
New solid phase microextraction fiber based on an eggshell membrane coating for determination of polycyclic aromatic hydrocarbons†
Mohammad Jabbari and
Habib Razmi*
Analytical Research Lab, Faculty of Sciences, Azarbaijan Shahid Madani University, P.O. Box: 53714-161, Tabriz, Iran. E-mail: H.Razmi@azaruniv.edu
First published on 5th September 2016
Abstract
In this work, we introduced a novel soluble eggshell membrane protein (SEP) coating for the fabrication of a solid phase microextraction (SPME) fiber for the first time. The SPME fiber based on SEP (SEP-fiber) was prepared using a glass tube by the simple dip-coating method. The eggshell membrane and its solubilized form, SEP, were characterized by scanning electron microscopy and Fourier transform infrared spectroscopy (FT-IR). The key parameters influencing the extraction efficiency were optimized. The performance of the SEP-fiber was evaluated by extraction of polycyclic aromatic hydrocarbons (PAHs) as model analytes prior to determination by HPLC-UV. Under the optimal conditions, the linearity responses for the PAHs were in the range 0.001–400 μg L−1 with determination coefficients (R2) of 0.996–0.999 and good repeatability (RSD ≤ 6.3%). The proposed method showed lower limits of detection of 0.2–47.3 ng L−1, which are better than those reported in the literature. The SEP-fiber exhibited good extraction capability and stability under acidic and basic conditions, and was easy to fabricate, cost effective and environmentally friendly, which makes it attractive when compared to other commercial SPME fibers. The SEP-fiber was successfully used for the analysis of spiked water samples demonstrating its capability in routine environmental analysis.
1. Introduction
In the last two decades, the solid phase microextraction (SPME) technique has attracted great attention because of its various advantages in comparison with other sample preparation methods.1,2 SPME is based on the distribution of analytes between the matrix or solution and a fiber coated with a stationary phase.3 It has been coupled with many analytical techniques, such as GC,4 HPLC,5,6 CE,7 UV8 and MS.9 The efficiency of the coated-fiber to adsorb the analytes from the samples is a key part of SPME, which controls the extraction efficiency.10 To date, a large number of organic and inorganic materials as SPME fiber coatings have been developed and commercialized.11,12 Organic polymer materials are the first kind of material used as SPME coatings. Because traditional SPME fibers utilize fused silica fiber as solid-phase carriers, organic polymer materials are easier to affix with fiber compared with inorganic materials. In addition, at high temperature, the thermal coefficient of expansions for polymer materials and fused-silica fibers are similar, so it is not easy for the coating to flake off from fibers. Furthermore, a great diversity of structures is another important reason why polymers are very popular.13 To this end, several commercial fused-silica based polymeric coating fibers have been developed, such as non-polar polydimethylsiloxane (PDMS), carboxen/PDMS, semi-polar PDMS/divinylbenzene (PDMS/DVB), polar polyacrylate (PA), carbowax/PDMS, and carbowax/templated resin (CW/TPR), etc. In spite of this, the commercial polymer coated SPME fibers are relatively expensive and difficult to prepare in a routine laboratory, with the disadvantages of relatively low operating temperature, stripping off from the substrate and swelling in organic solvents, which greatly limit their applications,14 so developing a low-cost SPME coating with easy preparation and higher potential for target compounds is useful.Recently, natural polymers such as chitosan, silk and eggshell membrane have attracted significant research interests for application in tissue engineering,15 fuel cell,16 drug delivery,17 separation sciences,18 and electrochemical sensors.19 The eggshell membrane is a light pink double-layered membrane inside the eggshell with a complex lattice network composed of stable and water insoluble protein fibers,19,20 which can be readily obtained almost anywhere as waste. The fibers of eggshell membrane are mainly composed of proteins (80–85%), of which ∼10% are collagens (types I, V and X) and 70–75% are other proteins and glycoproteins containing lysine-derived cross-links, amino acids, saccharides and lipids. Due to proteins and other biomolecules in its structure, the eggshell membrane has richly functional groups in surface including amine, amide, hydroxyl, sulfhydryl and carboxyl. Recently, the utilization of eggshell membrane as a solid substrate for solid phase extraction methods has attracted extensive attention due to having the high specific surface area, excellent chemical stability and high density of surface functional groups.18,21,22 Therefore, regarding to the good adsorption capability of eggshell membrane in solid phase extraction methods, it is expected to have potential application in the preparation of coating materials for SPME fiber. In order to use eggshell membrane for fabrication of SPME fiber, it should be solubilized for easy preparation of homogenized thin film with tunable thicknesses. Recently Yi and co-workers reported a convenient procedure for preparation of soluble eggshell membrane protein (SEP) which is soluble in common non-toxic solvents, but insoluble in water.23,24 To the best of our knowledge, up to now, the use of eggshell membrane for preparation of SPME fiber has not been reported.
The purpose of the present work is to develop a novel SPME fiber by using SEP as a coating material. The SEP was obtained by dissolving eggshell membrane in aqueous thioglycolic acid and acetic acid. Then a glass tube fiber was repeatedly immersed in a SEP solution to fabricate SEP-fiber with desirable thicknesses. To evaluate the extraction performance of the SEP-fiber, PAHs were selected as model analytes and analyzed by HPLC method in real water samples.
2. Experimental
2.1. Chemicals and reagents
Naphthalene (Naph) and anthracene (Ant) were supplied by Merck (Darmstadt, Germany); phenanthrene (Phen), fluoranthene (Flu) and pyrene (Pyr) were supplied by Fluka (Buchs, Switzerland). Standard solutions of each compound at a concentration of 100 mg L−1 were prepared in methanol and stored at 4 °C. All the working solutions were prepared by diluting these solutions by methanol. Thioglycolic acid (mercaptoacetic acid) and acetic acid were purchased from Merck (Darmstadt, Germany) and used without any further purification. All other chemicals were obtained from Merck (Darmstadt, Germany). For chromatographic analysis, methanol and acetonitrile of HPLC grade (Merck) and deionized water were used throughout the experiment.2.2. Instruments
A Jasco HPLC system, consisted of a PU-1580 isocratic pump, a Rheodyne 7725i injector with a 20 μL loop (Rheodyne, Cotati, CA, USA) and a UV-1575 spectrophotometric detector was used in the experiment. The chromatographic system was controlled by HSS-2000 provided by Jasco using the LC-Net II/ADC interface. The data were processed using BORWIN software (version 1.50). An analytical column Perfectsil Target ODS-3 (250 mm × 4.6 mm ID, 5 μm) (MZ-Analysentechnik, Germany) with an ODS-3 precolumn (10 mm × 4.0 mm I.D., 5 μm), which was maintained at room temperature (25 ± 1 °C), was employed for separation.The shape of SEP-fiber and eggshell membrane were characterized by scanning electron microscope model Mira 3 Tescan (TESCAN Electron Microscopy Inc., Brno-Czech Republic). In addition, FT-IR spectra (4000–400 cm−1) were recorded on a Vector 22 (Bruker, Ettlingen, Germany) FT-IR spectrometer using the KBr pellet technique with a ratio sample/KBr of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 by mass.
:100 by mass.
2.3. Preparation of SEP
The raw eggshells were collected from our university campus breakfast shop. It was first washed thoroughly in tap water to completely remove the albumen from the eggshell membrane, and then rinsed with deionized water. The washed eggshells were immersed in a 10% acetic acid solution at 25 °C for 2 h in the beaker to easy separation of eggshell membrane from the calcareous shell. The eggshell membrane was cleaned with deionized water for several times until no indication of acid was detected. Then it was dried at 45 °C in an electric oven for 5 h. The dried membrane was further ground to prepare powders particles with a mortar and pestle.For preparation of SEP solution, a sample of the raw eggshell membrane (1 g, including eggshell membrane powder) was dispersed in 20 mL of 1.25 mol L−1 aqueous thioglycolic acid in the presence of 10% acetic acid at room temperature (25 ± 1 °C). The mixture was heated to and held at 90 °C for 10 h. During this time eggshell membrane powder was dissolved and a homogeneous solution of SEP was obtained. The concentration of SEP was 50 mg mL−1.
2.4. Preparation of SEP-fiber
A glass tube (o.d. ∼ 1 mm with one end closed) as a substrate was pretreated by dipping it in a 1 mol L−1 NaOH solution for 1 h to expose the maximum number of silanol groups on the surface. Then it was washed with deionized water and placed in 0.1 mol L−1 HCl solutions for 30 min to neutralize the excess NaOH and it was washed again and dried.Prior to coating on the glass tube, the prepared SEP solution was ultrasonicated for 30 min to obtain a homogeneous suspension. Then the 2 cm of the glass tube in length was coated by vertically immersing into the SEP suspension in a 10 mL plastic tube. After that, the glass tube was drawn out and dried in air for 5 min. This coating process was repeated until the desired thickness of the coating was obtained. The 2 cm of the glass tube in length was coated with SEP.
2.5. Preparation of sample solutions
The recovery studies were carried out using tap water, well water, river water and rain water samples. Tap water sample was collected freshly from our laboratory (Azarbaijan University of Shahid Madani, Tabriz, Iran) and well water came from deep ground water in Tabriz (Iran). The rain water sample was collected in our university campus in April 2015. River water (Sufi Chay river, Maragheh, Iran) was picked up a few days before analysis. All environmental water samples were collected in brown bottles and were filtered through 0.45 mm microporous membranes immediately after sampling, and stored in the dark place at 4 °C until analysis. Initial analysis confirmed that they were free of target analytes. Stock solutions of 100 mg L−1 Naph, Phen, Ant, Flu and Pyr were individually prepared by dissolving appropriate amounts of each compound in methanol. Quantification of the analytes was done by external calibration where series of standard solutions were prepared by dilution of the stock solution (100 mg L−1) and analysis by HPLC-UV to obtain linear calibration plots for each analyte based on the chromatographic peak areas.2.6. Chromatographic conditions
The isocratic mobile phase consisted of acetonitrile:water in the ratio of 80:20 v/v, flowing through the column at a constant flow rate of 1 mL min−1. The eluent was monitored using UV detection at a wavelength of 254 nm. The mobile phase was filtered through a 0.22 mm membrane-type GV filter (Millipore). A 40 kHz and 138 W ultrasonic water bath with controlled temperature (sonic bath model LBS2-FALC instruments SRL Treviglio, Italy) was applied for degassing the mobile phase.
2.7. SPME procedure
A volume of 25 mL of sample solution was introduced into a 30 mL glass vial with 1 cm magnetic stirring bar (PTFE). The SPME fiber was immersed into the sample solution and the extraction procedure was carried out at room temperature (25 ± 1 °C) for the optimized period of time, 50 min, at the appropriate agitation speed, 700 rpm. After extraction, the fiber was removed from the sample vial and was immediately inserted into the 1 mL acetonitrile, as desorption solvent. Then acetonitrile was evaporated to near dryness under a nitrogen stream and the volume of acetonitrile was adjusted to 100 μL, and 20 μL of the mixture was injected into the chromatographic system.3. Results and discussion
3.1. Characterizations of the SEP and SEP fiber
The eggshell membrane is insoluble in water under ordinary condition. The insolubility of eggshell membrane is mainly caused by the presence of a large number of crosslinks of disulfide bonds. The treatment of eggshell membrane in aqueous thioglycolic acid reduces the degree of crosslinking.25 The eggshell membrane was dissolved by reductive cleavage of disulfide bonds with aqueous thioglycolic acid at 90 °C in the presence of 10% acetic acid. After being treated with thioglycolic acid and acetic acid, the shape of the eggshell membrane fibers is gradually destroyed; it became blurry after treating for 5 h, after 10 h eggshell membrane dissolved completely and produced homogeneous suspension24 (S1).The morphology of eggshell membrane and SEP were characterized by SEM and the images are shown in Fig. 1A and B respectively. A network-like structure was observed on the cleaned eggshell membrane surface, which reveal that the raw eggshell membrane is a mesh-work composed of interlinked and coalescing fibers ranging from 1 to 3 μm in diameter. The eggshell membrane is a complex porous structure made up of proteins, glycolipids, and carbohydrates. These macromolecules contain a large number of functional groups –OH, –COOH, and –NH2.26 Unlike eggshell membrane the surface of SEP film has the flat and homogeneous structure (Fig. 1B). Thus, it can be seen that the SEP has a completely different surface morphology from the raw eggshell membrane. This demonstrates that the cleavage of the disulfide bonds is accompanied by destruction of the eggshell membrane fibers.
 | ||
| Fig. 1 SEM images of (A) raw eggshell membrane and (B) SEP. | ||
To investigate the surface characteristics of eggshell membrane and the SEP, FT-IR in the range 400–4000 cm−1 was studied (Fig. S2†). The FT-IR spectrum of eggshell membrane exhibits significant peaks at 3300–3500 cm−1 (ascribed to O–H and N–H stretching mode), 1630 cm−1 (ascribed to N–H bending mode), and 1350–1385 cm−1 (ascribed to C–N stretching mode and C–H bending), proving the presence of amines and amides in eggshell membrane while the band at 1138 cm−1 correspond to C–O stretching. The spectral pattern and number of bands for the eggshell membrane and SEP were approximately similar at all ranges and the small band at 3133 cm−1 in the FT-IR spectrum of the SEP, strongly associated with the presence of –SH groups. This result indicated that the hydroxyl (–OH) groups of SEP have been esterified by thioglycolic acid (SHCH2COOH).26
Also, the morphology of the cross section of the SEP-fiber was examined with SEM. The SEM images in Fig. 2A and B indicated that the product is actually a thin film with average thickness around 67 μm. The thickness of the individual fiber sorbent layer was constant in the whole length due to the use of homogeneous SEP suspension in fiber preparation step. The SEP-fiber can be used to extraction of PAHs from aqueous sample by SPME method. According to the previously published report,27 the extraction ability was attributed to π interactions between the PAHs and the functionalized groups (amine, amide, hydroxyl, sulfhydryl) of the SEP. Fig. 2C shows a photograph of SEP-fiber illustrating a white and solid thin layer of SEP coated homogeneously on the glass tube.
 | ||
| Fig. 2 SEM images of cross section of SEP-fiber in two magnitude (A and B); and (C) shows photographic picture of the prepared fiber coated with SEP. | ||
3.2. Optimization of SPME procedure
There are some factors affecting the extraction performance of the coated fiber, such as fiber stability, coating thickness, extraction time, stirring rate, desorption time, desorption solvent, salt effect and pH effect. Before validating the analytical method, these factors were investigated and optimized. The chromatographic peak area, which is related to the amount of analytes, was used to evaluate the extraction efficiency under different experimental conditions.The stability of SPME fiber is crucial for practical application. Damaging the fiber coating was mainly caused by its exposure to acidic or basic solution and strong mechanical stirring during extraction processes. In order to evaluate the stability, swelling and adhesion behavior of the coating, the tip of the novel SEP-fiber was individually dipped in HCl (0.1 mol L−1), NaOH (0.1 mol L−1), NaCl (1 mol L−1), methanol, acetone, acetonitrile and n-hexane solutions for 4 h under room temperature (25 ± 1 °C). No visible swelling of the coating was observed in these aqueous and organic solvents. After this, the fiber was washed with deionized water and dried at 40 °C. Then, the fiber was used for the extraction experiments. The peak areas for the analytes obtained before and after the treatment of SEP-fiber were compared to evaluate its stability (Table S1†). The ratio of the fiber extraction efficiency after treatment to that of before treatment remained almost unchanged. Also, long term stability of the SEP-fiber was investigated and the SEP-fiber stored at room temperature (25 ± 1 °C) for three months showed the same extraction efficiency as a freshly prepared fiber.
The effect of coating thickness on the extraction is one of the most important parameters in the SPME procedure. The effect of coating time on the extraction procedure was investigated and the results are shown in Fig. 3. The SEP-fiber was fabricated in desirable thickness by repeating the dipping procedure from 1 till 14 times. As shown in Fig. 3, the extraction efficiency, increased with coating thickness, due to increase in surface area and adsorptive capacity. In the cases of naphthalene and phenanthrene extraction efficiency reached maximum value after 7 dipping times. The maximum responses for all analytes were observed at dipping time 9, at which the average thickness was about 67 μm. It was observed that when the coating time exceeds 9, the peak areas of the analytes remained almost the same.
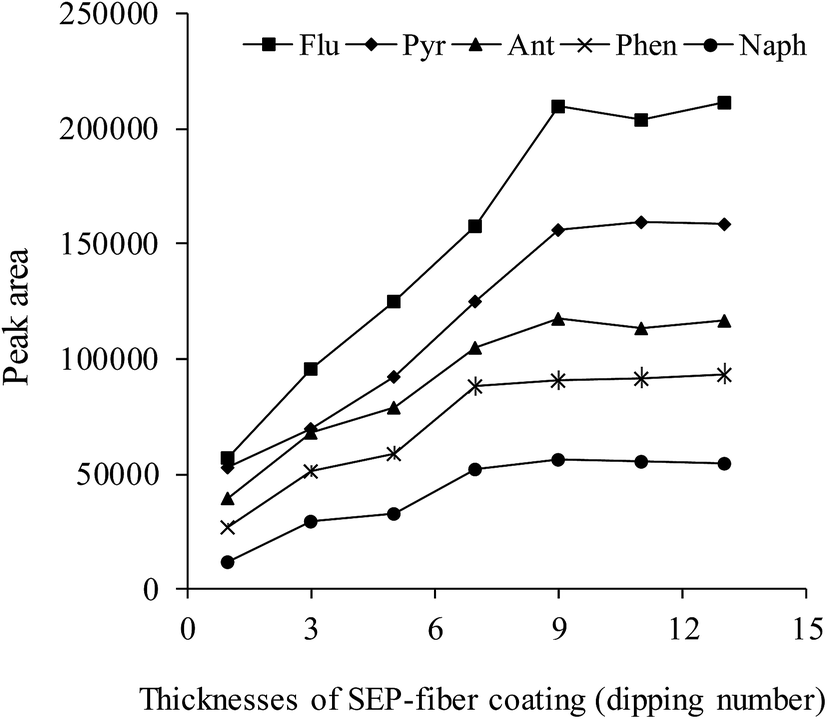 | ||
| Fig. 3 The influence of SEP-fiber coating thicknesses on extraction efficiency. Extraction conditions: concentrations of containing 40.0 μg L−1 Naph, 2.0 μg L−1 Ant, 4.0 μg L−1 Phen and 20.0 μg L−1 Pyr and Flu; extraction time, 50 min; desorption time, 5 min; desorption solvent acetonitrile. | ||
SPME is an equilibrium-based technique, and there is a direct relationship between the extraction amount and the extraction time.28 In this work, the effect of extraction time was examined in the range 10–90 min. As shown in Fig. S3† the peak areas of the compounds increase with an extraction time from 10 to 50 min and then remain almost constant. This phenomenon indicates that the adsorption is a dynamic equilibration process, and once the equilibrium is achieved, a further extension of the extraction time had almost no effect on the extraction efficiency. The rate-determining step of the equilibration process is diffusion of the analyte across a thin stationary, aqueous layer at the coating–aqueous phase boundary. The mass transport of the analytes to the surface of SEP coating, that is not pore structure, is slow. Because of this, relative long equilibrium times and long extraction time for all analytes was achieved by SEP coating. Based on the above experimental result, the extraction time of 50 min was selected.
Stirring rate is another important factor that affects the extraction performance.29 Fast agitation of the sample solution could be employed to enhance the extraction efficiency since agitation effectively accelerate the mass transfer of an analyte from the water sample to the fiber.3 In this study, the effect of the sample agitation rate was evaluated between 0 and 1100 rpm. Fig. S4† shows that the extraction efficiency increased as the stirring rate increased to 700 rpm and then remained almost unchanged. Therefore, 700 rpm was chosen for the next experiments.
Desorption of the analytes from the fiber is a crucial part in the whole SPME process. Complete desorption of the adsorbed analytes from the fiber will improve the sensitivity. In this work, organic solvents and desorption time used for SPME procedure were optimized. Different HPLC compatibles solvents, including methanol, acetonitrile, hexane and acetone have been used to desorb the analytes from SEP-fiber. Fig. S5A† showed that acetonitrile gives the highest desorption efficiency. So acetonitrile was selected as the desorption solvent.
Desorption time is of great importance for obtaining a satisfactory desorption efficiency for the analytes. Desorption time was studied in the range from 2 to 20 min (Fig. S5B†). The results indicated that the desorption efficiency almost kept invariable over the range 2–20 min. So the desorption time of 5 min was selected.
The addition of salt to the sample solution can decrease the solubility of the analytes and enhance their availability for the extraction due to the salting-out effect, but on the other hand, it can also increase the viscosity of the solution, which may reduce the extraction capability and the diffusion coefficient.30 In the present study, the effect of the salt addition on the extraction efficiency was studied with the use of NaCl. Fig. S6A† illustrates the peak area versus the salt concentration being changed in the range from 0 to 20% w/v. As shown, extraction efficiency of PAHs decreased with the increase in NaCl content. In order to obtain the best extraction efficiency, salt was not added into the sample solution for further studies.
pH is one of the important factors affecting the adsorption behavior in the SPME format because it determined the charge density of adsorbent and the present state of analytes in solution as ionic or molecular state.30 Therefore, the influence of the pH values on the extraction efficiency of PAHs was studied by adjusting solution pH between 3 and 12. As can be seen from Fig. S6B,† peak area for all the analytes was remarkable when the solution pH was below 4.0, and decreased with increase in pH up to 6. Only slight changes in recovery were observed when solution pH was changed from 6 to 9. Then peak areas increased again in alkaline pH values of 9 till 12. The high adsorption capacity of PAHs at higher alkaline and acidic condition suggested that specific adsorption–enhancement interaction occurred. The pH effect can be attributed to the fact that SEP contains some functional groups such as –NH2, –CONH2, –OH, and –COOH groups, which can be protonated or deprotonated relying on the pH value of the sample solution.23 Also, according to the literature, the isoelectric point (IEP) of eggshell membrane is 5.6 ± 0.2.30 Hence, the large peak area in acidic conditions can be attributed to the fact that the surface of SEP was positively charged when the pH level was below its IEP. Also further increases in pH value from 9 would make the SEP surface negatively charged.23,31,32 In acidic and basic conditions, the increased peak area might be related to the attractive interactions between SEP and PAHs. At a pH range from 6 to 9, the eggshell membrane surface became neutral or slightly negatively charged, leading to the reduced assembling of PAHs onto SEP compared to acidic and alkaline condition and thus decreased the PAHs recoveries.33 However, detailed information regarding these interactions remains unknown and deserves further study. From these results it was decided to adjust the pH of water samples at 3 for the subsequent assays.
3.3. Evaluation of analytical performance
The analytical characteristics of the proposed method under the conditions mentioned above were evaluated by determining the linear response range, determination coefficients, limits of detections (LODs), limits of quantifications (LOQs), and fiber-to-fiber and day-to-day deviations. Analytical data obtained are summarized in Table 1. The calibration graphs were constructed by plotting the analytes chromatographic peak area against the corresponding concentrations with triplicate measurements. Linear response ranges for the five PAHs were in the range 0.001–400 μg L−1 with determination coefficients (R2) of higher than 0.996. In order to determine the LOD and LOQ, the concentrations corresponding to signal-to-noise (S/N) ratio of three and ten have been considered. Determination of the S/N ratio was performed by comparing measured signals from samples with known low concentrations of analyte (Naph 0.25 μg L−1, Phen 0.005 μg L−1, Ant 0.001 μg L−1 Flu 0.025 μg L−1 and Pyr 0.1 μg L−1) with those of blank samples, at the same time windows where the analytes were detected. The results listed in Table 1 show that the value of LOD and LOQ for the five target PAHs (the concentration in solution was 40.0 μg L−1 Naph, 2.0 μg L−1 Ant, 4.0 μg L−1 Phen and 20.0 μg L−1 Pyr and Flu) range from 0.2 to 47.3 ng L−1 and 0.7 to 155.4 ng L−1, respectively. The SEP-fiber showed RSD% values in the range of 3.9–6.3% (n = 3) and 5.4–7.1% (n = 6) for fiber-to-fiber and day-to-day extraction experiments, respectively. This demonstrates the good precision and reproducibility of the method for the determination of PAHs.| Compound | Linear range (μg L−1) | R2 | LOD (ng L−1) | LOQ (ng L−1) | RSD (%) | |
|---|---|---|---|---|---|---|
| Fiber-to-fiber (n = 3) | Day-to-day (n = 6) | |||||
| Naph | 0.25–400 | 0.999 | 47.3 | 155.4 | 3.9 | 5.4 |
| Ant | 0.001–10 | 0.997 | 0.2 | 0.7 | 6.2 | 7.1 |
| Phen | 0.005–15 | 0.999 | 0.8 | 2.7 | 5.3 | 5.5 |
| Flu | 0.025–40 | 0.997 | 6.2 | 21.1 | 5.8 | 6.5 |
| Pyr | 0.1–40 | 0.996 | 15.1 | 49.2 | 6.3 | 6.8 |
| Analyte | Spiked (μg L−1) | Tap water (n = 3) | River water (n = 3) | Well water (n = 3) | Rain water (n = 3) |
|---|---|---|---|---|---|
| RRa (RSD)% | RR (RSD)% | RR (RSD)% | RR (RSD)% | ||
| a RR: relative recovery.b ND: not detected. | |||||
| Naph | 0 | NDb | ND | ND | ND |
| 20 | 85.5 (5.4) | 84.1 (5.2) | 105.4 (3.2) | 91.1 (6.2) | |
| 40 | 91.2 (3.8) | 88.7 (6.9) | 97.2 (5.1) | 92.7 (5.2) | |
| Phen | 0 | ND | ND | ND | ND |
| 1 | 88.0 (6.9) | 91.0 (4.4) | 87.2 (4.8) | 85.02 (4.6) | |
| 2 | 86.5 (5.6) | 91.0 (6.1) | 85.5 (6.2) | 90.5 (5.5) | |
| Ant | 0 | ND | ND | ND | ND |
| 2 | 95.5 (4.2) | 87.5 (4.6) | 84.0 (7.9) | 94.5 (5.8) | |
| 4 | 90.0 (5.8) | 95.5 (5.1) | 87.2 (7.4) | 80.2 (4.6) | |
| Flu | 0 | ND | ND | ND | ND |
| 4 | 92.7 (6.1) | 95.5 (4.8) | 90.2 (7.9) | 88.7 (6.7) | |
| 8 | 98.1 (4.7) | 98.7 (6.4) | 81.2 (7.6) | 92.2 (7.2) | |
| Pyr | 0 | ND | ND | ND | ND |
| 4 | 93.7 (6.7) | 94.5 (5.4) | 102.7 (5.5) | 95.2 (7.5) | |
| 8 | 92.6 (7.5) | 96.3 (6.2) | 103.6 (6.3) | 93.6 (5.3) | |
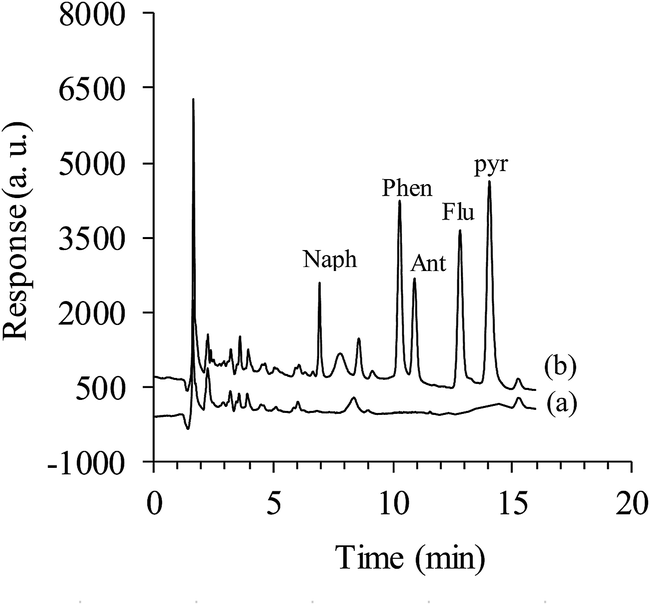 | ||
| Fig. 4 The typical chromatograms of (a) Sufi Chay river water sample and (b) Sufi Chay river water sample spiked with the PAHs at each concentration level of 20.0 μg L−1 Naph, 1.0 μg L−1 Ant, 2.0 μg L−1 Phen and 4.0 μg L−1 Pyr and Flu. | ||
| Fiber type and thickness | Analytical technique | LR (μg L−1) | LOD (ng L−1) | Extraction time (min) | Ref. |
|---|---|---|---|---|---|
| a Polyacrylate.b Polydimethylsiloxane.c Polydimethylsiloxane–divinylbenzene.d Soluble eggshell membrane protein. | |||||
| PAa – 85 μm | GC-FID | 0.1–100 | 30–590 | 90 | 34 |
| PDMSb – 100 μm | GC-MS | 10–10000 |
200–1500 | 30 | 35 |
| PDMS – 100 μm | HPLC-FID | 1–300 | 1–5 (μg L−1) | 30 | 36 |
| PDMS – 100 μm | HPLC-FLD | 0.01–1 | 1–6 | 60 | 37 |
| PDMS – 100 μm | GC-MS | 0.01–10 | 1–29 | 45 | 38 |
| PDMS/DVBc – 65 μm | GC-FID | 0.1–200 | 30–1000 | 30 | 39 |
| PDMS/DVB – 65 μm | GC-MS | 0.1–10 | 16–75 | 60 | 40 |
| SEPd – 67 μm | HPLC-UV | 0.001–400 | 0.2–47.3 | 50 | This work |
4. Conclusion
In this work, we reported the use of SEP as SPME fiber coating for extraction and determination of PAHs as model analytes in aqueous samples by HPLC-UV for the first time. Under the optimized conditions, this method demonstrated a wide range of linearity, low LOD and satisfactory precision. However, it also suffers from some disadvantages, such as short operation lifetime, because of the flaws in the surface of SEP-fiber after three extractions. The results suggest that the developed SPME method was suitable for the analysis of the PAHs in real water samples and the SEP can be a promising alternative coating for SPME fibers, which may have great potential for the extraction of more organic analytes in different samples.Acknowledgements
The authors gratefully acknowledge the Research Council of Azarbaijan Shahid Madani University. This work was supported by the Azarbaijan Shahid Madani University under Grant [No. 401/287].References
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- C. Dietz, J. Sanz and C. Cámara, J. Chromatogr. A, 2006, 1103, 183–192 CrossRef CAS PubMed.
- S. Wang, S. Lv, Z. Guo and F. Jiang, Microchim. Acta, 2014, 181, 427–433 CrossRef CAS.
- H. Zhang and H. K. Lee, J. Chromatogr. A, 2011, 1218, 4509–4516 CrossRef CAS PubMed.
- H. Heidari, H. Razmi and A. Jouyban, J. Chromatogr. A, 2012, 1245, 1–7 CrossRef CAS PubMed.
- M. R. Hadjmohammadi, M. Peyrovi and P. Biparva, J. Sep. Sci., 2010, 33, 1044–1051 CrossRef CAS PubMed.
- A.-L. Nguyen and J. H. T. Luong, Anal. Chem., 1997, 69, 1726–1731 CrossRef CAS PubMed.
- B. L. Wittkamp, S. B. Hawthorne and D. C. Tilotta, Anal. Chem., 1997, 69, 1197–1203 CrossRef CAS.
- Z. Mester, J. Lam, R. Sturgeon and J. Pawliszyn, J. Anal. At. Spectrom., 2000, 15, 837–842 RSC.
- H.-L. Xu, Y. Li, D.-Q. Jiang and X.-P. Yan, Anal. Chem., 2009, 81, 4971–4977 CrossRef CAS PubMed.
- L. Xu, J. Feng, J. Li, X. Liu and S. Jiang, J. Sep. Sci., 2012, 35, 93–100 CrossRef CAS PubMed.
- A. Kumar, Gaurav, A. K. Malik, D. K. Tewary and B. Singh, Anal. Chim. Acta, 2008, 610, 1–14 CrossRef CAS PubMed.
- G. Jiang, M. Huang, Y. Cai, J. Lv and Z. Zhao, J. Chromatogr. Sci., 2006, 44, 324–332 CAS.
- V. K. Ponnusamy and J.-F. Jen, J. Chromatogr. A, 2011, 1218, 6861–6868 CrossRef CAS PubMed.
- F. Croisier and C. Jérôme, Eur. Polym. J., 2013, 49, 780–792 CrossRef CAS.
- J. Ma and Y. Sahai, Carbohydr. Polym., 2013, 92, 955–975 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- H. Abdolmohammad-Zadeh and Z. Talleb, Microchim. Acta, 2014, 181, 1797–1805 CrossRef CAS.
- R. Mohammad-Rezaei, H. Razmi and S. Dehgan-Reyhan, Colloids Surf., B, 2014, 118, 188–193 CrossRef CAS PubMed.
- W. Chen, B. Li, C. Xu and L. Wang, Biosens. Bioelectron., 2009, 24, 2534–2540 CrossRef CAS PubMed.
- Y. Zhang, W. Wang, L. Li, Y. Huang and J. Cao, Talanta, 2010, 80, 1907–1912 CrossRef CAS PubMed.
- X. Zou and Y. Huang, Anal. Methods, 2013, 5, 6486–6493 RSC.
- F. Yi, Z.-X. Guo, L.-X. Zhang, J. Yu and Q. Li, Biomaterials, 2004, 25, 4591–4599 CrossRef CAS PubMed.
- F. Yi, J. Yu, Z.-X. Guo, L.-X. Zhang and Q. Li, Macromol. Biosci., 2003, 3, 234–237 CrossRef CAS.
- T. D. Lehman and B. J. Ortwerth, Biochim. Biophys. Acta, 2001, 1535, 110–119 CrossRef CAS.
- X. Z. Cheng, C. J. Hu, K. Cheng, B. M. Wei and S. C. Hu, Adv. Mater. Res., 2010, 113–114, 22–26 CrossRef.
- X. Liu, X. Lu, Y. Huang, C. Liu and S. Zhao, Talanta, 2014, 119, 341–347 CrossRef CAS PubMed.
- S. Zhang, Z. Du and G. Li, Anal. Chem., 2011, 83, 7531–7541 CrossRef CAS PubMed.
- S. Saha, R. Mistri and B. C. Ray, Anal. Bioanal. Chem., 2013, 405, 9265–9272 CrossRef CAS PubMed.
- X. Ma, J. Wang, Q. Wu, C. Wang and Z. Wang, Food Anal. Methods, 2014, 7, 1381–1386 CrossRef.
- W. Wang, B. Chen, Y. Huang and J. Cao, J. Chromatogr. A, 2010, 1217, 5659–5664 CrossRef CAS PubMed.
- W. Wang, B. Chen and Y. Huang, J. Agric. Food Chem., 2014, 62, 8051–8059 CrossRef CAS PubMed.
- J. Wang, Z. Chen and B. Chen, Environ. Sci. Technol., 2014, 48, 4817–4825 CrossRef CAS PubMed.
- R.-a. Doong, S.-m. Chang and Y.-c. Sun, J. Chromatogr. A, 2000, 879, 177–188 CrossRef CAS PubMed.
- R.-a. Doong, S.-m. Chang and Y.-c. Sun, J. Chromatogr. Sci., 2000, 38, 528–534 CAS.
- M. R. Negrão and M. F. Alpendurada, J. Chromatogr. A, 1998, 823, 211–218 CrossRef.
- P. Popp, C. Bauer, M. Möder and A. Paschke, J. Chromatogr. A, 2000, 897, 153–159 CrossRef CAS PubMed.
- A. J. King, J. W. Readman and J. L. Zhou, Anal. Chim. Acta, 2004, 523, 259–267 CrossRef CAS.
- M.-C. Wei and J.-F. Jen, Talanta, 2007, 72, 1269–1274 CrossRef CAS PubMed.
- D. Martin and J. Ruiz, Talanta, 2007, 71, 751–757 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10815f |
| This journal is © The Royal Society of Chemistry 2016 |