
Ribierre‡
*a
aDepartment of Physics, CNRS-Ewha International Research Center, Ewha Womans University, Seoul 120-750, Korea. E-mail: ribierre@opera.kyushu-u.ac.jp
bCenter for Length, Division of Physical Metrology, Korea Research Institute of Standards and Science (KRISS), 267 Gajeong-ro, Yuseong-gu, Daejeon, 305-340, South Korea
cSorbonne Universités, UPMC Univ Paris 06, CNRS, Institut Parisien de Chimie Moléculaire, UMR 8232, Chimie des Polymères, 4 Place Jussieu, 75005 Paris, France. E-mail: fabrice.mathevet@upmc.fr
dAdvanced Materials Engineering and Modelling Group, Wroclaw University of Science and Technology, 50-370 Wroclaw, Poland
eInstitut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504, CNRS – Université de Strasbourg, 23 Rue du Loess, 67034 Strasbourg, France
fAix Marseille Université, CNRS, CINaM UMR 7325, Campus de Luminy, Case 913, 13288 Marseille, France
gElements Chemistry Laboratory, RIKEN, Wako 351-0198, Japan. E-mail: pjpandre@riken.jp
First published on 6th June 2016
Abstract
The photophysical properties of donor–acceptor (D–A) and donor–acceptor–donor (D–A–D) liquid crystalline dyads and triads based on two different discotic mesogens are examined in thin films by steady-state optical spectroscopy and subpicosecond transient absorption measurements. In these systems, triphenylene and perylene bisimide units are covalently linked by flexible decyloxy chain(s) and act as an electron donor (D) and acceptor (A), respectively. These discotic liquid-crystalline systems form well-separated D and A π-stacked columnar structures in thin films. The absorption spectra of the films indicate an aggregation of the perylene bisimide and triphenylene moieties along the columns. Steady-state photoluminescence measurements show a strong fluorescence quenching that is mainly attributed to a photo-induced charge transfer process taking place between the triphenylene and perylene bisimide units. Subpicosecond transient absorption measurements show that the photoinduced charge transfer (CT) states in the dyad and triad films are formed within 0.3 ps and recombine on a 150–360 ps time scale. In addition, a correlation between the dynamics of the charge recombination process and the spacing distances between D and A units can be established in the dyad and triad films. This study provides important information on the relationship between molecular packing and the charge transfer properties in such self-organized D and A columnar nanostructures.
1. Introduction
Control of supramolecular self-assemblies in organic semiconducting thin films is a critical issue for improving the performance of organic optoelectronic devices.1–11 In this context, liquid crystalline (LC) molecular semiconductors, able to self-assemble into functional nanostructures, have attracted a great deal of attention.12–18 In particular, self-organization of electron donor (D) and acceptor (A) moieties into highly ordered molecular architectures is extremely promising for organic solar cell applications.19–24 Possible control of the energy and the charge transfer processes together with the existence of percolation pathways for both holes and electrons via the formation of well-defined, nanosegregated donor–acceptor domains are indeed of critical importance for enhancing photovoltaic performances. Among the variety of LC donor–acceptor (D–A) materials developed so far,25–28 covalently linked electron D–A dyad and triad molecules are of interest as their self-assembly can produce highly ordered well-separated structures of well-stacked D and A π-molecules.29–46 In such supramolecular systems, excitons are photogenerated close to the D–A interface and pathways for hole and electron transport are well defined, providing a suitable configuration for efficient formation of excited charge transfer (CT) states and ambipolar charge transport. While the power conversion efficiency of organic solar cells based on a LC nematic donor material blended with a fullerene derivative has recently reached 9.3%,47 the highest efficiency reported in devices based on D–A liquid crystals is about 2.7%.32,48 This strongly suggests that new LC D–A materials must be developed and that a better understanding of the structure–property relationship is needed in these systems to improve their optoelectronic and photovoltaic performance.Here, we study the photophysical properties of organic thin films based on LC D–A dyad and D–A–D triad systems composed of triphenylene electron donor and perylene bisimide electron acceptor units covalently linked by flexible decyloxy chain(s). Their chemical structures are displayed in Fig. 1a. The synthesis and the characterization of the structural properties in the solid-state of these compounds have been previously reported.49 It was shown that these discotic LC dyad and triad systems spontaneously self-organize into columnar nanostructures, with D and A moieties segregated into either alternated stacks within mixed columns or in distinct columns. In parallel, we demonstrated that conformational effects taking place in solution due to the flexible aliphatic chain connecting the donor and acceptor units lead to an intricate interplay between intramolecular electronic energy transfer and photoinduced electron transfer processes.50 Here, we examined the photophysical processes of these dyad and triad molecular systems in thin films using steady-state and time-resolved optical spectroscopy techniques, and a special attention is brought to the relation between charge dynamics and structural properties of the films. The overall results provide new important insights into photoinduced charge transfer processes and the structure–property relationship in self-organized discotic D–A materials. This study should be taken into account in future works devoted to the design and the development of novel D–A molecular or supramolecular systems with optimized charge separation and recombination dynamics.
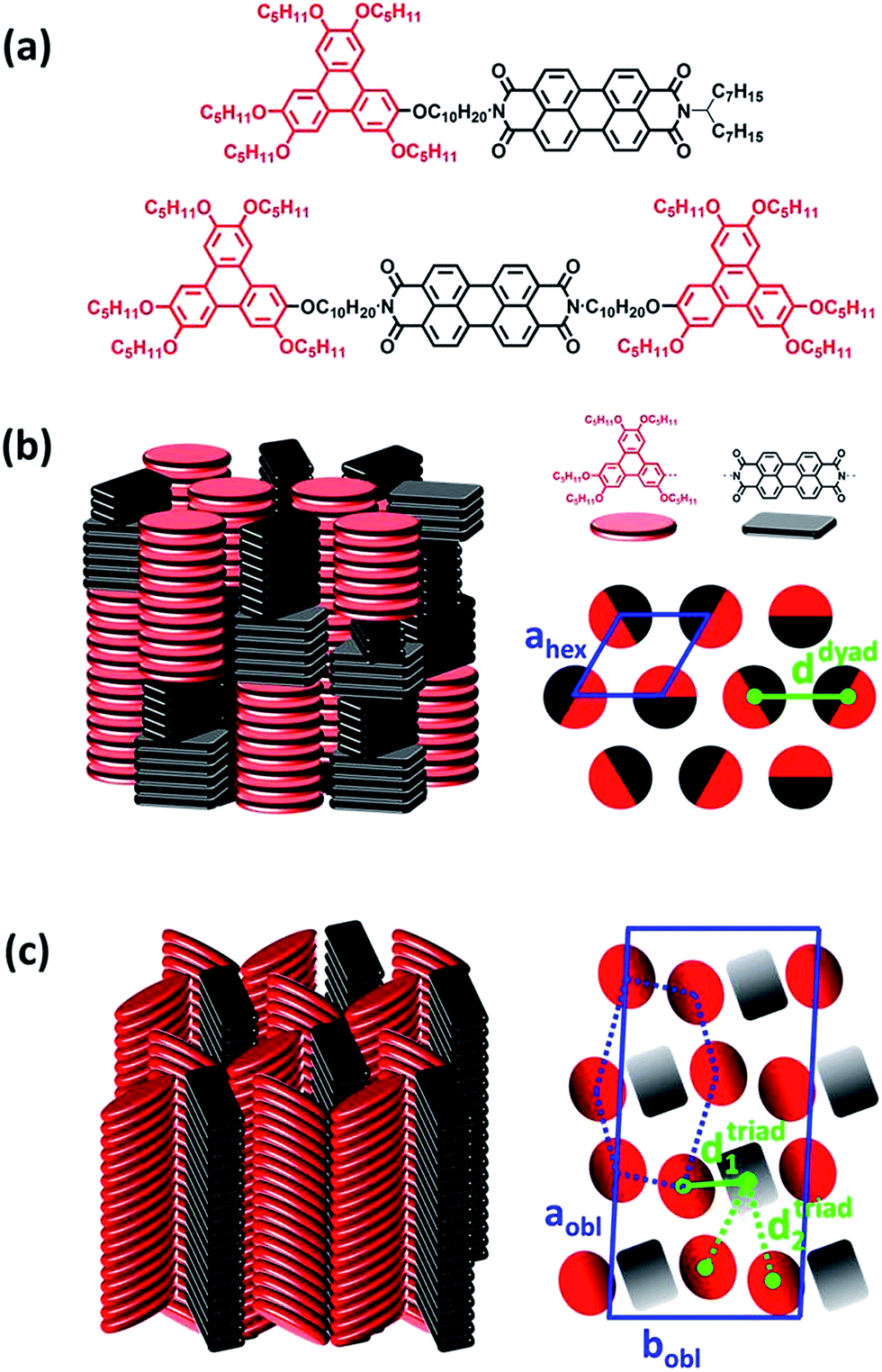 | ||
| Fig. 1 (a) Chemical structures of the dyad and triad molecules. (b) Schematic representation of (left) the self-organization within the Colhex mesophases of the D–A dyad and (right) the hexagonal lattice (blue lozenge) formed by undifferentiated columns. The distance ddyad ≈ 1.9 nm (green line) corresponds to the average center-to-center spacing distance between D and A columns. (c) Schematic representation of (left) the self-organization within the Colobl mesophases of the D–A–D triad after annealing and (right) the oblique lattice (blue parallelogram) formed by intermingled distinct columns located at the nodes of distorted hexagonal lattices (blue dashed distorted hexagon). The distances dtriad1 ≈ 1.4 and dtriad2 ≈ 2.5 nm (respectively green solid and dashed line) correspond to the two average center-to-center spacing distances between D and A columns coexisting within the triad columnar arrangement. | ||
2. Experimental
The synthesis and the purification procedure of the dyad and triad molecules used in this study have been previously published.49 For photophysical measurements, thin films were prepared onto pre-cleaned fused silica substrates by spin-coating from chloroform solutions. Film annealing was then performed in nitrogen-filled glove box or in vacuum at 130 °C for 3 hours. Typical film thicknesses for both dyad and triad films were measured by a Dektak profilometer and were found to be around 40 nm. For the concentration dependence of the absorption and steady-state fluorescence spectra in a poor solvent, diluted dyad solutions were prepared in methylcyclohexane at various concentrations ranging from 1.1 × 10−3 to 2.1 × 10−6 M and held in a quartz cuvette at ambient temperature.UV-vis absorption spectra of the solutions and thin films were recorded on a Hitachi U-3310 spectrophotometer. Steady-state fluorescence spectra of the spin-coated films were obtained using a Varian Cary Eclipse spectrofluorimeter. The pump and probe pulses in the femtosecond transient absorption measurements were produced by an OPA (OTOPAS-Prime, Spectra Physics) optically-pumped by a Ti:Sapphire regenerative amplifier (Spitfire Pro XP, Spectra Physics) at 800 nm. A Mai Tai laser based on a mode-locked titanium-doped sapphire laser (Tsunami) pumped by a continuous wave Nd:YVO4 laser (Millennia) was used as a seeding laser for the regenerative amplifier. In our experiments, the wavelength of the pumping beam was 325 nm and was obtained by 4th harmonic generation using two BBO crystals after the OPA output operating at 1300 nm. The power of the pump beam was adjusted using a set of neutral density filters. A white light continuum generated using a 2 mm thick sapphire crystal photo-excited by half of the 800 nm regenerative amplifier output was used for the probe beam. A delay line was used to control the time delay between the pump and the probe beams. The pump beam was modulated by a mechanical chopper at 220 Hz and the differential transmission ΔT/T of the probe beam was recorded as a function of the delay time using a photodiode and a lock-in detection. Note that the films were in ambient atmosphere during the measurements.
3. Results and discussion
3.1 Self-organization properties of the discotic liquid-crystalline D–A dyad and D–A–D triad in solid-state
The structural and self-organization properties of these dyad and triad in the solid-state have been previously investigated using different experimental techniques such as polarized optical microscopy, differential scanning calorimetry, temperature dependent X-ray diffraction and grazing incidence X-ray scattering (GIXS).49 It was demonstrated that both D–A dyad and D–A–D triad architectures present liquid crystalline properties and spontaneously self-organize into columnar mesophases at room temperature, with D and A moieties segregated into either alternated stacks within mixed columns or in well distinct columns. A schematic representation of the molecular packing of dyad and triad is displayed in Fig. 1. In case of the dyad, the triphenylene and perylene bisimide units alternate into different groups of stacked mesogens of same nature along a columnar axis long of about 30 units. The permutation of entire groups of columns preserves the segregation of D and A neighbouring columns at the local range, but ultimately leads to a hexagonal arrangement (Colhex) of apparent undifferentiated columns where the average center-to-center spacing distance (d) between D and A columns is around ddyad ≈ 1.9 nm as shown in Fig. 1b.Regarding the molecular packing of the triad, this compound also presents a columnar organization which however differs from the dyad arrangement in forming similar intermingled distinct D and A columns but located at the nodes of distorted hexagonal lattices with more specifically a long range correlated oblique lattice (Colobl) with tilted triphenylene and perylene diimide units within their columns after annealing the fluid state of the mesophase (Fig. 1c). This original organization is due to the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of triphenylene to perylene units in the triad which is favorable to the differentiation of columns as the major component can occupy the nodes of the distorted honeycomb lattice and the minor component, the centers of the pseudo-hexagonal cells, as displayed in Fig. 1b. Thus, in contrast to the dyad case, two average center-to-center spacing distances (d1 and d2) between D and A columns coexist in bulk that can be evaluated from the lattice geometry to be around dtriad1 ≈ 1.4 and dtriad2 ≈ 2.5 nm. It is worth noticing that the distribution of alternate stacks of the different mesogens along the columns in case of the dyad leads to an undifferentiation of the columns at the macroscopic scale but it has little consequence on the close packing of the neighbouring blocks. Overall, the structural properties as described above point out that the D–A–D triad, showing differentiated D and A columns, constitutes a quite promising molecular self-assembly for optoelectronic applications.
:1 stoichiometry of triphenylene to perylene units in the triad which is favorable to the differentiation of columns as the major component can occupy the nodes of the distorted honeycomb lattice and the minor component, the centers of the pseudo-hexagonal cells, as displayed in Fig. 1b. Thus, in contrast to the dyad case, two average center-to-center spacing distances (d1 and d2) between D and A columns coexist in bulk that can be evaluated from the lattice geometry to be around dtriad1 ≈ 1.4 and dtriad2 ≈ 2.5 nm. It is worth noticing that the distribution of alternate stacks of the different mesogens along the columns in case of the dyad leads to an undifferentiation of the columns at the macroscopic scale but it has little consequence on the close packing of the neighbouring blocks. Overall, the structural properties as described above point out that the D–A–D triad, showing differentiated D and A columns, constitutes a quite promising molecular self-assembly for optoelectronic applications.
3.2 Absorption and steady-state fluorescence emission spectra in solution and in self-organized thin films
As displayed in Fig. 2a, at the lowest concentration of 2.1 × 10−6 M, the methylcyclohexane (MCH) solution of the dyad shows absorption spectra of the perylene bisimide unit similar to that previously measured in dichloromethane (DCM) solution,50 indicating that there are no intermolecular interactions between dyad molecules. When increasing the dyad concentration in MCH, the molar absorption coefficient of the perylene bisimide moiety decreases and the absorption band profile becomes broader and less structured, in relation with the formation of aggregates. No clear spectral shift is associated to the aggregation process contrarily to other perylene derivatives,46 but an isobestic point nevertheless appears at about 530 nm, above which the absorption of the concentrated solution is higher, which reveals the interactions between loosely aggregated dyad molecules at higher concentration. The associated broader absorption band and hypochromic effect moreover indicate that aggregates are not ideal and that some disorder is induced by the presence of triphenylene-containing side chains.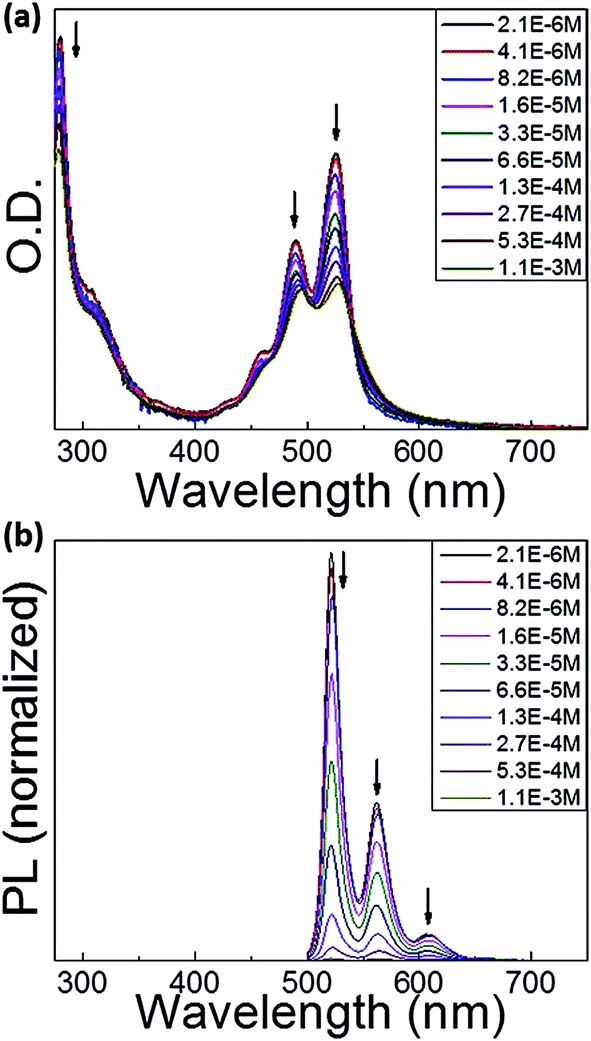 | ||
| Fig. 2 (a) Concentration-dependent absorption spectra of the dyad in methylcyclohexane solution at room temperature. Concentration varied from 1.1 × 10−3 to 2.1 × 10−6 M. Arrows show the changes when increasing the concentration. (b) Concentration-dependent fluorescence spectra of the dyad in methylcyclohexane solution at room temperature. Excitation wavelength was 450 nm. Concentration varied from 1.1 × 10−3 to 2.1 × 10−6 M. Arrows show the changes when increasing the concentration. | ||
The absorption band of the perylene bisimide unit of dyad in a thin film is 15 nm red-shifted compared to the spectrum in diluted DCM solution (Fig. 3). The less defined vibronic structure is consistent with increased molecular interactions in the aggregates.41,51,52 In particular the ratios between the (0, 0) and (0, 1) vibronic peaks located in films at 527 and 491 nm are smaller than those measured in DCM solution.50 A similar trend is observed for the triad derivative. Peak-to-peak ratio values of 1.37 and 0.91 respectively measured in dyad and triad films, should then be compared to the 1.6 value found in DCM solution. These spectral changes are the direct signature of strong molecular π–π stacking interactions between the perylene bisimide units in the solid state. This is consistent with the X-ray diffraction patterns showing the presence of a scattering signal around dπ = 3.5 Å,49 characteristic of the van der Waals contact distance between aromatic planes along columns.
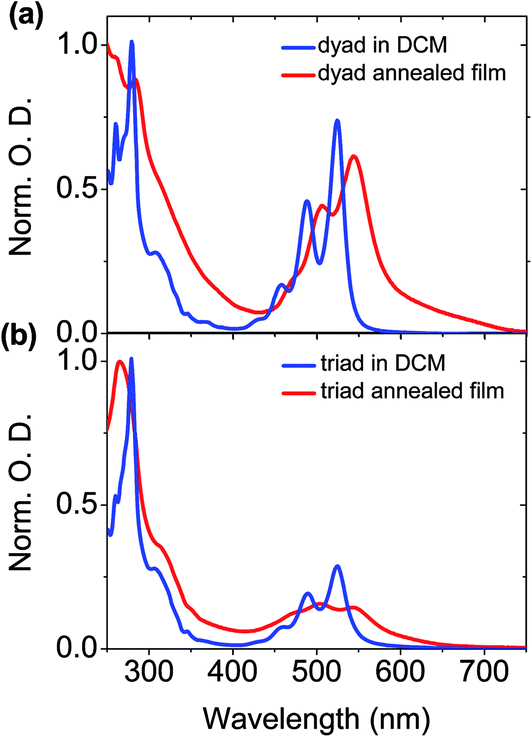 | ||
| Fig. 3 Normalized absorption spectra of the (a) dyad and (b) triad in DCM solution and in neat film. | ||
In dyad films, D and A moieties form stacks of few tens of units alternating along single columns. As a consequence, the structure consists of a hexagonal arrangement of unique type of columns, mixing in-plane lying D and A rings. As such, the face-to-face stacking of the perylene bisimide chromophores is consistent at first glance with H-type aggregation. Yet, the thin film absorption spectrum is red-shifted as compared to that measured in DCM solution.53 The triad compound also self-organizes into columnar mesophases at room temperature. However, in this latter case, the organization consists of an oblique arrangement of distinct D and A columns. Within both types of columns, the discotic cores are tilted with respect to the columnar axes within both D and A columns, and therefore imply a sliding of the aromatic planes with the respect to each other. Such a structural feature corresponds to J-type aggregation, which is consistent with the red-shift of the perylene bisimide absorption band observed in thin films (Fig. 3).
To explain the behaviour of the absorption spectrum of the dyad in film, it should be mentioned that the presence of sterically demanding swallow aliphatic tails should prevent the formation of perfect face-to-face stacking of perylene bisimide moieties, as sketched in Fig. 1b. A possible local organization should rather include a slight shift/slide of the perylene units, while maintaining a normal orientation within columns. Thus, in both triad and dyad, we might have the formation of J-aggregates, in good agreement with the bathochromic shift of the absorption in the solid-state. While the red-shift in the triad is correlated at longer range in relation with the macroscopic tilt angle, the J-aggregates in the dyad would correspond to the perylene bisimide stacks into which the moieties are slightly and randomly shifted the one from the others. Such a positional disorder between neighbouring dyes can provide indeed some allowness to the optical transition into the lower energy state of the Davydov excitonic coupling.54
With regards to the triphenylene units, the vibronic structure of the absorption spectrum of the dyad in solid state is not as well-defined as the one in DCM solution (Fig. 3). This suggests an aggregation of the triphenylene moieties in the film.55,56 In contrast, film and diluted DCM solution of the triad show similar features regarding the absorption bands of the triphenylene units. Triphenylene units are therefore poorly interacting along the differentiated columns of the triad as along the mixed dyad columns, which is easily understandable considering the peculiar geometry and stacking behaviour of the species. The triphenylene units are indeed genuine discs which naturally pile up to form cylindrical columns as evidenced by the previous X-ray diffraction experiments.49 Their aggregation process is therefore not constrained by the shape averaging to cylinder associated to the alternation of column segments at lattice nodes in the dyad. The aliphatic periphery connected to these units moreover naturally promotes untilted piles with optimal face-to-face superimposition,57 while the geometrical constrains between differentiated column shapes in the triad force tilt angles of 30°, which are deleterious for strong π-stacking interactions.49 In consequence, the comparison of solid state and solution absorption spectra proved to be a fruitful complement to the structural analysis as it provides the molecular justification of the geometrical concepts developed for the description of mesomorphism.
Perylene bisimide derivatives are known to exhibit strong fluorescence in diluted solution with photoluminescence quantum yield (PLQY) close to unity.58,59 In the dyad and triad investigated in this study, emission of perylene bisimide moieties grafted to donor moieties was observed to be much lower in diluted DCM solution, with PLQY of 38 and 11% for dyad and triad, respectively.50 This feature was attributed to a quenching of the emission due to a photo-induced intramolecular charge transfer taking place between interacting triphenylene and perylene bisimide moieties. Fig. 2b displays the concentration dependence of the fluorescence of the dyad in MCH. At low concentrations, the fluorescence spectra show the emission band of isolated perylene bisimide chromophores with a well-resolved vibronic structure. The fluorescence intensity gradually decreases upon increasing the solution concentration, indicating an increase of the fluorescence quenching due to aggregation. We did not observe the emergence of a new broad band at 600–850 nm in dyad and triad, which was observed in similar experiments carried out in perylene bisimide dyes.60 In the solid state, the emission from the perylene bisimide units is found to be extremely weak indicating a strong quenching of the photoluminescence. This is attributed to both the aggregation of perylene bisimide and the photo-induced charge transfer process between triphenylene and perylene bisimide units.
3.3 Subpicosecond transient absorption measurements in self-organized films
Subpicosecond transient absorption spectroscopy measurements in dyad and triad films, were carried out to clarify the mechanism of the exciton quenching, and to gain insight into the photophysical properties in the solid-state to ultimately provide new crucial information on the structure–property relationships in liquid crystalline D–A organic materials. The transient absorption spectra of the dyad and triad films, generated by pumping at 325 nm, are displayed in Fig. 4. Shortly after the photoexcitation, the negative transient absorption responses (ΔA < 0; ΔT/T > 0) at wavelengths shorter than 575 nm are associated with the ground-state bleaching of the perylene bisimide moieties and follow the same features as those of the linear absorption spectra reported in Fig. 2.50 A positive transient absorption feature (ΔA > 0; ΔT/T < 0) above 575 nm is observed in both materials. According to previous works examining the photophysical properties of perylene bisimide-based D–A organic materials in thin films, this feature can be attributed to the absorption of perylene bisimide anions formed through photoinduced charge separation process.38,41,50,61–63 We therefore attribute the large positive −ΔT/T signal with a maximum at ca. 725 nm to the absorption of the perylene bisimide radical anions and thus to the formation of a photo-induced CT state in both dyad and triad films. It is worth noting that the charge transfer process following the photoexcitation of the triphenylene units should lead to a contribution from the triphenylene cations in the transient absorption spectra. However, neither in thin film, nor in solution, we could observe such a feature which could be expected to appear at wavelengths shorter than 550 nm.64 This contribution should thus overlap with that of the perylene moiety in the dyad/triad bleaching band, but at a range of wavelength unavailable for our set-up. It is also worth noting that the triplet–triplet absorptions of neither TriPh65 nor PerDi66,67 could be detected in such systems, which suggests that the charge transfer dynamics precludes such phenomenon.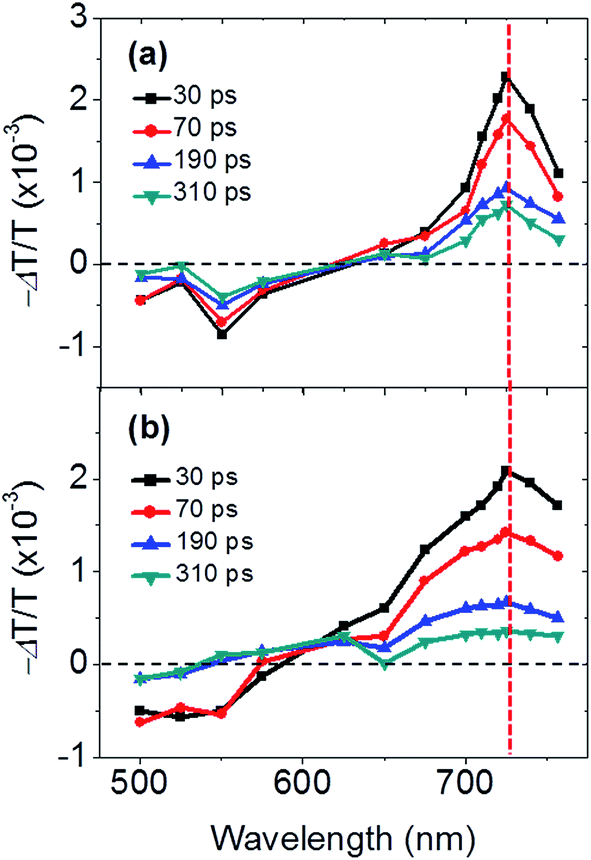 | ||
| Fig. 4 Transient absorption spectra measured in (a) dyad and (b) triad films. Pump wavelength was 325 nm where both the triphenylene and the perylene bisimide units absorb. The negative −ΔT/T at short wavelengths is due to the ground-state bleaching of the perylene bisimide moieties while the positive −ΔT/T at longer wavelengths is attributed to the absorption of perylene bisimide anions allowing us to examine the formation and recombination kinetics of the photo-induced CT states. | ||
To examine the dynamics of the charge separation and charge recombination processes and to extract their characteristic time constants, the temporal evolution of the transient absorption responses obtained at 725 nm in dyad and triad films are shown in Fig. 5. The rise time and decay time constants fitted from these data are summarized in Table 1. The rise times of the photo-induced radical perylene bisimide anions were determined by fitting the experimental data using a single exponential function saturating at a constant value yielding, for the dyad and triad films, values of 0.23 ± 0.05 ps and 0.27 ± 0.05 ps, respectively. These results indicate that the charge separation process exhibits similar dynamics in dyad and triad films. Noticeably, the charge transfer in film is of the same order of magnitude as the one measured in DCM solution.50 A decay time constant of the excited radical perylene bisimide anions was measured to be 234 ± 5 ps in the dyad film by fitting the decay with a single exponential decay function. In contrast, a sum of two exponential decay functions was required to describe well the kinetics observed in the triad films. The results indicate that about 82% of the photo-induced CT states recombine in about ∼151 ± 11 ps. The 18% remaining CT state population decays slower with a time constant of ∼358 ± 32 ps. These results demonstrate that, similarly to the solution case,50 the formation of the CT states in the dyad and triad films is significantly faster, by about three orders of magnitude, than its recombination process.
 | ||
| Fig. 5 (a) Ultrafast kinetics onset curves and their fits obtained at 725 nm probe wavelength with a 325 nm pumping in dyad and triad films. The blue dashed curve corresponds to the instrument response function of our experimental setup. (b) Kinetics decay curves and their fits obtained at 725 nm probe wavelength with a 325 nm pumping in dyad and triad films. | ||
Taking into account the uncertainties of the measured time constants, these results suggest that the dynamics of the recombination of the excited CT states in the dyad and triad films can be directly correlated to their structural properties. For instance, one can notice (see Fig. 1 and Table 1) that, whilst a single decay time constant is associated with a single D–A intercolumn spacing (ddyad = 1.9 nm) in the dyad films, two time constants were measured in the triad films, which display two different characteristic spacings between D and A columns (dtriad1 = 1.4 nm and dtriad2 = 2.5 nm). In addition, the measured decay time constants tend to decrease with shorter D and A units spacing. Because the D and A units are identical in the dyad and triad films, a simple Marcus theory analysis can be used to determine how D/A distance should affect the CT rate constants. In that framework, the rate constant for charge recombination, kCR, shows an exponential decay with the distance d between donor and acceptor units such as kCR ∝ exp[−βd], where β corresponds to a barrier height for electron tunnelling.43,68 In Fig. 6, kCR was plotted as a function of d, assuming that the three measured rates are associated with the spacing distances ddyad, dtriad1 and dtriad2. As expected, kCR is found to decay exponentially with the D/A spacing distance for β = 0.8 nm−1. This result validates our hypothesis stating that the dual exponential decay observed in the triad film is due to the presence of two different D/A distances and that the longer decay time constant arises from larger distance. However, because D and A moieties form stacks alternating along single columns, it should be noticed that there is in fact another characteristic spacing distance (dπ = 0.35 nm) corresponding to a face-to-face stacking between the perylene diimide and triphenylene units within a single column, which should be taken into account. Making an extrapolation from the fit shown in Fig. 6, this fourth distance dπ should lead to a time constant of about 65 ps. X-ray diffraction studies have previously shown that the triphenylene and the perylene bisimide units alternate in dyad film into different groups of stacked mesogens of same nature along a columnar axis long of about 30 units.49 In case of the triad films, this number of units increases up to at least 170. This implies that only about 3% and less than 0.5% of the D and A units in the dyad and triad films respectively, are located at an interface where the photo-induced electron transfer could occur within a single column. As a consequence, the population of CT states located at this interface with the D/A spacing of 0.35 nm is too small to be detected using our experimental setup. In parallel, we observed only one characteristic time constant for the formation of the CT state in the triad film, which at first sight does not seem to be consistent with the two spacings found in this system. This is likely due to the fact that the two very fast separation processes cannot be discriminated with our experimental setup. In addition, we cannot completely exclude the possibility that charge separation would only occur in the triad films via the most efficient and fast channel, which would be correlated to the shortest spacing of 1.4 nm between donor and acceptor columns.
 | ||
| Fig. 6 Logarithmic plot of the rate constant for the charge recombination, kCR, as a function of the donor–acceptor distance in the dyad/triad films. The solid line corresponds to the fit obtained with an exponential distance dependence using β = 0.8 nm−1. | ||
On the one hand and as mentioned above, the previous investigation of the photophysical properties of the dyad and triad systems in diluted DCM solutions demonstrated an energy transfer between the triphenylene and the perylene bisimide units occurring in competition with photo-induced CT state formation.50 On the other hand, our film measurements, and especially, the kinetics of the transient absorption responses at 725 nm, provide clear evidence that excitons generated either on triphenylene or on perylene bisimide moieties in the films are rapidly quenched via the formation of a photo-induced CT state. Indeed, no clear signature of an energy transfer from the D to the A units could be seen in the kinetics of the transient absorption data in contrast to the results previously obtained in solution. It should be emphasized, however, that the solution and films situations are different. While the molecular packing in dyad and triad thin films is frozen into D–A columnar nanostructures with well-defined characteristic spacings, the molecular conformations of the dyad and triad molecules in solution fluctuate due to the flexible non-conjugated bridge connecting the D and A units. Depending on the molecular conformations adopted by the dyad and triad molecules during the exciton lifetime of the photo-excited triphenylene units, either intramolecular photoinduced charge transfer or energy transfer becomes the dominant deactivation process. In the case of dyad and triad thin films, our study demonstrates that the molecular packing in the D–A columnar nanostructures is only favourable to the photoinduced charge transfer process, implying that the kinetics of the energy transfer in the solid-state are too slow and the process is therefore too inefficient to be observed.
Compared to most of liquid-crystalline D–A dyads/triads reported so far,29–46 the spacing between the discotic D and A units controlled by the aliphatic periphery is larger in the present molecular architecture. On the one hand, small spacing is generally preferred in order to ensure the fast and efficient formation of photoinduced CT states. On the other hand, the molecular design investigated herein provides more freedom to the perylene bisimide and triphenylene moieties to self-assemble and to form well-defined D and A columnar structures in the triad system. In addition, the formation of the CT states occurs in the range of 0.2–0.3 ± 0.05 ps for both dyad and triad films. This is comparable or even faster than the time constants reported for the formation of intramolecular excited CT states in a variety of D–A systems using perylene bisimide as electron acceptor.38,41,61,62 To the best of our knowledge, there are only two reports investigating the photophysical properties of liquid-crystalline D–A dyad/triad molecules self-assembled into distinct donor and acceptor nanosegregated domains in thin films.41,69 In particular, T. Roland et al. investigated the photophysical properties of a siloxane-functionalized D–A–D triad based on two identical bisthiophene derivatives attached to a central perylene bisimide unit without any non-conjugated flexible bridges. In the lamellar thin films, the formation of both intra- and inter-molecular CT states was found to occur in less than 0.1 ps. On the one hand, faster formation of bisthiophene-perylene bisimide CT states in films than in solution was explained by a strong intermolecular excitonic coupling and the possible delocalization of the CT states over neighbouring triad molecules.70 On the other hand, it was also found that around 80% of the excited CT states recombine in this material in less than 60 ps for both solution and films. Even if we cannot directly compare the dynamics found in this previous work with our results, since both the donor units and the supramolecular architecture are different, the role played by the molecular architecture of the D–A dyad/D–A–D triad on the charge transfer process can be discussed. The larger spacing between D and A moieties in our systems, due to the insulating continuum of molten alkyl chains between adjacent columns, presumably leads to slower formation of the CT states. Nevertheless, the dynamics observed in our films still remain significantly faster than 1 ps. Another important result is that it appears in our systems that the larger the distance between D and A units the slower the charge carrier recombination processes. These findings provide important insights into the dynamics of charge transfer in self-organized D–A materials and their structure–property relationship. In such architectures, the role of the length, the number and the distribution of alkyl chains grafted to the D and A moieties on their self-organization and their solid-state photophysical properties appear as one of the key issues for the chemical design of novel D–A systems.
4. Conclusions
We investigated the photophysical properties of liquid-crystalline donor–acceptor discotic dyad and triad systems based on triphenylene and perylene bisimide units in thin films. It was previously established that these molecules self-organize into columnar one-dimensional nanostructures in the solid-state. To examine the role of the molecular aggregation on the absorption and steady-state fluorescence spectra, their concentration-dependence was measured when the molecules are dispersed in a poor solvent such as methylcyclohexane. These results provided useful insights to interpret the absorption of the dyad and triad neat films. Photoluminescence and subpicosecond transient absorption measurements in films demonstrated a complete quenching of the singlet excitons through a very effective photo-induced charge transfer process. Following the photo-excitation, we found that photo-induced charge transfer states were formed in both dyad and triad in about 0.2–0.3 ps. The charge recombination process was significantly slower with characteristic time constants in the range between 150 and 360 ps.The dynamics of the formation and recombination of the charge transfer states were found to correlate well with the structural properties of the dyad and triad systems. These results provide thus important and useful information on the structure–property relationships in LC donor–acceptor organic materials. In particular, the design of molecular assemblies with long D/A spacing distance should be favoured as it appears to be beneficial to longer CT state lifetime while not impacting significantly the CT formation time. In addition, the fast formation of the charge transfer states and the relatively slow charge recombination process evidenced in these dyad and triad derivatives are promising for the molecular design of future self-organized donor–acceptor nanostructures with enhanced photovoltaic, photophysical and charge transport properties.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was carried out in the framework of CNRS International Associated Laboratory “Functional Nanostructures:Morphology, Nanoelectronics and Ultrafast Optics” (LIA NANOFUNC) and Leading Foreign Research Institute Recruitment Program through the National Research Foundation of Korea (NRF-2010-00453). The authors also acknowledge the International Research Network (GDRI, CNRS) on “Functional Materials for Organic Optics, Electronics and Devices” (FUNMOOD). JCR acknowledges the support from the Basic Science Researcher Program and the Quantum Metamaterials Research Center (QMMRC) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (grants 2011-0008650, 2012-0000543). KJL, JHW, YX, EK, PA, JWW and JCR acknowledge the support from the Ministry of Science, ICT & Future Planning, Korea (2015001948, 2014M3A6B3063706). PA would like to thank the Canon Foundation in Europe for supporting his Fellowship. LMM acknowledges the support from Polish National Science Centre grants DEC-2013/09/N/ST5/02464 and DEC-2013/09/B/ST5/03417. The authors are also extremely grateful to Dr Jean-Yves Bigot, Dr Mircea Vomir and Dr Marie Barthelemy for their great contribution to setup the femtosecond pump-probe experiment used in this study.References
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef PubMed.
- F. Cacialli, J. S. Wilson, J. J. Michels, C. Daniel, C. Silva, R. H. Friend, N. Severin, P. Samori, J. P. Rabe, M. J. O'Connell, P. N. Taylor and H. L. Anderson, Nat. Mater., 2002, 1, 160–164 CrossRef CAS PubMed.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685 CrossRef CAS.
- W. Pisula, A. Menon, M. Stepputat, I. Lieberwirth, U. Kolb, A. Tracz, H. Sirringhaus, T. Pakula and K. Müllen, Adv. Mater., 2005, 17, 684–689 CrossRef CAS.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, Adv. Funct. Mater., 2008, 18, 1329–1339 CrossRef CAS.
- J. C. Ribierre, S. Watanabe, M. Matsumoto, T. Muto and T. Aoyama, Appl. Phys. Lett., 2010, 96, 083303 CrossRef.
- J. C. Ribierre, L. Zhao, S. Furukawa, T. Kikitsu, D. Inoue, A. Muranaka, K. Takaishi, T. Muto, S. Matsumoto, D. Hashizume, M. Uchiyama, P. André, C. Adachi and T. Aoyama, Chem. Commun., 2015, 51, 5836–5839 RSC.
- J. C. Ribierre, Y. Yokota, M. Sato, A. Ishizuka, T. Tanaka, S. Watanabe, M. Matsumoto, A. Muranaka, S. Matsumoto, M. Uchiyama and T. Aoyama, RSC Adv., 2014, 4, 36729 RSC.
- R. H. Kim, H. J. Kim, I. Bae, S. K. Hwang, D. B. Velusamy, S. M. Cho, K. Takaishi, T. Muto, D. Hashizume, M. Uchiyama, P. André, F. Mathevet, B. Heinrich, T. Aoyama, D. E. Kim, H. Lee, J. C. Ribierre and C. Park, Nat. Commun., 2014, 5, 3583–3595 Search PubMed.
- C. T. Howells, K. Marbou, H. Kim, K. J. Lee, B. Heinrich, S. J. Kim, A. Nakao, T. Aoyama, S. Furukawa, J.-H. Kim, E. Kim, F. Mathevet, S. Mery, I. D. W. Samuel, A. Al Ghaferi, M. S. Dahlem, M. Uchiyama, S. Y. Kim, J. W. Wu, J.-C. Ribierre, C. Adachi, D.-W. Kim and P. André, J. Mater. Chem. A, 2016, 4, 4252–4263 CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761–1764 CrossRef CAS PubMed.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H. W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 419, 862 CrossRef CAS.
- H. Iino and J.-I. Hanna, Adv. Mater., 2011, 23, 1748–1751 CrossRef CAS PubMed.
- L. Mazur, A. Castiglione, K. Ocytko, F. Kameche, R. Macabies, A. Ainsebaa, D. Kreher, B. Heinrich, B. Donnio, S. Sanaur, E. Lacaze, J.-L. Fave, K. Matczyszyn, M. Samoc, J. W. Wu, A.-J. Attias, J.-C. Ribierre and F. Mathevet, Org. Electron., 2014, 15, 943–953 CrossRef CAS.
- W. Ma, C. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- X. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579–583 CrossRef CAS PubMed.
- S. E. Shaheen, C. J. Brabec, N. S. Sariciftci, F. Padinger, T. Fromherz and J. C. Hummelen, Appl. Phys. Lett., 2001, 78, 841–843 CrossRef CAS.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS PubMed.
- A. R. Mallia, P. S. Salini and M. Hariharan, J. Am. Chem. Soc., 2015, 137, 15604–15607 CrossRef CAS PubMed.
- D. Ley, C. X. Guzman, K. H. Adolfsson, A. M. Scott and A. B. Braunschweig, J. Am. Chem. Soc., 2014, 136, 7809–7812 CrossRef CAS PubMed.
- W. S. Shin, H.-H. Jeong, M.-K. Kim, S.-H. Jin, M.-R. Kim, J.-K. Lee, J. W. Lee and Y.-S. Gal, J. Mater. Chem., 2006, 16, 384–390 RSC.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS PubMed.
- D. Käfer, A. Bashir, X. Dou, G. Witte, K. Müllen and C. Wöll, Adv. Mater., 2010, 22, 384–388 CrossRef PubMed.
- Z. Chen, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137–140 CrossRef CAS PubMed.
- L. F. Dössel, V. Kamm, I. A. Howard, F. d. r. Laquai, W. Pisula, X. Feng, C. Li, M. Takase, T. Kudernac, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2012, 134, 5876–5886 CrossRef PubMed.
- H. Wonneberger, C.-Q. Ma, M. A. Gatys, C. Li, P. Bäuerle and K. Müllen, J. Phys. Chem. B, 2010, 114, 14343–14347 CrossRef CAS PubMed.
- A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2002, 8, 3353–3361 CrossRef CAS.
- L. Bu, X. Guo, B. Yu, Y. Fu, Y. Qu, Z. Xie, D. Yan, Y. Geng and F. Wang, Polymer, 2011, 52, 4253–4260 CrossRef CAS.
- J. M. Mativetsky, M. Kastler, R. C. Savage, D. Gentilini, M. Palma, W. Pisula, K. Müllen and P. Samorì, Adv. Funct. Mater., 2009, 19, 2486–2494 CrossRef CAS.
- J. Cremer, E. Mena-Osteritz, N. G. Pschierer, K. Mullen and P. Bauerle, Org. Biomol. Chem., 2005, 3, 985–995 CAS.
- G. Balaji, T. S. Kale, A. Keerthi, A. M. Della Pelle, S. Thayumanavan and S. Valiyaveettil, Org. Lett., 2011, 13, 18–21 CrossRef CAS PubMed.
- T. Nishizawa, K. Tajima and K. Hashimoto, J. Mater. Chem., 2007, 17, 2440–2445 RSC.
- R. Deschenaux, B. Donnio and D. Guillon, New J. Chem., 2007, 31, 1064–1073 RSC.
- A. Petrella, J. Cremer, L. De Cola, P. Bäuerle and R. M. Williams, J. Phys. Chem. A, 2005, 109, 11687–11695 CrossRef CAS PubMed.
- C. C. You, P. Espindola, C. Hippius, J. Heinze and F. Würthner, Adv. Funct. Mater., 2007, 17, 3764–3772 CrossRef CAS.
- M. K. R. Fischer, T. E. Kaiser, F. Wurthner and P. Bauerle, J. Mater. Chem., 2009, 19, 1129–1141 RSC.
- T. Roland, J. Leonard, G. Hernandez Ramirez, S. Mery, O. Yurchenko, S. Ludwigs and S. Haacke, Phys. Chem. Chem. Phys., 2012, 14, 273–279 RSC.
- C. C. Hofmann, P. Bauer, S. A. Haque, M. Thelakkat and J. Köhler, J. Chem. Phys., 2009, 131, 144512 CrossRef PubMed.
- J. Daub, R. Engl, J. Kurzawa, S. E. Miller, S. Schneider, A. Stockmann and M. R. Wasielewski, J. Phys. Chem. A, 2001, 105, 5655–5665 CrossRef CAS.
- P.-O. Schwartz, L. Biniek, E. Zaborova, B. Heinrich, M. Brinkmann, N. Leclerc and S. Méry, J. Am. Chem. Soc., 2014, 136, 5981–5992 CrossRef CAS PubMed.
- N. Gorczak, S. Tarkuç, N. Renaud, A. J. Houtepen, R. Eelkema, L. D. A. Siebbeles and F. C. Grozema, J. Phys. Chem. A, 2014, 118, 3891–3898 CrossRef CAS PubMed.
- P. E. Hartnett, S. M. Dyar, E. A. Margulies, L. E. Shoer, A. W. Cook, S. W. Eaton, T. J. Marks and M. R. Wasielewski, Chem. Sci., 2015, 6, 402–411 RSC.
- K. Sun, Z. Xiao, S. Lu, W. Zajaczkowski, W. Pisula, E. Hanssen, J. M. White, R. M. Williamson, J. Subbiah, J. Ouyang, A. B. Holmes, W. W. H. Wong and D. J. Jones, Nat. Commun., 2015, 6, 6013 CrossRef CAS PubMed.
- J. Qu, B. Gao, H. Tian, X. Zhang, Y. Wang, Z. Xie, H. Wang, Y. Geng and F. Wang, J. Mater. Chem. A, 2014, 2, 3632–3640 CAS.
- Y. Xiao, X. Su, L. Sosa-Vargas, E. Lacaze, B. Heinrich, B. Donnio, D. Kreher, F. Mathevet and A.-J. Attias, CrystEngComm, 2016 10.1039/c6ce00365f.
- K. J. Lee, J. H. Woo, E. S. Kim, Y. Xiao, X. Su, L. Mazur, A.-J. Attias, F. Fages, O. Cregut, A. Barsella, F. Mathevet, L. Mager, J. W. Wu, A. D'Aléo and J.-C. Ribierre, Phys. Chem. Chem. Phys., 2016, 18, 7875–7887 RSC.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- R. Gómez, D. Veldman, R. Blanco, C. Seoane, J. L. Segura and R. A. J. Janssen, Macromolecules, 2007, 40, 2760–2772 CrossRef.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley, New York, 1970 Search PubMed.
- M. Bagui, T. Dutta, S. Chakraborty, J. S. Melinger, H. Zhong, A. Keightley and Z. Peng, J. Phys. Chem. A, 2011, 115, 1579–1592 CrossRef CAS PubMed.
- V. Duzhko, H. Shi, K. D. Singer, A. N. Semyonov and R. J. Twieg, Langmuir, 2006, 22, 7947–7951 CrossRef CAS PubMed.
- S. Zamir, R. Poupko, Z. Luz, B. Hueser, C. Boeffel and H. Zimmermann, J. Am. Chem. Soc., 1994, 116, 1973–1980 CrossRef CAS.
- D. Dotcheva, M. Klapper and K. Müllen, Macromol. Chem. Phys., 1994, 195, 1905–1911 CrossRef CAS.
- H. Quante, Y. Geerts and K. Müllen, Chem. Mater., 1997, 9, 495–500 CrossRef CAS.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem.–Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed.
- C. Huang, M. M. Sartin, M. Cozzuol, N. Siegel, S. Barlow, J. W. Perry and S. R. Marder, J. Phys. Chem. A, 2012, 116, 4305–4317 CrossRef CAS PubMed.
- L. X. Chen, S. Xiao and L. Yu, J. Phys. Chem. B, 2006, 110, 11730–11738 CrossRef CAS PubMed.
- T. Kircher and H. G. Lohmannsroben, Phys. Chem. Chem. Phys., 1999, 1, 3987–3992 RSC.
- S. I. Klink, L. Grave, D. N. Reinhoudt, F. C. J. M. van Veggel, M. H. V. Werts, F. A. J. Geurts and J. W. Hofstraat, J. Phys. Chem. A, 2000, 104, 5457–5468 CrossRef CAS.
- B. R. Henry and M. Kasha, J. Chem. Phys., 1967, 47, 3319–3327 CrossRef CAS.
- A. A. Rachford, S. Goeb and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 2766–2767 CrossRef CAS PubMed.
- A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CAS.
- M. N. Paddon-Row, Acc. Chem. Res., 1994, 27, 18–25 CrossRef CAS.
- B.-R. Gao, J.-F. Qu, Y. Wang, Y.-Y. Fu, L. Wang, Q.-D. Chen, H.-B. Sun, Y.-H. Geng, H.-Y. Wang and Z.-Y. Xie, J. Phys. Chem. C, 2013, 117, 4836–4843 CAS.
- M. Polkehn, H. Tamura, P. Eisenbrandt, S. Haacke, S. Méry and I. Burghardt, J. Phys. Chem. Lett., 2016, 7, 1327–1334 CrossRef CAS PubMed.
Footnotes |
| † Contributed equally to this work. |
| ‡ Present Address: Center for Organic Photonics and Electronics Research (OPERA), Kyushu University, Fukuoka, Japan. |
| This journal is © The Royal Society of Chemistry 2016 |