
Two new fluorinated copolymers based on thieno[2,3-f]benzofuran for efficient polymer solar cells†
Dingjun Hea,
Lixia Qiua,
Zhiguo Zhangb,
Yongfang Lib,
Chunyue Pana and
Yingping Zou*a
aCollege of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China. E-mail: yingpingzou@csu.edu.cn
bBeijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 22nd June 2016
Abstract
Two new copolymers named TBFPF-BT and TBFPF-BO, composed of a fluorine substituted thieno[2,3-f]benzofuran donor unit and benzothiadiazole/benzooxadiazole acceptor unit, have been synthesized as the donor materials for polymer solar cell (PSC) applications. Both polymers presented wide absorptions (300–800 nm) in the UV-Vis region. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels were −5.28/−3.60 eV and −5.38/−3.69 eV for TBFPF-BT and TBFPF-BO, respectively. PSCs with the blends of TBFPF-BT/TBFPF-BO and PC71BM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, w/w) as photoactive layers exhibited relatively low photovoltaic performances. After optimizing with 1% 1,8-diiodooctane (DIO) as additive, dramatic changes in short-circuit current density (Jsc) and fill factor (FF) were seen, with PCE up to 6.80% and 5.98% under AM 1.5, 100 mW cm−2 for TBFPF-BT and TBFPF-BO, respectively. Compared to TBFPF-BO, the higher PCE mainly benefits from the higher hole mobility and better morphology for the TBFPF-BT and PC71BM blend film.
:2, w/w) as photoactive layers exhibited relatively low photovoltaic performances. After optimizing with 1% 1,8-diiodooctane (DIO) as additive, dramatic changes in short-circuit current density (Jsc) and fill factor (FF) were seen, with PCE up to 6.80% and 5.98% under AM 1.5, 100 mW cm−2 for TBFPF-BT and TBFPF-BO, respectively. Compared to TBFPF-BO, the higher PCE mainly benefits from the higher hole mobility and better morphology for the TBFPF-BT and PC71BM blend film.
Introduction
During the past several decades, bulk heterojunction structure (BHJ) polymer solar cells (PSCs), composed of electron-donating conjugated polymers and electron-accepting fullerene derivatives as the active layer to absorb sunlight, have attracted considerable attention in the research community.1,2 Tremendous efforts have been devoted to pushing the development of PSCs, which is recognized as one of the most efficient and direct methods to take full advantage of solar energy and address the energy crisis. Due to their unique advantages, such as lightweight, low cost, solution processibility and flexibility, many researchers are paying attention to materials innovation and device optimization to improve the power conversion efficiency (PCE) and significant progress has been made in this field. PCEs have experienced a dramatic increase from below 1% in the 1990s to, in recent years, over 10% for single junction3–6 and ∼12% for multi-junction structure BHJ PSCs.7,8 Currently, despite the achievement of such desirable results, there is still a great challenge for commercial applications. The design of new conjugated polymers and delving into the relationship between structure and properties to improve PCE and device stability seem still necessary. Empirically, utilizing novel donor materials may be the most effective method to pursue a higher PCE. Simultaneously, donor materials should possess basic features: (1) broad and strong absorption band in the visible and near-infrared region to match well with solar spectrum; (2) low-lying HOMO energy level for assuring a high open-circuit voltage (Voc); (3) a coplanar molecular structure and good crystalline properties to enhance charge transport for a high fill factor (FF); (4) excellent solubility for solution process.9In recent years, many groups have extensively studied the symmetrical two-dimensional (2D) building block for photovoltaic materials due to its high planarity, enlarged π-conjugated system and relatively deep energy levels which were beneficial for short-circuit current density (Jsc) and Voc.10,11 Among these 2D-conjugated structures, most efforts were devoted to benzodichalcogenophene (BDC) based derivatives, especially focusing on the well-known benzodithiophene (BDT) and benzodifuran (BDF) units. Hou and his coworkers have reported a series of 2D-conjugated copolymers based on BDT core by replacing the alkoxyl group with the alkylthienyl or alkythiothienyl group in the 4, 8 positions of BDT, and made great contributions to maximizing PCEs.12–14 Huo et al. reported a polymer based on a BDF core, named PBDF-T1, delivering a high PCE of 9.43% after optimizing the concentration of 1,8-diiodooctane (DIO), as far as we know, which was the state-of-the-art efficiency about BDF based polymers.15 Meanwhile, our group has also reported different kinds of copolymers based on BDT and BDF units combining with substituted alkoxyphenyl or alkythienyl groups as the conjugated side chains, some of them exhibited prominent photovoltaic properties with a PCE up to 8.6% for BDT-based polymers and 6.6% for BDF-based polymers, respectively.16–18 In contrast to well investigated symmetric structure, less attention has been paid on the asymmetric structures. Copolymers with non-symmetric structure probably possess superior photovoltaic properties as well.4,19 For example, thieno[3,4-b]thiophene (TT) unit, coupled with alkythienyl-substituted BDT (PTB7-Th) in a regiorandom pattern as the electron donor material demonstrated a fairly satisfying PCE over 10% and isoindigo (ID) unit, copolymerization with dithienocarbazole (P(IID1F-DTC)) also presented a high PCE of 7.1%.20 Replacing one of the five-member rings of BDT or BDF with furan or thiophene ring, we successfully constructed an asymmetric BDC derivative: thieno[2,3-f]benzofuran (TBF). As with the random packing mode of TT based polymer chains, TBF-based polymers displayed the similar chain's packing behavior. In our previous work, similar to BDT and BDF units, alkythienyl and alkoxyphenyl were employed to substitute the 4 and 8 positions of TBF unit, polymers with TBF core as donor materials, named PTBFTDTBT, PTBFP-BT and PTBFP-BO were reported with PCE up to 6.4%, 6.02% and 4.41%, respectively, without any post-treatment.21,22 TBF unit was not only successfully applied in constructing conjugated polymers and but also selected as the central building block for photovoltaic small molecules.23 Recently, zhang et al. firstly reported a TBF-based small molecule blended with N2200 acceptor as the active layer for fullerene-free OSC application with a PCE up to 3.74%.24 Such promising results encourage us TBF unit worthy to being deeply investigated. In light of the development of molecular design, incorporating fluorine atom onto the polymer backbone has evolved to be a common strategy to obtain a desirable PCE due to its advantages of tuning the electrochemical properties without disturbing the molecular planarity, and the intermolecular interactions to increase charge mobility of copolymers.13,25 Generally, fluorine-containing polymer has a relatively high Voc in contrast to the non-fluorinated one.26,27 The raised Voc of fluorinated copolymers should be ascribed to the decrease of HOMO energy level with strengthening intra/interchain dipole–dipole interactions. On the other hand, polymer chain's torsional angle along the backbone was minimized via noncovalent interactions of F⋯S, O⋯F. So the planarity and interchain ordering of polymer were improved by introduction of fluorine without losing solution processability.26,28,29 Certainly, molecular design is not the unique approach to improving PCEs, the device optimization, including interface modification and solvent additives, plays a critical role to realizing a proper energy level matches and suitable morphologies for high PCEs.30–32
Regarding of the reasons mentioned above and aiming to boosting PCEs from TBF-based polymers, the well-known acceptor units, 4,7-di(5-bromothiophen-2-yl)-5,6-dioctyloxybenzo[c][1,2,5]thiadiazole (BT) and 4,7-di(5-bromothiophen-2-yl)-5,6-dioctyloxybenzo[c][1,2,5]oxadiazole (BO), were selected to copolymerizing with the donor unit. Two new TBF-based conjugated polymers with fluorine substituted alkoxylphenyl as the conjugated side chain, named TBFPF-BT and TBFPF-BO, were synthesized. The properties of the two polymers were characterized in detail. Both polymers are soluble in the common solvents and have a broad absorption band ranging from 300 to 730 nm in the CHCl3 solution and extending to 800 nm in the film state. The molecular weight of TBFPF-BT and TBFPF-BO are 38.5 kDa and 32.3 kDa, respectively. The hole mobilities are 2.70 × 10−5 cm2 V−1 s−1 for TBFPF-BT and 2.55 × 10−5 cm2 V−1 s−1 for TBFPF-BO determined through space charge limited current (SCLC) method. Both polymers show high fill factors (FF) reaching up to 0.70 and 0.65 for TBFPF-BT and TBFPF-BO, respectively. Using a conventional device structure without any post-treatment, PCEs of 6.80% for TBFPF-BT and 5.98% for TBFPF-BO are obtained.
Experimental section
Materials
Thieno[2,3-f]benzofuran-4,8-dione, monomer M2 and M3 were prepared according to the previous literature with some modifications. Toluene was dried over Na/benzophenoneketyl prior to use. Anhydrous tetrahydrofuran (THF) was purchased from J&K, and other reagents were all commercially available from Alfa Asia and TCI Chemical Co. as ACS-grade quality, and used as received without any further purification unless indicated otherwise. All compounds were synthesized following the procedures described in Scheme 1.33–35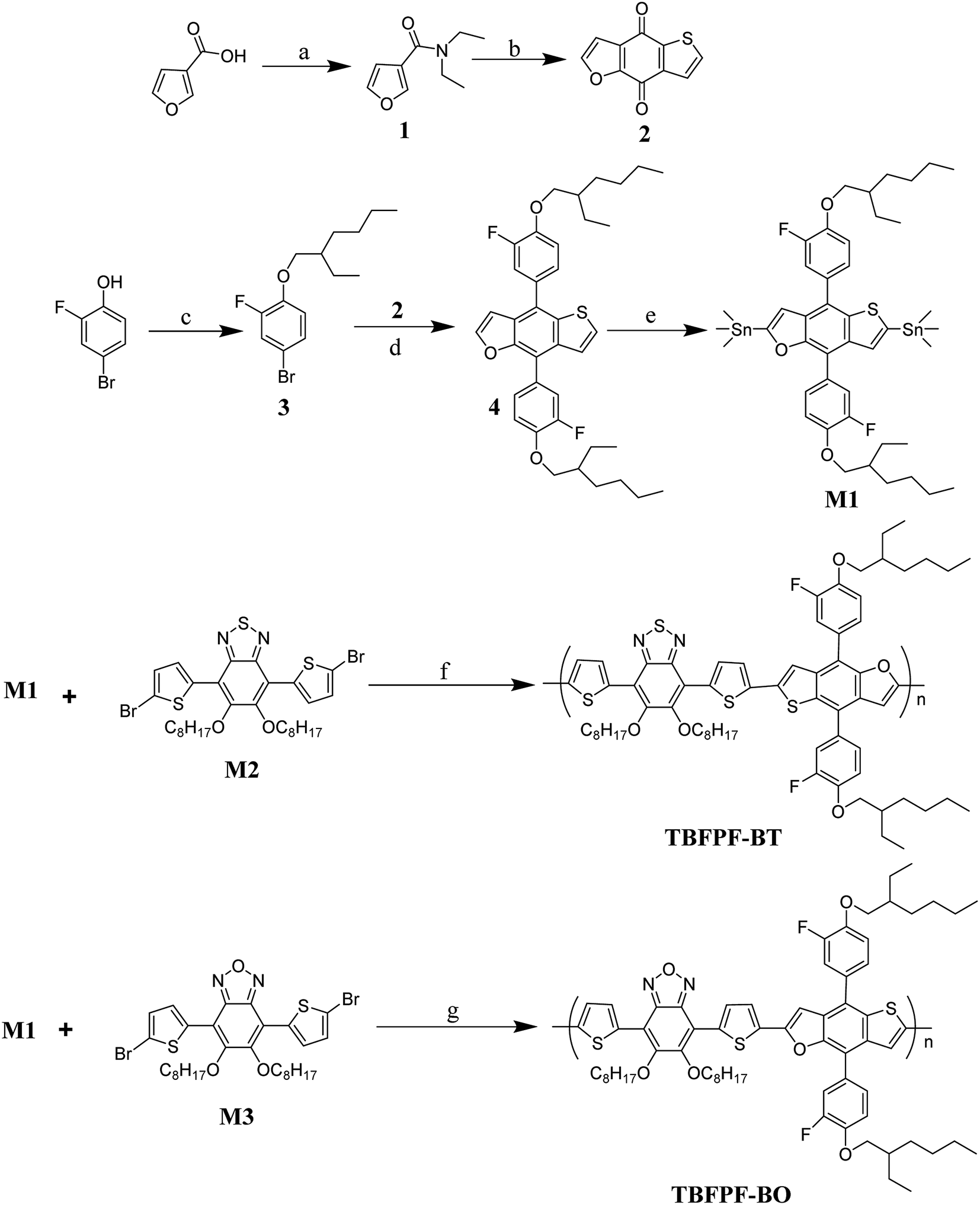 | ||
| Scheme 1 Synthesis route of monomer and polymers. Synthetic conditions, (a) SOCl2, NHEt2, CH2Cl2; (b) thiophene-3-carbaldehyde, n-BuLi, H2O; (c) DMF, K2CO3; (d) Mg, I2, THF; (e) n-BuLi, Sn(CH3)3Cl, THF; (f and g) toluene, Pd(PPh3)4, 110 °C. | ||
Measurements and characterization
1HNMR and 13CNMR were measured on Bruker DMX-400 and DMX-500 spectrometers in CDCl3 solution and the chemical shifts were recorded in ppm units with TMS as the internal standard. 19FNMR was used to characterize fluorine-containing compounds under the same conditions of 1HNMR. The UV-Vis absorption spectra of polymers in CHCl3 solution and thin film states were performed in a Shimadzu UV-2600 spectrophotometer. CHI650D electrochemical workstation were utilized for measuring cyclic voltammetry curves of the two polymers at a scan rate of 50 mV s−1 in 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) acetonitrile solution as electrolyte. Among the three-electrode system, the glassy carbon, platinum wire and Ag/AgCl electrode were used as working electrode, counter electrode and reference electrode, respectively. All measurements were calibrated against the internal standard of ferrocene (Fc), the ionization potential (IP) value of which is −4.8 eV for the Fc/Fc+ redox system. The HOMO energy levels were estimated from the onset oxidation point of CV curves. Number (Mn) and weight average (Mw) molecular weights were determined by gel permeation chromatography (GPC) on a Waters GPC 2410 using THF as eluent calibrated with polystyrene standards. A Perkin-Elmer TGA-7 was used for polymers' thermogravimetric analysis (TGA) at a heating rate of 10 K min−1 under a nitrogen atmosphere. Hole mobilities were calculated by space charge limited current (SCLC) method using ITO/PEDOT:PSS/active layer/Au device configuration. Atomic force microscopy (AFM) was employed to investigate the morphologies of polymer/PC71BM blend films spin-coated on silicon substrates in tapping mode.Fabrication and characterization of PSCs
Organic photovoltaic cells with a device configuration of glass/indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/polymer:PC71BM blend films/ZrAcac/Al were prepared. Patterned ITO glass (sheet resistance 10 Ω sq−1) was commercially available from CSG Holding Co. Ltd. (China). Prior to device fabrication, the patterned ITO-coated glass precleaned by a surfactant and then underwent a wet-cleaning process inside an ultrasonic bath, beginning with deionized water and followed by acetone and isopropyl alcohol. Then it was treated using an ultraviolet ozone chamber (Jelight Company, USA) for 20 min. PEDOT:PSS (Baytron PVP AI 4083, Germany) was filtered through a 0.45 mm filter and a thin layer (30 nm) was spin coated at 3000 rpm for 40 s onto the ITO glass substrate and then baked at 150 °C for 15 min. A mixture of TBFPF-BT/TBFPF-BO:PC71BM in ortho-dichlorobenzene (o-DCB) with different blend ratios and DIO concentrations was spin coated at 1000 rpm for 40 s on the top of PEDOT:PSS layer to form a photosensitive layer. The thickness of the photosensitive layer was ranged from 70 to 120 nm measured by an Ambios Technology XP-2 profilometer. Successively, the electron-transporting layer of ZrAcac (purchased from J&K chemical company) was simply prepared by spin-coating its ethanol solution (1 mg mL−1) atop the photoactive layer at 3000 rpm for 30 s at room temperature without thermal annealing or any other post treatment. Finally, an aluminum layer (100 nm) was deposited by thermal evaporation method under the vacuum condition of 5 × 10−5 Pa. The active layer area of the devices was 4.70 mm2. J–V curves were recorded on a Keithley 2400 Source Measure Unit. Photovoltaic performance was tested under an Air Mass 1.5 Global (AM 1.5 G) solar simulator (Class AAA solar simulator, Model 94063A, Oriel) with an irradiation intensity of 100 mW cm−2. The light intensity at each wavelength was calibrated using a standard single crystal Si photovoltaic cell. All the fabrication and characterizations were conducted in a glove box at inert atmosphere.
Monomer synthesis
1H NMR (400 MHz, CDCl3) δ 7.71 (s, 1H), 7.40 (dd, J = 3.4, 1.7 Hz, 1H), 6.58 (s, 1H), 3.55–3.39 (m, 4H), 1.20 (td, J = 7.1, 2.1 Hz, 6H).
1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 1.8 Hz, 1H), 7.71 (d, J = 5.0 Hz, 1H), 7.65 (d, J = 5.0 Hz, 1H), 6.97 (d, J = 1.8 Hz, 1H).
1H NMR (500 MHz, CDCl3, ppm): δ: 7.24 (dd, 1H), 7.19 (dd, 1H), 6.86 (t, 1H), 3.91 (d, 2H), 1.78 (m, 1H), 1.59–1.32 (m, 8H), 0.95 (m, 6H).
1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 2.3 Hz, 1H), 7.55–7.45 (m, 6H), 7.17 (t, J = 8.6 Hz, 2H), 6.88 (d, J = 2.3 Hz, 1H), 4.04 (d, J = 5.8 Hz, 4H), 1.86 (dd, J = 12.3, 6.1 Hz, 2H), 1.46–1.26 (m, 16H), 1.03–0.94 (m, 12H).
19F NMR (376 MHz, CDCl3) δ −133.66 (s, 1F), −134.31 (s, 1F).
1H NMR (500 MHz, CDCl3) δ 7.59 (s, 1H), 7.57–7.50 (m, 4H), 7.17 (td, J = 8.8, 4.5 Hz, 2H), 7.01–6.98 (m, 1H), 4.08–4.02 (m, 4H), 1.87 (td, J = 11.9, 6.0 Hz, 2H), 1.60–1.38 (m, 16H), 1.04–0.93 (m, 12H), 0.48–0.33 (m, 18H).
13C NMR (125 MHz, CDCl3) δ 130.86, 126.17, 124.86, 118.14, 116.86, 116.36, 114.84, 114.51, 77.27, 77.02, 76.76, 71.98, 39.67, 39.33, 30.49, 29.11, 23.86, 23.07, 14.10, 11.14, −8.33, −9.02.
19F NMR (471 MHz, CDCl3) δ −133.97 (s, 1F), −134.96 (s, 1F).
Anal. calcd for (C44H60F2O3SSn2) (%):C, 55.96; H, 6.40; F, 4.02; O, 5.08; S, 3.39; Sn, 25.14; found (%): C, 55.92; H, 6.37; S, 3.41.
General procedures for polymerization
The polymer synthesis was carried out in a common Stille cross-coupling polymerization using Pd(PPh3)4 as catalyst. The general procedures were described as following.In the 50 mL two-necked flask, M1 and M2/M3 were dissolved in 15 mL of toluene. The solution was flushed with Ar for 20 min to remove O2 and a catalyst amount of Pd(PPh3)4 (15 mg) was added into the flask. The reaction system was flushed with Ar again, and then the solution was warmed up to 110 °C and stirred for 24 h. After cooled to room temperature, the reaction mixture was poured into 200 mL of methanol and stirred for 30 min to precipitate out the crude polymer. The further purification was conducted by a Soxhlet extraction with methanol, hexane and chloroform successively. The chloroform fraction was collected and evaporated to afford the target polymers.
GPC (THF): Mn = 38.5 kDa; Mw = 53.4 kDa; PDI = 1.38. Anal. calcd for (C68H80F2N2O5S4)n (%): C, 69.64; H, 6.82; F, 3.24; N, 2.39; O, 6.82; S, 10.92; found: C, 69.61; H, 6, 87; N, 2.43; S, 10.90.
GPC (THF): Mn = 32.3 kDa; Mw = 51.6 kDa; PDI = 1.59. Anal. calcd for (C68H80F2N2O6S3)n (%): C, 70.61; H, 6.92; F, 3.28; N, 2.42; O, 8.31; S, 8.30; found (%): C, 70.63; H, 6.97; N, 2.37; S, 8.33.
Result and discussion
Polymerization result
TBFPF-BT and TBFPF-BO were synthesized according to the typical Stille coupling reaction with high yield and the procedures were described in Scheme 1. The crude copolymers were precipitated in methanol and filtered to collect the residue for further purification with Soxhlet extractions using methanol, hexane and chloroform successively to obtain pure polymers. Elemental analysis and NMR technique were utilized to identify the structures of monomers and copolymers and NMR spectra are shown in Fig. S1–S9.† Molecular weights determined by GPC using THF as eluent were 38.5 kDa (Mn) and 32.3 kDa (Mn) for TBFPF-BT and TBFPF-BO with corresponding polydispersity indices (PDI) of 1.38 and 1.59, respectively. All the corresponding parameters of polymers were listed in Table 1.| Polymers | Mn (kDa) | Mw (kDa) | PDI | Yield (%) | Td (°C) |
|---|---|---|---|---|---|
| TBFPF-BT | 38.5 | 53.4 | 1.38 | 88 | 335 |
| TBFPF-BO | 32.3 | 51.6 | 1.59 | 83 | 318 |
Thermal stability
Thermal stability is an essential parameter for optoelectronic applications. Herein, the copolymer's thermal stability was investigated through thermogravimetric analysis (TGA) with a heating rate of 10 K min−1 under an inert atmosphere, as shown in Fig. 1. TGA analysis of TBFPF-BT and TBFPF-BO show that both of them possess good thermal stability. The decomposition temperatures, calculated from the onset points of 5% weight loss, are 335 and 318 °C for TBFPF-BT and TBFPF-BO, respectively, which is desirable for device fabrication. The related thermal stability data were summarized in Table 1.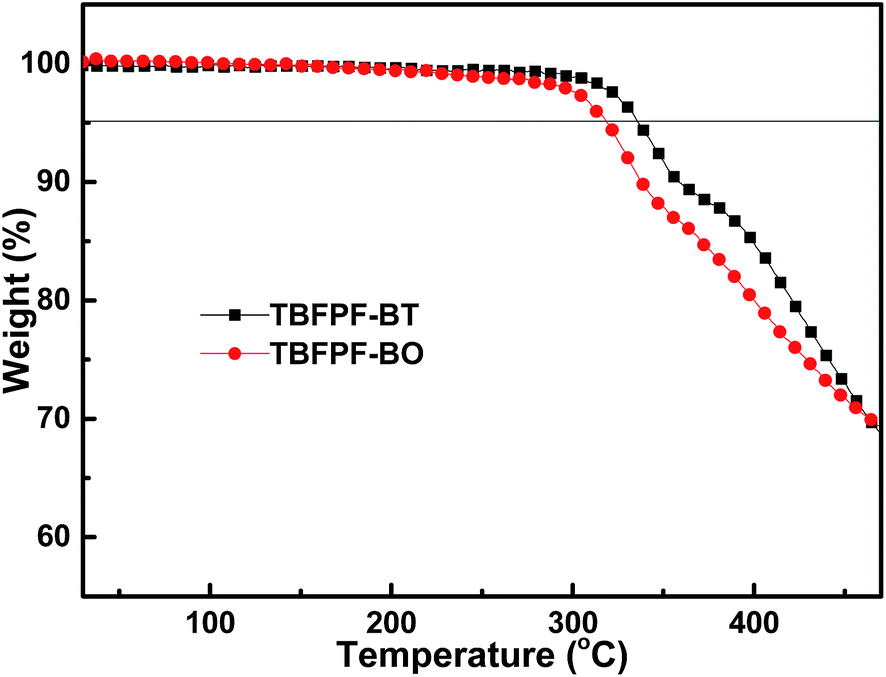 | ||
| Fig. 1 TGA plots of polymers with a heating rate of 10 K min−1. | ||
Optical properties
The optical properties of TBFPF-BT and TBFPF-BO were characterized in UV-Vis spectrophotometer and the absorption spectra in CHCl3 solution and film state at room temperature are shown in Fig. 2. The related optical parameters for two copolymers are summarized in Table 2. The two copolymers exhibit broad absorption with similar profile that matches well with the sunlight irradiation spectrum both in solution and film state, which indicates that these polymers should be an excellent absorber. In dilute CHCl3 solution, TBFPF-BT and TBFPF-BO display two well-defined main absorption peaks, one intense band at higher energies covers from 350 to 450 nm resulting from π–π* transitions and the other one at lower energies range from 500 to 700 nm which should be attributed to intramolecular charge transfer (ICT) between donor and acceptor units in the copolymer backbone.36 The thin-film absorption exhibit similar optical property to that in solution. Compared to the maximum absorption peak in solution, the thin film maximum absorptions of two polymers slightly redshifted about 8 nm and 5 nm, respectively, implying stronger intermolecular interactions exist in the thin film state than that in solution.37 In addition, obvious vibronic shoulder peaks at lower energies (∼640 nm) were observed in thin film, which further suggests significant order in the polymer structure in film state.38 The optical band gaps (Eoptg) of copolymers deducing from the onset point of absorption edge (λedge) are 1.68 eV for TBFPF-BT and 1.69 eV for TBFPF-BO according to the equation: Eoptg = 1240/λedge. | ||
| Fig. 2 UV-Vis absorption spectra of TBFPF-BT and TBFPF-BO (a) in dilute CHCl3 solution and (b) film state cast from CHCl3 solution. | ||
| Polymer | Solutiona | Filmb | Eoptg (eV) | Energy level | ||
|---|---|---|---|---|---|---|
| λmax (nm) | λmax (nm) | λonset (nm) | HOMOc (eV) | LUMOd (eV) | ||
| a UV-Vis absorption measurement from the dilute CHCl3 solution.b Films on quartz cast from chloroform solution.c EHOMO = −e(Eox + 4.4) (eV).d ELUMO = EHOMO + Eoptg (eV). | ||||||
| TBFPF-BT | 580 | 588 | 735 | 1.68 | −5.28 | −3.60 |
| TBFPF-BO | 578 | 583 | 730 | 1.69 | −5.38 | −3.69 |
Electrochemical properties
In order to investigate the electrochemical properties of copolymers, CV measurement is taken to estimate the energy levels of HOMO and LUMO of both copolymers. The CV curves are illustrated in Fig. 3 and the corresponding electrochemical data are summarized in Table 2. From the CV curves, the clear oxidation peaks were observed for both copolymers and the onset oxidation potentials recorded for TBFPF-BT and TBFPF-BO are 0.88 V and 0.98 V, respectively, where the unit of Eox is V vs. Ag/AgCl. Hence, according to the equation: EHOMO = −e(Eox + 4.4) (eV), the HOMO values are −5.28 eV for TBFPF-BT and −5.38 eV for TBFPF-BO. In comparison with TBFPF-BT, polymer with BO unit as the acceptor exhibits a slightly deeper HOMO energy level (∼0.1 eV) than that with BT unit as acceptor, which may be due to the electronegativity discrepancy between oxygen atom from BO and sulfur atom from BT unit (O, 3.5; S, 2.5). Because Voc was roughly determined by the energy difference between HOMO of donor and LUMO of PCBM acceptor.39 So the deeper HOMO level of TBFPF-BO ensure a higher Voc and good air stability which are beneficial for photovoltaic performance.40 The CV result suggests that apart from manipulating the backbone structures, fine-tuning of the molecular energy levels can be realized by changing the electron-withdrawing acceptor units of the D–A polymers. The LUMO energy levels of both copolymers were therefore calculated from equation: ELUMO = EHOMO + Eoptg to be −3.60 eV and −3.69 eV for TBFPF-BT and TBFPF-BO, respectively.41 Besides, according to the previous literature, the band offset between donor's LUMO level and LUMO level of PCBM acceptor must overcome the exciton binding energy (∼0.3 eV).42 TBFPF-BT and TBFPF-BO provided LUMO offsets are 0.4 eV and 0.31 eV, respectively. Such LUMO offsets are close to the generally accepted minimum requirement of driving force (∼0.30 eV) for efficient exciton separation into free charge carriers.43 | ||
| Fig. 3 (a) Plots of TBFPF-BT and TBFPF-BO films on a platinum electrode determined by the cyclic voltammetry measurements in an acetonitrile solution containing 0.1 M Bu4NPF6 at a scan rate of 20 mV s−1. (b) Diagram of energy levels of materials applied in the PSCs. | ||
Charge mobility
Charge carrier mobility is a significant factor to affect the performance of BHJ PSCs, in particular on Jsc and FF.44,45 Since PCBM has high enough electron transporting ability, the hole mobilities of copolymers are crucial for organic solar cells. Therefore, to investigate the hole mobilities of two polymers, SCLC was applied to characterize the J–V characteristics with a hole-only device structure of ITO/PEDOT:PSS/active layer/Au. The active layers of polymer and PC71BM are in optimized conditions with a weight ratio of 1:2. The SCLC could be approximately by the Mott–Gurney equation described below:
 | (1) |
 | (2) |
 | ||
| Fig. 4 J1/2–V curves of TBFPF-BT (a) and TBFPF-BO (b) determined by SCLC method under the optimized conditions. | ||
Photovoltaic properties
To explore the photovoltaic properties of TBFPF-BT and TBFPF-BO, BHJ organic solar cells were fabricated with a conventional sandwich configuration of ITO/PEDOT:PSS/TBFPF-BT (TBFPF-BO):PC71BM/ZrAcac/Al, as shown in Fig. 5c. The commercially available zirconium acetylacetonate (ZrAcac) was chosen as cathode interfacial layer (CIL),48 which recently demonstrated as an efficient CIL for PSCs by many groups due to its well-matched energy level with Al that is beneficial for efficient charge extraction, and inserted between photoactive layer and Al cathode. Both TBFPF-BT/PC71BM and TBFPF-BO/PC71BM were dissolved into o-DCB to make solution for spin-coating, and controlling different weight ratios of polymer donor to PC71BM acceptor to seek the optimized condition. The final weight ratio for high PCE was obtained as 1:2 under the illumination of AM1.5, 100 mW cm−2. The basic photovoltaic data are listed in Table S1.† TBFPF-BT blend exhibits a slightly increase in Jsc and FF resulting in an enhancing PCE of 4.27% compared to PBTFPFDTBO blend with a PCE of 4.07%. In addition, solvent additive has evolved into a common pathway to improve the device performance. To further optimize photovoltaic performances based on the above weight ratio, different v/v ratio of DIO was employed. Fig. 4a shows the typical current density–voltage (J–V) curves of TBFPF-BT and TBFPF-BO in optimized conditions and the corresponding parameters are summarized in Table 3. The other data with 0.5–5% ratio of DIO are shown in Table S2.† As depicted in Fig. 5a, we found that after treated with 1% DIO, TBFPF-BT based device shows an evident enhancement in Jsc from 7.64 mA cm−2 to 12.12 mA cm−2 and a slightly increase in FF from 0.67 to 0.71, respectively. Thus, a PCE up to 6.8% can be obtained. Similar to TBFPF-BT, device based on TBFPF-BO also presents a moderate PCE of 5.98%, which was mainly benefited from the improvement of Jsc (7.8 to 10.4 mA cm−2) and FF (0.57 to 0.65). Both copolymers demonstrate high FFs above 0.65, which indicates that the devices have relatively balanced charge transport and low serial resistance.49 Compared with TBFPF-BO, TBFPF-BT exhibits a larger Jsc and FF, which is attributed to its higher hole mobility. However, TBFPF-BO demonstrates superior Voc than that of TBFPF-BT, which is derived from the deeper HOMO energy level.
 | ||
| Fig. 5 (a) J–V curves of PSCs based on TBFPF-BT:PC71BM (1:2, w/w) and TBFPF-BO:PC71BM (1:2, w/w) with/without 1% DIO as additive under an irradiation intensity of AM1.5, 100 mW cm−2. (b) EQE spectra of TBFPF-BT:PC71BM (1:2, w/w) and TBFPF-BO:PC71BM (1:2, w/w). (c) Schematic diagram of device structure used in this work. | ||
| Active layer | Voc (V) | Jsc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| a Record on a conventional device configuration: ITO/PEDOT:PSS/polymer:PC71BM (1:2, w/w)/ZrAcac/Al with 1% v/v DIO as additive under illumination of AM1.5, 100 mW cm−2. |
||||
| TBFPF-BT:PC71BM |
0.80 | 12.12 | 71 | 6.80 |
| TBFPF-BO:PC71BM |
0.89 | 10.35 | 65 | 5.98 |
The accuracy of the photovoltaic measurements can be confirmed by the external quantum efficiency (EQE) of the devices prepared under optimum conditions shown in Fig. 5b. Two polymers presented similar plots of EQE spectrum with or without DIO in the whole response region. From EQE results, efficient photo-conversion efficiency was obtained in the wavelength range of 300–700 nm, with EQE values of 50–60% under the optimized conditions for both polymers. Meanwhile, devices of two polymers without DIO display relatively lower EQE values of 40–50% under the photo-response region, the different situations of phase separation, intra- and intermolecular stacking behaviors might be account for it. The integral current density values of two devices deducing from the EQE curves are well coincidence with those observed in J–V measurements.
Morphology
To further gain insight into the surface morphologies of active layers and better understand the original discrepancy in PCEs of TBFPF-BT and TBFPF-BO based PSCs, AFM was carried out in tapping mode. As illustrated in Fig. 6a and e, active layers of TBFPF-BT/PCBM and TBFPF-BO/PCBM presented relatively larger root-mean-square (RMS) values of 4.44 nm and 5.64 nm respectively before 1% v/v DIO was added as additive, and many small grains aggregated to form the larger domains were observed. However, upon 1% v/v of DIO incorporated into the blend films, images shown in Fig. 6c and g, TBFPF-BT/PCBM and TBFPF-BO/PCBM blend films exhibit smooth and uniform surface morphology with RMS of 2.97 nm and 3.95 nm, respectively. As well known that phase separation must meet the requirements of exciton dissociation and transporting distance (∼10 nm) at the donor–acceptor interface.50 Nevertheless, from the phase images Fig. 6b and f, the domain aggregation size of both blend films without DIO are too big to get efficient EQE. After the addition of DIO, domain aggregation seems alleviated for TBFPF-BT and TBFPF-BO blends, which are favorable for excitons to dissociate at the donor–acceptor interface resulting in higher Jsc, FF and EQE. Compared with TBFPF-BO, the morphology reveals that TBFPF-BT exhibited better film compatibility than TBFPF-BO to PC71BM in these blends, and thus leading to an increased in Jsc, FF and hole mobility, which is consistent with the above discussed photovoltaic properties. | ||
| Fig. 6 AFM height images (5 × 5 μm2): (a) TBFPF-BT:PC71BM (1:2) without DIO and (c) with DIO; (e) TBFPF-BO:PC71BM (1:2) without DIO and (g) with DIO. AFM Phase images (5 × 5 μm2) (b) TBFPF-BT:PC71BM (1:2) without DIO and (d) with DIO; (f) TBFPF-BO:PC71BM (1:2) without DIO and (h) with DIO. | ||
Conclusion
In conclusion, we designed two asymmetric low band gap TBF based polymers, TBFPF-BT and TBFPF-BO, and they were applied in PSC as photoactive layers. Both polymers exhibit broad absorption from 300 to 800 nm in the film state, the optical band gap from the onset absorption are 1.68 and 1.69 eV, respectively. Compared to TBFPF-BO, TBFPF-BT presented a higher hole mobility which is beneficial for enhancement in Jsc and FF. Moreover, the surface morphologies were investigated by AFM technique. After optimizing the morphologies with 1% v/v DIO, the large aggregation domain seemed disappeared and two polymer/PC71BM blend films showed favorable surface morphology and phase separation. Photovoltaic performance are investigated through a device architecture of ITO/PEDOT:PSS/polymer:PC71BM (1:2, w/w)/ZrAcac/Al. Under the optimized condition, the two polymers delivered anticipating PCEs of 6.8% with high FF = 0.71 for TBFPF-BT and 5.98% with FF = 0.65 for TBFPF-BO. Such preliminary results indicate that TBF based polymers might be promising candidates for efficient PSCs.
Acknowledgements
This work was supported by NSFC (No. 51173206), Project of Innovation-driven Plan in Central South University, China (2016CX035) and State Key Laboratory of Powder Metallurgy, Central South University, Changsha, China.References
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293–5300 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- Z. Zheng, S. Zhang, M. Zhang, K. Zhao, L. Ye, Y. Chen, B. Yang and J. Hou, Adv. Mater., 2015, 27, 1189–1194 CrossRef CAS PubMed.
- H. Yao, W. Zhao, Z. Zheng, Y. Cui, J. Zhang, Z. Wei and J. Hou, J. Mater. Chem. A, 2016, 4, 1708–1713 RSC.
- C. C. Chen, W. H. Chang, K. Yoshimura, K. Ohya, J. You, J. Gao, Z. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670–5677 CrossRef CAS PubMed.
- H. Zhou, Y. Zhang, C. K. Mai, S. D. Collins, G. C. Bazan, T. Q. Nguyen and A. J. Heeger, Adv. Mater., 2015, 27, 1767–1773 CrossRef CAS PubMed.
- T. Jiang, J. Yang, Y. Tao, C. Fan, L. Xue, Z. Zhang, H. Li, Y. Li and W. Huang, Polym. Chem., 2016, 7, 926–932 RSC.
- L. Huo, L. Ye, Y. Wu, Z. Li, X. Guo, M. Zhang, S. Zhang and J. Hou, Macromolecules, 2012, 45, 6923–6929 CrossRef CAS.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed. Engl., 2011, 50, 9697–9702 CrossRef CAS PubMed.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- L. Ye, S. Zhang, W. Zhao, H. Yao and J. Hou, Chem. Mater., 2014, 26, 3603–3605 CrossRef CAS.
- L. Huo, T. Liu, B. Fan, Z. Zhao, X. Sun, D. Wei, M. Yu, Y. Liu and Y. Sun, Adv. Mater., 2015, 27, 6969–6975 CrossRef CAS PubMed.
- J. Yuan, Y. Zou, R. Cui, Y.-H. Chao, Z. Wang, M. Ma, Y. He, Y. Li, A. Rindgen, W. Ma, D. Xiao, Z. Bo, X. Xu, L. Li and C.-S. Hsu, Macromolecules, 2015, 48, 4347–4356 CrossRef CAS.
- B. Liu, B. Qiu, X. Chen, L. Xiao, Y. Li, Y. He, L. Jiang and Y. Zou, Polym. Chem., 2014, 5, 5002 RSC.
- J. Yuan, L. Xiao, B. Liu, Y. Li, Y. He, C. Pan and Y. Zou, J. Mater. Chem. A, 2013, 1, 10639 RSC.
- J. W. Jo, J. W. Jung, E. H. Jung, H. Ahn, T. J. Shin and W. H. Jo, Energy Environ. Sci., 2015, 8, 2427–2434 Search PubMed.
- Y. Deng, J. Liu, J. Wang, L. Liu, W. Li, H. Tian, X. Zhang, Z. Xie, Y. Geng and F. Wang, Adv. Mater., 2014, 26, 471–476 CrossRef CAS PubMed.
- B. Qiu, R. Cui, J. Yuan, H. Peng, Z. Zhang, Y. Li and Y. Zou, Phys. Chem. Chem. Phys., 2015, 17, 17592–17600 RSC.
- L. Fan, R. Cui, X. Guo, D. Qian, B. Qiu, J. Yuan, Y. Li, W. Huang, J. Yang, W. Liu, X. Xu, L. Li and Y. Zou, J. Mater. Chem. C, 2014, 2, 5651 RSC.
- B. Qiu, J. Yuan, Y. Zou, D. He, H. Peng, Y. Li and Z. Zhang, Org. Electron., 2016, 35, 87–94 CrossRef CAS.
- Z. Tang, B. Liu, A. Melianas, J. Bergqvist, W. Tress, Q. Bao, D. Qian, O. Inganas and F. Zhang, Adv. Mater., 2015, 27, 1900–1907 CrossRef CAS PubMed.
- G. Li, X. Gong, J. Zhang, Y. Liu, S. Feng, C. Li and Z. Bo, ACS Appl. Mater. Interfaces, 2016, 8, 3686–3692 Search PubMed.
- P. Verstappen, J. Kesters, W. Vanormelingen, G. H. L. Heintges, J. Drijkoningen, T. Vangerven, L. Marin, S. Koudjina, B. Champagne, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, J. Mater. Chem. A, 2015, 3, 2960–2970 RSC.
- J. W. Jo, J. W. Jung, H.-W. Wang, P. Kim, T. P. Russell and W. H. Jo, Chem. Mater., 2014, 26, 4214–4220 CrossRef CAS.
- M. A. Uddin, T. H. Lee, S. Xu, S. Y. Park, T. Kim, S. Song, T. L. Nguyen, S.-j. Ko, S. Hwang, J. Y. Kim and H. Y. Woo, Chem. Mater., 2015, 27, 5997–6007 CrossRef CAS.
- N. Wang, Z. Chen, W. Wei and Z. Jiang, J. Am. Chem. Soc., 2013, 135, 17060–17068 CrossRef CAS PubMed.
- A. K. K. Kyaw, D. H. Wang, C. Luo, Y. Cao, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Adv. Energy Mater., 2014, 4, 1301469–1301477 CrossRef.
- J. A. Bartelt, J. D. Douglas, W. R. Mateker, A. E. Labban, C. J. Tassone, M. F. Toney, J. M. J. Fréchet, P. M. Beaujuge and M. D. McGehee, Adv. Energy Mater., 2014, 4, 1301733–1301743 CrossRef.
- L. A. Perez, K. W. Chou, J. A. Love, T. S. van der Poll, D. M. Smilgies, T. Q. Nguyen, E. J. Kramer, A. Amassian and G. C. Bazan, Adv. Mater., 2013, 25, 6380–6384 CrossRef CAS PubMed.
- Y. Aeschi, H. Li, Z. Cao, S. Chen, A. Amacher, N. Bieri, B. Özen, J. Hauser, S. Decurtins and S. Tan, Org. Lett., 2013, 15, 5586–5589 CrossRef CAS PubMed.
- P. Ding, C. C. Chu, B. Liu, B. Peng, Y. Zou, Y. He, K. Zhou and C. S. Hsu, Macromol. Chem. Phys., 2010, 211, 2555–2561 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- B. Kim, H. R. Yeom, M. H. Yun, J. Y. Kim and C. Yang, Macromolecules, 2012, 45, 8658–8664 CrossRef CAS.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979–986 CrossRef CAS.
- L. Huo, T. Liu, X. Sun, Y. Cai, A. J. Heeger and Y. Sun, Adv. Mater., 2015, 27, 2938–2944 CrossRef CAS PubMed.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- M. Liu, Y. Liang, P. Chen, D. Chen, K. Liu, Y. Li, S. Liu, X. Gong, F. Huang, S.-J. Su and Y. Cao, J. Mater. Chem. A, 2014, 2, 321–325 RSC.
- Z. Guo, D. Lee, R. D. Schaller, X. Zuo, B. Lee, T. Luo, H. Gao and L. Huang, J. Am. Chem. Soc., 2014, 136, 10024–10032 CrossRef CAS PubMed.
- N. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS PubMed.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- P. W. M. Blom, V. D. Mihailetchi, L. J. A. Koster and D. E. Markov, Adv. Mater., 2007, 19, 1551–1566 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- C. Cui, Z. He, Y. Wu, X. Cheng, H. Wu, Y. Li, Y. Cao and W.-Y. Wong, Energy Environ. Sci., 2016, 9, 885–891 Search PubMed.
- Z. Tan, S. Li, F. Wang, D. Qian, J. Lin, J. Hou and Y. Li, Sci. Rep., 2014, 4, 4691 CrossRef PubMed.
- B. Liu, X. Chen, Y. He, Y. Li, X. Xu, L. Xiao, L. Li and Y. Zou, J. Mater. Chem. A, 2013, 1, 570–577 RSC.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. a. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1HNMR, 13CNMR and 19FNMR spectra of compounds and polymers are shown in Fig. S1–S9. The photovoltaic data of different weight ratio and DIO ratio are shown in Tables S1 and S2, respectively. Hole mobilities of blend films without DIO are presented in Fig. S10. See DOI: 10.1039/c6ra09791j |
| This journal is © The Royal Society of Chemistry 2016 |