
Hybrid cross-linked hydrogels based on fibrous protein/block copolymers and layered silicate nanoparticles: tunable thermosensitivity, biodegradability and mechanical durability†
Niloofar Eslahi‡
a,
Abdolreza Simchi*ab,
Morteza Mehrjooc,
Mohammad Ali Shokrgozarc and
Shahin Bonakdarc
aDepartment of Materials Science and Engineering, Sharif University of Technology, Azadi Avenue, P.O. Box 11365/8639, Tehran, Iran. E-mail: simchi@sharif.edu; Fax: +98 21 6600 5717; Tel: +98 21 6616 5261
bInstitute for Nanoscience and Nanotechnology, Sharif University of Technology, Azadi Avenue, P.O. Box 11365/8639, Tehran, Iran
cNational Cell Bank of Iran, Pasteur Institute of Iran, P.O. Box 13169/43551, Tehran, Iran
First published on 23rd June 2016
Abstract
Stimuli responsive polymer hydrogels have sparked a considerable interest for biomedical applications. In this work, we present a novel injectable thermal responsive hydrogel based on mineral nanoclay (LAPONITE®), wool-derived fibrous protein (keratin), triblock copolymer (Pluronic), and chitosan biopolymer with potential applications for articular cartilage tissue engineering. Genipin was utilized as a natural crosslinker. The formation of polymer conjugates between the components is confirmed by FTIR and 1H NMR spectroscopy. The nanocomposite hydrogel contains interconnected pores within the hydrogel network and the size of the pores is found to reduce at higher crosslinking density and on incorporation of LAPONITE® up to 6 wt%. A decrease in the swelling ratio and biodegradation after chemical crosslinking with genipin and addition of nanoclay is also observed. The ability of the hydrogels to undergo in situ crosslinking and rapid gelation under physiological conditions is shown. Evaluation of the viscoelastic properties of the hydrogels in simulated physiological conditions, i.e. elastic modulus (G′) and viscous modulus (G′′), indicates a significant enhancement of the properties through incorporation of the nanoclay mineral, which could be as high as 6 fold depending on the LAPONITE® concentration. To examine the in vitro cytotoxicity of the hydrogels for biomedical applications, an MTT assay using chondrocyte cells was performed. The cell attachment and viability were assessed as well. It can be concluded that the developed hydrogels are biocompatible (>90%) with good cell adhesion depending on their formulation and microstructure. The superior physico-mechanical properties of the hydrogels along with their cytocompatibility and the ability to encapsulate live cells at physiological conditions suggest that they have a high capacity to be used as cartilage scaffolds.
1. Introduction
Articular cartilage is a flexible and connective tissue in which the chondrocytes are sparsely distributed into a highly organized extracellular matrix (ECM) rich in proteoglycans, glycosaminoglycans (GAGs), and collagen fibers.1 The repair of cartilage damage, which occurs as a result of injury, disease, trauma and tumor, is one of the most challenging clinical problems in orthopedics.2 The low repair capacity of cartilage is attributed to the lack of chondrocyte mobility in the ECM and the absence of progenitor cells and vascular networks.3 Among different scaffolds being developed for cartilage regeneration,4–7 hydrogels are of great interest due to their interesting properties that can mimic the physiochemical and biological properties of the native hydrated cartilage ECM.8,9Injectable hydrogels, which are in situ formed after injection at the defect site, have received much attention in recent years.10–12 The injectable nature of these hydrogels provides the attractive feature of facile and homogenous cell distribution within any defect size or shape prior to gelation in good alignment with the surrounding tissue by a simple minimally invasive injection procedure.11,13 Thermosensitive hydrogels as specific injectable biomaterials can undergo reversible sol–gel transitions upon exposure to physiological temperature.14 A popular class of thermogelling systems is those based on the commercially available triblock copolymer (PEO–PPO–PEO) known as Pluronic with admirable biocompatibility.15 The amphiphilic character of Pluronic (Pl) allows it to form thermoreversible gels. With increasing concentration above the critical micelle concentration (CMC) or rising temperature above the lower critical solution temperature (LCST), the copolymer molecules can self-assemble into micelles in aqueous solutions through hydrophobic interactions between the PPO moieties within the polymer.16–18 Hydrogels based on Pluronic F127 (PEO99–PPO65–PEO99) have been increasingly applied to deliver drugs, growth factors or cells to promote the regeneration of injured tissues.19–21 However, Pl hydrogels are mechanically weak and rapidly dissolve out under physiological environments,22,23 which significantly limits their use as load bearing tissue matrix. To overcome such constraints, physical or chemical modification by different polymers have been investigated.24 For instance, Zhao et al.25 designed a heparin/Pl 407 hydrogel with a gelation temperature of ∼38 °C and maximum elastic modulus of 10 kPa; Leroy et al.26 synthesized PLA/Pl copolymers with enhanced Young's moduli (∼500 MPa) and yield strain (3% to 4%); Lee et al.27 introduced thermo-sensitive and injectable hyaluronic acid/Pl composite hydrogels with excellent tissue-adhesion properties and superior in vivo gel stability.
Chitosan (CH) is a cationic polysaccharide composed of glucosamine and N-acetylglucosamine residues which is derived from chitin found in the exoskeleton of shellfish. This biopolymer has a number of beneficial biological properties such as low toxicity, low immunogenicity, biodegradability, biocompatibility, and modifiable functional groups that all have made it promising candidate for various pharmaceutical and biomedical applications.13,28,29 Since CH has similar structure to GAG, it is a promising biomaterial to modulate the morphology of chondrocytes, and thus promoting differentiation and stimulating cartilage regeneration.30–32 Moreover, the stiffness of CH can provide enhancement in the mechanical strength of thermo-reversible hydrogels.33 Several studies have grafted Pl onto CH to produce a water-soluble thermosensitive copolymer as an injectable hydrogel for cell/drug delivery and tissue engineering applications.16,17,34–37 These conjugate hydrogels (PlCH) have shown an improved mechanical properties and biocompatibility.17 However, the stability of the conjugated hydrogels may not be sufficient enough for cartilage reconstruction in practice.
Preparing ideal scaffolds for tissue engineering requires mimicking natural environments in the ECM containing specific peptide motifs that bind to receptors of cell surface and stimulate adhesion and spreading of chondrocyte cells.38,39 Keratin (K) is the major structural fibrous protein found in outer covering such as hair, wool, feathers, nails and horns of mammals, reptiles, and birds.40 It has been reported that keratin-based biomaterials support cellular adhesion, proliferation and migration.41,42 Like other naturally derived protein biomaterials, keratin possesses amino acid sequences (i.e., RGD) that may interact with integrin and promote cell attachment and binding. In addition, the ability of this protein to self-assemble into various physical shapes has been exploited for the development of new biomaterials particularly for tissue engineering applications.43–45 For example, Xu et al.46 produced 3D nanofibrous scaffolds from feather keratin having intrinsic water stability for cartilage tissue engineering due to their structural similarity to native ECM.
Recently, a wide range of nanoparticles have been incorporated into polymer hydrogels in order to control their phase behavior and improve their mechanical properties.47,48 Interactions of nanoparticles with the matrix affect the stability of the resulting hydrogels. Among a variety of nanoparticles, LAPONITE® (LP), consisting of synthetic silicate nanoparticles with an average diameter of 30 nm and a thickness of 1 nm, has been used to synthesize nanocomposite polymer hydrogels with enhanced mechanical properties.49,50 It was found that exfoliated LP particles may act as multifunctional crosslinkers in forming composite hydrogels, and the polymer chains were anchored to the particles to assemble a network structure.51,52 Wu et al.53 overcame the rapid dissolution properties of Pluronic copolymer by synergistic combination of nanoparticles gelation characteristics with polymer thermo-sensitivity. The same researchers also synthesized mechanically tough nanocomposite hydrogels by photo-crosslinking Pl diacrylate in the presence of LP nanoparticles. The resulting hydrogels had high elongations and improved toughness when compared to their polymeric hydrogel counterparts.52,54
In this study, the thermosensitive PlCH hydrogel was crosslinked with keratin by employing genipin (GP) to prepare cell supporting scaffolds for articular cartilage repair. Genipin is obtained from geniposide, an iridoid glucoside isolated from the fruits of Genipa americana and Gardenia jasminoides Ellis and applied in herbal medicine due to its anti-inflammatory, antiphlogistic, neurogenic, hemostatic, and low cytotoxicity features.55 This natural biocompatible crosslinking reagent has recently attracted interest in the fabrication of biopolymers because of its ability to crosslink chitosan and proteins.56–59 Considering the biocompatibility and reinforcing effects of LP nanoparticles along with the thermosensitive properties associated with the PlCHK hydrogels, the complementary properties of each constituent has been utilized in this study to design novel tunable nanocomposite hydrogels capable of promoting cartilage tissue regeneration. For this purpose, different amounts of LP nanoparticles were incorporated into the hydrogels, which were then crosslinked with GP at concentrations of 1–15 mM. The gelation behavior of the hydrogels in simulated physiological environment is shown. The physicochemical and mechanical properties of the injectable hydrogels are also reported. To evaluate the capacity of the developed nanocomposites for biomedical applications, their in vitro biological performance is presented as well.
2. Experimental
2.1 Materials
Pluronic F127 (PEO99–PPO65–PEO99) and chitosan (medium molecular weight, 190–310 kDa, 75–85% deacetylated) were purchased from Sigma-Aldrich (USA). Succinic anhydride, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (ECD), N-hydroxysuccinimide (NHS), 1,4-dioxane, triethylamine (TEA), and 4-dimethylaminopyridine (DMAP) were obtained from Merck (Germany). Genipin (GP), methyl-2-hydroxy-9-(hydroxymethyl)-3-oxabicyclonona-4,8-diene-5-carboxylate, was provided from Challenge Bioproducts Ltd. (Taiwan). Medical grade hectorite-type synthetic silicate powder (LAPONITE® RD) with a chemical composition of Na+0.7[(Si8Mg5.5Li0.3)O20(OH)4]−0.7, was purchased from Southern Clay Products Inc. (USA). Keratin was extracted from New Zealand Merino wool (20 μm fineness) by sulphitolysis reaction. The particle size distribution of LP nanoparticles and the details of protein extraction and characterization were explained in ESI.† All other chemicals were of analytical grade and used without further purification.2.2 Synthesis of thermosensitive copolymers
Pluronic was grafted onto chitosan chain through EDC/NHS chemistry.17,37 The carboxylation of terminal hydroxyl groups in Pl (12 g) was performed by using succinic anhydride (250 mg), DMAP (260 mg), and TEA (300 μL) in 60 mL dioxane during stirring at room temperature for 24 h. The solvent was then removed by employing a rotary evaporator and the residue was filtered and precipitated in cold diethyl ether twice. The precipitate was dried under vacuum overnight to attain carboxylated Pluronic (yield ∼ 85%). The carboxylated copolymer (10 g) was grafted to CH (1 g) by EDC (0.77 g) and NHS (0.46 g) in 50 mL of phosphate buffer solution (0.01 M at pH = 5.5) at room temperature for 24 h. The conjugation process occurred through forming the amide bond between amine groups in CH and the carboxyl groups of copolymer using EDC and NHS as coupling agents. The product was dialyzed against distilled water using a dialysis tubing cellulose membrane (Sigma, USA, Mw cut-off 14 kDa) and finally lyophilized to obtain conjugated PlCH. The grafting efficiency (%) was calculated by:60
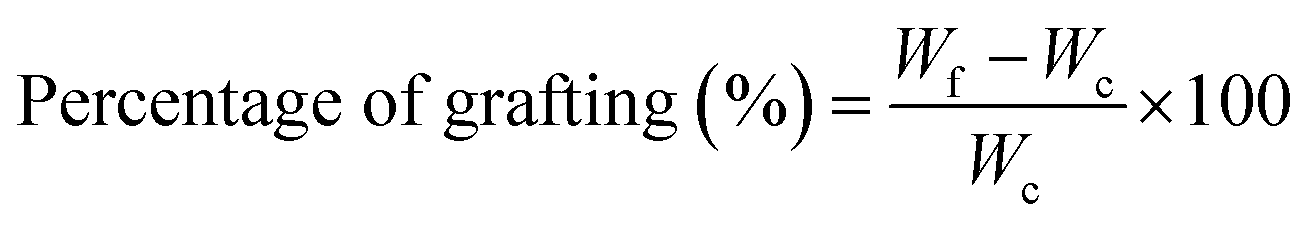 | (1) |
2.3 Protein crosslinking and preparation of nanocomposite hydrogels
The conjugated PlCH (0.5 g) was homogenously mixed with keratin (0.05 g) in 5 mL phosphate buffer solution (0.01 M PBS with a pH of 7.4) at 4 °C for 2 h. Different amounts of LP nanoparticles (0, 3, 6% wt) and GP (1, 2.5, 5, 10 and 15 mM) was added into the mixture and incubated at 37 °C to allow gel formation. The prepared hydrogels were subsequently cured at 37 °C for 24 h for further studies.2.4 Materials characterizations
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio) in PBS were utilized as controls.
:1 weight ratio) in PBS were utilized as controls.
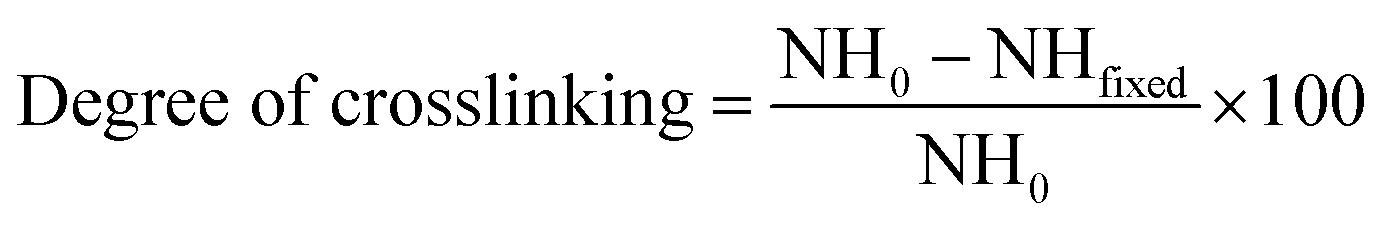 | (2) |
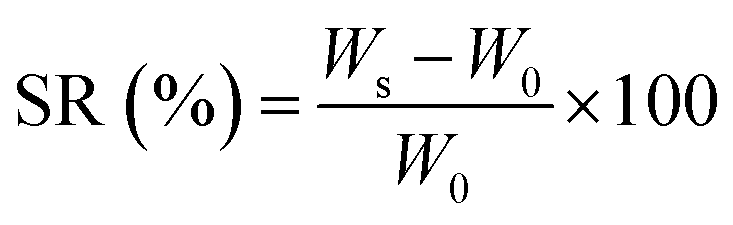 | (3) |
Mass erosion/degradation rates were also determined similarly at different time intervals up to 30 days. All experiments were performed in triplicate.
2.4.7.1 Isolation of chondrocytes. Articular chondrocytes (CHONs) were isolated from knee joints of New Zealand white rabbits according to the Ethical Committee of National Cell Bank of Iran (NCBI). The cartilaginous tissue was cut off from the joint, washed several times with antibiotic containing medium and then minced to small pieces using a scalpel blade. The obtained slices were predigested in 0.25% trypsin–EDTA solution (Sigma, USA) for 1 h and then transferred to the collagenase type II solution (0.08 mg mL−1, Sigma, USA) for 12 h in an incubator. The resulting cell suspension was passed through a 40 μm filter. Cells were then harvested by centrifugation at 1500 rpm for 5 min and cultured in DMEM (GIBCO, Scotland)/Ham's F12 supplemented with 10% fetal bovine serum (FBS) (Seromed, Germany) in an incubator at 37 °C, 5% CO2.64
2.4.7.2 MTT assay. Prior to cell culture studies, hydrogels were sterilized by an immersion in 70% ethanol solution overnight, washed twice in sterile ultrapure water, air dried in a sterile environment, and then kept under UV for 1 h. Cytotoxicity testing of the hydrogels was evaluated according to ISO 10993-5 using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma, USA) assay. To prepare the extracts for each hydrogels, the samples were incubated at 37 °C in 1 mL of RPMI 1640 culture medium (Sigma) supplemented with 10% (w/w) fetal bovine serum (FBS, GIBCO, Scotland) for 3, 7 and 14 days. Pure culture medium (RPMI and FBS) kept under similar conditions was used as the control. Cells were seeded into 96-well plates at a density of 1 × 104 cells per mL. The culture medium (100 μL) was replaced with 3, 7, and 14 day extracts after 24 h. The extracts were eliminated after another 24 h and 100 μL of a 0.5 mg mL−1 MTT solution was added to each well and were incubated at 37 °C for additional 4 h. The solution was then removed carefully and 150 μL isopropanol (Sigma, USA) was subsequently added to each well to dissolve the MTT formazan purple crystals. Absorbency of the solution was measured at 545 nm using an ELISA Reader (Stat Fax-2100, USA) and the relative viability (%) normalized by control group was calculated. It should be noted that culture medium with no sample and dexamethasone (10−5 M) were considered as negative and positive controls, respectively.65
2.4.7.3 Cell attachment. The morphology of CHONs seeded on the hydrogels was observed by SEM (VEGA TS5136M, TESCAN, USA). For this purpose, cells with a density of 1 × 105 cells per a 50 μL of culture medium were cultured on each hydrogel sample and incubated at 37 °C for 4 hours. Then, un-attached cells were washed four times with DMEM culture medium. To prepare the samples for SEM analysis, the constructs were fixed with 4% formalin solution, dehydrated in serial alcohol solutions and sputter coated with gold.
2.4.7.4 Live/dead assay. Cell viability within the constructs was observed using acridine orange–propidium iodide (AO/PI) staining. Chondrocytes were mixed with hydrogels and then incubated at 37 °C to allow it to gel. The hydrogels containing 106 cells per mL were stained after 7 days in vitro culturing and incubated for 15 minutes in the dark followed by imaging using a confocal electron microscopy (Leica TCs SP5 II, USA) in green (live) and red (dead) channels respectively.59,64
3. Results and discussion
3.1 Conjugating and crosslinking of polymer hydrogels
The FTIR and 1H NMR spectroscopy measurements were carried out to confirm the formation of PlCH conjugate. The FTIR spectra of different samples are shown in Fig. 1a. The absorbing bands located at ∼1108 and 1243 cm−1 are ascribed to the C–O–C stretching of aliphatic ether group and twisting vibration of –CH2 in Pl, respectively.66 The peak at 1732 cm−1, which is assigned to the carboxyl (COOH) stretching vibration, determines successful carboxylation of the Pl copolymer by means of succinic anhydride. The broad absorption band appeared in the spectrum of CH in the range of 3100 to 3600 cm−1 with a maximum at 3448 cm−1 reveals overlapping of the O–H and N–H stretching vibrations of functional groups engaged in hydrogen bonds. The peak around 2883 cm−1 is assigned to aliphatic groups of carbohydrate ring. The bands at 1152, 1098 and 1031 cm−1 correspond to the bridge oxygen (C–O–C) stretching bands as characteristics of glycosidic linkage in the saccharide structure. The spectrum of CH also shows the distinctive absorption bands at 1639 cm−1 and 1596 cm−1 which are related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching in amide I and –NH2 bending in non-acetylated 2-aminoglucose primary amine, respectively. The peak at 1558 cm−1 is associated with the N–H bending vibration in amide II group.67 The spectrum of the conjugated PlCH exhibits a peak at ∼1650 cm−1 which could be associated to the CO stretching vibration of the amide formed between the carboxylated Pl and amine groups of CH. The diminished peak at 1596 cm−1 is presumably due to the loss of primary amine (–NH2) groups to the secondary (–NH) ones when chitosan is grafted to the activated Pl.36 Meanwhile, the peak intensity of amide I is intensified as compared with pristine CH that supports the formation of amide bonds and copolymer conjugation.34 The disappearance of the peak corresponding to the carboxylic groups at 1732 cm−1 also indicates that all the functional groups of Pl are consumed.37
O stretching in amide I and –NH2 bending in non-acetylated 2-aminoglucose primary amine, respectively. The peak at 1558 cm−1 is associated with the N–H bending vibration in amide II group.67 The spectrum of the conjugated PlCH exhibits a peak at ∼1650 cm−1 which could be associated to the CO stretching vibration of the amide formed between the carboxylated Pl and amine groups of CH. The diminished peak at 1596 cm−1 is presumably due to the loss of primary amine (–NH2) groups to the secondary (–NH) ones when chitosan is grafted to the activated Pl.36 Meanwhile, the peak intensity of amide I is intensified as compared with pristine CH that supports the formation of amide bonds and copolymer conjugation.34 The disappearance of the peak corresponding to the carboxylic groups at 1732 cm−1 also indicates that all the functional groups of Pl are consumed.37
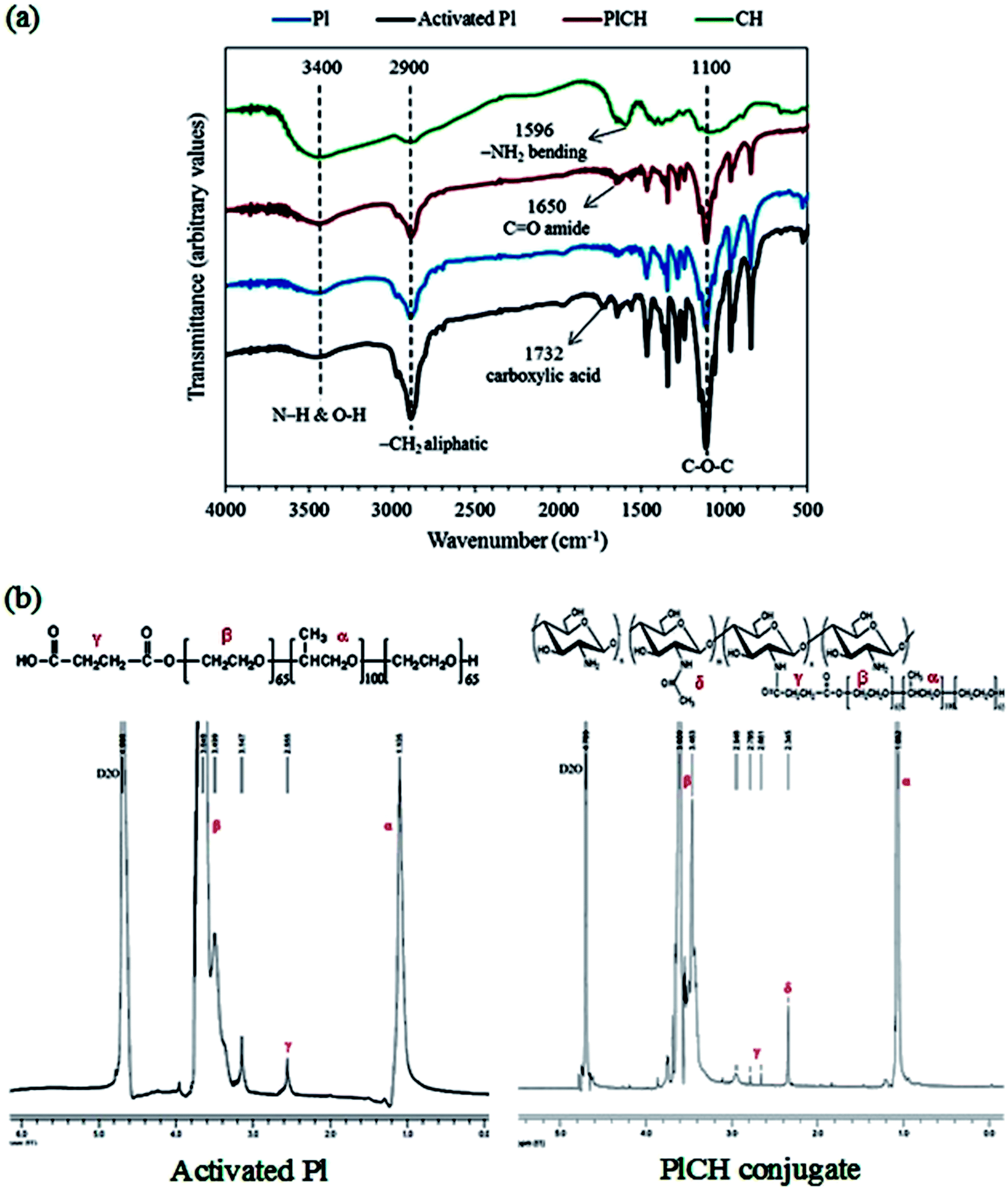 | ||
| Fig. 1 (a) FTIR and (b) 1H NMR spectra of activated PL and PlCH conjugate. | ||
To further support formation of amide bonds, 1H NMR was performed. The spectra of activated Pl in Fig. 1b presents signals for the ethylene protons (CH2–CH2) of PEO segments and methyl protons (OCH2CH(CH3)O) of PPO segment at δ ∼ 3.5–3.8 and 1.10 ppm, respectively. There is also a weak resonance peak at δ ∼ 2.6 ppm, which corresponds to the methylene protons (CH2CH2COOH) of the succinic groups. This observation supports conjugation of carboxyl to terminal hydroxyl groups of Pl.34,36 After grafting with CH, the major peaks of activated Pl along with the resonance peaks at δ ∼ 2.3 and 2.9 ppm of CH are visible. These peaks are ascribed to the methyl proton of partially acetylated groups and the proton on the carbon bearing amino groups of CH, respectively.16 The splitting of the methylene resonance peak (δ ∼ 2.6 ppm) into two peaks at δ ∼ 2.66 and 2.78 ppm determines reaction between CH and the activated Pl through an amide bond formed by EDC/NHS chemistry.36,37
The FTIR spectra of the crosslinked hydrogels with the fibrous protein (keratin) are shown in Fig. 2a. The broad absorption band within 3200 and 3600 cm−1 region is attributed to the stretching vibration of N–H and O–H bonds. Those peaks appeared around 2900 cm−1 are related to C–H stretching modes of aliphatic groups.68 The spectrum of keratin shows characteristic absorption bands assigned mainly to the amide bonds in the range of 1200 to 1700 cm−1, as described in details in ESI.† As for the composite hydrogel, the characteristic peaks of PlCH as well as K are visible. The intensified peaks at 1500–1700 cm−1 are attributed to the presence of fibrous protein with amino acid groups. The interactions between K and PlCH probably exist through hydrogen bonding between amino groups of CH and functional groups of K. After chemical crosslinking by GP, the location and intensity of characteristics peaks of amide type I and II are changed. For instance, the absorbance at 1555 cm−1, which corresponds to N–H bending of amide II either in CH or K, is decreased drastically. A reduction in the intensity of peaks in the 1240–1470 cm−1 regions is also noticed. Moreover, the CO stretching vibration of amide I band is shifted to lower wavenumbers from 1650 to 1640 cm−1. This observation might indicate the formation of secondary amide linkages as a result of crosslinking reactions between primary amino groups of CH/K with the ester groups of GP.
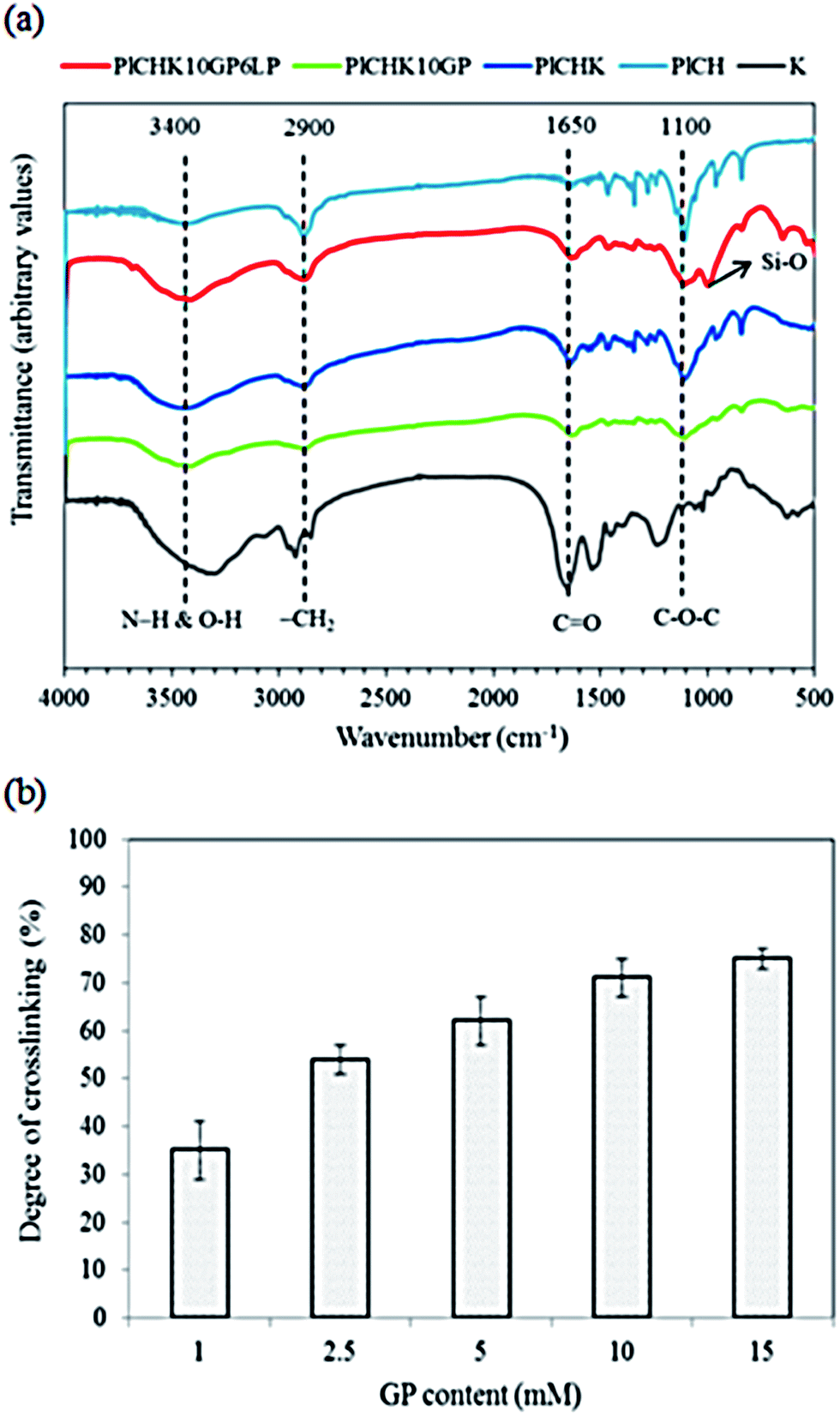 | ||
| Fig. 2 (a) FTIR spectra of crosslinked hydrogels with keratin. (b) Calculated degree of cross-linking as a function of GP concentration. | ||
After introducing of LP nanoparticles, a new peak at about 1000 cm−1 corresponding to the Si–O stretching vibration of the silicate nanoparticles is appeared.69 The stretching vibration of Si–OH groups of LP nanoparticles overlaps with the stretching vibration of hydroxyl groups of the conjugated copolymer in the range of 3400–3600 cm−1. Meanwhile, hydrogen bonding between amino and carboxyl groups of polymer with oxygen containing groups of LP is inevitable. Generally, there is no obvious difference between the spectra of the hydrogels after addition of LP nanoparticles, implying that the chemical structure of the hydrogels is preserved upon incorporation of LP nanoparticles. In other words, exfoliation of silicate nanoparticles within the hydrogel induces physical crosslinking without affecting its chemical structure.
To determine the extent of crosslinking reactions, NHN assay was performed (Fig. 2b). The crosslinking degree rises with increasing GP concentration as a result of a higher extent of reaction. The ester groups of GP interact with the amino groups of PlCH and K, which leads to the formation of amide bonds, as shown in Scheme 1. GP crosslinking mechanism consists of two reactions involving different sites on GP molecule. The first step is the nucleophilic attack of the genipin C3 carbon atom from a primary amine group to form an intermediate aldehyde group. The resulting secondary amine reacts with the aldehyde group to form a heterocyclic compound. The following step is a nucleophilic substitution reaction that involves the replacement of the ester group on the GP molecule by a secondary amide linkage.70 The reaction is complicated by the oxygen radical-induced polymerization of GP that happens in a heterocyclic compound formation which gives the solution a blue color.71 This dark bluish color change during the formation of crosslinked hydrogels does not pose a problem for cartilage reconstruction. On the other hand, it might be useful as a landmark for injection. Previous studies showed that GP could efficiently crosslink CH much faster than proteins such as gelatin and silk fibroin.56,62 It has also been shown that GP preferentially reacts with the amino acids lysine and arginine of certain proteins.58,72
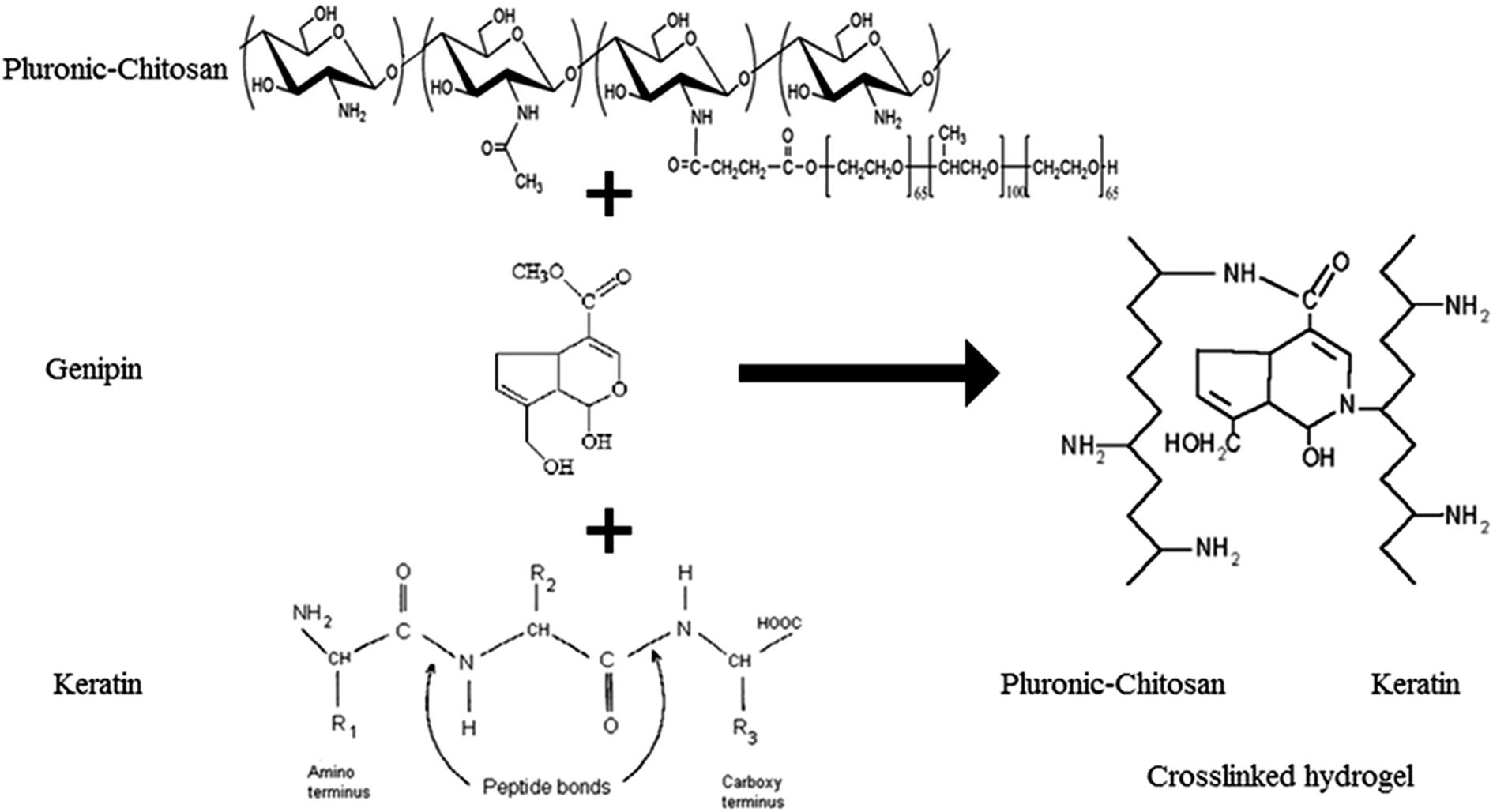 | ||
| Scheme 1 Proposed crosslinking reaction of PlCH with keratin using GP. | ||
3.2 Thermosensitive behavior of hydrogels
A suitable injectable hydrogel should be a free-flowing liquid with low viscosity under non-physiological conditions to allow reproducible administration. After injection, it should undergo a rapid phase transition to form a strong gel capable of withstanding shear forces at physiological conditions.24 The sol–gel transition was first characterized by monitoring the flow property as a function of temperature. The results are shown in Table 1. The polymers show a reversible phase transition from sol to gel with increasing temperature from 10 to 40 °C. All hydrogels are flowing solutions below 20 °C. Pl and Pl + CH blend begin to form a gel-like system at 22 and 25 °C, respectively. Complete gelation of conjugated PlCH system occurs at 32 °C. This temperature shift is favorable in practical applications. The obtained gels were transparent with good consistency without any sliding or shifting when flipping the vials over. It was found that a concentration of 17% (weight fraction of the solid polymer in PBS) was sufficient for the conjugated PlCH to exhibit sol–gel transition; however, a higher concentration (20%) was required for Pl and blended Pl + CH to attain viscoelastic gels upon heating. The thermoreversible behavior of Pl is ascribed to the micelle formation at a temperature above LCST or a concentration above CMC as a result of PPO block dehydration.16,18 The formation of highly ordered structures and micelle entanglements due to the hydrophobic interactions of the PPO group and dehydrated chitosan are proposed as the driving force in gel formation.17 Grafting hydrophilic polymers such as chitosan onto Pluronic would increase the hydrophilicity of the system. This leads to an increase in the sol–gel temperature, since the hydrophobic interactions are compensated at higher temperatures by enhanced polymer–water interactions.73| Sample | Gelation temperature (°C) | Minimum polymer concentration required for gelation (wt%) |
|---|---|---|
| Pl | 22 | 20 |
| Pl + CH | 25 | 20 |
| PlCH | 32 | 17 |
| PlCHK | 35 | 15 |
| PlCHK10GP | 37 | 12 |
| PlCHK10GP3LP | 35 | 10 |
The sol–gel phase transition behavior of conjugated Pl–CH can be tuned after addition of K, GP and LP through synergistic combination of these materials with copolymer thermo-sensitivity. Table 1 shows that K and GP shift the sol–gel transition to around the body temperature while the polymer concentration required for gelation is reduced. For instance, the gelation temperature of the crosslinked hydrogel at a concentration of 12% in the presence of 10 mM GP is 37 °C. The results also determine that addition of LP nanoparticles decreases the phase transition temperature and the polymer concentration necessary for gelation. At 3 wt% LP, the gelation occurs at 35 °C for 10% polymer concentration. Thermogelling in Pluronic/LP mixture has been investigated by other researchers and it was found that sol-to-gel transition could occur at much lower concentrations of the copolymer.54,74,75 Sun et al.74 attributed the onset of thermogelling to depletion flocculation of the LP nanoparticles into a network by spherical micelles of Pluronic. The more hydrophobic PPO segments preferentially adsorbed onto the charged LP surfaces, providing a platform for delayed gelation through hydrophobic interactions.53 The amount of adsorption and the thickness of layer depend on the polymer hydrophobicity, which is strongly affected by temperature.76,77
3.3 Rheological studies
For non-invasive introduction of hydrogels in vivo, it is important to study their rheological properties under physiological conditions. Gelation temperature of the conjugated hydrogel crosslinked with 10 mM GP was determined by oscillatory rheometry through measuring the storage modulus (G′) and loss modulus (G′′) versus temperature at angular frequency of 1 Hz. The obtained results are shown in Fig. 3a. The values of moduli are gradually increased as the temperature rises to 30 °C. At higher temperatures, the gelation process significantly increases the moduli. The higher growth rate of the storage modulus compared to the loss modulus indicates development of gel structure and stiffening.64 The phase transition of Pluronic-based hydrogels above its LCST occurs through hydrophobic interactions between the PPO moieties which leads to the formation of micellar networks.34,60 With increasing temperature, the number of intermolecular hydrogen-bonding interactions is reduced and water surrounding the CH/K chains is removed.17 The hydrophobic interactions of the dehydrated biopolymers as well as Pluronic micellar aggregation in aqueous solution are suggested to be the main driving force behind the formation of the composite hydrogels.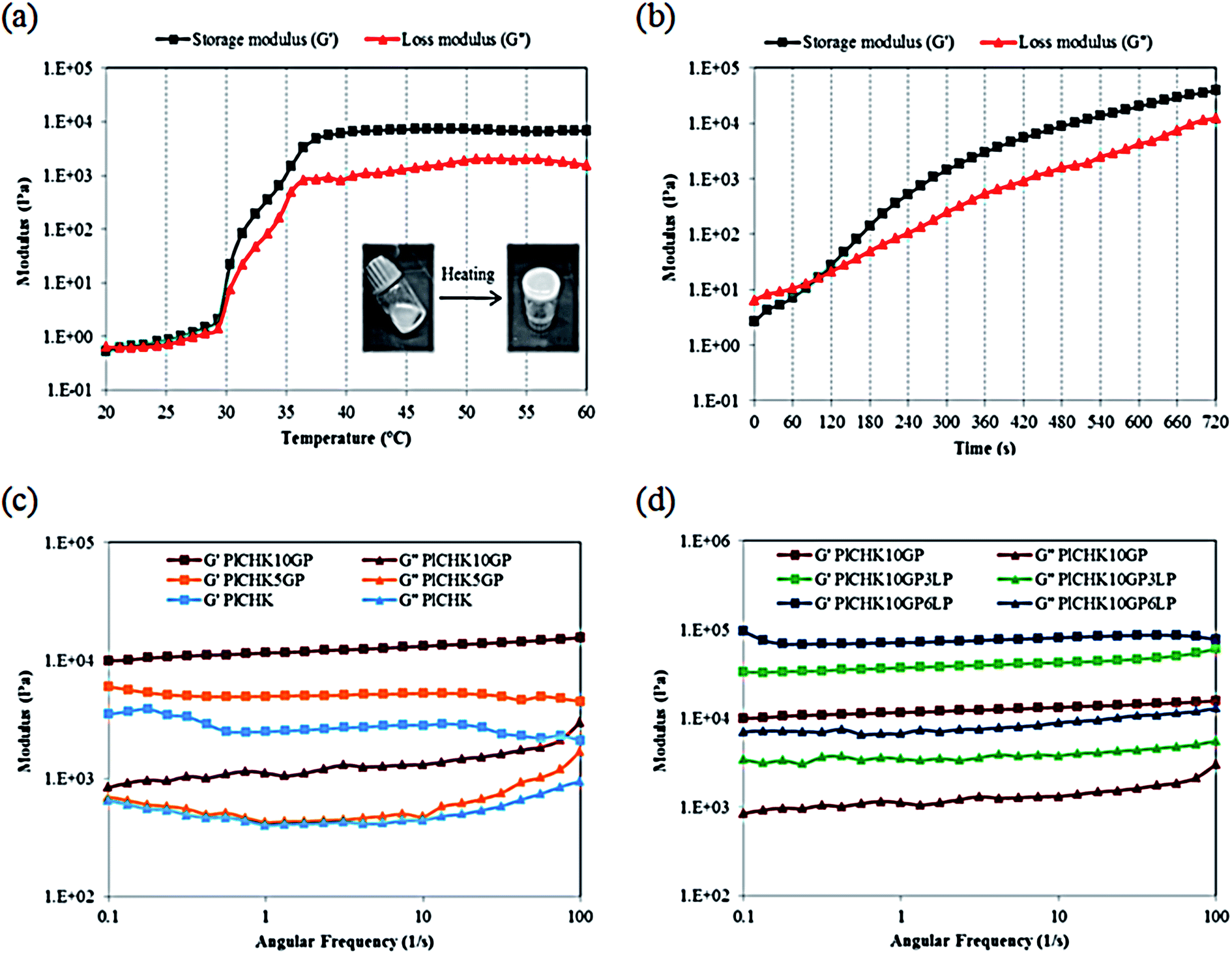 | ||
| Fig. 3 Rheological characterization of hydrogels via (a) temperature sweep, (b) time sweep, (c and d) frequency sweep. | ||
In addition to the thermogelling behavior, gelation time is another key aspect of injectable hydrogel design. Hydrogels should remain fluid during injections and surgical procedures, but once injected, they should transform into gel quickly.59 The formation of hydrogels can be monitored from the development of the viscoelastic functions of the material at the gel point where the transition from the liquid-like to the solid-like behavior occurs. Therefore, gel point is determined from the crossover of the storage modulus and the loss modulus.63 The evolution of G′ and G′′ moduli of the crosslinked hydrogel at 37 °C was monitored as a function of time during gel formation (Fig. 3b). Prior to gelation, the loss modulus G′′ is higher than the elastic modulus G′, which indicates a liquid-like behavior and dominant viscous properties in the beginning of the reaction. At longer times, the increment rate of G′ is more than G′′. This implies that the liquid-like system is transformed into a more solid gel with dominant elastic properties. This transformation is related to the rate and number of crosslinks formed upon heating.78 When chemical crosslinkage is introduced, the physical chain entanglements of the polymeric chains are gradually replaced by a permanent covalent network that favors the increase in G′ over time. With the progress of reaction, the loss and storage moduli intersect at the gel point.58 In fact, GP can act as an in situ covalent crosslinker upon gelation.63,79 It is noteworthy that the sol–gel transition in the crosslinked hydrogels is irreversible as a consequence of the formation of covalent bonds. From the time sweep test results, the gelation time is measured approximately 2 min for the composite hydrogel. This short gelation time is suitable for injection of the hydrogel.
Frequency sweep experiments were also carried out to investigate the viscoelastic properties of the cured systems. The preformed hydrogels were subjected to oscillatory shear tests with frequency sweep from 0.1 to 100 Hz in the linear viscoelastic region. The results are shown in Fig. 3c and d. The storage modulus exhibits almost a frequency independent feature, which reveals the formation of stable crosslinked networks.80 The curves are characteristic for a solid-like material with G′ > G′′ over the measured range of frequencies. As shown in Fig. 3c and Table 2, the amount of crosslinking and thus the strength is proportional to the concentration of GP. For instance, the storage modulus is enhanced from 2.5 to 11.6 kPa with the addition of 10 mM GP. These results determine the role of GP in providing chemically crosslinked networks, by which the mechanical performance and viscoelastic features can be tailored to meet the demands of specific applications. It is also found that the viscoelastic performance of the hydrogels can be improved by LP nanoparticles (Fig. 3d and Table 2). The addition of clay nanoparticles not only increases the elastic modulus of hydrogels by more than three folds, but also enhances the loss modulus significantly. This observation suggests that LP nanoparticles impart viscous properties to the hydrogel network. The interactions between the nanoparticles and the polymer chains affect the mechanical properties of the hydrogels depending on the polymer molecular weight.54 Several studies suggest that in Pluronic/LAPONITE® blends, both PPO and PEO absorb to the clay sheets, although PPO has higher affinity to the platelet surfaces than PEO.75,76 If the polymer chains are long enough to bridge between adjacent LP platelets, physically crosslinked hydrogels with gum-like consistency will be formed.52,54 In total, the enhanced crosslinking density (by means of GP chemical crosslinking) as well as the presence of exfoliated silicate layers improves the shear moduli of the nanocomposite hydrogels.
| Sample | Storage modulus (Pa) | Loss modulus (Pa) |
|---|---|---|
| PlCHK | 2500 ± 34 | 425 ± 11 |
| PlCHK5GP | 4970 ± 28 | 402 ± 8 |
| PlCHK10GP | 11600 ± 52 |
1110 ± 24 |
| PlCHK10GP3LP | 37000 ± 47 |
3470 ± 27 |
| PlCHK10GP6LP | 71300 ± 36 |
6680 ± 19 |
3.4 Microstructure of hydrogels
In addition to tuning mechanical properties, the microstructure of hydrogels is also important as this controls mass transport, aiding the delivery of biological moieties and regeneration of tissues within the hydrogel matrix.52 Fig. 4 shows representative SEM images of some selected hydrogels. The dehydrated cross-sections represent homogeneous and porous network structure with irregular shapes and pore sizes ranging from 5 to 100 μm, which is suitable for cartilage tissue engineering.57 The observed structural integrity indicates a good compatibility between the components in the composite hydrogels. The interior microstructures of the hydrogels in Fig. 4 show that the pore size decreases slightly with increasing the polymer concentration in the hydrogel due to an increase in the number of hydrophobic moieties supporting 3D network structure.17 Evidently, the addition of GP as a natural crosslinker leads to a denser structure with smaller pores due to the chemical crosslinking reactions.78 This finding reflects the difference in the density of crosslinking which is much higher in the covalently crosslinked hydrogels.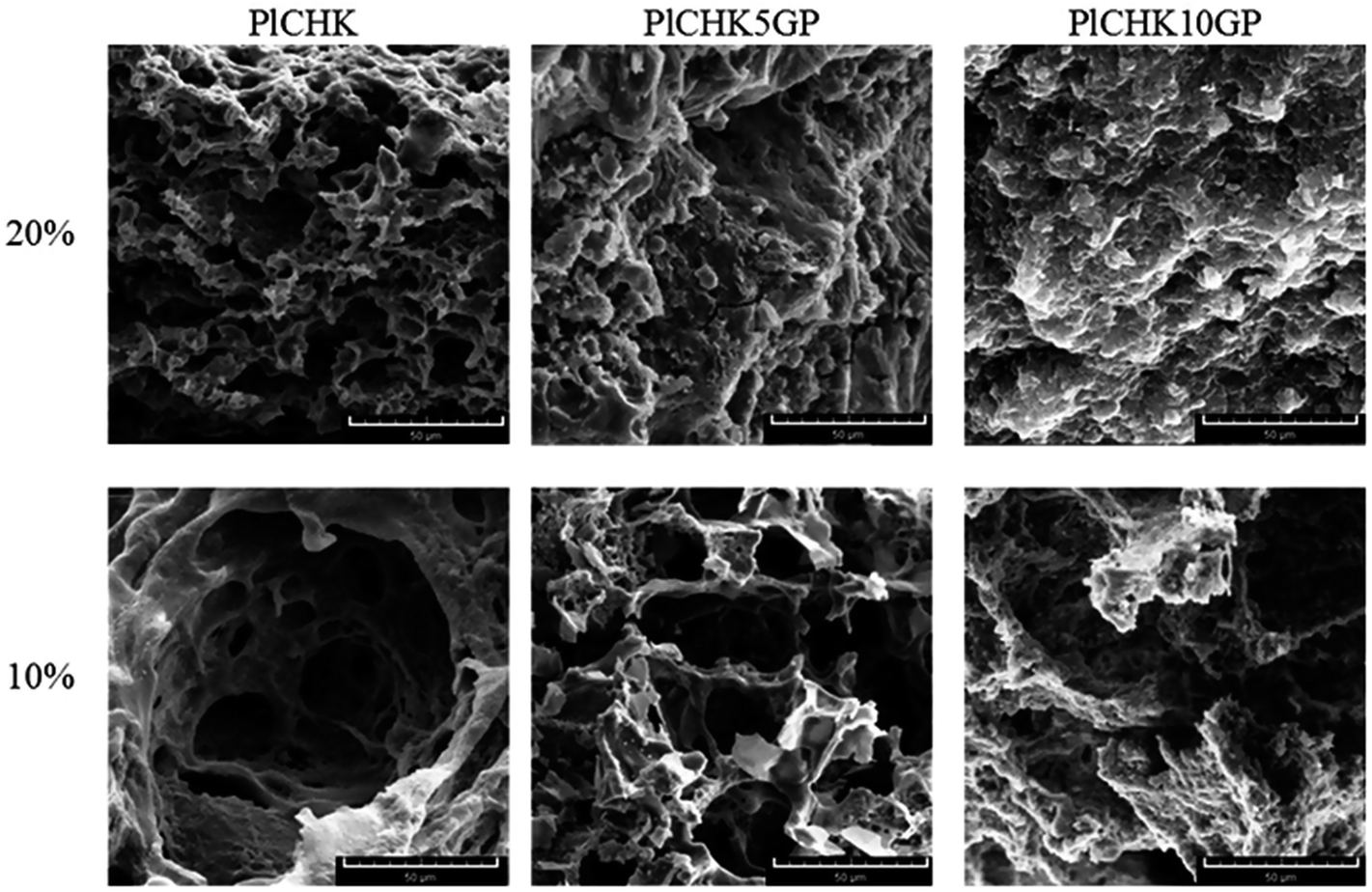 | ||
| Fig. 4 SEM images of uncrosslinked and crosslinked hydrogels with 5 and 10 mm GP at polymer concentration of 10 wt% and 20 wt%. The scale bar is 50 μm. | ||
The effect of LP concentration on hydrogels morphology is also shown in Fig. 5. A decreasing trend in the pore size with increasing the LP content is noticeable. Interestingly, at 6 wt% LP, a honeycomb-like porous structure is formed. Gaharwar et al.52 showed a similar trend for PEG hydrogels containing 5% silicate. Meanwhile, no large agglomeration of nanoparticles in the hydrogels is noticed, which implies the good dispersion of LP in the polymer matrix. Therefore, the porous structure of the hydrogels can be manipulated by changing the polymer concentration and its composition.
 | ||
| Fig. 5 Cross-section morphology of lyophilized crosslinked hydrogels containing (a) 0%, (b) 3% and (c) 6% LP nanoparticles; (d–f) after 10 days of incubation in PBS at 37 °C. | ||
Here it should be noted that, in cartilage tissue repair, the scaffolds must structurally be stable in the body environment while providing open channels for mass flow. In order to examine the microstructure of the hydrogels in simulated body condition, they were incubated in PBS at 37 °C for 10 days. Representative SEM images are shown in Fig. 5d–f. As seen, the interconnected pore channels form an open-cell structure suitable for cartilage repair. In cartilage tissue engineering, a wide range of pore sizes between less than 10 μm to almost 500 μm has been reported for the secretion of chondrogenic factors.81 The hydrophilic nature of hydrogels enables complete wetting of the entire matrix by the culture medium, which allows the cells to be entrapped through the interconnected pores required for differentiation and proliferation within the cartilage tissue.81
TEM micrographs of nanocomposite hydrogel containing 6% LP in Fig. 6 exhibit network-like structure of polymeric hydrogel with uniformly dispersed LP nanoparticles. The clay platelets with a length of ∼20–30 nm and a thickness of ∼1–2 nm are fully exfoliated and homogenously distributed within the hydrogel. In agreement with previous studies,82,83 no severe agglomeration of LP is also noticeable, which leads to the improved mechanical properties of the nanocomposite hydrogels.
 | ||
| Fig. 6 TEM images of nanocomposite hydrogel containing 6% LP nanoparticles. Arrows indicate single LP platelets. | ||
3.5 Swelling and degradation
The swelling characteristics of hydrogels can affect their biological performance and mechanical properties as well as the diffusion rate of fluids. These properties are generally influenced by many factors such as crosslinking density, gel composition, and porosity.62 The swelling ratio of the hydrogels after 24 h immersion in PBS is shown in Fig. 7a. The hydrogels display significant water absorption capability owing to their hydrophilic nature and highly porous structure. The uncrosslinked hydrogel exhibits the highest equilibrium swelling ratio of 352%, but swelling is decreased at higher GP concentrations. A similar trend is observed for the nanocomposite hydrogels containing LP nanoclay.52 The higher the crosslinking degree, the lower is the water adsorption capacity of the composite hydrogels since the number of available hydrophilic groups is reduced. Additionally, the pore sizes are smaller in nanocomposite hydrogels, which decrease the water uptake.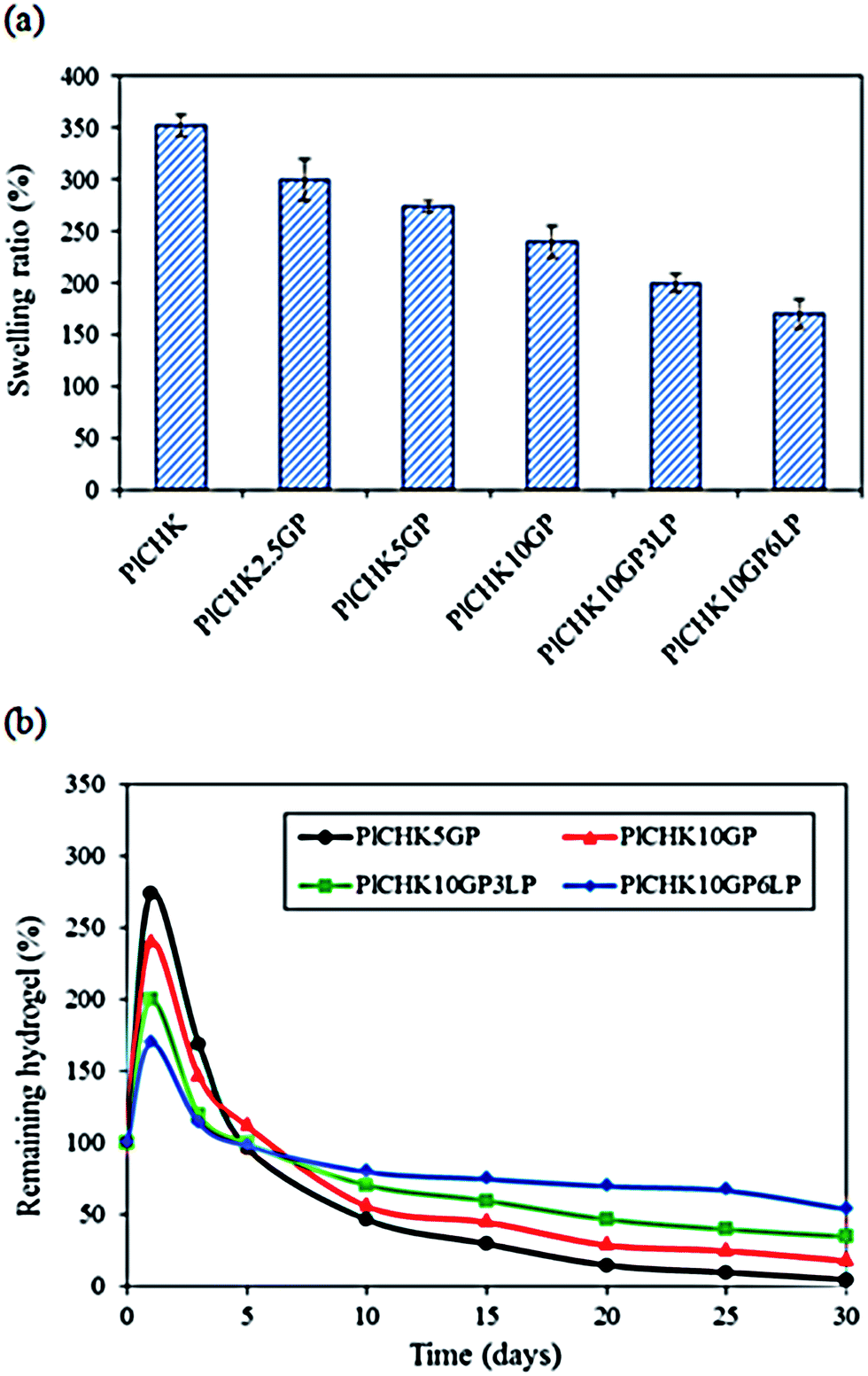 | ||
| Fig. 7 (a) Swelling ratio of the hydrogels. (b) In Vitro biodegradation after different times of incubation in pbs at 37 °C. | ||
Due to the long repair time of cartilage, it is crucial that the engineered scaffolds have the ability to maintain a durable ECM-like structure under physiological environment until tissue regeneration.23 We have found that uncrosslinked hydrogels are degraded quickly after 48 h of incubation (data not shown) because of their high swelling ratio and solubility in aqueous media. Therefore, we used GP to induce covalent crosslinking and promote hydrogels stability.63 The degradation profile for the % weight remaining of hydrogels after different times of incubation in PBS is shown in Fig. 7b. The initial rise is attributed to the water uptake and swelling. It was found that degradation of the hydrogels was significantly retarded by the addition of GP and LP, owing to more compact and stable hydrogel structures. In fact, intra- and inter-molecular networks hindered the degradation of the hydrogels through dissolution of the hydrophilic polymer chains/segments into the aqueous phase.58,62 The nanocomposite hydrogel containing 6% LP was the most stable scaffold among the examined materials. The presence of charged silicate nanoparticles can interact with the polymer molecules and act as an effective physical crosslinking agent. On the other hand, GP provides chemical crosslinking which improves the hydrogel stability under physiological environment. Thus, the combined physico-chemical crosslinking caused long-term stability (>30 days) of the hydrogels.
3.6 In vitro biological performance
Ideal hydrogel biomaterials for tissue engineering are preferentially biodegradable, while providing a favorable microenvironment for cell adhesion to promote tissue regeneration.9,11 The cytotoxicity of hydrogels was evaluated by MTT assay. Fig. 8a shows the high viability (>95%) of CHONs incubated on the hydrogels up to 14 days compared with the control. This observation reflects the biocompatibility of the crosslinked hydrogels and confirms nontoxicity of GP treatment. Both CH and K are well known as biocompatible and biodegradable biopolymers that have been widely studied due to their ability to support cell growth and integrate with surrounding ECM.9,44 Pluronic F127 has been also approved by FDA (Food and Drug Administration) for use as food additives and pharmaceutical ingredient.36 Genepin is a plant extracted crosslinker with anti-inflammatory, anti-fibrotic, neurogenic, and hemostatic properties.79 It is 10000-fold less cytotoxic than glutaraldehyde and has been utilized as a therapeutic agent.63 The experimental results have shown that the nanocomposite hydrogels exhibit slightly higher cytocompatibility than the polymeric hydrogels, although statistical differences were not found. While LP nanoparticles are biocompatible,84 their dissolution in the physiological solution and release of inorganic ions (i.e., Na+, Si(OH)4, Mg2+, Li+) could improve cell viability.52,85,86 Particularly, divalent cations such as magnesium play significant roles in cellular adhesion to biomaterial surfaces that are mediated mainly by adhesion proteins belonging to the integrin family. Orthosilicate acid (Si(OH)4) has also been shown to stimulate collagen I formation and osteoblastic differentiation.87
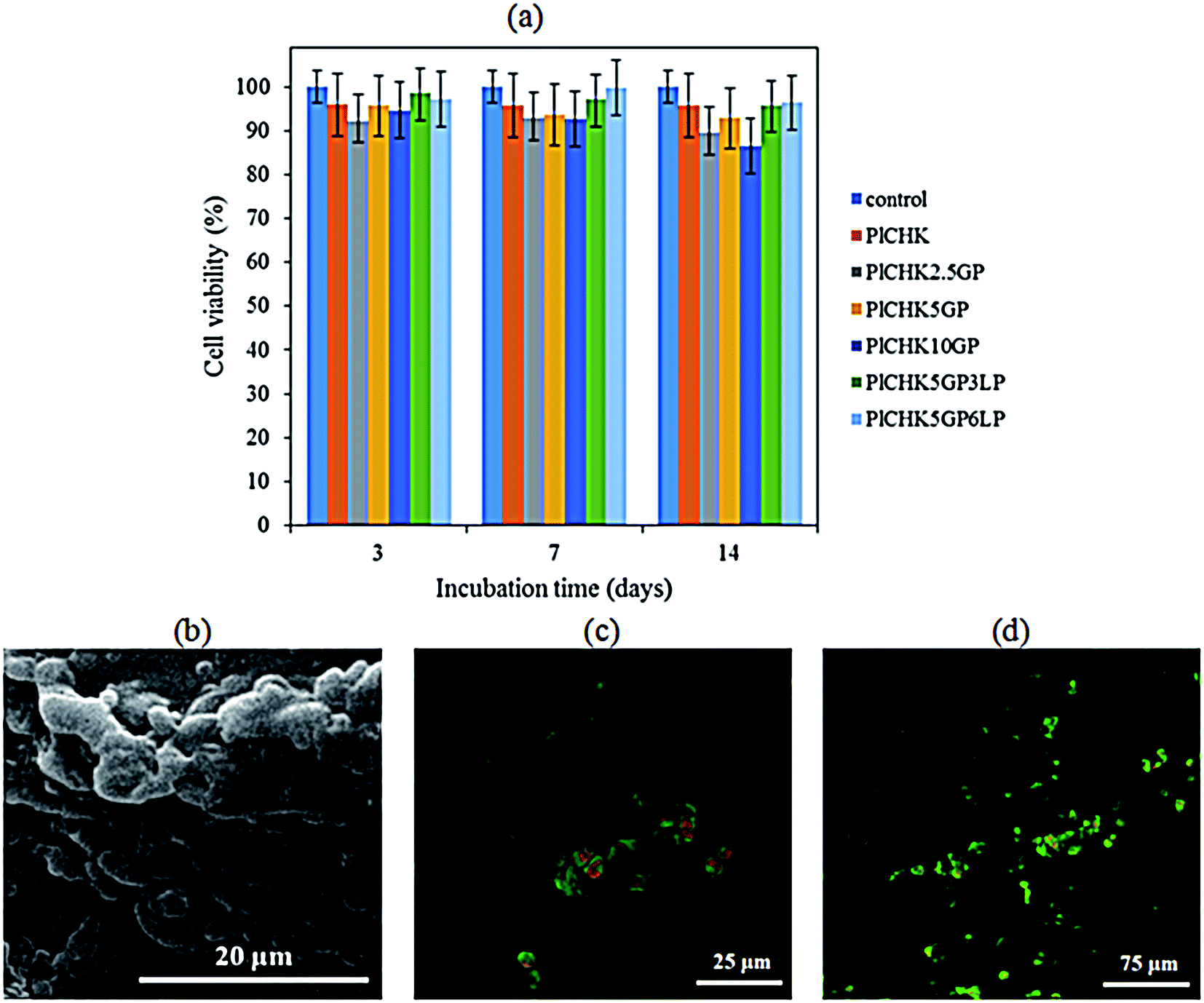 | ||
| Fig. 8 (a) MTT results of hydrogels in three incubation time intervals. (b) SEM images of CHONs attached to the surface of the crosslinked nanocomposite hydrogels containing 6% LP. (c) & (d) Fluorescent images of CHONs encapsulated within hydrogels (PlCHK5GP and PlCHK5GP6LP, respectively) after 7 days in vitro culturing with AO/PI staining. The green stains represent the live cells while the red stains represent the dead cells. | ||
Fig. 8b show SEM images of CHONs incubated onto the crosslinked nanocomposite hydrogels containing 6% LP. The cells readily attached on the surface of the hydrogels and formed adhesion contacts, indicating a desirable cellular attachment required for proliferation. Besides, the spherical morphology of CHONs is maintained due to the 3D structure of the hydrogels. It has been reported that the morphology as well as phenotype of the chondrocytes is changed from spherical to spindle in a monolayer culture. This morphology can be changed to spherical following culture in 3D hydrogels.64,88 Results of live/dead staining in Fig. 8c and d illustrate that CHONs remained alive during encapsulation and gelation process within hydrogel. There are more viable cells in the nanocomposite hydrogel containing hydrophilic charged silicate nanodiscs (LP) as compared to the pristine crosslinked hydrogel. The surface chemistry, hydrophilicity and porous structure of hydrogels influence their biocompatibility, attachment, spreading and migration of cells.86,89 Previous attempts to induce and tune cell adhesion on polymer surfaces include the incorporation of cell recognition motifs, such as RGD peptide sequence.38,39 Other methods are surface patterning to control cell adhesion, e.g. surface modification with hydrophilic or hydrophobic polymers or growth factors.33,90 We have shown that incorporation of biocompatible LP nanoparticles in the hydrogels could enhance cellular adhesion and proliferation as a viable strategy to maintain chondrogenic induction. Similar results showing the positive effect of silicate nanoparticles on the cell attachment and viability have also been reported.52,86 Nevertheless, it should be noted that in vivo assays are required to evaluate the technical feasibility of the thermoresponsive hydrogels for cartilage tissue engineering applications which will be presented in our future study.
4. Conclusions
A novel injectable nanocomposite hydrogel was designed for potential cartilage tissue engineering applications. Pluronic F127 copolymer was conjugated with chitosan and crosslinked with keratin using genepin to attain hydrogels with tunable viscoelastic, physical and biological properties. LAPONITE® nanoparticles were also incorporated into the hydrogels as a reinforcing agent. Results revealed that a more homogenous hydrogel network with better stability and stiffness was attained after GP crosslinking. Moreover, physicochemical properties of the hydrogels such as crosslinking degree, swelling ratio, biodegradation, and porosity could be tuned by changing the components concentration and composition. The microstructure of the fabricated hydrogels showed uniformly distributed and interconnected pores within the range of 5–100 μm. The pore size decreased with higher crosslinking density as well as LP nanoparticles incorporation. TEM images revealed good dispersion and exfoliation of LP clay platelets in the nanocomposite hydrogel. Oscillatory shear experiments suggested that the silicate nanoparticles physically interacted with the covalently crosslinked polymer networks and imparted viscoelasticity to the hydrogels. The addition of LP improved the elastic modulus and biostability of the polymeric hydrogels up to 6 folds without hampering the in vitro cell viability. Results of cytotoxicity assay also showed that the fabricated scaffolds were not harmful to the cultured cells and not only kept the cell viability at normal level (>90%), but also enhanced cell proliferation. Incorporation of silicate nanoparticles into the hydrogels improved CHONs attachment and bioactivity. In conclusion, the introduced nanocomposite hydrogels are potentially applicable in regenerative medicine to provide structural support to the reconstructing region in load-bearing cartilage tissue.Acknowledgements
The authors thank funding support from the Grant Program of Sharif University of Technology (No. G930305) and the National Elites Foundation of Iran. They also acknowledge the personnel of Cell Bank of Iran Pasteur Institute for their cooperation and useful consultation on biological assay.References
- Q. G. Wang, N. Hughes, S. H. Cartmell and N. J. Kuiper, Eur. Cells Mater., 2010, 19, 86–95 CrossRef CAS PubMed.
- E. A. Makris, A. H. Gomoll, K. N. Malizos, J. C. Hu and K. A. Athanasiou, Nat. Rev. Rheumatol., 2015, 11, 21–34 CrossRef CAS PubMed.
- R. Vasita and D. S. Katti, Int. J. Nanomed., 2006, 1, 15–30 CrossRef CAS PubMed.
- R. Stoop, Injury, 2008, 39(1), S77–S87 CrossRef PubMed.
- L. Kock, C. C. van Donkelaar and K. Ito, Cell Tissue Res., 2012, 347, 613–627 CrossRef CAS PubMed.
- A. O. Oseni, C. Crowley, M. Z. Boland, P. E. Butler and A. M. Seifalian, in Tissue Engineering for Tissue and Organ Regeneration, ed. D. Eberli, InTech, 2011, 12, DOI:10.5772/22453.
- K. A. Athanasiou, E. M. Darling and J. C. Hu, Articular Cartilage Tissue Engineering, Morgan & Claypool Publishers, 2010 Search PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS PubMed.
- B. Balakrishnan and R. Banerjee, Chem. Rev., 2011, 111, 4453–4474 CrossRef CAS PubMed.
- Y. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- H. Tan and K. G. Marra, Materials, 2010, 3, 1746–1767 CrossRef CAS.
- A. A. Amini and L. S. Nair, Biomed. Mater., 2012, 7, 024105 CrossRef PubMed.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, M. Karperien, C. A. van Blitterswijk, Z. Y. Zhong and J. Feijen, Biomaterials, 2009, 30, 2544–2551 CrossRef CAS PubMed.
- M. S. Kim and H. J. Chun, Tissue Eng. Regener. Med., 2011, 8, 117–123 Search PubMed.
- J. Escobar-Chávez, M. López-Cervantes, A. Naik, Y. Kalia, D. Quintanar-Guerrero and A. Ganem-Quintanar, J. Pharm. Pharm. Sci., 2006, 9, 339–358 Search PubMed.
- H. J. Chung, D. H. Go, J. W. Bae, I. K. Jung, J. W. Lee and K. D. Park, Curr. Appl. Phys., 2005, 5, 485–488 CrossRef.
- K. M. Park, S. Y. Lee, Y. K. Joung, J. S. Na, M. C. Lee and K. D. Park, Acta Biomater., 2009, 5, 1956–1965 CrossRef CAS PubMed.
- B. Chu, Langmuir, 1995, 11, 414–421 CrossRef CAS.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed.
- K. Derakhshandeh, M. Fashi and S. Seifoleslami, Drug Des., Dev. Ther., 2010, 4, 255–262 CrossRef CAS.
- E. A. Yapar and Ö. Ýnal, Trop. J. Pharm. Res., 2013, 11, 855–866 Search PubMed.
- M. Elluru, H. Ma, M. Hadjiargyrou, B. S. Hsiao and B. Chu, Polymer, 2013, 54, 2088–2095 CrossRef CAS.
- J. B. Lee, J. J. Yoon, D. S. Lee and T. G. Park, J. Biomater. Sci., Polym. Ed., 2004, 15, 1571–1583 CrossRef CAS PubMed.
- H.-R. Lin, K. Sung and W.-J. Vong, Biomacromolecules, 2004, 5, 2358–2365 CrossRef CAS PubMed.
- Y. Z. Zhao, H. F. Lv, C. T. Lu, L. J. Chen, M. Lin, M. Zhang, X. Jiang, X. T. Shen, R. R. Jin, J. Cai, X. Q. Tian and H. L. Wong, PLoS One, 2013, 8, e73178 CrossRef CAS PubMed.
- A. Leroy, C. Pinese, C. Bony, X. Garric, D. Noël, B. Nottelet and J. Coudane, Mater. Sci. Eng., C, 2013, 33, 4133–4139 CrossRef CAS PubMed.
- Y. Lee, H. J. Chung, S. Yeo, C.-H. Ahn, H. Lee, P. B. Messersmith and T. G. Park, Soft Matter, 2010, 6, 977–983 RSC.
- W. Cui, L. Cheng, H. Li, Y. Zhou, Y. Zhang and J. Chang, Polymer, 2012, 53, 2298–2305 CrossRef CAS.
- M. J. Moura, H. Faneca, M. P. Lima, M. H. Gil and M. M. Figueiredo, Biomacromolecules, 2011, 12, 3275–3284 CrossRef CAS PubMed.
- Y. Hong, H. Song, Y. Gong, Z. Mao, C. Gao and J. Shen, Acta Biomater., 2007, 3, 23–31 CrossRef CAS PubMed.
- H. Sá-Lima, S. G. Caridade, J. F. Mano and R. L. Reis, Soft Matter, 2010, 6, 5184–5195 RSC.
- H. Park, B. Choi, J. Hu and M. Lee, Acta Biomater., 2013, 9, 4779–4786 CrossRef CAS PubMed.
- J. S. Choi and H. S. Yoo, J. Biomater. Sci., Polym. Ed., 2013, 24, 210–223 CAS.
- K. M. Park, J. W. Bae, Y. K. Joung, J. W. Shin and K. D. Park, Colloids Surf., B, 2008, 63, 1–6 CrossRef CAS PubMed.
- V. Kumar, P. K. Gupta, V. K. Pawar, A. Verma, R. Khatik, P. Tripathi, P. Shukla, B. Yadav, J. Parmar and R. Dixit, J. Biomater. Tissue Eng., 2014, 4, 210–216 CrossRef CAS.
- W. Zhang, K. Gilstrap, L. Wu, R. B. KC, M. A. Moss, Q. Wang, X. Lu and X. He, ACS Nano, 2010, 4, 6747–6759 CrossRef CAS PubMed.
- Z. Liu and P. Yao, Polym. Chem., 2014, 5, 1072–1081 RSC.
- K. M. Park, Y. K. Joung, K. D. Park, S. Y. Lee and M. C. Lee, Macromol. Res., 2008, 16, 517–523 CrossRef CAS.
- S. Q. Liu, Q. Tian, L. Wang, J. L. Hedrick, J. H. P. Hui, Y. Y. Yang and P. L. R. Ee, Macromol. Rapid Commun., 2010, 31, 1148–1154 CrossRef CAS PubMed.
- J. G. Rouse and M. E. Van Dyke, Materials, 2010, 3, 999–1014 CrossRef.
- S. Wang, F. Taraballi, L. P. Tan and K. W. Ng, Cell Tissue Res., 2012, 347, 795–802 CrossRef CAS PubMed.
- A. Tachibana, S. Kaneko, T. Tanabe and K. Yamauchi, Biomaterials, 2005, 26, 297–302 CrossRef CAS PubMed.
- H. Lee, K. Noh, S. Lee, I.-K. Kwon, D.-W. Han, I.-S. Lee and Y.-S. Hwang, Tissue Eng. Regener. Med., 2014, 11, 255–265 CrossRef CAS.
- G. D. Mogosanu, A. M. Grumezescu and M. C. Chifiriuc, Curr. Drug Targets, 2014, 15, 518–530 CrossRef CAS PubMed.
- A. Vasconcelos and A. Cavaco-Paulo, Curr. Drug Targets, 2013, 14, 612–619 CrossRef CAS PubMed.
- H. Xu, S. Cai, L. Xu and Y. Yang, Langmuir, 2014, 30, 8461–8470 CrossRef CAS PubMed.
- P. Schexnailder and G. Schmidt, Colloid Polym. Sci., 2009, 287, 1–11 Search PubMed.
- A. K. Gaharwar, N. A. Peppas and A. Khademhosseini, Biotechnol. Bioeng., 2014, 111, 441–453 CrossRef CAS PubMed.
- C.-W. Chang, A. van Spreeuwel, C. Zhang and S. Varghese, Soft Matter, 2010, 6, 5157–5164 RSC.
- K. Haraguchi, Polym. J., 2011, 43, 223–241 CrossRef CAS.
- S. K. Mujumdar and R. A. Siegel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6630–6640 CrossRef CAS PubMed.
- A. K. Gaharwar, C. P. Rivera, C.-J. Wu and G. Schmidt, Acta Biomater., 2011, 7, 4139–4148 CrossRef CAS PubMed.
- C. J. Wu and G. Schmidt, Macromol. Rapid Commun., 2009, 30, 1492–1497 CrossRef CAS PubMed.
- C.-J. Wu, A. K. Gaharwar, B. K. Chan and G. Schmidt, Macromolecules, 2011, 44, 8215–8224 CrossRef CAS.
- H.-J. Koo, K.-H. Lim, H.-J. Jung and E.-H. Park, J. Ethnopharmacol., 2006, 103, 496–500 CrossRef CAS PubMed.
- L. P. Yan, Y. J. Wang, L. Ren, G. Wu, S. G. Caridade, J. B. Fan, L. Y. Wang, P. H. Ji, J. M. Oliveira, J. T. Oliveira, J. F. Mano and R. L. Reis, J. Biomed. Mater. Res., Part A, 2010, 95, 465–475 CrossRef PubMed.
- S.-M. Lien, W.-T. Li and T.-J. Huang, Mater. Sci. Eng., C, 2008, 28, 36–43 CrossRef CAS.
- S. S. Silva, A. Motta, M. T. Rodrigues, A. F. Pinheiro, M. E. Gomes, J. F. Mano, R. L. Reis and C. Migliaresi, Biomacromolecules, 2008, 9, 2764–2774 CrossRef CAS PubMed.
- G. Jalani, D. H. Rosenzweig, G. Makhoul, S. Abdalla, R. Cecere, F. Vetrone, L. Haglund and M. Cerruti, Macromol. Biosci., 2015, 15, 473–480 CrossRef CAS PubMed.
- H. J. Chung, J. W. Bae, H. D. Park, J. W. Lee and K. D. Park, Macromol. Symp., 2005, 224, 275–286 CrossRef CAS.
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CrossRef CAS PubMed.
- W. Xiao, W. Liu, J. Sun, X. Dan, D. Wei and H. Fan, J. Bioact. Compat. Polym., 2012, 27, 327–341 CrossRef.
- D. Macaya, K. K. Ng and M. Spector, Adv. Funct. Mater., 2011, 21, 4788–4797 CrossRef CAS.
- F. Mirahmadi, M. Tafazzoli-Shadpour, M. A. Shokrgozar and S. Bonakdar, Mater. Sci. Eng., C, 2013, 33, 4786–4794 CrossRef CAS PubMed.
- S. Bonakdar, S. H. Emami, M. A. Shokrgozar, A. Farhadi, S. A. H. Ahmadi and A. Amanzadeh, Mater. Sci. Eng., C, 2010, 30, 636–643 CrossRef CAS.
- Y.-l. Su, J. Wang and H.-z. Liu, Macromolecules, 2002, 35, 6426–6431 CrossRef CAS.
- J. Ostrowska-Czubenko and M. Gierszewska-Drużyńska, Carbohydr. Polym., 2009, 77, 590–598 CrossRef CAS.
- N. Eslahi, F. Dadashian, N. Hemmati Nejad and M. Rabiee, J. Appl. Polym. Sci., 2014, 131 DOI:10.1002/APP.40294.
- P. Li, N. H. Kim, H. Siddaramaiah and J. H. Lee, Composites, Part B, 2009, 40, 275–283 CrossRef.
- V. Chiono, E. Pulieri, G. Vozzi, G. Ciardelli, A. Ahluwalia and P. Giusti, J. Mater. Sci.: Mater. Med., 2008, 19, 889–898 CrossRef CAS PubMed.
- M. F. Butler, Y. F. Ng and P. D. Pudney, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3941–3953 CrossRef CAS.
- F.-L. Mi, Biomacromolecules, 2005, 6, 975–987 CrossRef CAS PubMed.
- C.-C. Chen, C.-L. Fang, S. A. Al-Suwayeh, Y.-L. Leu and J.-Y. Fang, Int. J. Pharm., 2011, 415, 119–128 CrossRef CAS PubMed.
- K. Sun and S. R. Raghavan, Langmuir, 2010, 26, 8015–8020 CrossRef CAS PubMed.
- I. Boucenna, L. Royon, P. Colinart, M.-A. Guedeau-Boudeville and A. Mourchid, Langmuir, 2010, 26, 14430–14436 CrossRef CAS PubMed.
- A. Nelson and T. Cosgrove, Langmuir, 2005, 21, 9176–9182 CrossRef CAS PubMed.
- R. De Lisi, M. Gradzielski, G. Lazzara, S. Milioto, N. Muratore and S. Prévost, J. Phys. Chem. B, 2008, 112, 9328–9336 CrossRef CAS PubMed.
- J. Xu, S. Strandman, J. X. Zhu, J. Barralet and M. Cerruti, Biomaterials, 2015, 37, 395–404 CrossRef CAS PubMed.
- H. W. Sung, Y. Chang, I. L. Liang, W. H. Chang and Y. C. Chen, J. Biomed. Mater. Res., 2000, 52, 77–87 CrossRef CAS PubMed.
- M. J. Moura, M. M. Figueiredo and M. H. Gil, Biomacromolecules, 2007, 8, 3823–3829 CrossRef CAS PubMed.
- S.-M. Lien, L.-Y. Ko and T.-J. Huang, Acta Biomater., 2009, 5, 670–679 CrossRef CAS PubMed.
- A. Klein, P. G. Whitten, K. Resch and G. Pinter, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 1763–1773 CrossRef CAS.
- J. Du, S. Xu, S. Feng, L. Yu, J. Wang and Y. Liu, Soft Matter, 2016, 12, 1649–1654 RSC.
- Y. Li, J. L. Santos, D. Maciel, H. Tomás and J. Rodrigues, J. Controlled Release, 2011, 152, e55–e57 CrossRef CAS PubMed.
- P. J. Schexnailder, A. K. Gaharwar, I. Bartlett, L. Rush, B. L. Seal and G. Schmidt, Macromol. Biosci., 2010, 10, 1416–1423 CrossRef CAS PubMed.
- A. K. Gaharwar, P. J. Schexnailder, B. P. Kline and G. Schmidt, Acta Biomater., 2011, 7, 568–577 CrossRef CAS PubMed.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757–2774 CrossRef CAS PubMed.
- N. Koushki, A. A. Katbab, H. Tavassoli, A. Jahanbakhsh, M. Majidi and S. Bonakdar, RSC Adv., 2015, 5, 9089–9096 RSC.
- P. Thevenot, W. Hu and L. Tang, Curr. Top. Med. Chem., 2008, 8, 270 CrossRef CAS PubMed.
- R. J. Wade, E. J. Bassin, W. M. Gramlich and J. A. Burdick, Adv. Mater., 2015, 27, 1356–1362 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08563f |
| ‡ Present address: Department of Textile Engineering, Science and Research Branch, Islamic Azad University, P.O. Box 14515/775, Tehran, Iran. |
| This journal is © The Royal Society of Chemistry 2016 |