
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of the function of oxygen moieties on graphene oxide and reduced graphene oxide in the nucleation and growth of silver nanoparticles†
Kaja Spilarewicz-Stanek,
Aneta Kisielewska,
Joanna Ginter,
Karolina Bałuszyńska and
Ireneusz Piwoński*
University of Lodz, Faculty of Chemistry, Department of Materials Technology and Chemistry, Pomorska 163, 90-236 Lodz, Poland. E-mail: irek@uni.lodz.pl
First published on 15th June 2016
Abstract
The goal of the presented investigation was to study the differences in the decoration of graphene sheets, having various amounts of oxygen containing functional groups, with silver nanoparticles (AgNPs). The reduction of graphehe oxide (GO) was performed with the use of ascorbic acid (AA), leading to partially-reduced graphene oxide (PRGO) and reduced graphene oxide (RGO). The reduction process was monitored and confirmed by Raman Spectroscopy and Fourier Transform – Infrared Spectroscopy (FT-IR). The level of oxygen functional groups in the respective types of graphene was controlled by the duration of the reduction reactions. One-step synthesis of silver nanoparticles (AgNPs) was performed with no additional reducing agents in dark conditions in situ, i.e. directly on the graphene sheets deposited on a silicon wafer. It was proved that the presence of oxygen moieties on the graphene surface provides reactive sites for the spontaneous chemical reduction of Ag+ ions. It was also demonstrated that the amount of oxygen moieties on the surface of graphene plays an important role in the nucleation and growth of AgNPs. Moreover, it was found that the number of AgNPs investigated by Scanning Electron Microscopy (SEM) and Atomic Force Microscopy (AFM) increases with the amount of oxygen moieties, while their size decreases. Finally, it was found that the intensity of Raman bands characteristic of graphene are strongly increased for samples with AgNPs, due to surface enhancement Raman scattering (SERS).
1. Introduction
Nanocomposites consisting of graphene and metal nanoparticles (NPs) are novel materials, which find numerous potential applications as photocatalysts,1 sensors,2 electrodes,3 surface-enhanced Raman scattering (SERS) materials,4,5 antimicrobial materials6,7 and flexible, transparent, conductive films.8 Graphene oxide (GO) and reduced graphene oxide (RGO) are two main types of graphene derivatives, however, GO is a main product of graphene preparation in numerous methods. Chemically derived graphene sheets such as GO are attractive building blocks (and main constituents, next to metallic nanoparticles) for the preparation of novel, highly advanced nanocomposites. Due to this fact, the preparation of nanocomposites built of graphene in various forms in combination with NPs with controllable size and shape is highly desirable.GO is a two-dimensional monolayer nanomaterial with a honeycomb structure built of carbon atoms related to pristine graphene but modified by an oxidation process. In consequence, the surface of GO contains chemical functional groups carrying oxygen. The chemical structure of GO can be considered as a random arrangement of oxidized regions with oxygen functional groups, combined with non-oxidized areas where most of carbon atoms have sp2 hybridization.9 Carboxyl and carbonyl groups are localized on sp2 hybridized carbon atoms placed at the edges of GO sheet, while hydroxyl and epoxy groups are localized on sp3 hybridized carbon atoms placed on the basal plane.10 However, the presence of oxygen moieties damages the conjugated structure and captures π-electrons, which leads to a decrease of both carrier mobility and carrier concentration.9 In consequence, the free movement of electrons along the sheet is locally blocked by attached groups and lattice defects, which causes lower conductivity in comparison with pristine graphene.
In order to obtain a structure similar to pristine graphene, GO has to be reduced to RGO. During the reduction process, most oxygen containing functional groups are removed from the surface of the carbon lattice leading to recovery of the conjugated network.9 There are numerous reduction methods, which differ in the efficiency of the elimination of oxygen containing groups, defect formation and reconstruction of carbon atoms in sp2 hybridization. One of the most common deoxygenation processes is a chemical reduction with the use of hydrazine.10,11 Unfortunately, hydrazine is not only a highly toxic compound but also detrimental to human health.12 It seems that a suitable substitute to hydrazine is an environmental-friendly ascorbic acid (AA), giving comparable results of reduction compared to hydrazine and causing a safer reduction process. This method gives highly reduced graphene oxide, but does not eliminate all functional groups from the plane.13 This impediment can be advantageous for the synthesis of metal nanoparticles on the surface of RGO.
However, the presence of defects and residual oxygenated groups on GO and RGO sheets can be used as nucleation sites for the growth of metallic nanostructures.11,14 Oxygen moieties have also been used as anchors for the adsorption of positive metal ions (e.g. Ag+). Moreover, the oxygen moieties can also stabilize AgNPs after growth.6
A convenient method in the preparation of metallic NPs on graphene is the use of inorganic salts as precursors. The most common method for the preparation of metallic NPs on graphene is the mixing of GO (or RGO) water suspensions with a metal precursor in the presence of a reducing agent, such as sodium borohydride,15 ethylene glycol,16,17 sodium citrate14 or hydroquinone.6 The disadvantage in the use of these compounds as reducing agents to gain metallic particles is that some of these substances (such a NaBH4 and hydroquinone) can also simultaneously reduce GO, or strongly interact with GO, affecting the mechanism of AgNPs growth.18 Some improvements in the dispersion and growth of NPs on the graphene sheets can be achieved by the simultaneous use of a reducing agent and illumination during the synthesis.19
Other authors reported that a reducing agent is dispensable in the preparation of graphene–NPs composites.20–22 Illumination, supported with alkali agents (NaOH or KOH) playing the role of catalyst in the synthesis, was used. The proposed mechanism of this process20 is based on the reducing nature of GO under moderate alkaline conditions. Otherwise, the addition of the excess of alkali might lead to the formation of metal hydroxide, and interfere with the process of photo-induced growth.21
The above presented methods of AgNPs growth on graphene were typically performed in water suspensions. However, similar preparations can be also carried out in ethanol. For example, GO was suspended in ethanol and AgNPs were achieved on its surface by the reduction of AgNO3 using ultraviolet C (UV-C) irradiation.23 In order to confirm that using this approach the photochemical reduction was complete and the whole Ag+ ions were reduced, two additional attempts were made with the use of an additional reducing agent (dimethylformamide). The similar morphology of the received materials proved that no additional reduction took place with the use of a reducing agent. This experiment was evidence for the direct chemical reduction of a metal precursor in the presence of GO and RGO stimulated by light.23
In spite of numerous reports concerning the reduction of metal ions on graphene, only limited data are available relating to the direct reduction of NPs on GO and RGO without adding any auxiliary reducing agent and without light stimulation. For example, Chen et al. reported that GO itself can act as the reducing agent in the synthesis of PdNPs, performed by simply mixing GO and K2PdCl4 aqueous solution in an ice bath for 30 min under vigorous stirring.24 Interestingly, it was also reported that similar nanocomposites can be obtained under dark conditions, suggesting that no significant light effect or other power source in the PdNPs–GO formation process is needed. Similar results presented by Kong et al., demonstrated that Au3+ can be spontaneously reduced to AuNPs on the RGO sheets in the absence of reducing agents under ambient condition.25
Investigations of graphene-NPs nanocomposites are usually conducted in suspensions. Zhou et al. proposed an alternative method for the preparation of such composites, using a substrate for deposition and stabilization of graphene sheets prior to synthesis.26 GO was adsorbed on 3-aminopropyltriethoxysilane-modified Si–SiOx substrate, resulting in a uniform distribution of graphene sheets. RGO was also achieved by reduction in a hydrazine vapor environment. The obtained RGO coatings were next used in the synthesis of AgNPs by immersion in an aqueous solution of AgNO3 under heating. It was found that AgNPs were grown spontaneously, without adding any reducing agent. This result proved that Ag+ ions can be directly reduced on GO and RGO sheets.
There are a few developed methods concerning the preparation of GO/RGO–NPs nanocomposites, but it is still unclear which parameters define the nucleation and growth of metal nanoparticles on graphene. There are also virtually no reports on the direct synthesis of AgNPs on single graphene sheets without adding any reducing agent under dark conditions, which could be proof of the spontaneous reactions between Ag+ ions and oxygen containing functional groups.
In this paper we present the preparation of AgNPs/graphene composites under dark conditions through spontaneous reduction of Ag+ ions on different graphene types having various amount of oxygen moieties on their surface. In order to investigate the kinetics of growth of AgNPs on graphene sheets having various amount of oxygen containing functional groups, three graphene forms were prepared: graphene oxide (GO), partially-reduced graphene oxide (PRGO) and reduced graphene oxide (RGO). Moreover, we demonstrate that well separated and distributed graphene planar sheets deposited on silicon substrate can be achieved using a simple dip-coating method. The choice of this type of substrate allowed the reliable study of the AgNPs decoration of graphene sheets. To our knowledge, studies of NPs growth on partially reduced graphene oxide, and its comparison with the growth of NPs on GO and RGO have not yet been reported, and are novel elements in the field of the investigation of graphene-based materials. The goal of our study was also to discover how AgNPs grown on various graphene forms affect the intensity of signals in Raman spectroscopy.
2. Experimental
2.1. Materials
Single-sided polished silicon wafers Si (100) were supplied by ITME (Institute of Electronic Materials Technology, Warsaw, Poland). Graphene oxide (GO) dispersed in water with a concentration of 2 mg ml−1 was purchased from Sigma-Aldrich. L+ ascorbic acid (puriss. p.a. Riedel – de Haën) and ammonia with a concentration of 25% (CHEMPUR), were used for the reduction of GO. Silver nitrate (99.85%, POCH, currently Avantor Performance Materials, Poland) was used as a solution in ethanol (POCH, pure, min. 99.7%) at a concentration of 0.1 mM. Commercially reduced graphene oxide (RGO*, Aldrich) was used as a reference in our investigations of the reduction process. All reagents were of analytical grade and used as received.2.2. Deposition of GO sheets on silicon wafer
GO was deposited from water suspensions on silicon wafers Si (100) with the use of a dip-coating method. In this method, silicon wafers were immersed into the 0.2 mg ml−1 GO suspension with the use of a computer-controlled dip-coater (NIMA Technology LB, UK) operating at an immersion and withdrawal velocity of 25 mm min−1. The resulting coatings were heated to 90 °C for 1 h to stabilize them and to smooth the sheets.2.3. Reduction of graphene oxide
Ascorbic acid (AA) water solution with a concentration of 2 mM was prepared and alkalized by ammonia to obtain pH 10 in agreement with previous reported studies.13 Part of the previously prepared GO layers on Si wafers were immersed into a Petri dish containing AA solution and heated to 90 °C for 15 min and 1 h for the preparation of PRGO and RGO respectively. Samples were rinsed with water and dried in air.2.4. In situ synthesis of silver nanoparticles on GO, PRGO and RGO sheets
Synthesis of AgNPs on GO, PRGO and RGO was the second step of nanocomposite preparation. Silicon wafers with deposited GO, PRGO and RGO sheets were immersed in an ethanol solution of silver nitrate having the concentration of 0.1 mM, and put in a dark-room for 5 minutes at room temperature. After AgNPs synthesis all samples were rinsed with water and dried in air.2.5. Raman spectroscopy and FT-IR spectroscopy examination
Raman measurements were performed on an NTEGRA SPECTRA SOLAR Raman spectrometer (NT-MDT) at 632.8 nm wavelength incident laser light.Attenuated total reflection Fourier transform infrared (ATR-FT-IR) spectra were recorded by Bruker Vertex 70 spectrophotometer equipped with ATR accessory and DTGS detector. For the purpose of FT-IR measurements, all steps of the preparation of nanocomposites were identical to those presented in paragraphs 2.3 and 2.4. The modification concerns only the choice of the substrate and the deposition technique – the graphene-based nanocomposites were deposited on the aluminum foils using the sessile drop technique.
2.6. Scanning electron microscopy and atomic force microscopy surface imaging
A Field Emission Scanning Electron Microscope (FEI NovaNano SEM 450) equipped with Schottky gun operating in immersion mode using through lens detector (TLD) was applied in the imaging of the AgNPs supported on graphene surfaces. An energy-dispersive X-ray spectroscopy (EDX) was carried out to determine the elemental compositions of all types of graphene and graphene composites using EDAX TEAM equipped with an EDS Octane Pro detector.An atomic force microscope (AFM, Dimension Icon, Bruker) for surface imaging was also applied. The measurements were made in tapping mode using NSC35/Si3N4/AlBS (NT-MDT) cantilevers. The data from the AFM surface topography was analyzed using NanoScope Analysis 1.40 software (Bruker).
The number of AgNPs was calculated and their sizes were determined by measuring the diameters of each nanoparticle on the images. The particle size distribution histograms for the AgNPs were obtained from the measurements of at least 100–150 AgNPs.
3. Result and discussion
Low-magnification SEM and AFM images present the topography of separated, wrinkled GO sheets, deposited on the surface of silicon wafer with the use of a dip-coating method – Fig. 1. This type of substrate is very suitable for the imaging of graphene sheets and further monitoring of AgNPs growth because of its smoothness. It was found that the thickness of a single GO sheet measured by AFM was about 1.2 nm, which matches the results reported in previous ref. 20. | ||
| Fig. 1 Graphene oxide sheets imaged by SEM (A), AFM (B) and local surface profile (C). | ||
GO was next reduced to PRGO or RGO form to diminish the amount of oxygen containing functional groups localized on the surface of the GO sheets. In order to perform the deoxygenation process in mild conditions, the heating of the GO with AA was performed in an alkaline environment.13 The amount of oxygen containing functional groups can be adjusted by the time of reduction. The chemical changes occurring during the reduction of GO to RGO by AA were monitored with the use of FT-IR and Raman spectroscopies.
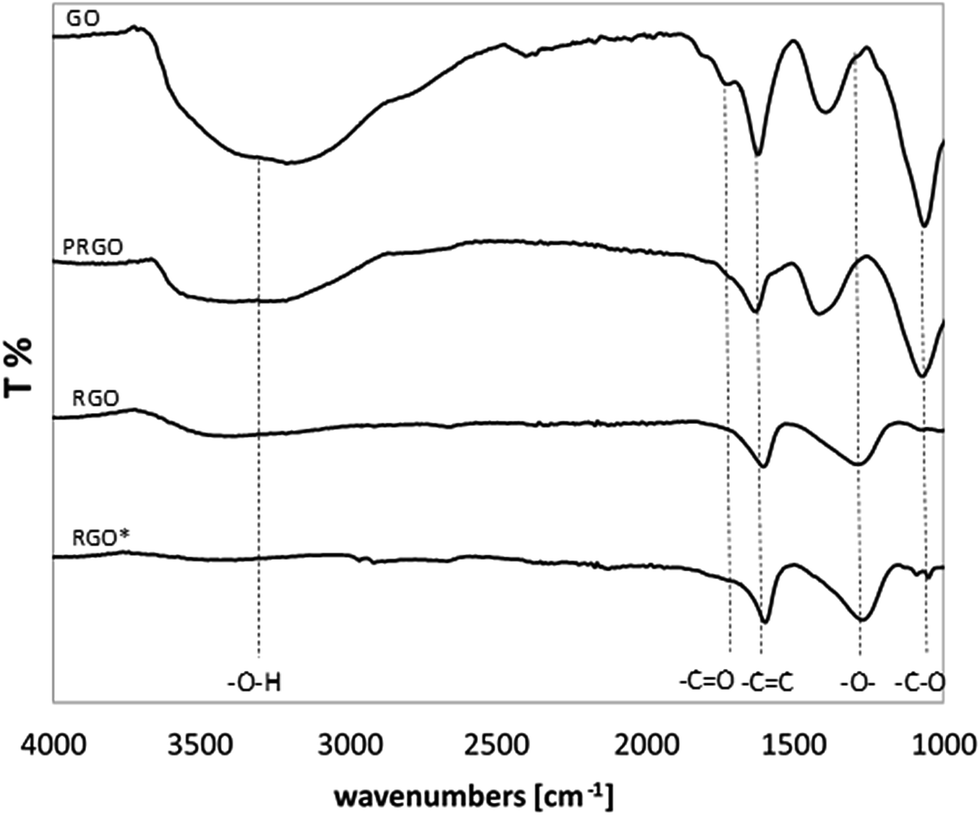
Fig. 2 presents the FT-IR spectra of GO, PRGO and RGO, as well as commercially available RGO* as a reference. The FT-IR spectrum of the GO shows typical absorption bands associated with the presence of various oxygen containing functional moieties on the GO surface. The broad peak in the range of 3000–3660 cm−1 is attributed to the O–H stretching vibrations of the C–OH groups and adsorbed water molecules.27–32 While the peak at 1722 cm−1 corresponds to the stretching vibrations from carbonyl and carboxylic groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O),30,33–36 the peak at 1620 cm−1 comses from O–H bending vibration of adsorbed water30,36,37 and also from the skeletal vibrations of aromatic CC sp2 hybridized carbon (unoxidized graphitic domains),29,33–35,38 the peak at 1388 cm−1 is assigned to the C–OH stretching vibrations of COOH groups,36 whereas the peak at 1220 cm−1 is attributed to the C–O–C breathing vibrations of epoxy groups30 and, finally, the peak at 1062 cm−1 corresponds to the stretching vibrations of C–O groups.27,32–35
O),30,33–36 the peak at 1620 cm−1 comses from O–H bending vibration of adsorbed water30,36,37 and also from the skeletal vibrations of aromatic CC sp2 hybridized carbon (unoxidized graphitic domains),29,33–35,38 the peak at 1388 cm−1 is assigned to the C–OH stretching vibrations of COOH groups,36 whereas the peak at 1220 cm−1 is attributed to the C–O–C breathing vibrations of epoxy groups30 and, finally, the peak at 1062 cm−1 corresponds to the stretching vibrations of C–O groups.27,32–35
 | ||
| Fig. 2 FT-IR spectra of GO, PRGO and RGO. | ||
In comparison with GO, the intensity of all the above mentioned absorption bands decreased for all samples reduced in AA for 15 min and 60 min. This decrease was even more significant when the time of reduction was extended. After 60 min of chemical reduction, the FT-IR spectrum of RGO shows three broad peaks at ca. 3400, 1595 and 1295 cm−1 corresponding to O–H stretching, aromatic CC stretching and O–H bending, and C–O–C stretching respectively, which is consistent with previous reports.32 In comparison with GO, the disappearance of the peak at 1722 cm−1 was also clearly visible, which indicates that the carbonyl group was completely removed upon chemical reduction. It is worth noticing that after 60 min reduction, the RGO spectrum is comparable to the spectrum of the commercial RGO* applied as a reference sample. In the case of RGO, the noticeable decrease in peak intensity at 3400 cm−1 indicates that a large fraction of the O–H groups was removed. However, the spectrum of the RGO* shows a lower level of O–H groups compared to the chemically reduced RGO.
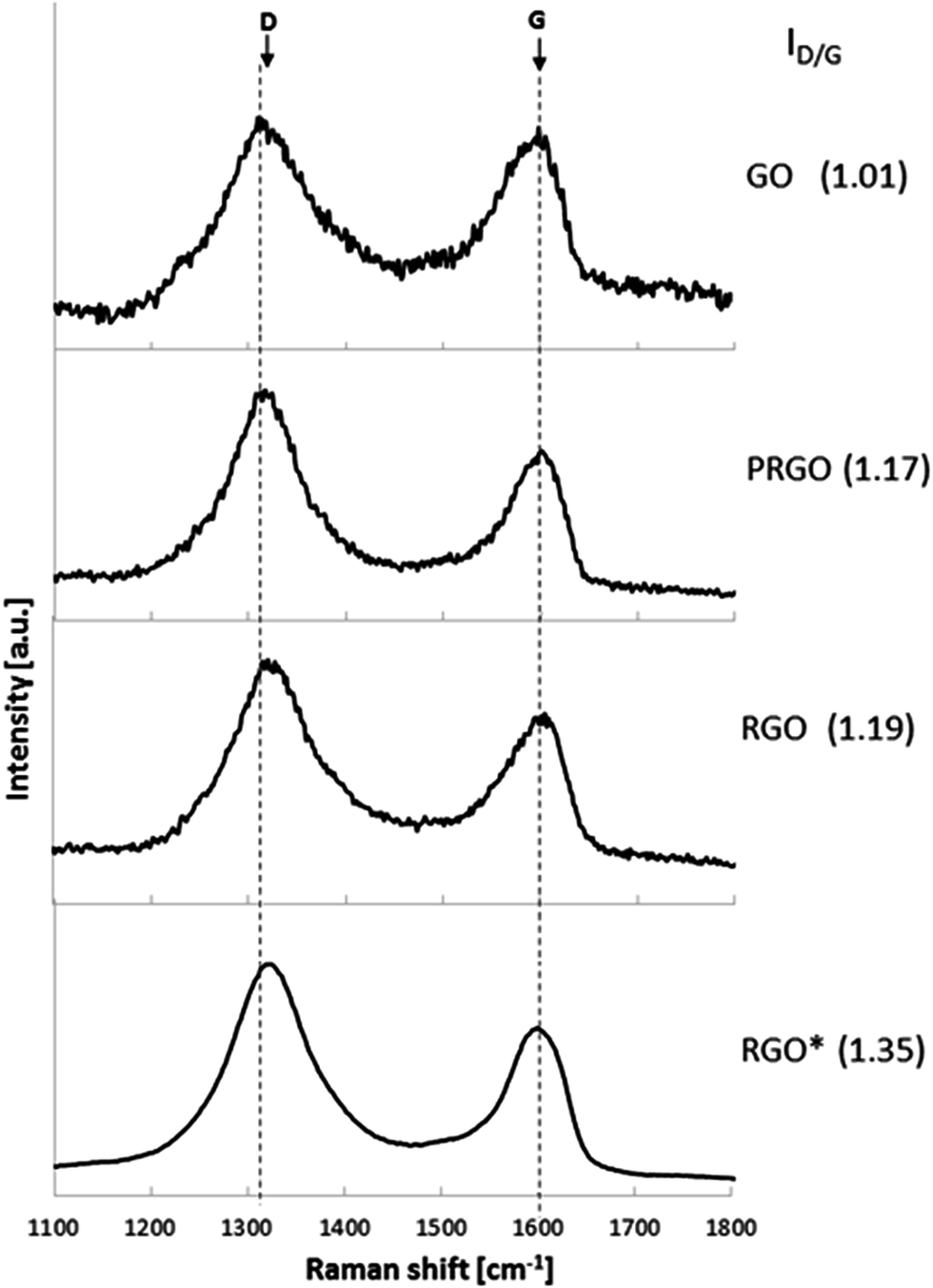
Further insight into the differences of the graphene structure after its reduction was performed using Raman spectroscopy. Fig. 3 shows the Raman spectra of GO, PRGO and RGO exhibiting various reduction level. Raman spectra of all investigated graphene structures exhibit two typical peaks, at around 1320 cm−1 and 1600 cm−1, which correspond to the D and G bands, respectively. The D band is assigned to the vibrations of carbon atoms with sp3 hybridization, whereas the G band is related to the vibration of sp2-bonded carbon atoms.31 In addition, the intensity ratio of the D band to the G band (ID/IG) indicates the degree of disorder and the change in average size of the sp2 domains.39 In comparison with GO, the intensities of the D band of PRGO and RGO increase, resulting in a higher ID/IG ratio than that of GO. Owing to the differences in reduction degree, GO, PRGO and RGO show different ID/IG ratios. An increase of the ID/IG ratio of the samples treated with a reducing agent for 60 min (1.19) and 30 min (1.17) in comparison with untreated GO (1.01) is caused by the appearance of defects, ripples and edges as a result of the AA reduction process. These observations indicate a higher level of defragmentation after reduction as well as a smaller size of sp2 domain.39 These results are in agreement with previously reported studies of the chemical reduction.32
 | ||
| Fig. 3 Raman spectra of GO, PRGO and RGO. | ||
The analysis of FT-IR and Raman spectra confirmed that after the one-hour-reaction of GO with AA under alkaline conditions the reduction was completed. It was also found that if the reduction was shortened to 15 min, the FT-IR indicated the elimination of approximately half of the initial number of oxygen containing functional groups in comparison with initial GO form. In this way, three model structures of graphene, having various amount of oxygen moieties, were obtained (labeled as GO, PRGO, RGO respectively) and subsequently used to study the kinetics of the growth of AgNPs.
Graphene sheets having various amounts of oxygen containing groups were used for further studies of AgNPs growth, which is, however, complicated due to two reasons. Firstly, RGO layers have a tendency to form irreversible agglomerates during the reduction process induced by van der Walls and stacking interactions between individual graphene sheets.40 Secondly, the number and size of metal nanoparticles which grow on graphene depend on the number of graphene layers.41 With a view to overcoming these difficulties, the dip-coating procedure was applied which enabled us to obtain single and separated sheets of GO randomly distributed on the surface, which was subsequently reduced to RGO. Graphene layers prepared in such a way can be considered as a model system, enabling the study of AgNPs growth accurately. In order to eliminate the risk of simultaneous reduction of GO during the growth of AgNPs, the reduction of Ag+ ions was realized using redox reactions occurring between oxygen groups on the graphene sheet and adsorbed Ag+ ions without adding any additional reducing agent. In this way, all changes observed during the nucleation and growth of AgNPs on GO, PRGO and RGO depend only on their chemical structure (the amount of oxygen containing groups).
The SEM image presented in Fig. 4A, and low-magnification image (Fig. 4B), show representative topography of RGO flake, indicating that AgNPs grown on its surface were anchored only to the flakes. Further analysis and morphology of the obtained graphene–AgNPs nanocomposites were examined by SEM and AFM imaging, presented in Fig. 5 & 6. In the high-magnification images, separate AgNPs can be observed, typically visible as white objects grown on graphene sheets. It was found that the size, shape and density of AgNPs grown on each type of graphene form, having various amount of oxygen moieties, were significantly different, indicating various mechanisms of their growth.
 | ||
Fig. 4 SEM image (magnification 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×) presenting the morphology of AgNPs on RGO flakes (A) taken from a low-magnification image (2500×) presenting the general view of RGO flakes with AgNPs deposited on silicon wafer (B). AgNPs were grown only at RGO. 000×) presenting the morphology of AgNPs on RGO flakes (A) taken from a low-magnification image (2500×) presenting the general view of RGO flakes with AgNPs deposited on silicon wafer (B). AgNPs were grown only at RGO. | ||
 | ||
| Fig. 5 Surface morphology and distribution of AgNPs on GO, PRGO and RGO imaged by SEM. | ||
 | ||
| Fig. 6 Surface morphology and distribution of AgNPs on GO, PRGO and RGO imaged by AFM. | ||
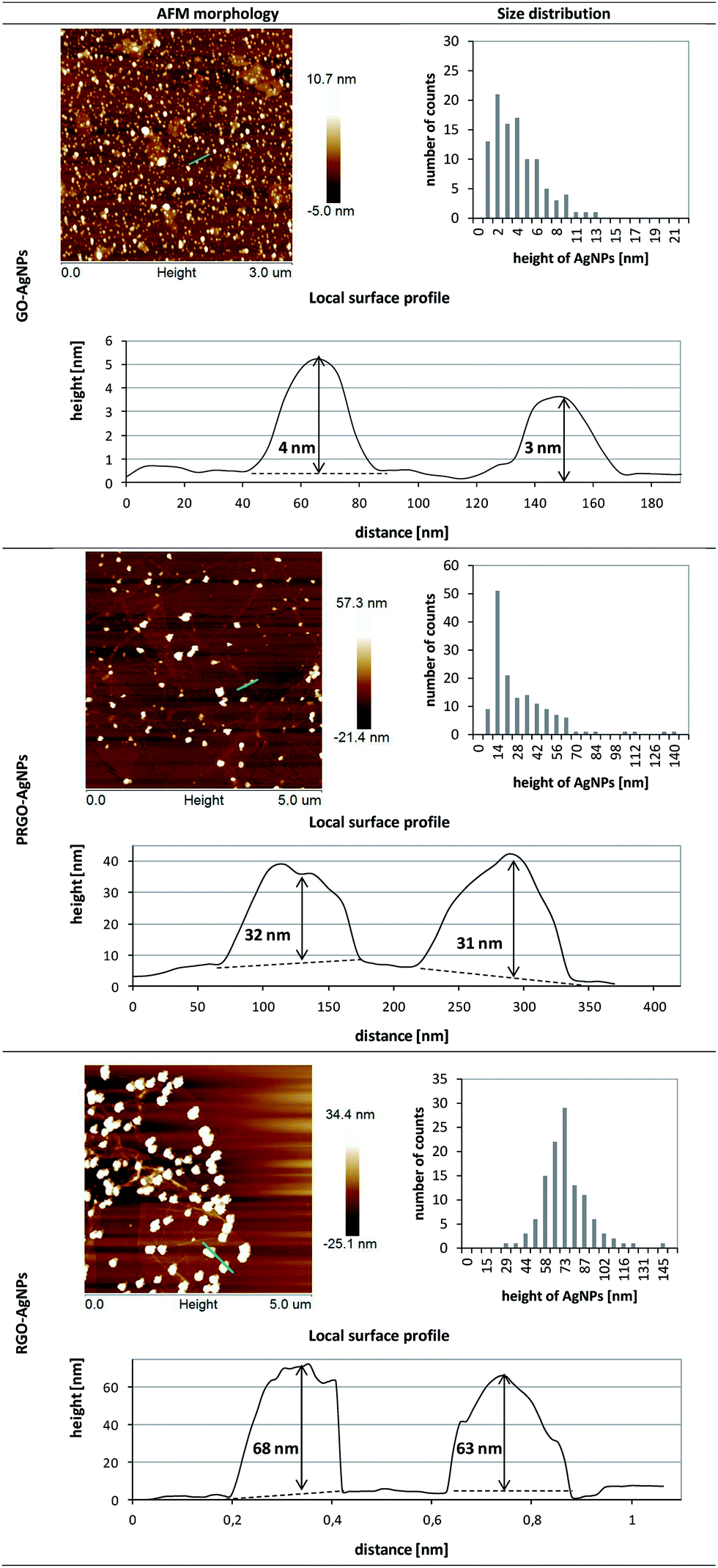
It was shown by the imaging of each type of graphene–AgNPs composite that they exhibit very similar morphologies at randomly chosen locations, indicating the homogenous distribution of AgNPs within each individual type of composite. However, there were considerable differences in the size and distribution of AgNPs between various types of composites prepared using GO, PRGO and RGO. Therefore, for further investigation of AgNPs formation, and especially for statistical calculations, various magnifications of SEM images were chosen for each type of nanocomposite individually, in order to have a comparable number of AgNPs on each image. This operation was also necessary to examine only planar areas of flakes, excluding wrinkles, and get a comparable set of AgNPs for plotting their size distribution. The acquired images were also used to calculate such parameters as the diameter and number of AgNPs as well as their surface density (the surface area occupied by nanoparticles related to one μm2). These parameters are gathered in Table 1.
| Sample | Surface density [AgNP/μm2] | Average size of AgNPs [nm] | Size range [nm] | ||
|---|---|---|---|---|---|
| SEM | AFM | SEM | AFM | ||
| GO–AgNPs | 203 | 204 | 3 ± 2 | 4 ± 2 | 1–15 |
| PRGO–AgNPs | 15 | 12 | 33 ± 13 | 24 ± 15 | 1–140 |
| RGO–AgNPs | 4 | 5 | 72 ± 8 | 72 ± 9 | 45–145 |
Fig. 5 shows the SEM images of GO–AgNPs, PRGO–AgNPs and RGO–AgNPs. It can be seen that AgNPs grown on GO were single-entity, quasi-spherical and uniformly dispersed on the sheet surface. The average size of AgNPs was 3 ± 2 nm. The maximal diameter did not exceed 20 nm. The surface density of obtained AgNPs was very high – 203 AgNPs/μm2.
AgNPs grown on RGO exhibited different sizes and surface density – Fig. 5. AgNPs were much larger than on GO and had an anisotropic shape – Fig. 4A. The mean width size of AgNPs located on RGO sheet was 72 ± 8 nm. However, the maximal recorded size reached 140 nm, while AgNPs smaller than 28 nm were not observed. The surface density of the obtained AgNPs was on the level of 4 AgNPs/μm2, which was 40 times less than the surface density of AgNPs on GO.
In contrast to GO and RGO, AgNPs grown on PRGO were not homogenous in their shape and surface density. Moreover, their morphology was a combination of topographies typical for AgNPs grown on both GO and RGO surfaces. The size of AgNPs on PRGO was less than 20 nm and shapes were quasi-spherical, while other AgNPs having more than about 20 nm had anisotropic forms. It means that the non-complete reduction leads to an asymmetry in the nucleation sites, which strongly affect the shape of AgNPs. The average size of AgNPs for the examined surface of PRGO–AgNPs was 33 ± 13 nm, while the surface density of AgNPs located on PRGO was 15 AgNPs/μm2 – Table 1.
The formation of AgNPs on graphene sheets was also confirmed by AFM measurement – Fig. 6. The height of the AgNPs on the GO sheets was 4 ± 2 nm and the surface density of the obtained AgNPs was 204 AgNPs/μm2. The examination of RGO–AgNPs composite revealed that the average height of AgNPs was 72 ± 9 nm, while the surface density was 5 AgNPs/μm2. Interestingly, AFM analysis of PRGO–AgNPs displayed complex morphology. The AgNPs size distribution presented in the histogram clearly showed a number of populations of AgNPs, exhibiting mainly small and medium sizes, with larger ones having irregular shapes. The surface density of AgNPs grown on PRGO was 12 AgNPs/μm2.
It is worth noting that the results obtained within each type of composite revealed comparable sizes and surface densities of AgNPs achieved in both SEM and AFM surface imaging, except for PRGO–AgNPs – Table 1. This difference might be a result of the shorter time of GO reduction compared to RGO, leading to local differences in the removal of oxygen containing functional groups in the graphene and their various surface densities. These fluctuations in the non-homogenous distribution of oxygen moieties were also visible in further examination of PRGO–AgNPs surface, which revealed that various amounts of AgNPs having particular sizes can occur locally in randomly chosen areas. Therefore, a homogenous size range and surface density distributions for PRGO–AgNPs composite were not observed over the whole area. In another words, due to the short reduction time, PRGO sheets exhibited areas with larger and lower amount of oxygen moieties, which, in consequence, affected the nucleation and growth of AgNPs on these reaction sites. Because of these local differences in the removal of oxygen containing groups during the reduction process, the topography of some areas on the same PRGO–AgNPs sheet was a combination of topographies acquired for GO and RGO, including certain areas being closer to GO–AgNPs surface, while the topography of the rest of the sheet was closer to the RGO–AgNPs surface.
The experiential correlation found between the size/density of AgNPs and the density of oxygen functional groups in GO and RGO is a characteristic feature in the preparation of AgNPs–GO and AgNPs–RGO composites. Similar observations were made by Zhou et al.,26 who reported that the much higher density of AgNPs with smaller sizes (6.0 ± 3.6 nm) were formed on the GO surface carrying a greater amount of oxygen containing groups compared to the lower-density and bigger heterogeneous AgNPs formed on RGO (ranging from tens of nanometers to 1 μm), with a low number of these groups. The results obtained for GO–AgNPs were also in clear agreement with data previously reported by Martinez-Orozco et al.23 AgNPs having an average size of 10 nm (in the size range of 0–20 nm) and 15 nm (in the size range of 0–30 nm) for 1 wt% and 2 wt% quantity of metallic silver in the nanocomposite were obtained, respectively. The AgNPs were a little larger than the AgNPs obtained in our experiments, which can be explained by the use of UV-C illumination and the longer time of synthesis, which enabled the further growth of AgNPs.
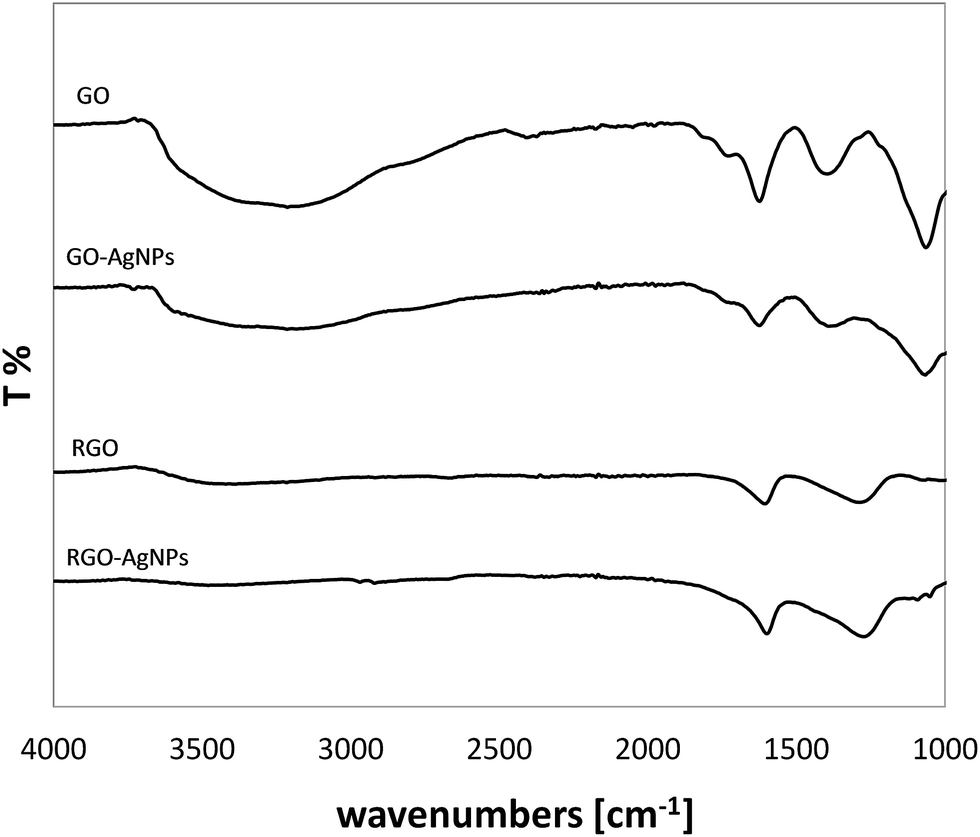
With a view to confirming the chemical changes occurring during AgNPs growth on GO and RGO, FT-IR analysis was conducted. Fig. 7 shows the FT-IR spectra of GO–AgNPs and RGO–AgNPs nanostructures as well as GO and RGO without AgNPs as reference samples. FT-IR spectroscopy provided evidence of the presence of various types of oxygen functional groups such as O–H, CO, C–O, –O–, C–OH on the GO, which could be located on the basal planes and edges of the GO sheets.9,10 It was found that after treatment of GO with silver ions, and the consequent growth of AgNPs, a noticeable decrease in the intensity of all absorption bands corresponding to the oxygen containing functional groups can be observed. These findings demonstrate the strong interaction between silver ions and oxygen functional groups through the formation of a coordination bond or electrostatic attraction.19 These results are in agreement with previously reported studies of NPs generation, where GO was indicated as a factor playing a triple role: as a carrier of reduction centers, stabilizer and reducing agent.20 In the case of RGO, it seems that mostly hydroxyl groups were involved in the interaction with silver ions, since the intensity O–H band decreased after RGO modification with AgNPs.
 | ||
| Fig. 7 FT-IR spectra of GO–AgNPs, RGO–AgNPs nanostructures and reference samples GO and RGO, respectively. | ||
In the present study, the influence of the AgNPs grafted to GO, PRGO and RGO on Raman signal enhancement was also investigated. Two competitive phenomena occurring during the decoration of graphene-based nanomaterials with AgNPs can be observed. The first one is associated with the reduction of graphene during AgNPs growth, whereas the second phenomenon is connected only to AgNP growth. Both phenomena strictly depend on the GO, PRGO and RGO structures. An increase of the ID/IG ratio of the GO–AgNPs sample (1.20) in comparison with untreated GO (1.01) is caused by the partial reduction and fragmentation of GO, leading to the increase of sp3 domains. This result is in agreement with the previously reported studies of GO reduction during AgNPs decoration presented by Lan et al.19 In the case of RGO, the situation is slightly different. The number of oxygen containing functional groups on the RGO flakes is low. Therefore, further reduction and fragmentation of RGO is limited. In such a system, the main phenomenon occurring on RGO is the growth of AgNPs, mainly on oxygen containing groups associated with sp3 domains, leading to a further reduction of their number and causing a slight decrease of the ID/IG ratio – from 1.19 to 1.14 for RGO and RGO–AgNPs, respectively. The ID/IG ratios recorded for PRGO and PRGO–AgNPs are similar, and slightly increased from 1.17 (PRGO) to 1.18 (PRGO–AgNPs). This suggests that the processes of PRGO reduction and the growth of AgNPs are in equilibrium and occur simultaneously.
It is well-known that composites with noble metal nanoparticles as surface-enhanced Raman scattering (SERS) substrates are used for the detection of organic compounds in nanomolar concentrations.42 Two origins of SERS effect can be distinguished: electromagnetic – assigned to the excitation of localized surface plasmons of noble metal nanoparticles, and chemical enhancement – associated with the formation of charge-transfer complexes.43 As shown in Fig. 8, the intensities of both D and G bands in the Raman spectra on each type of graphene with AgNPs are enhanced in comparison with those without AgNPs, but with different strengths – Fig. 8. The differences in G signal enhancement in Raman occur in composites of graphene with AgNPs in the following order: RGO–AgNPs < PRGO–AgNPs < GO–AgNPs – Fig. 8D. This enhancement was the highest for GO containing large amount of small AgNPs – Fig. 8A. The Raman spectra for GO is presented in Fig. 3. With the aim of comparing SERS activity, the ratio of the peak areas of D and G bands for graphene–AgNPs (AG–AgNPs) to the peak areas of D and G bands for graphene (AG) for each sample was calculated and shown in Fig. 8D. For GO–AgNPs composites, an 87-fold enhancement of Raman signal was achieved in comparison with the GO sample. This finding could be explained by the size of AgNPs as well as the distance between them.37 An enhancement of Raman signal is especially visible for GO–AgNPs composite where AgNPs had small sizes and were located close together – Fig. 5, 6 and Table 1. However, the distance between AgNPs which decorate RGO surfaces was too large and there was no signal enhancement. In PRGO, the distance between AgNPs seemed to be also appropriate, but the population of the largest AgNPs suppressed this effect, providing only a slight enhancement of the Raman signal. Moreover the non-homogenous AgNPs size distribution and various distances between them caused a different contribution to the Raman signal enhancement, giving considerable data spreading, visible as large error bar – Fig. 8D. This correlates well with the SEM and AFM results. Taking into account these parameters, SERS effect depends not only on the size of nanoparticles but also on surface density of AgNPs (i.e. the distance between them).
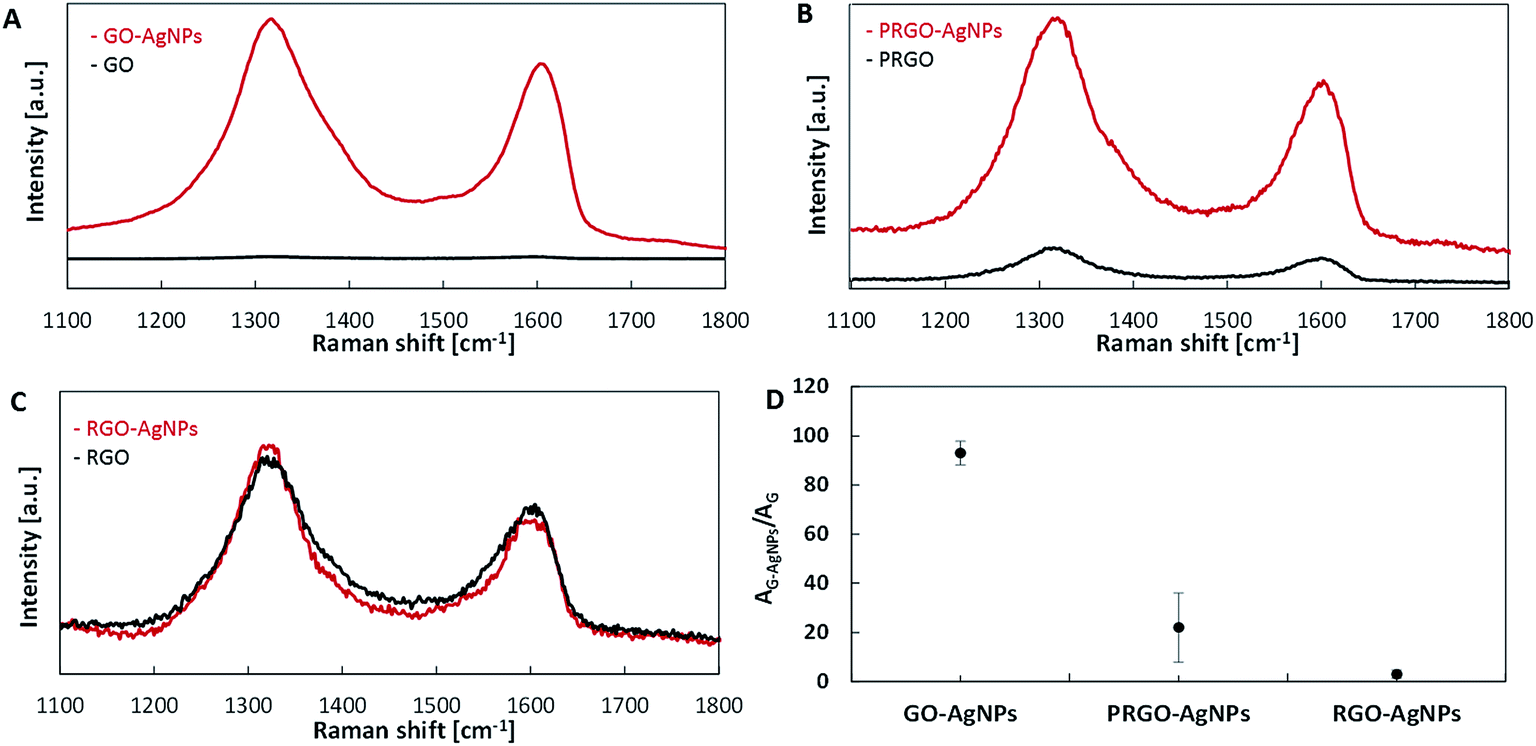 | ||
| Fig. 8 Raman spectra of GO–AgNPs (A), PRGO–AgNPs (B), RGO–AgNPs (C) and relative Raman intensities (D). | ||
The last part of the measurements was devoted to the investigation of the chemical composition of each type of graphene-based material with the use of the EDX method. As expected, the presence of carbon and oxygen was revealed in the GO, PRGO and RGO samples – Fig. 9A. Besides these elements, an additional low-intensity peak indicating the presence of silver can be found in the GO–AgNPs, PRGO–AgNPs and RGO–AgNPs nanocomposites – Fig. 9B. The concentration of silver is highest for GO–AgNPs composites, which corresponds well with SEM and AFM imaging, showing large amounts of small AgNPs for this type of composite. Although EDX measurement has a qualitative character, some estimation of the relative amount of constituents detected in the samples can be made assuming that the reference peak for carbon equals 100%. It is worth noting that the oxygen/carbon relative intensity ratio follows the graphene reduction steps. The comparison of the areas of oxygen to carbon peaks demonstrate that the relative concentration of oxygen is highest for GO, and becomes gradually lower for PRGO and RGO. Similar changes can be observed for graphene nanocomposites containing silver, which also has its input in the additional reduction of graphene during AgNPs nucleation and growth. This is clearly visible in the comparison of relative oxygen to carbon ratios calculated on the basis of peak areas characteristic for oxygen and carbon, gathered in Fig. 10. Besides the lowering tendency in oxygen content for GO, PRGO and RGO samples, the highest difference in oxygen concentration upon the growth of AgNPs can be noticed for the oxidized form of graphene (GO) – i.e. in this sample, where the amount of accumulated silver was the highest. This observation indicates again, that silver not only causes the reduction of graphene but also tends to grow preferentially on surfaces rich in oxygen. Details concerning the intensity (peak areas) of carbon, oxygen and silver as well as the oxygen/carbon and silver/carbon ratios are summarized in Table 2 in the ESI.†
 | ||
| Fig. 9 EDX analysis of GO, PRGO, RGO (A), and GO–AgNPs, PRGO–AgNPs, RGO–AgNPs nanocomposites (B). | ||
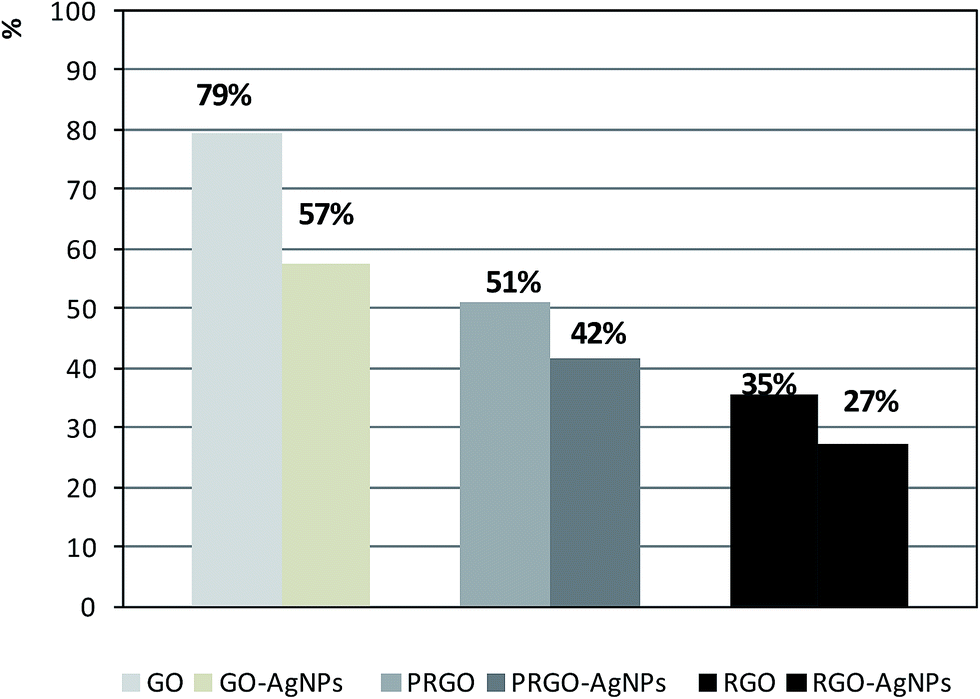 | ||
| Fig. 10 The comparison of relative oxygen to carbon ratios taken from EDX measurements for given type of sample. Percentages above each bar were calculated assuming the carbon peak area as 100% for each sample. | ||
The analysis of SEM/AFM images and FT-IR spectra enabled us to shed some light on the possible mechanisms of the nucleation and growth of AgNPs – Fig. 11. The nucleation strongly depends on the structure of the graphene form, especially on the number of oxygen containing functional groups which act as a nucleation sites.26,44 The high density of oxygen moieties present on GO promote more nucleation than growth, which results in a higher number of created AgNPs, as opposed to PRGO and then to RGO. As presented in FT-IR spectra, the most diminished intensity band belonging to oxygen moiety during AgNPs growth on GO was the hydroxyl group, in all cases. This may signal that the hydroxyl group is, most probably, the strongest nucleation center for the formation of AgNPs.
 | ||
| Fig. 11 Proposed mechanism of nucleation and growth of AgNPs on GO and RGO based on Lerf-Klinowski model. | ||
Besides hydroxyl groups, another factor governing the nucleation of AgNPs is the graphene structure. The quantity and size of aromatic conjugated domains has an influence on growth, since they act as electron-donating sources to reduce Ag+ ions. Reduction of GO provides a highly-restored π-conjugated network in RGO form. Zhou et al. reported that the largest size of these domains provides for the formation of larger sizes of nanoparticles on RGO than on GO surfaces.26 Nevertheless, in our Raman investigation a significant increase in the size of sp2 domains after reduction was not observed. Meanwhile, the tendency of the formation of the biggest AgNPs on the RGO rather than on the GO surface was still observed. This means that sp2 domains have an influence on not only the growth but also on the mobility of electrons within these domains. The results gained in this study also suggest that another important factor for understanding the mechanism of AgNPs growth on reduced GO forms is the local, irregular removal of oxygen moieties during the reduction process. AgNPs–PRGO morphology exhibited small areas decorated with a large quantity of small AgNPs next to larger areas with the presence of a few large AgNPs on the same graphene flake. This observation suggests that Ag+ ions have limited access to the areas with larger amounts of residual oxygenated groups, either due to steric effects or, simply, they are not adsorbed on areas without –OH groups. The above results show that the density and size of generated metallic nanoparticles can be modulated by changing the reduction time of GO, which can be subsequently used as a way to control the growth of AgNPs. A model scheme of AgNPs growth on GO and RGO surfaces is proposed in Fig. 11. The presented structure of GO is based on the widely accepted Lerf-Klinowski model of GO.9 In turn, the proposed structure of RGO is based on the mechanism of GO reduction by polyphenols described by Thakur et al.45 It can be also seen that AgNPs were grown mainly on hydroxyl groups localized on graphene flakes.
4. Conclusions
In the present study, the synthesis of AgNPs on graphene was performed. The novelty of this study was conducting AgNPs growth in dark conditions without additional reducing agents. Elucidation of the mechanisms in AgNPs decoration of three various forms of graphene, which differ in the quantity of oxygen functional groups, labeled as GO, PRGO and RGO, was presented. It was found that the kinetics of AgNPs growth on these types of graphene exhibited significant differences. It was confirmed that both the nucleation and growth of AgNPs on graphene sheets are governed mainly by differences in the number of oxygen moieties and also by differences in electron mobility in sp2/sp3 domains between the investigated forms of graphene. These two factors lead, in consequence, to the different density and size of AgNPs.The chemical changes in graphene structures arising as a result of the growing of AgNPs was proved with the FT-IR method. The quantity of oxygen containing functional groups are directly responsible for the nucleation and growth of AgNPs. GO has a greater ability to form many small AgNPs, due to the large number of oxygen moieties. In turn, graphene in reduced forms (PRGO and RGO) exhibits a lower number of residual oxygen containing groups acting as nucleation sites, which leads to the formation of a smaller number of large AgNPs and a larger distance between them. It was shown that expanding the distance between nanoparticles had a negative effect on the intensity of Raman signals, and, in consequence, reduced the SERS effect. Our study clearly demonstrated that GO was a better substrate to create SERS materials than reduced forms of GO, due to the large number of small nanoparticles and the small distances between them (hot spots).
The oxygen containing functional groups in GO also play a role of anchoring groups during deposition of graphene on flat substrates. They enable the preparation of stable suspensions in water, which allows for easy deposition of GO as thin coatings. We demonstrated that such depositions can be performed using a dip-coating method, leading to a uniform graphene covering with the possibility of distinguishing the edges of individual sheets using microscopy techniques. Moreover, another advantage of reduction performed on graphene deposited as coating is preventing the scrunching up of separate flakes. The obtained coatings can be considered as model materials to study the decoration of graphene with AgNPs, because of the suitably separated sheets. Consequently, the influence of graphene thickness on AgNPs size and density is limited.
Acknowledgements
The financial support of this work through: University of Lodz, Subsidy for scientific research contributing to the development of young researchers and PhD students, project number: B1511100000879.02 (2015) and the National Science Centre, Research grant no. 2014/13/B/ST8/03114; is gratefully acknowledged.References
- Q. Zhuo, Y. Ma, J. Gao, P. Zhang, Y. Xia, Y. Tian, X. Sun, J. Zhong and X. Sun, Inorg. Chem., 2013, 52, 3141–3147 CrossRef CAS PubMed.
- R. S. Dey and C. R. Raj, J. Phys. Chem. C, 2010, 114, 21427–21433 CrossRef CAS.
- Z. S. Wu, G. Zhou, L. C. Yin, W. Ren, F. Li and H. M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS.
- S. Murphy, L. Huang and P. V. Kamat, J. Phys. Chem. C, 2013, 117, 4740–4747 CrossRef CAS.
- P. Wang, D. Zhang, L. Zhang and Y. Fang, Chem. Phys. Lett., 2013, 556, 146–150 CrossRef CAS.
- Q. Bao, D. Zhang and P. Qi, J. Colloid Interface Sci., 2011, 360, 263–470 CrossRef PubMed.
- V. H. Nguyen, B. K. Kim, Y. L. Jo and J. J. Shim, J. Supercrit. Fluids, 2012, 72, 28–35 CrossRef CAS.
- H. W. Tien, Y. L. Huang, S. Y. Yang, J. Y. Wang and C. C. M. Ma, Carbon, 2011, 49, 1550–1560 CrossRef CAS.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- G. Choudhary and H. Hansen, Chemosphere, 1998, 37, 801–843 CrossRef CAS PubMed.
- M. J. Fernandez-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. M. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 CrossRef CAS.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Grácio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- H. Mehl, M. M. Oliveira and A. J. G. Zarbin, J. Colloid Interface Sci., 2015, 438, 29–38 CrossRef CAS PubMed.
- K. S. Subrahmanyam, A. K. Manna, S. K. Pati and C. N. R. Rao, Chem. Phys. Lett., 2010, 497, 70–75 CrossRef CAS.
- C. Xu, X. Wang and J. Zhu, J. Phys. Chem. C, 2008, 112, 19841–19845 CrossRef CAS.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- N. T. Lan, D. T. Chi, N. X. Dinh, N. D. Hung, H. Lan, P. A. Tuan, L. H. Thang, N. N. Trung, N. Q. Hoa, T. Q. Huy, N. V. Quy, T. T. Duong, V. N. Phan and A. T. Le, J. Alloys Compd., 2014, 615, 843–848 CrossRef CAS.
- R. Pasricha, S. Gupta and A. K. Srivastava, Small, 2009, 5, 2253–2259 CrossRef CAS PubMed.
- J. Chen, X. Zheng, H. Wang and W. Zheng, Thin Solid Films, 2011, 520, 179–185 CrossRef CAS.
- L. Yuan, L. Jiang, J. Liu, Z. Xia, S. Wang and G. Sun, Electrochim. Acta, 2014, 135, 168–174 CrossRef CAS.
- R. D. Martinez-Orozco, H. C. Rosu, S.-W. Lee and V. Rodriguez-Gonzalez, J. Hazard. Mater., 2013, 263, 52–60 CrossRef CAS PubMed.
- X. Chen, G. Wu, J. Chen, X. Chen, Z. Xie and X. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS PubMed.
- B. S. Kong, J. Geng and H. T. Jung, Chem. Commun., 2009, 16, 2174–2176 RSC.
- X. Zhou, X. Huang, X. Qi, S. Wu, C. Xue, F. Y. C. Boey, Q. Yan, P. Chen and H. Zhang, J. Phys. Chem. C, 2009, 113, 10842–10846 CrossRef CAS.
- H. Zhang, X. Fan, X. Quan, S. Chen and H. Yu, Environ. Sci. Technol., 2011, 45, 5731–5736 CrossRef CAS PubMed.
- M. S. A. S. Shah, K. Zhang, A. R. Park, K. S. Kim, N. G. Park, J. H. Park and P. J. Yoo, Nanoscale, 2013, 5, 5093–59101 RSC.
- N. T. Lan, D. T. Chi, N. X. Dinh, N. D. Hung, H. Lan, P. A. Tuan, L. H. Tang, N. N. Trung, N. Q. Hoa, T. Q. Huy, N. V. Quy, T. T. Duong, V. N. Phan and A. T. Le, J. Alloys Compd., 2014, 615, 843–848 CrossRef CAS.
- L. M. Pastrana-Martinez, S. Morales-Torres, V. Likodimos, P. Falaras, J. L. Figueiredo, J. L. Faria and A. M. T. Silva, Appl. Catal., B, 2014, 158, 329–340 CrossRef.
- J. Zhu, Y. Cao and J. He, J. Colloid Interface Sci., 2014, 420, 119–126 CrossRef CAS PubMed.
- H. Ding, S. Zhang, J.-T. Chen, X. P. Hu, Z. F. Du, Y. X. Qiu and D. L. Zhao, Thin Solid Films, 2015, 584, 29–36 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Grácio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Balkus Jr, ACS Catal., 2012, 2, 949–956 CrossRef CAS.
- Y. F. Zhao, Y. Xie, Z. Y. Sun, H. Y. Zhang, R. T. Tao, C. L. Huang and Z. M. Liu, Sci. China: Chem., 2012, 55, 1294–1302 CrossRef CAS.
- S. Wang, Y. Zhang, H. L. Ma, Q. Zhang, W. Xu, J. Peng, J. Li, Z. Z. Yu and M. Zhai, Carbon, 2013, 55, 245–252 CrossRef CAS.
- M. Zhu, P. Chen and M. Liu, ACS Nano, 2011, 5, 4529–4536 CrossRef CAS PubMed.
- E. Vasilaki, I. Georgaki, D. Vernardou, M. Vamvakaki and N. Katsarakis, Appl. Surf. Sci., 2015, 353, 865–872 CrossRef CAS.
- L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2009, 131, 1043–1049 CrossRef CAS PubMed.
- H. Zhou, C. Qiu, Z. Liu, H. Yang, L. Hu, J. Liu, H. Yang, C. Gu and L. Sun, J. Am. Chem. Soc., 2010, 132, 944–946 CrossRef CAS PubMed.
- I. V. Lightcap, S. Murphy, T. Schumer and P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 1453–1458 CrossRef CAS PubMed.
- A. M. Golsheikh, N. M. Huang, H. N. Lim and R. Zakaria, RSC Adv., 2014, 4, 36401–36411 RSC.
- H. Wang, J. T. Robinson, G. Diankov and H. Dai, J. Am. Chem. Soc., 2010, 132, 3270–3271 CrossRef CAS PubMed.
- S. Thakur and N. Karak, Carbon, 2012, 50, 5331–5339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10483e |
| This journal is © The Royal Society of Chemistry 2016 |