
Selective extraction of bioactive glycoprotein in neutral environment through Concanavalin A mediated template immobilization and dopamine surface imprinting
Xue Qu*ab,
Feifei Wangab,
Yi Sunab,
Yu Tianab,
Rui Chenab,
Xiaoyu Maab and
Changsheng Liu*ab
aKey Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai 200237, PR China. E-mail: quxue@ecust.edu.cn; liucs@ecust.edu.cn
bThe State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, PR China
First published on 25th August 2016
Abstract
Glycoproteins play important roles in various biological events. Extraction of specific bioactive glycoproteins from physiological fluids is highly required for clinical diagnosis and treatment. Concanavalin A (Con A) is a tetramer lectin protein that can specifically bind glycospecies containing sugar units, and this binding occurs in physiological environments. Inspired by this, we propose a sugar–lectin recognition based glycoprotein surface imprint, which is anticipated to selectively extract bioactive glycoproteins in physiological environments. The glycosylated biomarker, transferrin, was used as a model template. Transferrin was first immobilized on a Con A modified Fe3O4 surface via the sugar–lectin interaction at pH 7.4. Dopamine was then polymerized onto this surface for glycoprotein imprinting at the same pH, and continuous oxygen bubbling into the dopamine solution improved the polymerization speed. After removal of the template by boronate buffer, transferrin imprinted Fe3O4 was obtained. The subsequent binding experiment indicated that the transferrin imprint has pH dependent binding affinity towards this protein. The optimal transferrin binding capacity, as well as the maximum imprinting factor was observed at neutral pH. The isotherm adsorption study reveals that imprinted and non-imprinted material are both fitted with the Langmuir adsorption model. Single selective adsorption and competitive adsorption experiments show that the obtained surface imprint has selectivity towards template glycoprotein, and circular dichroism (CD) spectral analysis indicates that boronate buffer washing has no negative influence on the natural structure of transferrin and Con A. These results demonstrate that this novel glycoprotein surface imprint can work under physiological conditions, and the glycoprotein extraction method is mild enough to keep the bioactivity of these targets for further bio-application.
Introduction
Glycoproteins are proteins that contain oligosaccharide chains (glycans) covalently attached to polypeptide side-chains. In mammalian proteins, nearly 50% of the total proteins are glycoproteins.1 Glycoproteins play important roles in various biological events, such as molecular recognition, immune response, signal transduction and regulation of development.2–5 The occurrence of many diseases is closely associated with the improper expression of related glycoproteins.6 A variety of glycoproteins have been applied as disease biomarkers and therapeutic targets in clinical settings.7–9 Extraction of specific glycoproteins from physiological fluids with bioactivity is a significant requirement for clinical diagnosis and treatment.Molecular imprinting technology (MIT) is a smart strategy to create molecular recognition materials by polymerizing suitable functional monomers in the presence of template molecules.10 After removing the templates, the resultant recognition cavities within the molecularly imprinted polymers (MIPs) allow specific binding of the templates. So far, MIPs synthesized via the template pre-immobilized surface imprinting approach have been proved to specifically recognize and separate large biological targets such as proteins.11–14 This approach can provide clear cut imprinting cavities and also can enhance mass transfer, via presenting imprinting cavities at, or close to the polymer surface.
Conventionally, pre-immobilization of glycoproteins is achieved by using reversible covalent binding between boronic acid and cis-diol units that are attached to the glycoprotein.15 A basic pH is required to immobilize these glyco-species because most commercially available boronic acids are weak acids with a pKa of ca. 8–9. Chen et al. reported the first example of boronate mediated horseradish peroxidase immobilized surface imprinting by using magnetic Fe3O4@Au nanofibers as the substrate. The resultant MIPs exhibited high adsorption capacity and good specificity toward the template glycoprotein at pH 10.0.16 Liu's team developed a series of glycoprotein MIPs using this strategy and found that these imprints can be applied in a wide pH range, including neutral biological environments, such as human serum.17–21
Lectins are carbohydrate-binding functional proteins that are highly specific to sugar moieties in neutral environments. Concanavalin A (Con A) is a tetramer lectin protein and each Con A molecule can provide four sites for sugar binding. A variety of glycospecies, including monosaccharides, polysaccharides and glycoproteins, can bind to Con A via sugar–lectin interaction. Traditionally, Con A has been universally fixed onto various matrixes for affinity chromatography, with the aim to purify diverse glycospecies in biological samples.22–24 Although Con A is reportedly hard to be prepared and lacks storage stability, artificial blood glucose level monitors comprising dye-labeled Con A have been capable of working for 6–12 months in intradermal tissue.25–27 Our previous works also developed Con A involved functional assembly for biological applications.28,29
In this paper, we propose an alternative glycoprotein surface imprint that is anticipated to work in physiological environments, for which bio-inspired Con A mediated template immobilization, combined with subsequent dopamine polymerization is employed to in situ imprint glycosylated biomarker transferrin (TRF) at neutral pH. The proposed strategy is illustrated in Scheme 1. Nano-scaled Fe3O4 is employed as the solid substrate, due to its good biocompatibility, low toxicity and simple preparation. The monosaccharide mannose is first chemically grafted onto Fe3O4 to allow the surface modification of Con A via a mild sugar recognition process. Further, pre-immobilization of the template onto the Fe3O4 surface is achieved via the same lectin–sugar interaction. After that, dopamine polymerization occurring at the Fe3O4 surface initiates the surface imprinting of TRF. After the deposition of polydopamine film with an ideal thickness, the immobilized TRF is washed away by boronate buffer to generate TRF imprinted nanoparticles. Compared to the conventionally used sodium dodecyl sulfonate (SDS) incorporated acidic solution for protein template washing,10,30–33 boronate buffer is believed not to cause protein denaturation, like SDS surfactants.
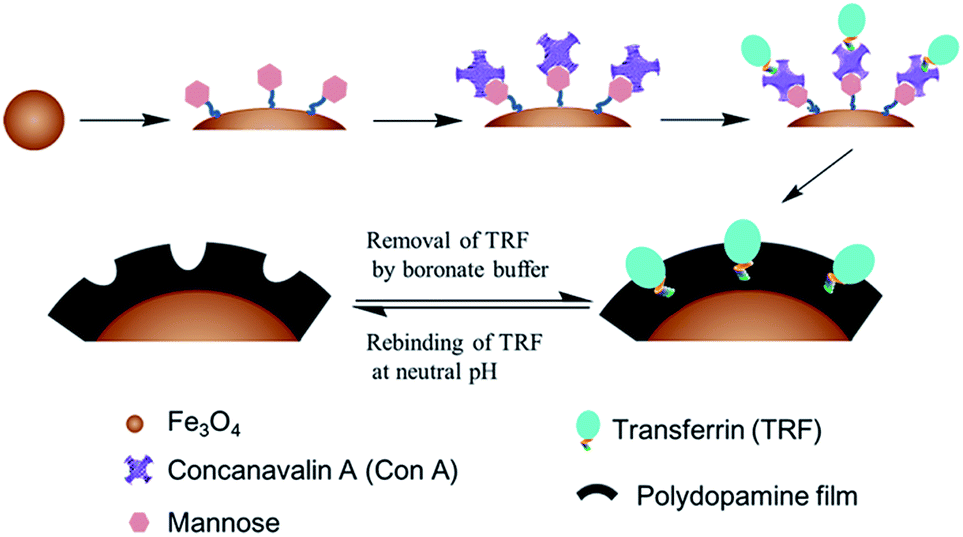 | ||
| Scheme 1 In situ surface imprinting of transferrin by Concanavalin A mediated template immobilization and dopamine surface imprinting at neutral pH. The generated glycoprotein imprint has the potential to work in physiological environments. Boronate buffer washing allows the protection of the bioactivity of extracted glycoprotein and surface fixed Concanavalin A. | ||
We envision that this Con A mediated glycoprotein imprinting can work at physiological conditions, and the glycoprotein extraction method is mild enough to keep the bioactivity of these targets, which facilitates the further bio-application of the extracted proteins. The proposed glycoprotein surface imprints have the potential to meet the emerging demands for identification and quantification of glyco-species in life sciences and clinical medicine.
Experimental
Materials
Dopamine was purchased from Adamas Chemical Company. 3-Aminopropyltriethoxysilane (APTES) was purchased from Aladdin Industrial Corporation. Bovine serum albumin (BSA) (Mw = 66 kDa, pI = 4.9), ovalbumin (OVA) (Mw = 44 kDa, pI = 4.7), ribonuclease B (RNaSe B) (Mw = 14.7 kDa, pI = 8.9) and hemoglobin (Hb) (Mw = 64 kDa, pI = 6.9) were purchased from Yuanye Biotech Ltd. (Shanghai, China). Human serum albumin (HSA) (Mw = 66 kDa, pI = 4.9) was purchased from Soledad Biological Technology Co. Ltd. (Beijing, China). FeCl3·6H2O, sodium acetate (NaOAc), ethylene glycol, diethylene glycol, tetrahydrofuran (THF), succinic anhydride were all purchased from Jingchun Reagent Co. Ltd. (Shanghai, China). Transferrin (TRF) (Mw = 77 kDa, pI = 4.6–5.6), Concanavalin A (Con A) (from Canavalia ensiformis, Jack bean) and the monosaccharide mannose were all purchased from Sigma. All other reagents were purchased from China Nation Medicines (Shanghai, China).Preparation of magnetic Fe3O4 nanoparticles
Fe3O4 magnetic nanoparticles were synthesized through a solvothermal reduction method according to a previous report.34,35 Briefly, FeCl3·6H2O (1.08 g) and NaOAc (4.0 g) were dissolved in ethylene glycol (14 mL) and diethylene glycol (26 mL). The obtained homogeneous yellow solution was moved to a Teflon-lined stainless steel autoclave, sealed and heated at 200 °C. After reaction for 16 h, the autoclave was cooled to room temperature. The obtained black Fe3O4 nanoparticles were washed several times with water and ethanol, respectively. Finally, the products were collected with a magnet and then dried under vacuum.Synthesis of –COOH modified Fe3O4 nanoparticles
The synthesis of carboxyl group modified Fe3O4 involved two steps, including the preparation of amine-functionalized magnetic particles, followed by further –COOH surface modification.36 (1) For the preparation of amine-functionalized magnetic particles (Fe3O4–NH2), 3 g of Fe3O4 nanoparticles were dispersed into 200 mL of ethanol/H2O mixture solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under ultrasonication at room temperature. The Fe3O4 suspension was adjusted to pH 4.0 by ethanoic acid, and mechanically stirred for 2 h under Ar protection. Then, 4 mL of APTES were added to the mixture to allow amine modification. The above mixture was stirred overnight at 60 °C under Ar protection. After that, the nanoparticles were washed several times with water and ethanol, and then dried in a vacuum. (2) For carboxyl surface modification, 1.2 g of succinic anhydride was dissolved in 60 mL of anhydrous THF. The resultant solution was then added drop by drop into a THF solution containing 300 mg of amine-functionalized Fe3O4. After reacting for 24 h, the product was collected and washed three times with THF, and then vacuum dried to obtain the COOH-functionalized Fe3O4 nanoparticles (Fe3O4–COOH).
:1) under ultrasonication at room temperature. The Fe3O4 suspension was adjusted to pH 4.0 by ethanoic acid, and mechanically stirred for 2 h under Ar protection. Then, 4 mL of APTES were added to the mixture to allow amine modification. The above mixture was stirred overnight at 60 °C under Ar protection. After that, the nanoparticles were washed several times with water and ethanol, and then dried in a vacuum. (2) For carboxyl surface modification, 1.2 g of succinic anhydride was dissolved in 60 mL of anhydrous THF. The resultant solution was then added drop by drop into a THF solution containing 300 mg of amine-functionalized Fe3O4. After reacting for 24 h, the product was collected and washed three times with THF, and then vacuum dried to obtain the COOH-functionalized Fe3O4 nanoparticles (Fe3O4–COOH).
Fabrication of Con A-modified Fe3O4 nanoparticles
The synthesis of Con A modified Fe3O4 involved two steps, including the chemical coupling of mannose onto Fe3O4–COOH, followed by the biospecific surface assembly of Con A. (1) For mannose coupling, the carboxyl groups on Fe3O4–COOH nanoparticles were first exposed to a MES buffered solution containing 10 mM of EDC and NHS. After 4 h of stirring, 15 mg of epichitosamine were added into the solution to allow mannose coupling. After 24 h of reaction, the product (Fe3O4-man) was collected, washed with buffer three times, and dried under vacumn. (2) For the biospecific surface assembly of Con A, 40 mL of 0.25 mg mL−1 Con A solution buffered by PBS at pH 7.4 were added into a suspension containing 200 mg of man-Fe3O4. After incubation for 4 h, the product (Fe3O4–Con A) was washed and collected for further application.Preparation of Fe3O4@TRF-MIPs and Fe3O4@NIPs
The TRF-imprinted particles were synthesized by dopamine surface polymerization. 80 mg of Fe3O4–Con A nanoparticles and 20 mg of TRF were dissolved into 40 mL of PBS (10 mM, pH 7.4). The mixture was stirred for 2 h at room temperature for TRF immobilization. After that, the particles were collected and exposed to a dopamine solution (1 mg mL−1) with continuous bubbling of oxygen. The polymerization of dopamine was performed for a predetermined time interval at room temperature. After that, an appropriate polydopamine film thickness was formed. Fe3O4 particles were collected and boronic acid buffer (BBS, 10 mM, pH = 9.6) was used to wash the particles to remove TRF templates. According to our previous experience,37 the thickness of the polydopamine can be well controlled by simply adjusting the time for polymerization. Here, we created the imprinting layer via a continuous polymerization for 9 h. The non-imprinted particles were prepared using an identical procedure, but without the addition of the protein template.Binding experiments
The binding tests were performed in PBS at 25 °C. Briefly, 10 mg Fe3O4@TRF-MIPs and Fe3O4@NIPs particles were suspended in 1.0 mL of protein template standard solutions with different initial concentrations ranging from 0.05 mg mL−1 to 1.0 mg mL−1. The samples were incubated on a rocking table for 12 h to reach equilibrium at room temperature. The concentration of free template protein in the supernatant was measured by BCA assay.In the selectivity experiments, BSA, Hb and OVA were chosen as reference proteins, with initial concentrations of 1.0 mg mL−1.
The adsorption capacity (Qe) of the template protein or competitive protein bound to the imprinted particles was calculated by the following formula:
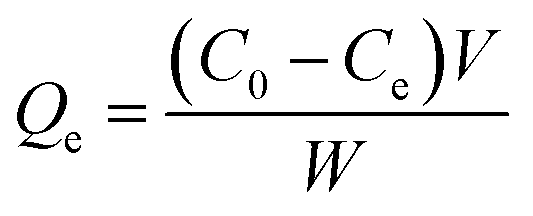 | (1) |
The imprinting factor (IF) was used to estimate the specificity of MIPs and calculated from the following equation:
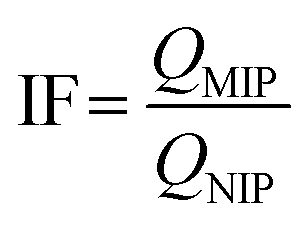 | (2) |
Characterization
The morphology and structure of nanoparticles were examined by scanning electron microscopy (S-4800, Hitachi, Japan) and transmission electron microscopy (JEM-2100F, JEOL, Japan). The infrared spectra were recorded on a Fourier transform infrared (FT-IR) spectrometer (Nicolet 6700, USA). Zeta potential was tested with ZETAsizer 3000HS (Malvern Instruments Ltd., UK). Fluorescence images were recorded by laser confocal microscopy (Nikon A1R, Nikon, Japan). The structures of the proteins were detected by far-UV circular dichroism spectroscopy (Model J-715, Tokyo, Japan). Electrophoretic analysis of proteins was performed using regular SDS-PAGE with 12% running and 5% stacking gels, according to the manual introduction (Bio-Rad, Hercules, CA, USA). Proteins were stained with Coomasie Brilliant Blue G250. UV-visible spectroscopy was carried out with a UV/Vis spectrometer (Sunnyvale, CA).Results and discussion
Fabrication of Con A-modified Fe3O4 nanoparticles
In this study, we attempted to use Con A to immobilize the glycoprotein template for subsequent surface imprinting. We first introduced Con A onto the Fe3O4 nanoparticles' surface according to the procedure illustrated in Fig. 1A.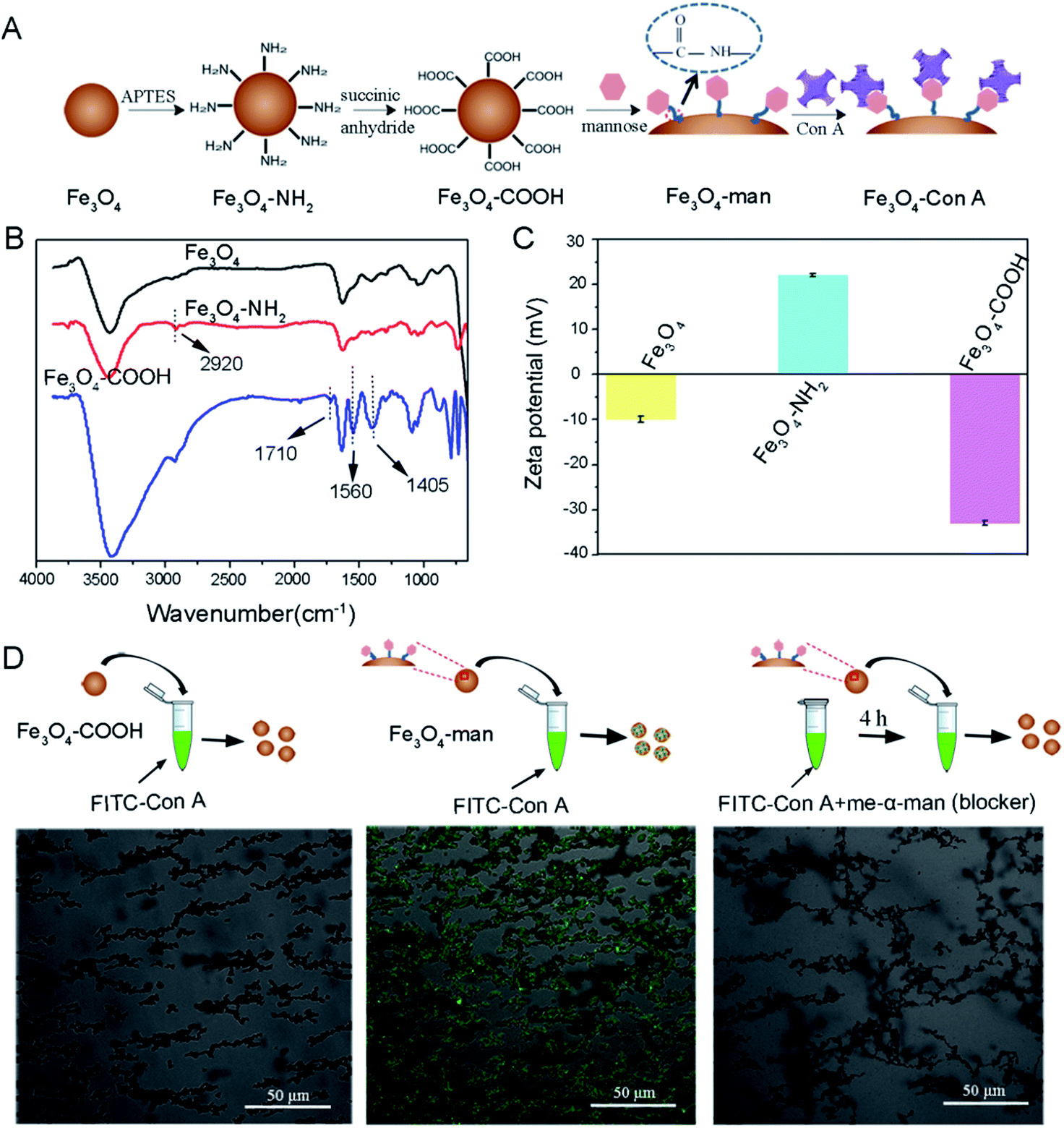 | ||
| Fig. 1 Fabrication and characterization of Con A-modified Fe3O4 nanoparticles. (A) Scheme for the fabrication process. (B) FT-IR spectra for Fe3O4, Fe3O4–NH2 and Fe3O4–COOH. (C) Zeta potential of Fe3O4, Fe3O4–NH2 and Fe3O4–COOH in water. (D) Confocal images of Fe3O4–COOH (left) and Fe3O4-man (middle) after exposure to FITC-Con A solution, and Fe3O4-man after exposure to FITC-Con A/Me-α-man solution (right). FITC-Con A was inhibited to bind onto the Fe3O4-man's surface by the biospecific blocking of free monosaccharide Me-α-man. | ||
Chemical evidence for the formation of –NH2 modified Fe3O4 (denoted as Fe3O4–NH2) and the further –COOH modified Fe3O4 (denoted as Fe3O4–COOH) was provided by FT-IR measurement. Fig. 1B shows the spectra of bare Fe3O4, Fe3O4–NH2 and Fe3O4–COOH nanoparticles, respectively. A new peak at about 2920 cm−1 appears for Fe3O4–NH2, compared to the bare Fe3O4, which may be assigned to the stretching vibration of the –C–H group that was incorporated during the surface aminolization process.38 The further derivatization of Fe3O4–NH2 with –COOH yielded two strong absorption peaks at 1560 and 1405 cm−1, which may correspond to the anti-symmetrical and symmetric vibration of the ester bond in the carboxylic acid group. A weak peak at 1710 cm−1 can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching on the carboxylic acid group.39
O stretching on the carboxylic acid group.39
Zeta potential analysis by dynamic light scattering (DLS) was further performed to provide complementary evidence for the formation of –NH2 modified Fe3O4 and the further –COOH modified Fe3O4. The result in Fig. 1C shows that the pristine Fe3O4 has a negatively charged surface with a zeta potential of −10.3 mV, which is because of the presence of coordinated water molecules. After the treatment with APTES, the zeta potential of the particles was increased to 22.4 mV. We assume that the appearance of amine groups on the Fe3O4 surface triggered the surface charge conversion. When we further modified the Fe3O4–NH2 nanoparticles with succinic anhydride, the zeta potential of the generated particles was observed to be converted again and became more negative, which indicates the successful introduction of –COOH onto the Fe3O4 surface, resulting in a negatively charged surface.
The introduction of Con A onto Fe3O4 nanoparticles was bridged by small mannose molecules. Epichitosamine was firstly modified onto Fe3O4–COOH via a classic amidation reaction and the resultant mannose containing surface facilitated Con A binding through sugar–lectin recognition. Visual evidence for the presence of Con A on Fe3O4 was provided by laser scanning confocal microscopy observation at λex = 488 nm in Fig. 1D. The left most image shows that –COOH terminated Fe3O4 nanoparticles after exposure to FITC labeled Con A solution have no fluorescence. As a contrast, mannose terminated Fe3O4 particles in the middle image were observed to give strong fluorescence after incubation with FITC-Con A. If mannose terminated Fe3O4 is added into a solution containing both FITC-Con A and Me-α-man, no fluorescence is observed in the right image. The ability of Me-α-man to suppress Con A–sugar interaction has been fully explained in our previous study;28 therefore these images demonstrate that Con A was bound to Fe3O4 particles via sugar–lection biorecognition and this binding could be interrupted by monosaccharide completion.
Generation of polydopamine imprinting film under neutral conditions
In order to use the synthetic Fe3O4@TRF-MIPs under physiological conditions, a neutral pH condition (i.e. pH 7.4) is preferred to initiate the protein imprinting process. Generally, self-polymerization of dopamine is facilitated at weak alkaline pH or using an oxidizing agent at neutral pH.40 We developed a simple method to grow polydopamine imprinting film at neutral pH by continuously bubbling O2 into the dopamine dissolved solution.Fig. 2A gives the visual evidence of dopamine polymerization at pH 7.4. The yellow-brown color generated from the dopamine solution indicates the formation of dopamine polymer. Compared to the air saturated solution, saturating the solution with O2 resulted in a much darker solution at the same time point, indicating the improvement of dopamine polymerization. With time elongation, more and more polymers were generated and the solution was observed to change to dark brown. We further performed UV-Vis measurement to monitor the formation of polydopamine quantitatively. Fig. 2B shows the absorbance of the dopamine solution (1 mg mL−1) at 420 nm as a function of time. It was found that continuously donating O2 significantly accelerated the precipitation of polydopamine at neutral pH.
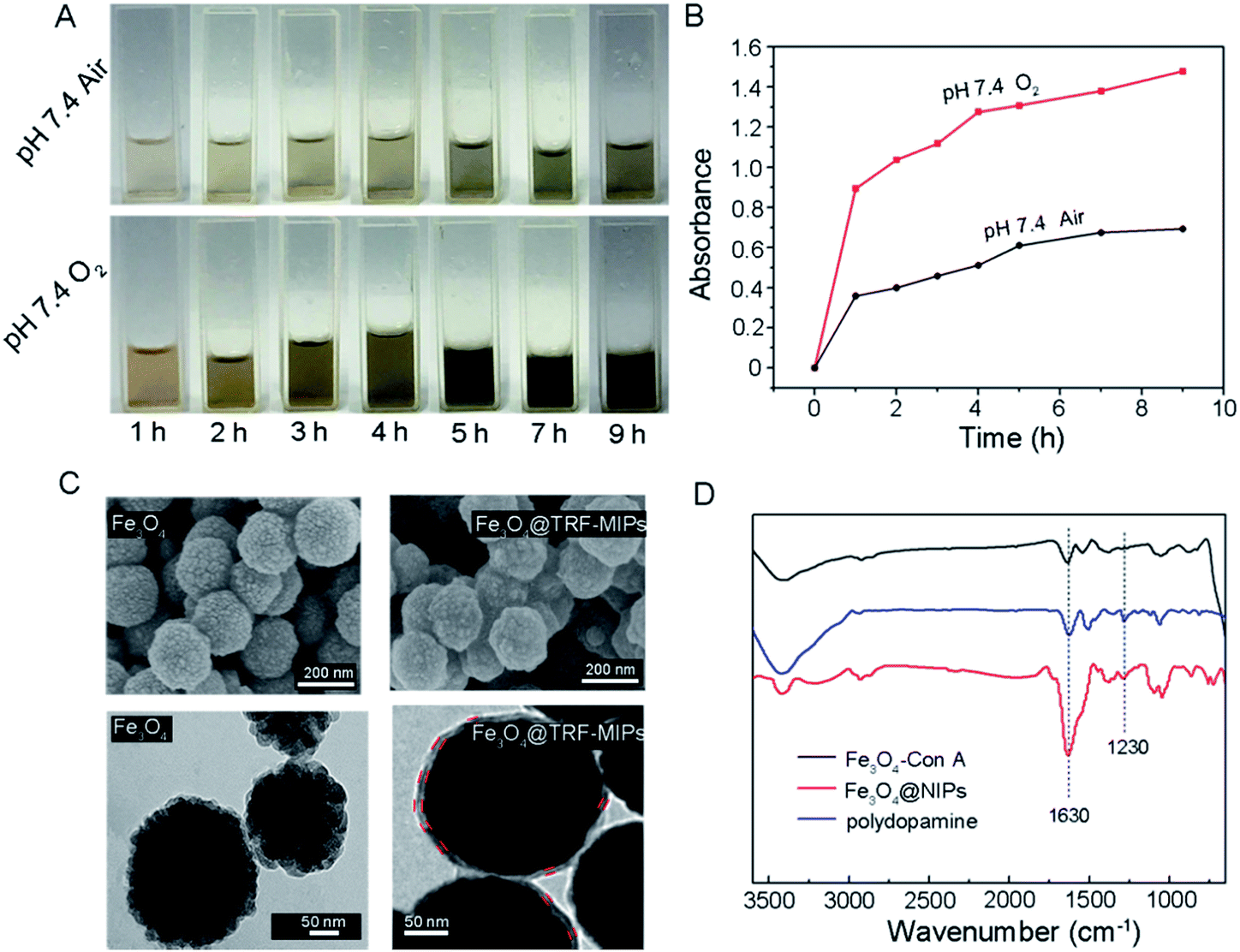 | ||
| Fig. 2 (A) Visual observations of dopamine polymerization in air or O2 environment at different time points. (B) UV-Vis absorption of the dopamine solution (1 mg mL−1) at 420 nm as a function of time in air or O2 environment. (C) SEM and TEM images of Fe3O4 and Fe3O4 with polydopamine film. The thickness of the polydopamine film was estimated by measuring the distance between paired parallel red lines. (D) FT-IR spectra for Fe3O4, polydopamine and Fe3O4@NIPs. | ||
We next prepared the TRF imprinted Fe3O4 nanoparticles and non-imprinted nanoparticles by inducing dopamine in situ self-polymerization at neutral pH. Experimentally, Con A modified Fe3O4 particles were pre-incubated with transferrin in PBS solution to allow the immobilization of templates, and then the nanoparticles were collected and exposed to a 1 mg mL−1 dopamine solution saturated with O2 for an allotted time. After the imprinted polydopamine film formation and protein template removal, core–shell imprinted nanoparticles were obtained [denoted as Fe3O4@TRF-MIPs]. The non-imprinted nanoparticles were synthesized following the same procedure, except for the addition of templates [denoted as Fe3O4@NIPs].
The surface morphology of bare Fe3O4 and Fe3O4 coated with polydopamine were observed by SEM and TEM, respectively (Fig. 2C). The bare Fe3O4 nanoparticles are roughly spherical and the average diameter of Fe3O4 is about 200 nm. TEM images show that the bare Fe3O4 nanoparticles are composed of tiny nanocrystals with sizes less than 10 nm. After 9 h of polymerization, the deposited polydopamine was observed on the surface of Fe3O4@TRF-MIPs. The polydopamine thickness was quantified by measuring at least 10 different particles to obtain an average of 8.5 ± 0.8 nm. Compared to the TRF protein size of 4.5 × 5.8 × 13.6 nm, the generated polydopamine covers 63% of template size in its longest dimension (i.e., 13.6 nm).18
FT-IR spectra provide direct evidence for the formation of polydopamine. The FT-IR spectra of Fe3O4–Con A and Fe3O4@NIPs are shown in Fig. 2D. Compared to Fe3O4–Con A, Fe3O4@NIPs present new absorption bands at 1230 cm−1, attributed to the stretching vibration of the phenolic hydroxyl. Moreover, the stronger peak at around 1630 cm−1 belongs to the stretching mode of the benzene ring.41 The characteristic absorption peaks are fitted with the spectra of polydopamine. These results confirm that dopamine was deposited onto the surface of the nanoparticles, and agree well with the observations by SEM and TEM.
Extraction of bioactive TRF by boronate buffered solutions
Here, we extracted the template from the nanoparticles by boronate buffered solutions (i.e. BBS), based on the competitive boronate–carbohydrate interaction.42 To evaluate the bioactivity of the eluted protein, we performed CD spectral measurements for the eluted TRF. Two other samples, of which the transferrin proteins were individually dissolved in PBS and SDS-HAc solution, served as controls. The far-UV region of the CD spectra between 250 and 190 nm, corresponding to the absorption of the peptide bond, are widely employed to analyze the secondary structures of proteins, i.e., α-helix, β-sheet, and random coil. The results are shown in Fig. 3A. The CD spectra of TRF in elution buffer (i.e. BBS) show the same pattern as that of a PBS solution of TRF. However, the SDS-HAc solution of TRF shows a quite different CD spectrum. The ratio of the CD intensity at 208 and 220 nm can give the quantitative evaluation for conformation changes of the protein.43 In our study, the values of Int208/220 of TRF protein in PBS, elution BBS and SDS-HAc solutions were 1.09, 1.08 and 1.37, respectively. The value of TRF in SDS/HAc is significantly higher than that in the former two solutions. These results indicate that the secondary structure of TRF in BBS is the same as that in PBS buffer, which implies that TRF bioactive structure can remain after BBS solution elution. However, the use of conventional SDS-HAc solution can denature the TRF protein.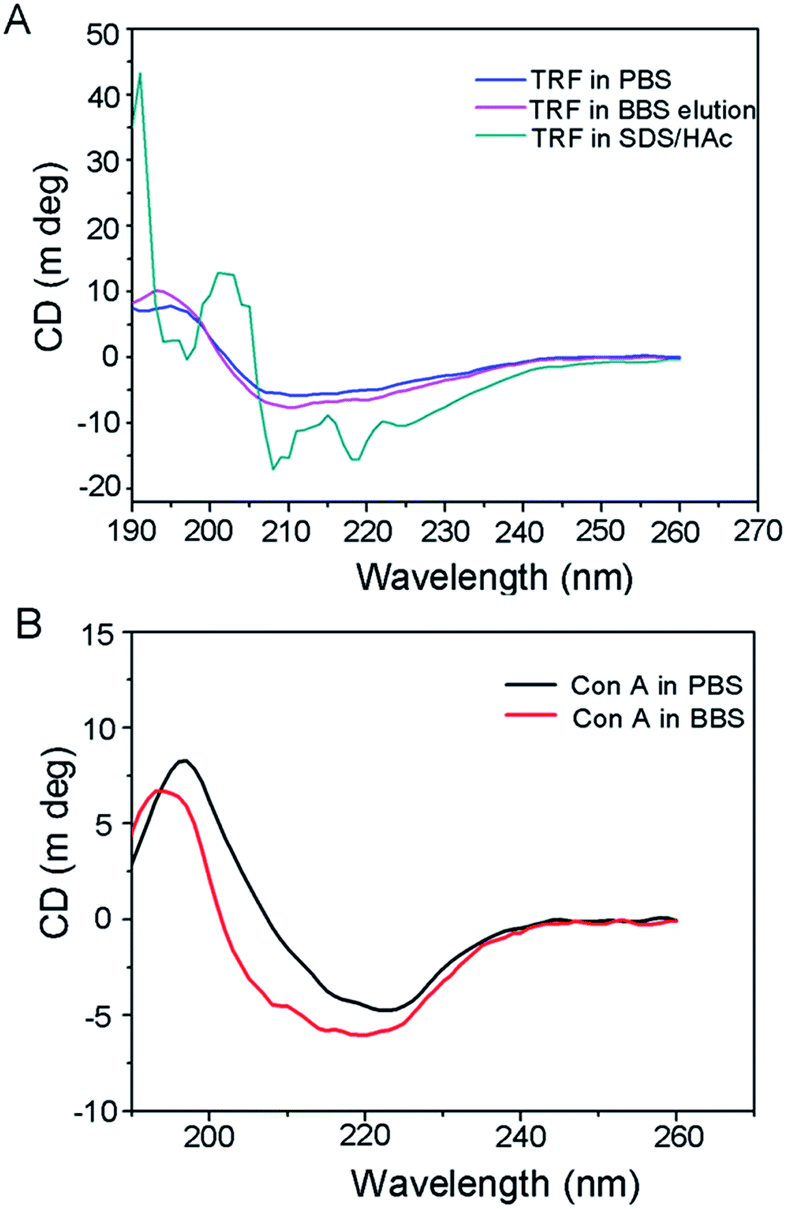 | ||
| Fig. 3 (A) CD spectra of TRF in PBS, SDS/HAc and the eluted TRF in BBS. (B) CD spectra of Con A dissolved in PBS and BBS. | ||
It has been reported that during the fabrication of protein imprints, SDS-HAc solution is a quite effective approach to removing the protein template to give the imprinted cavities.44,45 However, our study indicates that using a harsh SDS-HAc can damage the protein, and therefore does not facilitate the further application of the extracted targets, especially for some bio-assaying that requires the bioactive structure of the proteins. Alternatively, boronate buffer was proved to mildly extract glycoprotein and can thus replace the function of traditional SDS-HAc washing, as well as keep the bioactivity of glycoprotein targets.
For understanding the influence of BBS washing on the bio-function of immobilized Con A on Fe3O4, we also performed CD measurement of Con A that dissolves in BBS solution. The Con A PBS solution was used as a negative control. The results shown in Fig. 3B reveal that the CD curves of Con A in BBS have the same pattern as that in PBS solution, indicating that Con A protein can retain its bioactivity in BBS media. We can therefore infer that the BBS elution process would have no influence on its biofunction.
Adsorption isotherm
We performed the equilibrium binding experiment to study the adsorption behavior of TRF-imprinted and non-imprinted nanoparticles.Fig. 4 shows the adsorption isotherms measured at pH 7.4. The equilibrium adsorption capacities Qe for imprinted particles were observed to increase sharply at first, and then gradually reached saturation. The corresponding non-imprinted particles have lower binding capacities, compared to the imprinted particles, which suggests the successful creation of imprinted cavities for the TRF template.
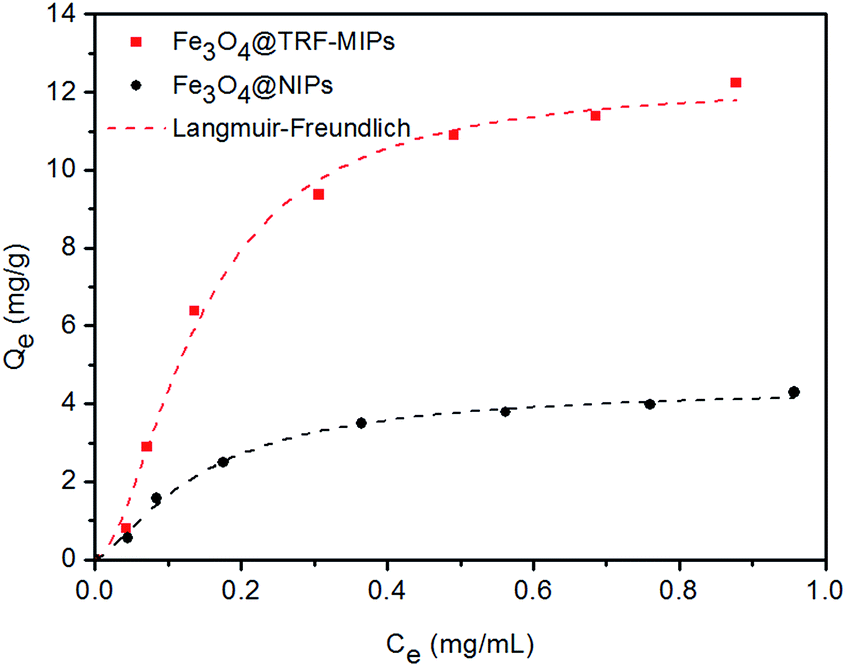 | ||
| Fig. 4 Adsorption isotherm of Fe3O4@TRF-MIPs and Fe3O4@NIPs. The experimental data were fitted to the Langmuir–Freudlich (LF) isotherm. | ||
In order to quantitatively describe the binding performance of imprinted particles, the Langmuir–Freundlich isotherm adsorption equation was applied to fit the curves, shown in Fig. 4. The Langmuir–Freundlich isotherm is expressed as the following equation:
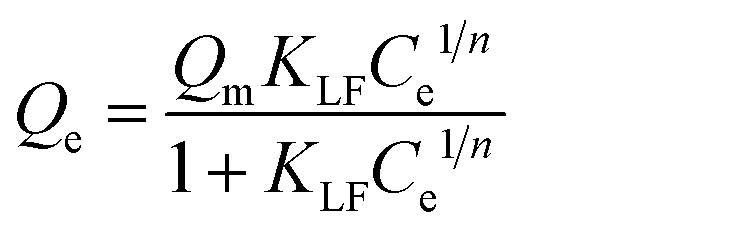 | (3) |
| Sample | Qm (mL mg−1) | KLF (mL mg−1) | n | R2 | IF |
|---|---|---|---|---|---|
| Fe3O4@TRF-MIPs | 12.28 | 30.38 | 1.75 | 0.993 | 2.76 |
| Fe3O4@NIPs | 4.44 | 15.05 | 1.40 | 0.994 |
The LF isotherm was observed to fit the binding behavior of the nanoparticles quite well and gives high correlation coefficients (R2). Fe3O4@TRF-MIPs have a higher association constant KLF of 30.38 mL mg−1, as well as a higher binding capacity Qm of 12.28 mg g−1, when compared to the non-imprinted samples, indicating the presence of imprinted cavities that improve the binding performance. The calculated imprinting factor IF of 2.76 was achieved.
Effect of pH on TRF adsorption
The effect of pH on the rebinding of TRF was studied for Fe3O4@TRF-MIPs and its control of non-imprinted particles. In this experiment, a certain amount of particles was incubated with 1 mg mL−1 of TRF proteins at pH 6.5, 6.8, 7.4, 8.0 and 8.5, respectively. Their ability to rebind the template protein was assayed for each buffer. Buffers with pH lower than 6.5 were not estimated in this study because of the concern of tetramer Con A dissociation.46Fig. 5 shows the equilibrium adsorption capacity Qe, and IF values for TRF rebinding. The pH environment has no significant influence on TRF binding to Fe3O4@NIPs particles. However, Fe3O4@TRF-MIPs demonstrated a pH dependent binding affinity towards TRF. The optimal TRF binding as well as maximum imprinting factor was observed at pH 7.4, instead of the pI value of TRF (5.6–6.6).
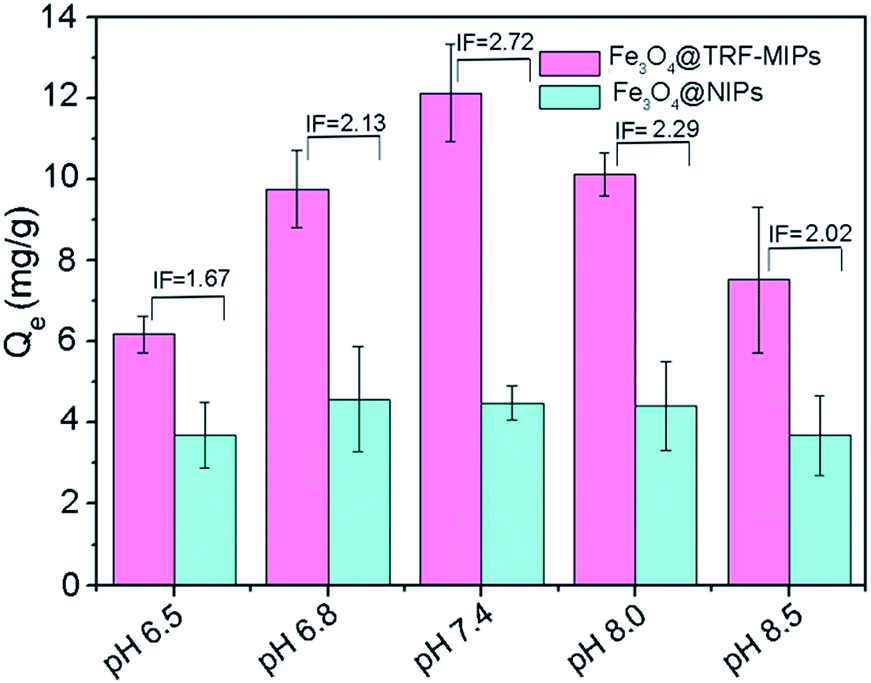 | ||
| Fig. 5 Binding capacities and IF values of Fe3O4@TRF-MIPs and Fe3O4@NIPs towards TRF at various pH values. | ||
This result is probably because the preparation of imprinted materials was performed at pH 7.4, at which the target protein has a specific conformation and charge distribution to guide adjacent functional monomers to aggregate and form a rigid polymer matrix. After the removal of templates, the generated imprinting cavities are believed to match the conformation and surface charges of TRF protein at pH 7.4 quite well. In other cases, when protein is dissolved in higher or lower pH solutions, the conformation and the surface charges of TRF might be changed and therefore cannot facilitate the specific binding towards TRF by imprinted particles. The following binding experiments were all carried out in PBS at pH 7.4.
Selective adsorption
We performed a single protein batch rebinding test to evaluate the rebinding selectivity of transferrin imprints. In this study, except for TRF (Mw = 77 kDa, pI = 5.9) three other proteins, including bovine serum albumin BSA (Mw = 66.4 kDa, pI = 4.9), hemoglobin HB (Mw = 64 kDa, pI = 6.9) and ovalbumin OVA (Mw = 44 kDa, pI = 4.7), were exposed to Fe3O4@TRF-MIPs or Fe3O4@NIPs individually and their binding capacities and IF values were quantified and listed in Fig. 6. The TRF-imprinted particles clearly showed the greatest affinity for TRF, followed in order by HB, BSA and OVA. The obviously preferential TRF adsorption over the glycoprotein of OVA demonstrates the presence of TRF matched imprinting cavities.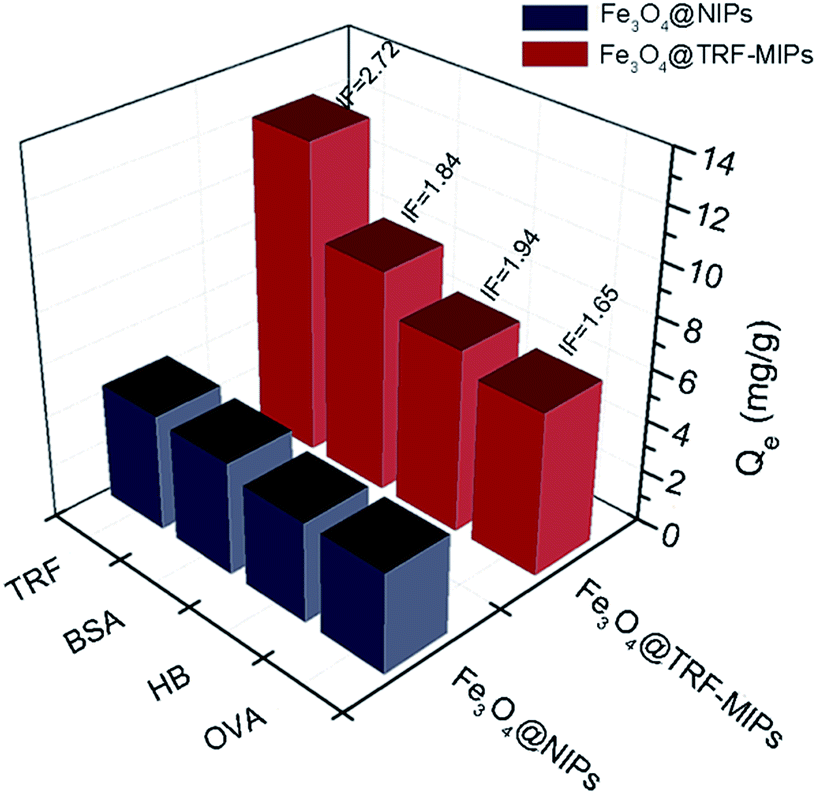 | ||
| Fig. 6 Binding capacities and IF values of Fe3O4@TRF-MIPs nanoparticles and Fe3O4@NIPs nanoparticles towards different proteins. | ||
SDS-PAGE
More robust evidence for template selective binding by Fe3O4@TRF-MIPs was provided by separating TRF from a spiked protein mixture. Non-glycosylated human serum albumin HSA (Mw = 66 kDa, pI = 4.9) and glycosylated RNase B (Mw = 14.7 kDa, pI = 8.9) that exist in the physiological fluid were used as the competitor models. The Fe3O4@TRF-MIPs was incubated with a mixture solution composed of equal amounts of TRF, HSA and RNase B (0.4 mg mL−1) for 12 h to obtain equilibrium. After that, the imprinted particles were separated and the proteins left in solution were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).As shown in Fig. 7, there are three distinctive bands in lane 2 for HSA, TRF and RNase B, respectively. After the protein mixture contacted Fe3O4@TRF-MIPs, the TRF band clearly faded and narrowed while the RNase B band remained unchanged. The band for HSA was observed to get slightly smaller, probably because of the similar molecular size, compared to TRF. It indicated the specific adsorption for the TRF template. When this trial switched to Fe3O4@NIPs, the three bands were almost unchanged (lane 3). The above results suggest that the imprinted particles contain imprinting cavities for specific TRF templates, and have the potential to selectively extract TRF from a complicated environment.
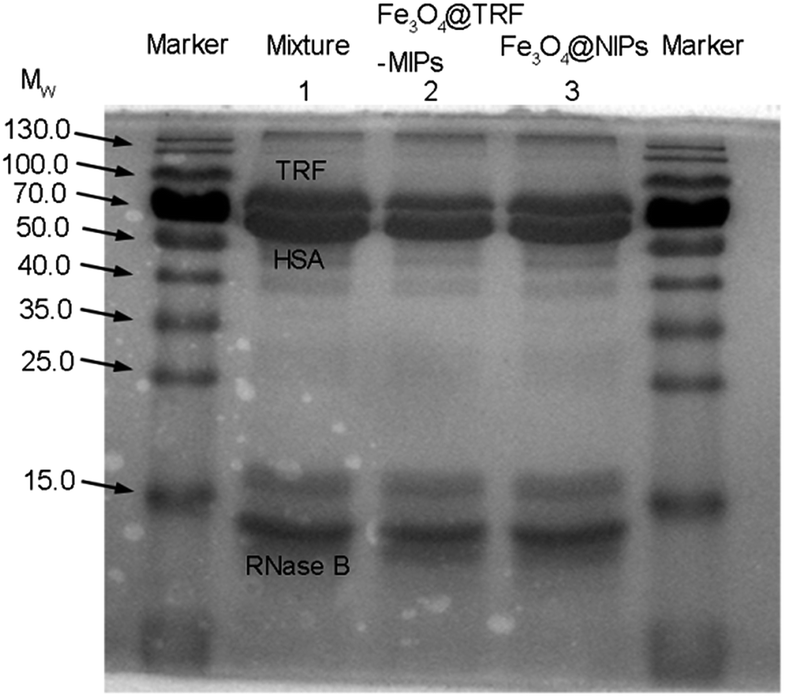 | ||
| Fig. 7 SDS-PAGE analysis of a tri-protein mixture after treatment with Fe3O4@TRF-MIPs and Fe3O4@NIPs. Lane 1: 0.4 mg mL−1 HRP, RNase B and HSA ternary solution; lane 2: remaining solution after the treatment by Fe3O4@TRF-MIPs; lane 3: remaining solution after the treatment by Fe3O4@NIPs. | ||
Table 2 gives the semi-quantitative description of protein specific extraction by TRF imprints. After the treatment with Fe3O4@TRF-MIPs, the free TRF, HSA and RNase B in the tertiary mixture were reduced by 32.54%, 25.62% and 18.4%, respectively, which indicates more TRF was extracted by TRF imprints. However, when the same trial was performed using non imprinted particles, the adsorption amount towards the three proteins was almost the same. Fe3O4@TRF-MIPs display obvious binding specificity towards TRF templates.
| Protein | Band relative intensity in lane 2 (after treatment with Fe3O4@TRF-MIPs) | Adsorbed protein by Fe3O4@TRF-MIPs | Band relative intensity in lane 3 (after treatment with Fe3O4@NIPs) | Adsorbed protein by Fe3O4@NIPs |
|---|---|---|---|---|
| TRF | 67.5% | 32.5% | 86.9% | 13.1% |
| HSA | 74.4% | 25.6% | 87.5% | 12.5% |
| RNase B | 81.6% | 18.4% | 86.3% | 13.7% |
Conclusions
We report a novel glycoprotein imprint by using Con A mediated template immobilization, combined with subsequent dopamine polymerization. The glycosylated biomarker, transferrin, was used as the model template. Boronate buffer was used for template extraction. CD spectra indicated that boronate buffer washing has no negative influence on the natural structure of transferrin and Con A. The imprinting performance, including binding capacities, binding constants, imprinting factors for the as-prepared imprint was estimated, and the rebinding selectivity towards transferrin in single or competitive conditions was also evaluated. The results indicate that the transferrin imprint has pH dependent binding affinity towards this protein. The optimal transferrin binding capacity, as well as the maximum imprinting factor, was observed at neutral pH. Imprinted and non-imprinted materials were both fitted with the Langmuir adsorption model. The obtained surface imprint has high selectivity towards the template glycoprotein. Our proposed novel glycoprotein surface imprint can work at physiological conditions, and the glycoprotein extraction method is mild enough to keep the bioactivity of these targets for further bio-application.Acknowledgements
We acknowledge the support from the National Basic Research Program of China (2012CB933600), the National Natural Science Foundation of China (51573047), Innovation Program of Shanghai Municipal Education Commission (14ZZ060), Shanghai Science and Technology Development Funds (14QA1401000) and the 111 project (B14018).Notes and references
- Y. Wang, Z. Chen, Y. Liu and J. Li, Nanoscale, 2013, 5, 7349–7355 RSC.
- W. Zhang, W. Liu, P. Li, H. Xiao, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2014, 53, 12489–12493 CAS.
- Y. Lu, Z. Bie, Y. Liu and Z. Liu, Analyst, 2013, 138, 290–298 RSC.
- Y. Ji, Z. Xiong, G. Huang and H. Zou, Analyst, 2014, 139, 4987–4993 RSC.
- S. Muniyan, N. K. Chaturvedi, J. G. Dwyer, C. A. Lagrange, W. G. Chaney and M. F. Lin, Int. J. Mol. Sci., 2013, 14, 10438–10464 CrossRef PubMed.
- N. Xia, D. Deng, L. Zhang, B. Yuan, M. Jing, J. Du and L. Liu, Biosens. Bioelectron., 2013, 43, 155–159 CrossRef CAS PubMed.
- A. Helenius and M. Aebi, Intracellular functions of N-linked glycans, Science, 2001, 291, 2364–2369 CrossRef CAS PubMed.
- A. H. Abuznait, C. Cain, D. Ingram, D. Burk and A. Kaddoumi, J. Pharm. Pharmacol., 2011, 63, 1111–1118 CrossRef CAS PubMed.
- L. P. Stepan, E. S. Trueblood, K. Hale, J. Babcook, L. Borges and C. L. Sutherland, J. Histochem. Cytochem., 2011, 59, 701–710 CrossRef CAS PubMed.
- Y. Li, M. Hong, Miaomiao, Q. Bin, Z. Lin, Z. Cai and G. Chen, J. Mater. Chem. B, 2013, 1, 1044–1057 RSC.
- T. Shiomi, M. Matsui, F. Mizukami and K. Sakaguchi, Biomaterials, 2005, 26, 5564–5571 CrossRef CAS PubMed.
- G. Fu, H. He, Z. Chai, H. Chen, J. Kong, Y. Wang and Y. Jiang, Anal. Chem., 2011, 83, 1431–1436 CrossRef CAS PubMed.
- L. Liu, J. Zheng, G. Fang and W. Xie, Anal. Chim. Acta, 2012, 726, 85–92 CrossRef CAS PubMed.
- S. Li, K. Yang, J. Liu, B. Jiang, L. Zhang and Y. Zhang, Anal. Chem., 2015, 87, 4617–4620 CrossRef CAS PubMed.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- Y. Li, M. Hong, Q. Bin, Z. Lin, Z. Cai and G. Chen, J. Mater. Chem. B, 2013, 1, 1044–1051 RSC.
- X. Bi and Z. Liu, Anal. Chem., 2014, 86, 12382–12389 CrossRef CAS PubMed.
- S. Wang, J. Ye, Z. Bie and Z. Liu, Chem. Sci., 2014, 5, 1135–1140 RSC.
- J. Ye, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 10386–10389 CrossRef CAS PubMed.
- Z. Bie, Y. Chen, J. Ye, S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2015, 54, 10211–10215 CrossRef CAS PubMed.
- X. Bi and Z. Liu, Anal. Chem., 2014, 86, 959–966 CrossRef CAS PubMed.
- J. Tang, Y. Liu, P. Yin, G. Yao, G. Yan, C. Deng and X. Zhang, Proteomics, 2010, 10, 2000–2014 CrossRef CAS PubMed.
- S. Feng, N. Yang, S. Pennathur and D. Lubman, Anal. Chem., 2009, 81, 3773–3783 CrossRef.
- B. F. Mann, A. K. Mann, S. E. Skrabalak and M. V. Novotny, Anal. Chem., 2013, 85, 1905–1912 CrossRef CAS PubMed.
- J. Schultz, S. Mansouri and I. Goldstein, Diabetes Care, 1982, 5, 245–253 CrossRef CAS PubMed.
- R. Ballerstadt, C. Evans, R. McNichols and A. Gowda, Biosens. Bioelectron., 2006, 22, 275–284 CrossRef CAS PubMed.
- R. Ballerstadt and J. S. Schultz, Anal. Chem., 2000, 72, 4185–4192 CrossRef CAS PubMed.
- J. Li, X. Qu, G. F. Payne, C. Zhang, Y. Zhang, J. Li, J. Ren, H. Hong and C. Liu, Adv. Funct. Mater., 2015, 25, 1404–1417 CrossRef CAS.
- C. Zhang, X. Qu, J. Li, H. Hong, J. Li, J. Ren, G. F. Payne and C. Liu, Adv. Healthcare Mater., 2015, 4, 1972–1981 CrossRef CAS PubMed.
- Y. Hao, R. Gao, D. Liu, G. He, Y. Tang and Z. Guo, Talanta, 2016, 153, 211–220 CrossRef CAS PubMed.
- X. Jia, M. Xu, Y. Wang, D. Ran, S. Yang and M. Zhang, Analyst, 2013, 138, 651–658 RSC.
- T. Zhou, K. Zhang, T. Kamra and L. Ye, J. Mater. Chem. B, 2015, 3, 1254–1260 RSC.
- Z. Lin, L. Sun, W. Liu, Z. Xia, H. Yang and G. Chen, J. Mater. Chem. B, 2014, 2, 637–643 RSC.
- S. Xuan, Y. X. J. Wang, J. C. Yu and K. Cham-Fai Leung, Chem. Mater., 2009, 21, 5079–5087 CrossRef CAS.
- S. Xuan, Y. X. Wang, J. C. Yu and K. C. Leung, Langmuir, 2009, 25, 11835–11843 CrossRef CAS PubMed.
- V. Mahalingam, S. Onclin, M. Péter, B. Ravoo and D. N. Reinhoudt, Langmuir, 2004, 20, 11756–11762 CrossRef CAS PubMed.
- G. Wu, J. Li, X. Qu, Y. Zhang, H. Hong and C. Liu, RSC Adv., 2015, 5, 47010–47021 RSC.
- X. Kan, Q. Zhao, D. Shao, Z. Geng and J. Zhu, J. Phys. Chem. B, 2010, 114, 3999–4004 CrossRef CAS PubMed.
- M. Zhang, X. He, L. Chen and Y. Zhang, J. Mater. Chem., 2010, 20, 10696–10704 RSC.
- X. Du, L. Li, J. Li, C. Yang, N. Frenkel, A. Welle, S. Heissler, A. Nefedov, M. Grunze and P. A. Levkin, Adv. Mater., 2014, 26, 8029–8033 CrossRef CAS PubMed.
- D. Liu, H. Wang, H. Jiang and D. Zhou, J. Appl. Polym. Sci., 2015, 133, 42734 Search PubMed.
- G. Gupta and C. R. Lowe, J. Mol. Recognit., 2004, 17, 218–235 CrossRef CAS PubMed.
- S. Li, Z. Peng and R. M. Leblanc, Anal. Chem., 2015, 87, 6455–6459 CrossRef CAS PubMed.
- D. M. Hawkins, D. Stevenson and S. M. Reddy, Anal. Chim. Acta, 2005, 542, 61–65 CrossRef CAS.
- J. Liu, Q. Deng, K. Yang, L. Zhang, Z. Liang and Y. Zhang, J. Sep. Sci., 2010, 33, 2757–2761 CrossRef CAS PubMed.
- D. Wah, A. Romero and J. Calvete, J. Mol. Biol., 2001, 310, 885–894 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |