
Transition-metal-free C–C bond cleavage and formation: efficient synthesis of 2,3-diarylimidazo[1,2-α]pyridines from 2-aminopyridines and alkynoates†
Zhengwang Chen*,
Yuelu Wen,
Guotian Luo,
Min Ye and
Qinghao Wang*
Key Laboratory of Organo-Pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University, Ganzhou, 341000, China. E-mail: chenzwang@126.com; wangqinghao2016@126.com; Fax: +86 797-8793670; Tel: +86 797-8793670
First published on 5th September 2016
Abstract
A highly efficient transition-metal-free cyclization reaction for the synthesis of 2,3-diarylimidazo[1,2-α]pyridines is described. A variety of substituted 2-aminopyridines and alkynoates are compatible under the standard conditions. This protocol is marked by the cleavage of a C–C bond and the formation of a new Csp2–Csp2 bond under transition-metal-free conditions.
The imidazo[1,2-α]pyridines (IPYs) are important nitrogen-containing heterocycles and have attracted intensive research interest from chemists due to their outstanding application value in the area of natural products and pharmaceuticals.1 Many substituted IPYs possess biological activities, such as antiviral, antifungal, anticancer, antibacterial, anti-inflammatory activities.2 Moreover, several compounds with an IPY moiety are commercially available drugs, including olprinone, alpidem, zolpidem, necopidem, zolimidine and saripidem.3 Consequently, much effort has been made for the preparation of IPYs in the past few decades.4 Among a variety of new synthetic methods, transition-metal-catalyzed reactions are vital tools for the synthesis of IPY derivatives.5 However, some transition-metal salts still have some drawbacks, such as high price, residual metallic impurities to the products, and generation of toxic waste. Therefore, the development of new strategies for constructing IPYs with expedient conditions and high efficiency under transition-metal-free conditions remains an extremely attractive task for organic chemists.6
The cleavage of unstrained C–C bond has attracted considerable attention and emerged as a extraordinary challenge over the past fewer decades. Recently, significant progress has been made for the construction of complicated molecules through C–C bond cleavage.7 Among these transformations, transition-metal salts are often used to activate the inert C–C bonds, and unattractive stoichiometric oxidants are always the necessary reagent. Hence, the discovery of new systems to selectively cleave C–C bond for organic synthesis under environmentally friendly manner is still appealing. 2,3-Diarylimidazo[1,2-α]pyridine derivatives also exhibit a broad range of biological activities.8 Owing to their remarkable activities in pharmaceutical and agrochemical area, several synthetic strategies have been developed for the preparation of 2,3-diarylimidazo[1,2-α]pyridines like condensation,9 multicomponent reactions10 and metal-catalyzed C–H functionalizations.11 In 2012, Liu's group reported an elegant example for the imidazopyridines through the copper(II)/iron(III) co-catalyzed intermolecular diamination of 2-aminopyridines with alkynes (Scheme 1a).12 Herein, we describe an expedient and efficient transition-metal-free intermolecular cyclization reaction to give 2,3-diarylimidazo[1,2-α]pyridines through C–C bond cleavage and formation manner (Scheme 1b). It is important to note that, there are rare examples to construct heterocycle molecules through C–C bond cleavage and the formation of Csp2–Csp2 bond under transition-metal-free conditions.
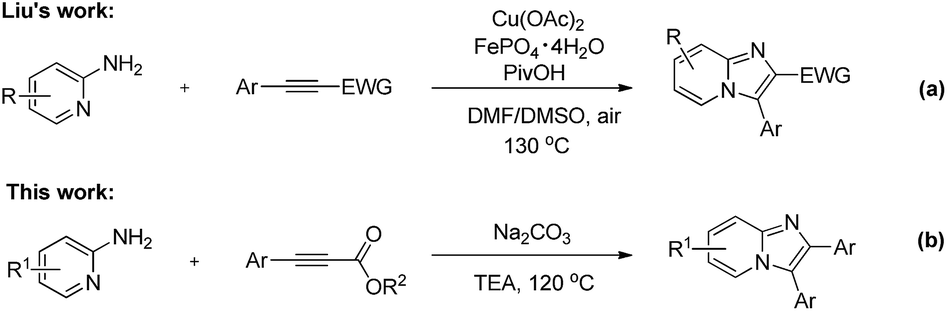 | ||
| Scheme 1 Intermolecular cyclization of 2-aminopyridines with alkynes. | ||
We commenced our study by screening the reaction between 2-aminopyridine 1a and ethyl phenylpropiolate 2a to obtain the optimal reaction conditions. As shown in Table 1, 2,3-diphenylimidazo[1,2-α]pyridine can be formed with a promising 33% yield in the presence of DBU in toluene at 120 °C for 20 h (Table 1, entry 1). The control experiment showed that 3a could not be obtained without addition of any base (Table 1, entry 2). Then a series of common solvents were tested, and we found that TEA could afford the corresponding product in 48% yield (Table 1, entries 3–8). In view of the lower conversion of the reaction, the amount of the 2-aminopyridine was investigated, an excess of 1a favored the transformation (Table 1, entries 9 and 10). We then examined the effect of base on this reaction, and showed that base played an important role in the outcome of the reaction. Among the organic and inorganic bases used, Na2CO3 was established as the base of choice for the reaction (Table 1, entries 11–17). Lower temperature disfavored the reaction (Table 1, entries 18 and 19). After some attempts, the optimal reaction conditions are as follows: 1a (0.3 mmol) and 2a (0.2 mmol) with Na2CO3 in TEA at 120 °C for 20 h.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Temperature | Yieldb (%) |
| a Reaction conditions: 2-aminopyridine (0.1 mmol), ethyl 3-phenylpropiolate (0.2 mmol), additive (0.5 equiv.) and solvent (1 mL) for 20 h.b Yields were determined by GC analysis with mesitylene as internal standard; isolated yields in brackets.c 2-Aminopyridine (0.2 mmol) was used.d 2-Aminopyridine (0.3 mmol) was used. TEA = triethylamine; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; DABCO = 1,4-diazabicyclo[2.2.2]octane; DMAP = N,N-4-dimethylaminopyridine; n.p. = no product. | ||||
| 1 | Toluene | DBU | 120 | 33 |
| 2 | Toluene | — | 120 | n.p. |
| 3 | Xylene | DBU | 120 | 27 |
| 4 | DMF | DBU | 120 | Trace |
| 5 | H2O | DBU | 120 | n.p. |
| 6 | DMSO | DBU | 120 | Trace |
| 7 | TEA | DBU | 120 | 48 |
| 8 | NMP | DBU | 120 | 15 |
| 9c | TEA | DBU | 120 | 57 |
| 10d | TEA | DBU | 120 | 68 |
| 11d | TEA | — | 120 | 55 |
| 12d | TEA | DMAP | 120 | n.p. |
| 13d | TEA | Pyridine | 120 | 75 |
| 14d | TEA | DABCO | 120 | 85 |
| 15d | TEA | K3PO4 | 120 | n.p. |
| 16d | TEA | Na2CO3 | 120 | 88 (85) |
| 17d | TEA | Cs2CO3 | 120 | n.p. |
| 18d | TEA | Na2CO3 | 110 | 70 |
| 19d | TEA | Na2CO3 | 130 | 87 |
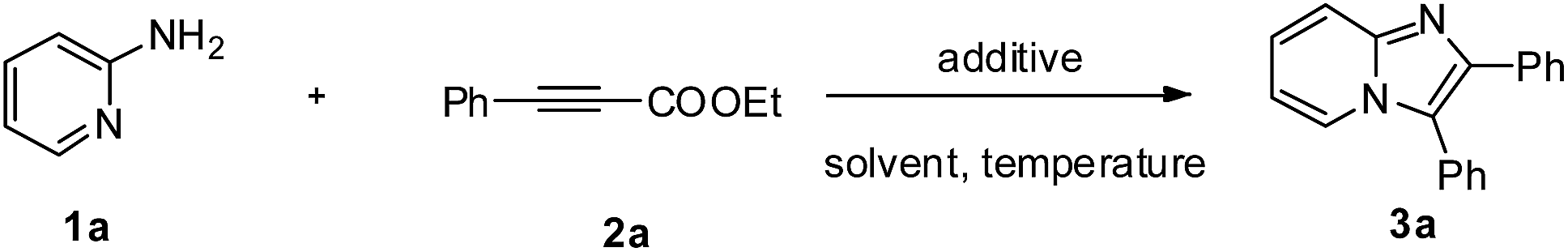
With the optimized reaction conditions established, we next concentrated on the generality of the cyclization reaction with regard to both reaction partners (Scheme 2). In general, both electron-rich and electron-deficient substitutions on the aromatic ring were effective in the standard conditions (Scheme 2, 3b–t). First, the effect of the groups on the 2-aminopyridine moiety was examined. The methyl group at the 3-, 4-, and 5-positions on the pyridine ring led to the corresponding products in good yields (Scheme 2, 3b–d). The substrate with strong electron-rich substitution methoxy group only afforded the product in low yield (Scheme 2, 3e). We were pleased to notice that carbon-halogen groups were well tolerated in the standard conditions. Especially, the aryl bromide and chloride could be further functionalized via cross-coupling reactions (Scheme 2, 3f–h). It is strange that the substrates containing Cl and Br substituents only afforded the products in moderate yields even in a prolonged reaction time (Scheme 2, 3g–h). Subsequently, the scope of the ethyl alkynoates by reacting with 2-aminopyridine were evaluated. Gratifyingly, the reaction proceeded effectively at a slightly elevated temperature. Substrates with either electron-donating groups or electron-withdrawing groups attached to the benzene ring could furnish the products in good yields (Scheme 2, 3i–q). The reaction conditions were compatible with alkyl, aryl, fluoro, chloro, bromo, cyano, and ester groups (Scheme 2, 3i–p). The challenging substrate, thienyl-containing substrate gave the desired product in 83% yield (Scheme 2, 3q). Finally, the reaction between substituted aminopyridines and ethyl alkynoates were tried, they also provided the corresponding products in satisfactory yields (Scheme 2, 3r–t). It is noteworthy that methyl alkynoate also gave the corresponding product in similar yield (Scheme 2, 3b). The structure of 3a was further confirmed by X-ray crystal diffraction measurements (Fig. 1).13
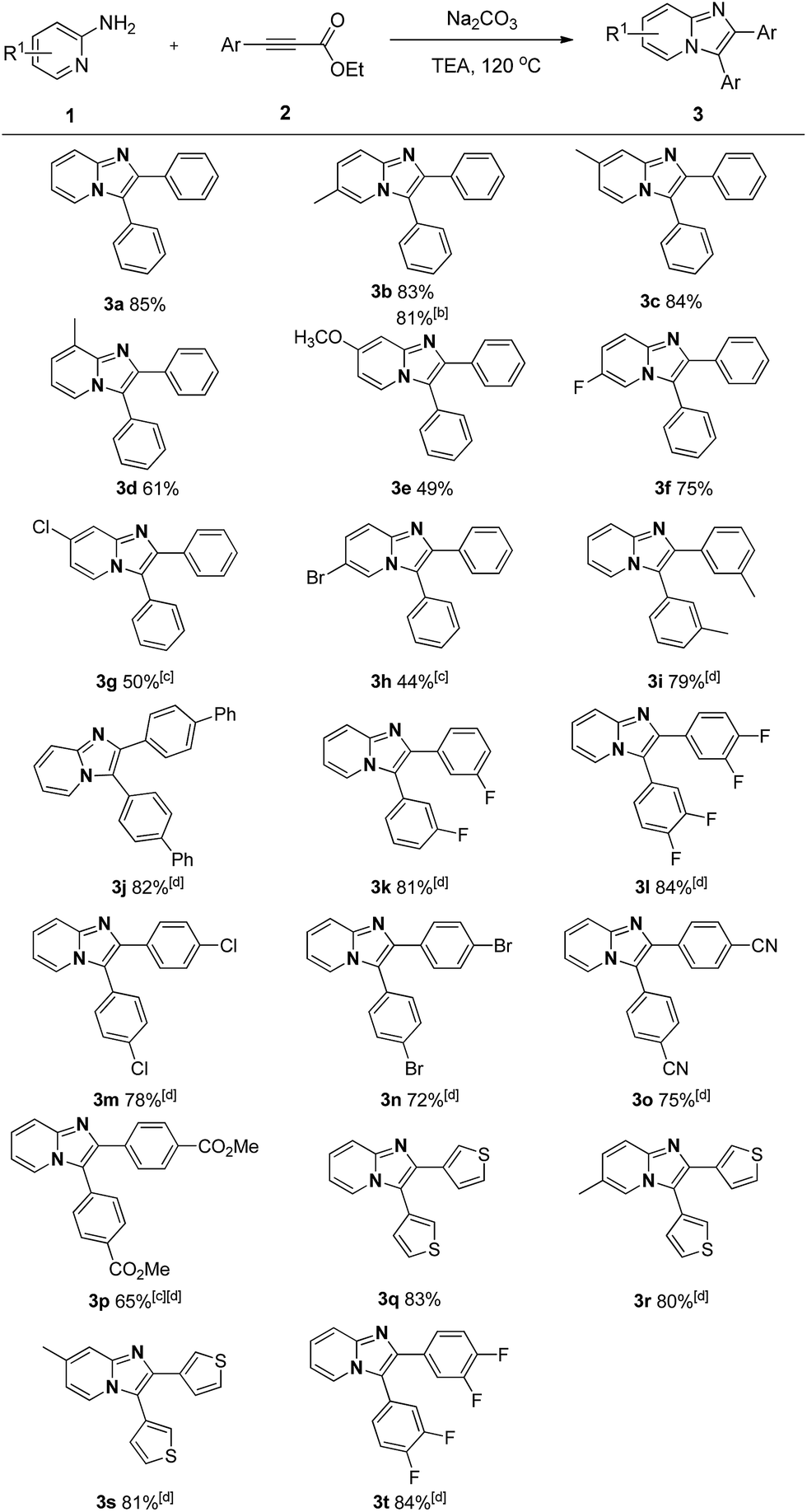 | ||
| Scheme 2 Intermolecular cyclization of 2-aminopyridines with alkynes. a Reaction conditions: 1 (0.3 mmol), 2 (0.2 mmol), Na2CO3 (0.5 equiv.) and TEA (1 mL) at 120 °C for 20 h; yields of isolated products are given. b Methyl 3-phenylpropiolate was used as substrate. c Reacted for 40 h. d Reacted at 140 °C. | ||
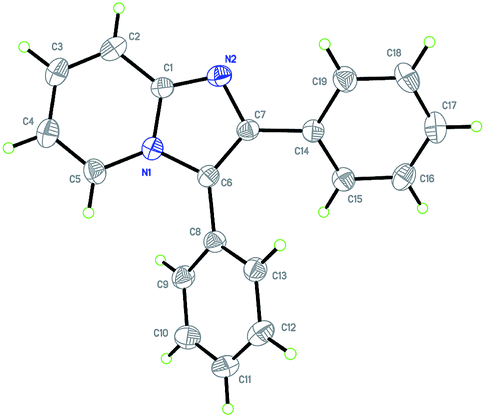 | ||
| Fig. 1 X-ray structure of 3a. | ||
Control experiments were conducted to better understand the transformation (see Scheme 1 in the ESI†). 2-Aminopyridine was used to react with A, B and C under standard conditions, respectively. However, these reactions did not give any 3a product. The results indicate that A, B and C are not the intermediates in the transformation. Addition of a free radical scavenger ethene-1,1-diyldibenzene or 2,6-di-tert-butyl-4-methylphenol (BHT), and no obvious inhibition was observed. These results suggested that this reaction did not proceed through a radical process.
Although detailed experimental evidence is still pending, a tentative mechanism is proposed in Scheme 3. First, 1a was transformed into intermediate E through intermolecular Michael addition. Then, intermediate F was generated by intramolecular [2 + 2] cycloaddition reaction. Finally, the product 3a was formed via ring-opening reaction with the cleavage of the C–C bonds, and the diethyl but-2-enedioate byproduct was afforded at the same time. Luckily, the diethyl but-2-enedioate byproduct can be detected in GC-MS spectrometer.
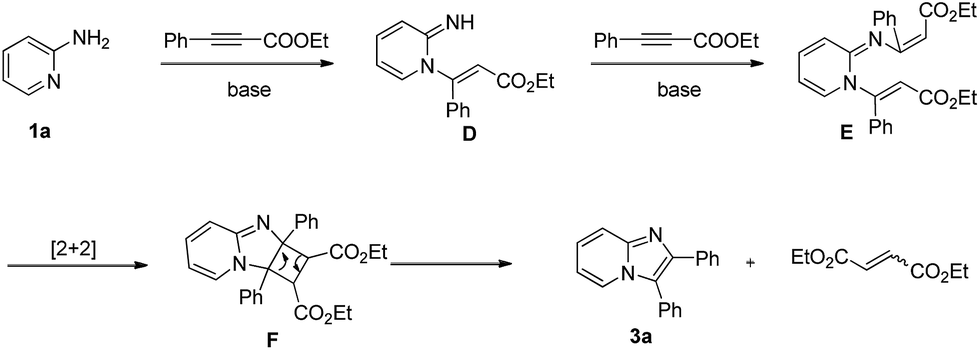 | ||
| Scheme 3 Possible reaction mechanism. | ||
In summary, we have developed a convenient and expedient transition-metal-free protocol for the formation of 2,3-diarylimidazo[1,2-α]pyridines. Both electron-rich and electron-deficient substituents on the aromatic rings are compatible in the standard conditions. The new approach serves not only as an efficient method to 2,3-diarylimidazo[1,2-α]pyridines but also as a rare example of a C–C bond cleavage and formation under simple transition-metal-free conditions. The scope, applications and further mechanistic research of this methodology are currently underway in our laboratory.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21202023 and 21462002), the Innovation Fund of Jiangxi Province (YC2014-S412) and the NSF of Jiangxi Provincial Education Department (GJJ151005).Notes and references
- (a) F. Couty and G. Evano, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 11, p. 409 Search PubMed; (b) C. Enguehard-Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888 CrossRef CAS PubMed.
- (a) K. S. Gudmundsson and B. A. Johns, Org. Lett., 2003, 5, 1369 CrossRef CAS PubMed; (b) M. A. Ismail, R. Brun, T. Wenzler, F. A. Tanious, W. D. Wilson and D. W. Boykin, J. Med. Chem., 2004, 47, 3658 CrossRef CAS PubMed; (c) K. S. Gudmundsson, J. D. Williams, J. C. Drach and L. B. Townsend, J. Med. Chem., 2003, 46, 1449 CrossRef CAS PubMed; (d) S. Ulloora, R. Shabaraya and A. V. Adhikari, Bioorg. Med. Chem. Lett., 2013, 23, 3368 CrossRef CAS PubMed.
- (a) S. Z. Langer, S. Arbilla, J. Benavides and B. Scatton, Adv. Biochem. Psychopharmacol., 1990, 46, 61 CAS; (b) K. Mizushige, T. Ueda, K. Yukiiri and H. Suzuki, Cardiovasc. Drug Rev., 2002, 20, 163 CrossRef CAS PubMed; (c) L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305 CrossRef CAS PubMed; (d) R. J. Boerner and H. J. Moller, Psychopharmakother, 1997, 4, 145 Search PubMed.
- For selective reviews, see: (a) A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555 RSC; (b) K. Pericherla, P. Kaswan, K. Pandey and A. Kumar, Synthesis, 2015, 47, 887 CrossRef CAS; (c) M. J. D. Pires, D. L. Poeira and M. M. B. Marques, Eur. J. Org. Chem., 2015, 7197 CrossRef.
- For selected examples, see: (a) T. B. Nguyen, M. Corbin, P. Retailleau, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2015, 17, 4956 CrossRef CAS PubMed; (b) H. Cao, X. Liu, J. Liao, J. Huang, H. Qiu, Q. Chen and Y. Chen, J. Org. Chem., 2014, 79, 11209 CrossRef CAS PubMed; (c) P. Kaswan, A. Porter, K. Pericherla, M. Simone, S. Peters, A. Kumar and B. Deboef, Org. Lett., 2015, 17, 5208 CrossRef CAS PubMed; (d) C. Wang, S. Lei, H. Cao, S. J. Liu, H. Deng and C. Yan, J. Org. Chem., 2015, 80, 12725 CrossRef CAS PubMed; (e) I. M. McDonald and K. M. Peese, Org. Lett., 2015, 17, 6002 CrossRef CAS PubMed; (f) H. Cao, S. Lei, N. Li, L. Chen, J. Liu, H. Cai, S. Qiu and J. Tan, Chem. Commun., 2015, 51, 1823 RSC; (g) I. V. Rassokhina, V. Z. Shirinian, I. V. Zavarzin, V. Gevorgyan and Y. A. Volkova, J. Org. Chem., 2015, 80, 11212 CrossRef CAS PubMed; (h) S. Lei, G. Chen, Y. Mai, L. Chen, H. Cai, J. Tan and H. Cao, Adv. Synth. Catal., 2016, 358, 67 CrossRef CAS; (i) Y. Gao, M. Yin, W. Wu, H. Huang and H. Jiang, Adv. Synth. Catal., 2013, 355, 2263 CrossRef CAS; (j) A. K. Bagdi, M. Rahman, S. Santra, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1741 CrossRef CAS; (k) S. Santra, A. K. Bagdi, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1065 CrossRef CAS.
- For selective review and examples, see: (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed; (b) C. Huo, J. Tang, H. Xie, Y. Wang and J. Dong, Org. Lett., 2016, 18, 1016 CrossRef CAS PubMed; (c) S. K. Lee and J. K. Park, J. Org. Chem., 2015, 80, 3723 CrossRef CAS PubMed; (d) X. Xiao, Y. Xie, S. Bai, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2015, 17, 3998 CrossRef CAS PubMed; (e) H. Cao, X. Liu, L. Zhao, J. Cen, J. Lin, Q. Zhu and M. Fu, Org. Lett., 2014, 16, 146 CrossRef CAS PubMed; (f) P. R. Adiyala, G. S. Mani, J. B. Nanubolu, K. C. Shekar and R. A. Maurya, Org. Lett., 2015, 17, 4308 CrossRef CAS PubMed; (g) Y. Xie, J. Wu, X. Che, Y. Chen, H. Huang and G.-J. Deng, Green Chem., 2016, 18, 667 RSC.
- For selective reviews, see: (a) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed; (b) H. Liu, M. Feng and X. Jiang, Chem.–Asian J., 2014, 9, 3360 CrossRef CAS PubMed; (c) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567 RSC; (d) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC; (e) T. Seiser and N. Cramer, Org. Biomol. Chem., 2009, 7, 2835 RSC; (f) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed.
- (a) A. Scribner, R. Dennis, S. Lee, G. Ouvry, D. Perrey, M. Fisher, M. Wyvratt, P. Leavitt, P. Liberator, A. Gurnett, C. Brown, J. Mathew, D. Thompson, D. Schmatz and T. Biftu, Eur. J. Med. Chem., 2008, 43, 1123 CrossRef CAS PubMed; (b) K. S. Gudmundsson and B. A. Johns, Bioorg. Med. Chem. Lett., 2007, 17, 2735 CrossRef CAS PubMed; (c) C. Enguehard-Gueiffier, F. Fauvelle, J.-C. Debouzy, A. Peinnequin, I. Thery, V. Dabouis and A. Gueiffier, Eur. J. Pharm. Sci., 2005, 24, 219 CrossRef CAS PubMed.
- (a) M.-A. Hiebel, Y. Fall, M.-C. Scherrmann and S. Berteina-Raboin, Eur. J. Org. Chem., 2014, 4643 CrossRef CAS; (b) H. S. Patel, J. A. Linn, D. H. Drewry, D. A. Hillesheim, W. J. Zuercher and W. J. Hoekstra, Tetrahedron Lett., 2003, 44, 4077 CrossRef CAS.
- Y. Wang, B. Frett and H. Li, Org. Lett., 2014, 16, 3016 CrossRef CAS PubMed.
- (a) H. Cao, H. Zhan, Y. Lin, X. Lin, Z. Du and H. Jiang, Org. Lett., 2012, 14, 1688 CrossRef CAS PubMed; (b) H. Huang, X. Ji, X. Tang, M. Zhang, X. Li and H. Jiang, Org. Lett., 2013, 15, 6254 CrossRef CAS PubMed; (c) S. Marhadour, M.-A. Bazin and P. Marchand, Tetrahedron Lett., 2012, 53, 297 CrossRef CAS.
- J. Zeng, Y. J. Tan, M. L. Leow and X.-W. Liu, Org. Lett., 2012, 14, 4386 CrossRef CAS PubMed.
- CCDC 1441068 contains the supplementary crystallographic data for this paper (compound 3a).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, copies of 1H and 13C NMR spectra for selected compounds. CCDC 1441068. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra19291b |
| This journal is © The Royal Society of Chemistry 2016 |