
Comparative structure activity relationship for heterogeneous phosphatase-like catalytic activities of one-dimensional Cu(II) coordination polymers†
Navid Hussaina,
Pooja Joshib,
Shah Raj Alib and
Vimal K. Bhardwaj*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab 140001, India. E-mail: vimalb@iitrpr.ac.in; Tel: +91-1881242271
bDepartment of Chemistry, D.S.B. Campus, Kumaun University, Nainital, Uttarakhand 263002, India
First published on 20th June 2016
Abstract
A group of one-dimensional Cu(II) coordination polymers {[Cu3(L1)(NO3)2(DMF)(H2O)]·3(DMF)}n (1), [Cu3(L1)(Cl)2(DMF)2]n (2), [Cu3(L2)(NO3)4(H2O)4]n (3) with different coordination environments, was chosen to investigate their potential in heterogeneous catalytic studies for the cleavage of phosphate ester bond. In complexes 1, 2 and 3 the trinuclear copper units are connected by μ2-O−, μ2-Cl and μ-[O–N–O] bridging modes that leads to the formation of sheet like or stacking layer arrangement of coordination polymers with different metal–metal distance. Moreover these complexes have different donor sites of ligands and exogenous ligands in their coordination environment. Therefore the role of different coordination environments of these complexes was explored for transesterification of DNA model substrate, 2-hydroxypropyl-p-nitrophenylphosphate (HPNP). The reaction between the copper(II) complexes and HPNP was investigated spectrophotometrically to understand mechanistic aspects of phosphate ester bond cleavage in HPNP. Kinetic experiments were performed to determine initial rate of reactions and the activity order follows: 1 > 3 >2. Various structural factors of complexes such as coordinating solvents, mode of bridging and metal–metal distance play a significant role in the catalytic activities. The maximum phosphatase like activity of complex 1 (kcat = 9.6 × 10−3 s−1) can be attributed to coordinated water molecules, shorter metal–metal distance and exposed metal centers.
Introduction
The syntheses of multinuclear metal complexes are of immense interest in heterogeneous catalysis.1 In this regard self-assembled coordination polymers have attracted much attention because of scientific interest and their potential applications in oxygenation, hydrolysis, transesterification and decomposition reactions.2 A variety of Cu(II) coordination polymers with different geometries exhibit significant role in catalytic applications.3 Among these applications, the cleavage of phosphate ester bonds either by hydrolysis or transesterification process,4 has received considerable attention due to important biochemical utility in genetic engineering. Phosphate esters are found as backbones in nucleotides, owing to good resistance toward cleavage under neutral conditions. The hydrolytic enzymes such as polymerases, recombinases and topoisomerases etc. contain transition metal ions, close to each other in their active sites.5 These metal ions act as Lewis acid sites and assist the cleavage of phosphate ester bonds by the cooperative action between two metal centers.6 Therefore many model transition metal complexes have been studied to mimic extraordinary catalytic activities of these enzymes.7 Model studies using self-assembled coordination polymers remain to be increasingly significant to explore the mechanistic pathways.8 In model studies of coordination polymers, the judicious choice of bridging anions or solvent is also a crucial factor to enhance their catalytic activities.9 Consequently, numerous phenoxo-bridged compounds of transition metal ions have been studied and are found to be very efficient in hydrolysis or transesterification reactions.10 The bent μ2-O bridging in such complexes forms a perfectly planar M2(μ2-O)2 type ring by reducing the M–O–M angle (≪180°) as shown in Scheme 1a.11 Lesser M–O–M angle reduces the M–M distance that further favours the cooperative action for catalytic studies of model complexes. In contrast to above linear μ2-L/μ-L type of bridging with higher M–L–M angle (∼180°) hinders the cooperative action as well as activity of complex due to increased M–M distance (Scheme 1b).9a,12 | ||
| Scheme 1 Two different bridging modes. | ||
In addition the homogenous catalytic activity of synthetic metal complexes toward phosphate ester bond cleavage, in solution has been reported by many researchers with different mechanisms.4,13 However, the heterogeneous catalysis is a new approach to develop more efficient, selective and reusable catalyst. There are very rare reports on heterogeneous catalytic studies for the cleavage of phosphate ester bonds. The insolubility of the material is an important tool to enhance its efficiency.14 Easy separation, efficient recycling and prevention of biomolecular self-destructive reactions are main advantages of heterogeneous catalysis over homogeneous catalysis. The coordination polymers are a new class of developing heterogeneous catalysts for various applications.15 To the best of our knowledge none of coordination polymers have ever been reported for heterogeneous phosphatase like activities.
Since metal–metal distance, exogenous ligands (nature and number of coordinated solvent molecules) and different donor sites of ligands (deprotonated phenolate and pyridine) can play a significant role in phosphatase like studies, we selected polymeric Cu(II) complexes viz. {[Cu3(L1)(NO3)2(DMF)(H2O)]·3(DMF)}n (1), [Cu3(L1)(Cl)2(DMF)2]n (2), [Cu3(L2)(NO3)4(H2O)4]n (3) (Fig. 1), used for photocatalytic activities in our earlier reports.16 These complexes are considered ideal candidates for comparative hetero-catalytic activities (phosphatase like activities) due to presence of different coordination environments and their insoluble nature in common organic solvents. All these complexes have different bridging modes (μ2-O−, μ2-Cl and μ-[O–N–O] in complexes 1, 2 and 3) that results in the formation of sheet like or stacking layer arrangement of coordination polymers with different metal–metal distance. Recently we have reported nickel(II) complexes of some Schiff bases and found them to exhibit reasonable phosphatase like and photocatalytic activities.9a,17 Thus we were inclined to perform heterogeneous catalytic studies of the above mentioned one-dimensional Cu(II) coordination polymers with different coordination environments. The hetero-catalytic activities of these complexes, towards phosphate ester bond cleavage were studied and structure activity relationship of these complexes with appropriate mechanisms is finally reported.
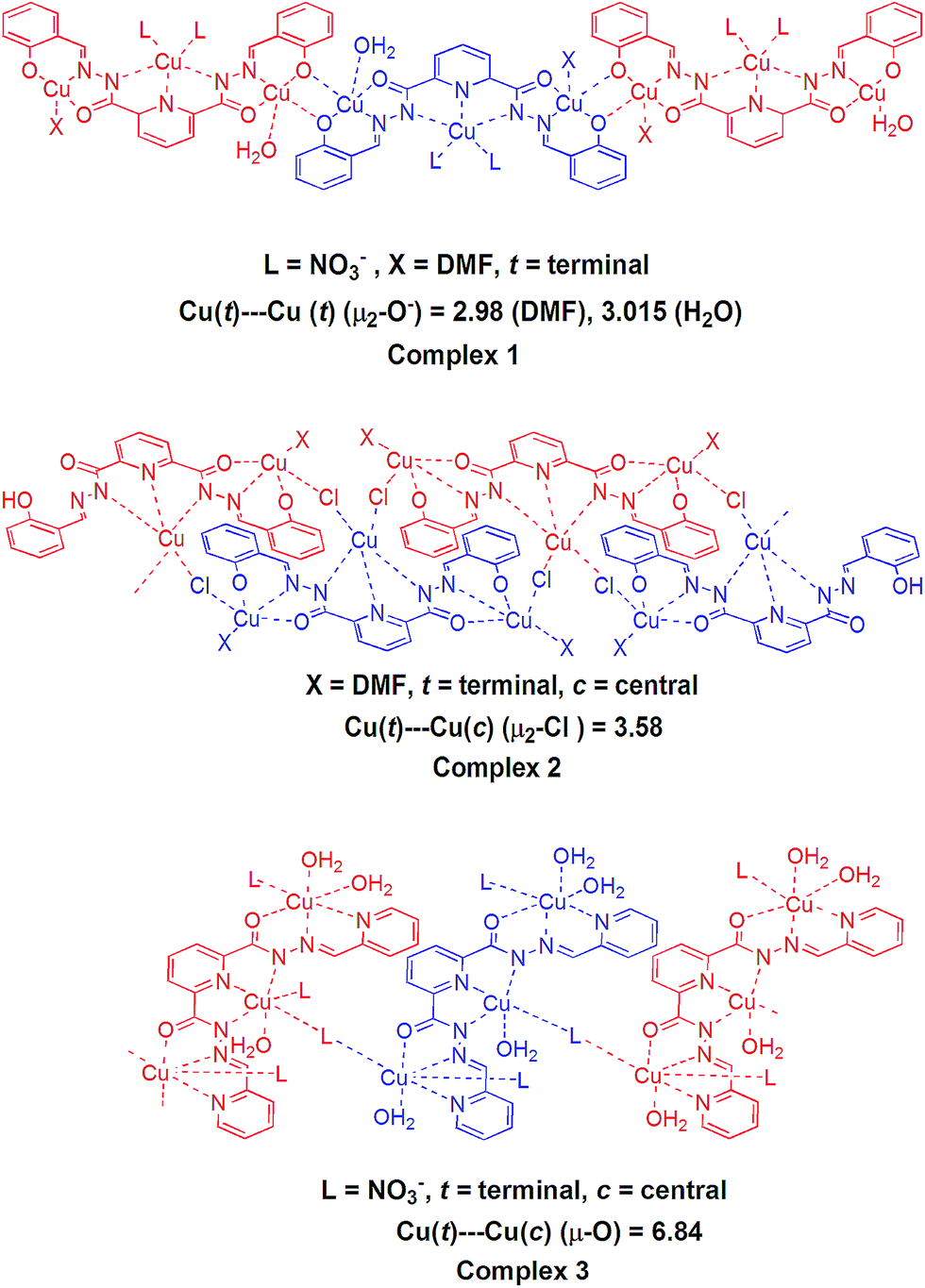 | ||
| Fig. 1 Coordination environment of the complexes 1, 2 and 3 showing phenoxo-bridged (μ2-O) 1D sheet, chloride bridged (μ2-Cl) one-dimensional zigzag stacking layered chain and μ-[O–N–O] bridged parallel top down structures, respectively. | ||
Results and discussion
Syntheses of complexes
Structurally characterized one-dimensional Cu(II) coordination polymers {[Cu3(L1)(NO3)2(DMF)(H2O)]·3(DMF)}n (1), [Cu3(L1)(Cl)2(DMF)2]n (2), [Cu3(L2)(NO3)4(H2O)4]n (3) were prepared by already reported procedure developed in our lab.16 The finally synthesized products were identified by their reported physicochemical properties.Phosphate ester bond cleavage studies and kinetics
It has been reported by many researchers that labile coordination site of transition metal complex favours the hydrolysis/transesterification of phosphate esters.18 Depending upon catalyst as well as substrate, different catalysis mechanism for different model systems have been reported by many researchers.19 Consequently the efforts were made to investigate the phosphatase like activities of synthesized complexes 1–3 with appropriate mechanism. The heterogeneous catalytic activities of all the three complexes 1–3 (50 μM) for transesterification of DNA model substrate 2-hydroxypropyl-p-nitrophenylphosphate (HPNP) (100 μM) were studied in 10% MeOH buffered solution. The relative catalytic efficiency of these model complexes was monitored by initial rate method spectrophotometrically. The increase in the absorption value of the liberated product (p-nitrophenolate ion, λmax = 400 nm) of HPNP transesterification reaction was measured as a function of time. The reaction rates (Vo, M s−1) of transesterification reaction were calculated by the initial rate method from the plots of [p-nitrophenolate] versus time. Vo divided by the concentration of HPNP gives first-order rate constants (kobs, s−1).The initial catalytic experiments were performed without adjusting the pH of solution and all the complexes showed very low or negligible activity. With increase in pH of solution there was increase in the activity of complexes 1 and 3 toward the transesterification of HPNP. The course of reactions with a solution of complexes 1–3 is shown in Fig. 2 and S1.†
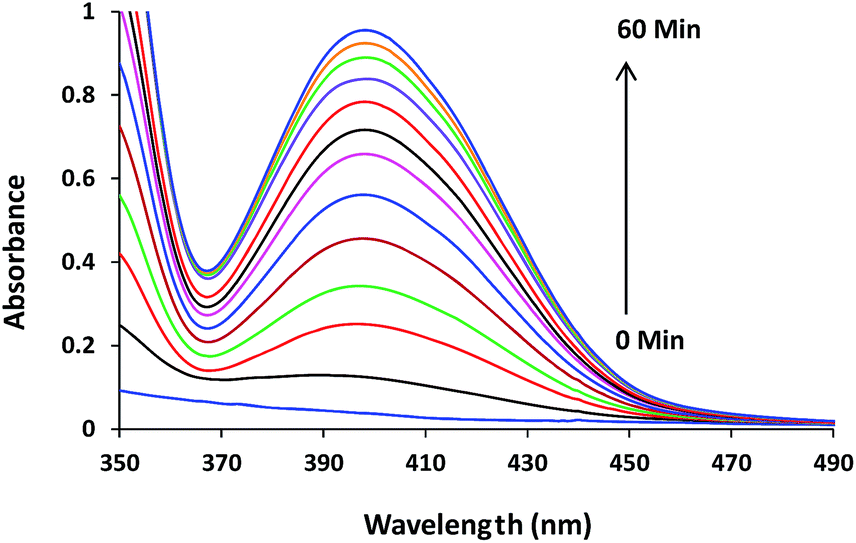 | ||
| Fig. 2 Absorption spectra for the transesterification of HPNP (100 μM) in the absence and presence of complex 1 (50 μM) in 10% MeOH recorded at an interval of 5 minutes at 30 °C. | ||
The comparative progress of catalytic reactions (i.e. increase in absorbance intensity corresponding to p-nitrophenolate) in the presence of complexes 1–3 at particular pH is shown in Fig. 3. As evident from the figure, a very low or negligible activity was shown by complex 2. Therefore all the further studies were performed for complexes 1 and 3 only.
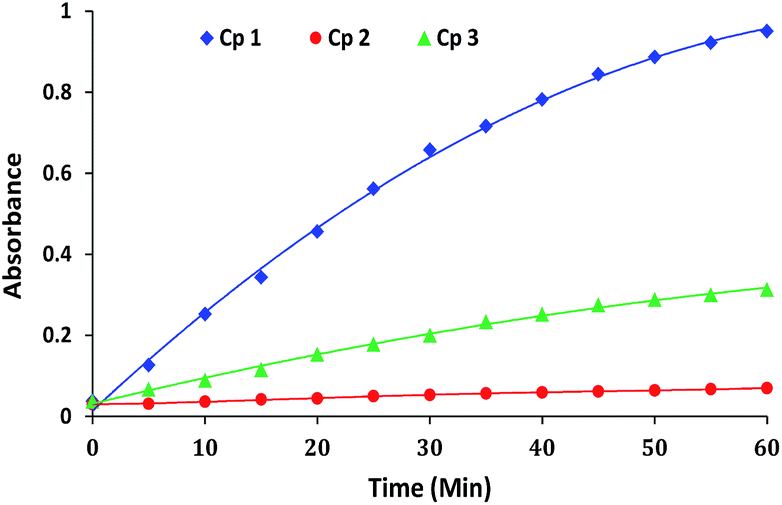 | ||
| Fig. 3 Comparative absorption profile due to the formation of p-nitrophenolate (λmax = 400 nm) on addition of HPNP to complex 1, 2 and 3 at pH 9.0, 9.0 and 8.5, respectively. [Complex] = 50 μM; [HPNP] = 100 μM in MeOH–H2O (10%, v/v) solution at 30 °C. | ||
The plot of rate constant (kobs) versus pH shows that the rate of transesterification for complexes 1 and 3 significantly depends upon the pH of the solution (Fig. 4). Complexes 1 and 3 showed a very low activity at pH 7 which maximize at pH 9.0 and 8.5 respectively, then decreased slightly. The nature of rate versus pH curve in both complexes displays acid–base equilibrium controlled kinetics. The pH titrations results infer that in a typical ester cleavage process for any complex to act as an efficient catalyst, the incoming substrate first binds efficiently to the catalyst. The substrate further experiences nucleophilic attack by the metal-coordinated solvent to undergo phosphate ester bond cleavage through intramolecular transesterification. As evident from Fig. 4, complexes 1 and 3 have maxima at different pH values (pH = 9.0 for complex 1 and pH = 8.5 for complex 3) because of different donor sites i.e. deprotonated phenolate and pyridine, respectively. Consequently pKa values for both the complexes were also determined by curve fitting programme (Origin 8.5). The dependence of rate of a reaction on pH (Fig. 4), was simulated following the Boltzmann equation: y = (A1 − A2)/{1 + exp(x − x0)/dx} + A2, where A1 = upper rate value; A2 = lower rate value; x = pH; x0 = pKa. The pKa1 values for both the complexes 1 and 3 were found as 8.34 ± 0.06 and 7.81 ± 0.04, respectively. A similar observation has also been reported by Greatti et al.20 The higher pKa value of 1 lowers the acidity of central metal ions that results into the saturation at higher pH. However in complex 3 the central metal ion is somewhat more acidic and achieves the maxima at lower pH. The decrease in activity at higher pH can be explained due to the presence of tightly bound Cu(II)-hydroxides species that causes a weak affinity of the catalyst for the substrate.21 All the further studies were performed at pH 9.0 and 8.5 for complexes 1 and 3, respectively.
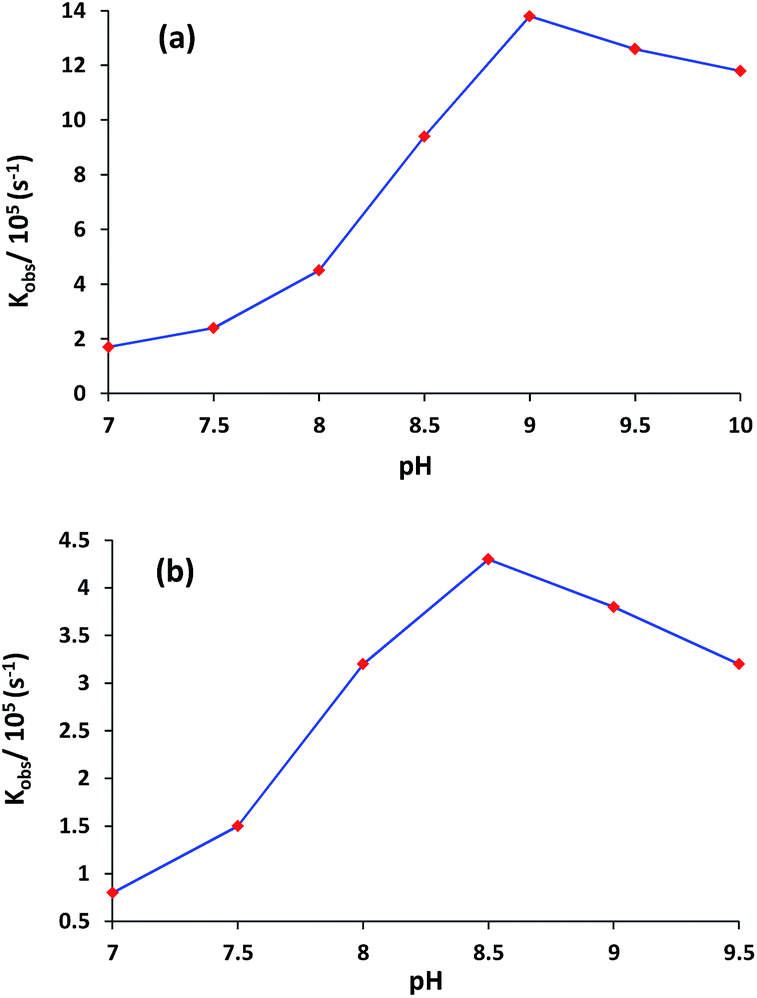 | ||
| Fig. 4 Dependence of the rate of HPNP transesterification on pH by complex {[Cu3(L1)(NO3)2(DMF)(H2O)]·3(DMF)}n (1) and [Cu3(L2)(NO3)4(H2O)4]n (3). [Complex] = 50 μM; [HPNP] = 100 μM in MeOH–H2O (10%, v/v) buffered solution at 30 °C. | ||
The results revealed that in complexes 1 {[Cu3(L1)(NO3)2(DMF)(H2O)]·3(DMF)}n and 3 [Cu3(L2)(NO3)4(H2O)4]n the deprotonation of metal coordinated water molecule at higher pH is responsible for their catalytic activity. The deprotonation of metal bound water molecule under basic conditions generates effective nucleophiles. Nucleophilic attack of such metal bound species to DNA model substrate HPNP results into to a cyclic transition state. This transition state finally favours P–O bond cleavage completing intramolecular transesterification process followed by the formation of product in the form of p-nitrophenolate ion (Scheme 2).
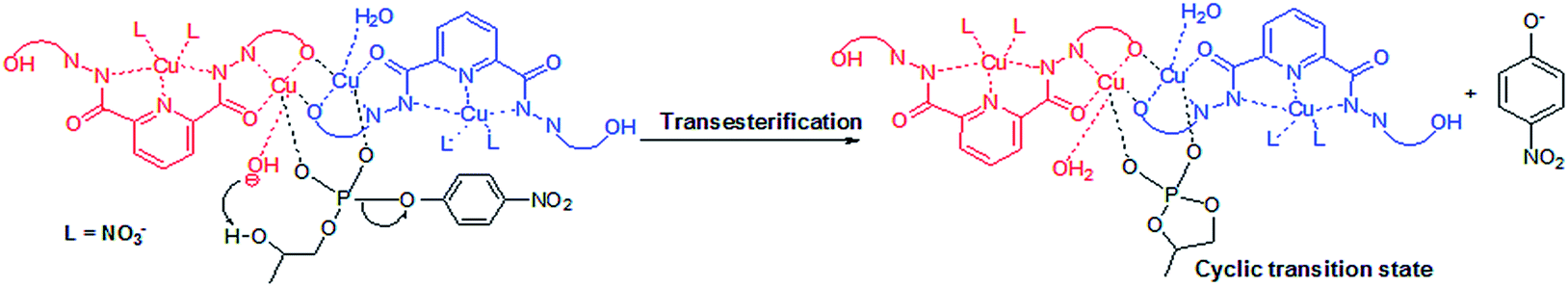 | ||
| Scheme 2 Mechanism of transesterification. | ||
The above perceptions are well supported in literature by similar behaviour observed for closely related systems where the role of coordinated solvents in catalytic mechanism has been explored.22 In the proposed catalytic mechanism, formation of cyclic transition state was further supported by 31P NMR spectroscopy for complex 1. 31P NMR signals of substrate (HPNP), glycero-1,2-cyclic phosphate (synthesized cyclic phosphate product)23 and substrate catalyst reaction mixture were compared. The substrate (HPNP) and synthesised cyclic phosphate showed a signal at −5.2 ppm and 17.5 ppm, respectively in D2O/DMSO-d6 mixture (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) as shown in Fig. S2a and b.†24 Complex 1 (0.25 mM) was added to a solution of HPNP (0.1 mM) in D2O/DMSO-d6 mixture (70:30), reaction mixture stirred and filtered to record final NMR. Appearance of a signal at 17.8 ppm confirmed the formation of cyclic phosphodiester during transesterification reaction (Fig. S2c†). The progress of catalytic reaction has also been monitored at different time intervals (after 15 min, 30 min and 1 h) with the help of 31P NMR spectroscopy Fig. S3.† The reaction mixture showed distinct transition from non-cyclic to cyclic transition state. As evident from figure, reaction mixture showed the appearance of NMR signal at 17.8 ppm corresponding to formation of cyclic phosphate with simultaneous disappearance of a signal at −5.2 ppm corresponding to non-cyclic substrate (HPNP). After 1 h all the substrate was converted into cyclic product.
:30) as shown in Fig. S2a and b.†24 Complex 1 (0.25 mM) was added to a solution of HPNP (0.1 mM) in D2O/DMSO-d6 mixture (70:30), reaction mixture stirred and filtered to record final NMR. Appearance of a signal at 17.8 ppm confirmed the formation of cyclic phosphodiester during transesterification reaction (Fig. S2c†). The progress of catalytic reaction has also been monitored at different time intervals (after 15 min, 30 min and 1 h) with the help of 31P NMR spectroscopy Fig. S3.† The reaction mixture showed distinct transition from non-cyclic to cyclic transition state. As evident from figure, reaction mixture showed the appearance of NMR signal at 17.8 ppm corresponding to formation of cyclic phosphate with simultaneous disappearance of a signal at −5.2 ppm corresponding to non-cyclic substrate (HPNP). After 1 h all the substrate was converted into cyclic product.
The role of complexes in transesterification reaction was confirmed by a blank experiment. A very low spontaneous rate of HPNP transesterification (kobs ∼ 10−8 s−1) was observed in the absence of any complex. Therefore the rates of catalytic reaction (kobs) were also studied at variable concentrations of complexes 1 and 3. The kinetics data for both the complexes show first order dependence on catalyst concentration with saturation at higher concentration (Fig. 5). In order to investigate the influence of substrate concentration on rates, the complexes 1 and 3 were treated with different concentrations of HPNP (50–500 μM). Initially a first-order dependence of the substrate concentration was observed. At higher concentrations first order rate kinetics gradually deviated from unity due to saturation in kinetics, for both complexes (Fig. 6 and S4†). The dependence of rate on the substrate concentration and then saturation suggests a catalyst-substrate binding to be an initial step in a catalytic process followed by pre-equilibrium between complex and substrate.
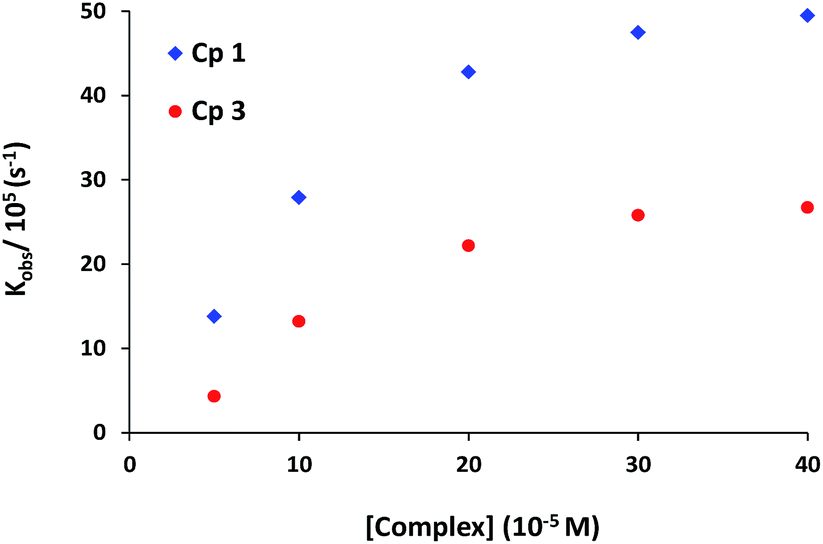 | ||
| Fig. 5 Dependence of the reaction rate on the concentration of the complexes for HPNP transesterification. [Complex] = 50–400 μM; [HPNP] = 100 μM in MeOH–H2O (10%, v/v) buffered solution at 30 °C. | ||
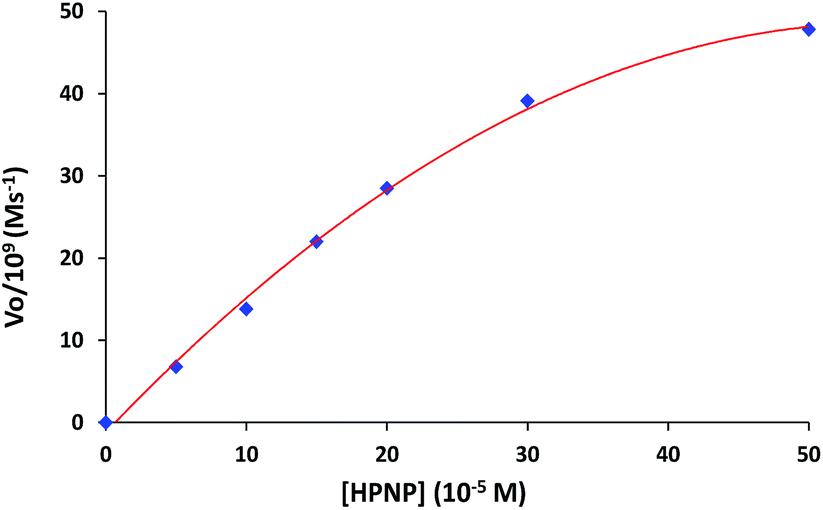 | ||
| Fig. 6 Dependence of rate of reaction on substrate concentration (50–500 μM) for complex 1 (50 μM) at 30 °C in 10% MeOH. | ||
The rates of reactions for different substrate concentrations were fitted to the Michaelis–Menten equation and linearized by means of Lineweaver–Burk plot (Fig. 7) to calculate kinetic parameters (Table 1) for these complexes. Complex 1 exhibits higher activity (kcat = 9.6 × 10−3) in comparison to complex 3 (kcat = 1.6 × 10−3). Km is the substrate concentration with which half the maximum rate is attained, it measures the affinity of the substrate for the enzyme.
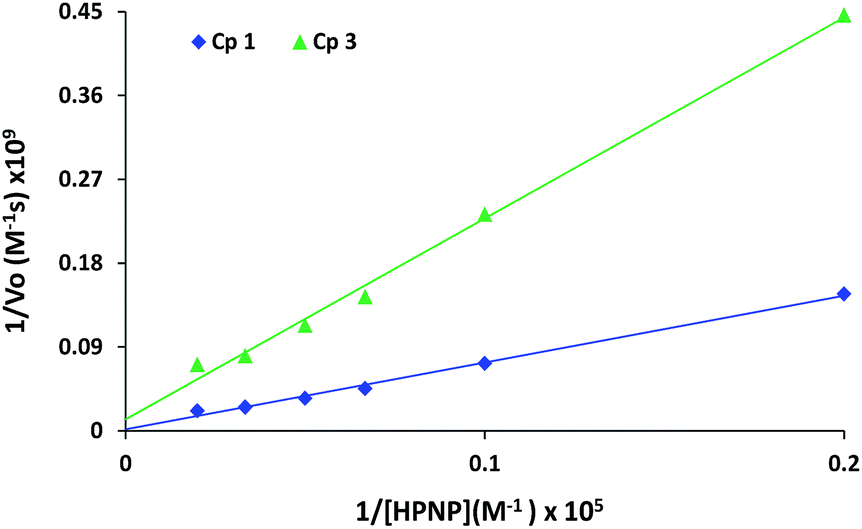 | ||
| Fig. 7 Lineweaver–Burk plot for the HPNP transesterification by complexes 1 and 3. | ||
| Cp. | kobs (s−1) | Vmax (M s−1) | Km (M) | Turnover number or kcat (s−1) | Ea (M−1 s−1) | fb | Catalyst selectivityc |
|---|---|---|---|---|---|---|---|
| a E = kcat/Km (catalytic efficiency).b f = kcat/knc (catalytic factor), where knc = 2.3 × 10−8 s−1 (uncatalyzed reaction constant).c Catalyst selectivity = [desired product]/[reactant] at saturation. | |||||||
| 1 | 1.4 × 10−4 | 4.8 × 10−7 | 3.3 × 10−3 | 9.6 × 10−3 | 2.9 | 4.17 × 105 | 5.1 × 10−1 |
| 3 | 4.3 × 10−5 | 8.3 × 10−8 | 1.7 × 10−3 | 1.6 × 10−3 | 0.94 | 6.95 × 104 | 1.7 × 10−1 |
Structure activity relationship
The experimental results mentioned above show that the complexes 1 and 3 were found to be active toward the phosphatase like activity. A negligible activity was found for complex 2. Complex 1 exhibited highest phosphatase-like activity with 4.2 × 105 times greater rate enhancement, in comparison to uncatalysed reaction (knc = 2.3 × 10−8 s−1). A medium activity was observed for complex 3 (6.8 × 104 times greater rate enhancement in comparison to uncatalysed reaction). To rationalize the observed relative performance of catalysts, various structural factors that affect the structure–activity relationship were investigated.As shown in Fig. 1, in the coordination environment of complex 1, one of the terminal copper atoms contains two bridged oxygen atoms (μ2-O) from the phenolic ring of the ligand and single oxygen atom from coordinated DMF. In the other terminal copper atom the coordination environment is same except that fifth coordination site occupied by H2O instead of DMF. This μ2-O bridging in 1D chain of complex forms a planar M2(μ2-O)2 type ring that is responsible for the decreased M–O–M angle (≪180°). Lesser M–O–M angle minimizes the terminal Cu(t)–Cu(t)– distance by 2.98 Å in the former case and 3.01 Å in the later (Fig. 1). The topological analysis of 1D polymeric complex by TOPOS,25 suggests (312·49)·(36)2 Schläfli point symbol (Fig. 8). The whole 1D framework can be represented as a (3,8) 2-nodal connected net.
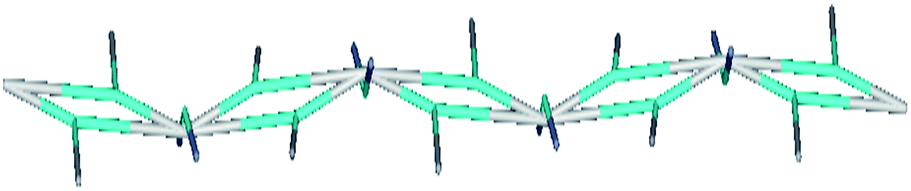 | ||
| Fig. 8 Schematic representation of topological net of 1. | ||
In complex 2, the central pentacoordinated square pyramidal copper atom of one trinuclear unit is coordinated by two oxygen atoms, one nitrogen and one chlorine atom equatorially from the ligands and axially by oxygen from DMF. However no H2O molecule is present in the coordination environment of the metal center. The central copper atom is connected to two terminal copper atoms of different neighbouring units through chloride bridging (Fig. 1). The intramolecular distance of chloride bridged terminal and central copper atoms i.e., Cu(t)⋯Cu(c) is 3.58 Å. The top-bottom μ2-Cl bridging mode between adjacent trinuclear units results in the formation of one-dimensional zigzag stacking layered chain structure. From the viewpoint of structural topology, the extended point symbol for this network is (3·4·5)·(3·42·52·7) with 2-nodal (3,4) connected net (Fig. 9).
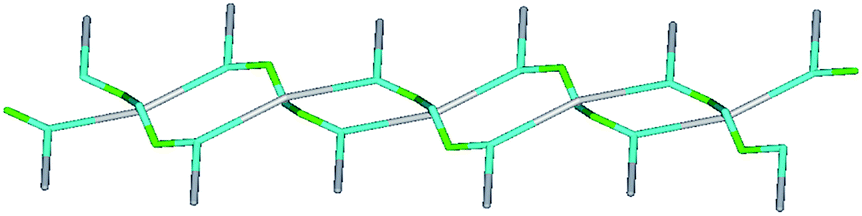 | ||
| Fig. 9 Schematic representation of topological net of 2. | ||
In trinuclear unit of complex 3, central octahedral copper atom is coordinated axially by two oxygen atoms of the ligands and equatorially by three nitrogen atoms of ligand and one oxygen atom from H2O molecule. The terminal copper atom is occupied by two oxygen atoms from H2O molecule axially and three nitrogen atoms from ligand equatorially along with one oxygen from coordinated H2O molecule. The nitrate ion is involved in coordination with the central copper of one unit and terminal copper of the other unit through two oxygen atoms in μ-[O–N–O] bridging mode in a parallel top down arrangement that results in a 1D chain structure. The Cu(t)⋯Cu(c) distance in each of above mentioned bridging (nitrate bridging) unit is 6.84 Å as shown in Fig. 1. To understand the nature of this framework, topology analysis is provided by TOPOS. Thus, the whole 1D framework can be represented as a 2-nodal (2,3) connected net with (3) point symbol (Fig. 10).
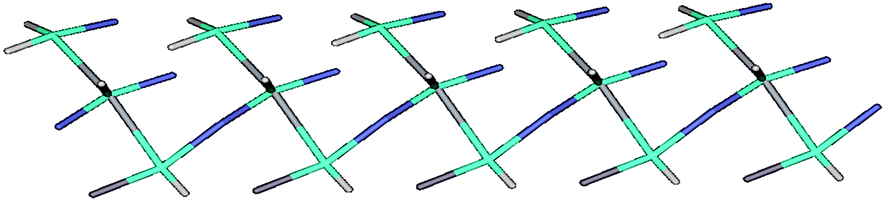 | ||
| Fig. 10 Schematic representation of topological net of 3. | ||
Above mentioned structural factors such as type of exogenous ligand, mode of bridging and metal–metal distance are known to play a key role in relative activities of complexes.26 The coordinated water molecules in both the complexes 1 and 3 act as nucleophile to catalyse the phosphate ester bond cleavage. However in complex 1 the favourable metal–metal distance (3.015 Å) between solvent coordinated copper centers is observed, that further enhances the activity, owing to the fact that a metal–metal distance of 2.9–3.2 Å is favourable for the cooperative action.27 Moreover sheet like arrangement of adjacent trinuclear unit assisted by phenoxo bridging increases the cooperative effect and is appropriately exposed for the nucleophilic attack (as supported by TOPOS). On the contrary, the stacking layer arrangement due to nitrate bridging (μ-[O–N–O]) in complex 3 accounts for larger metal–metal distance (6.85 Å) decreasing the cooperative effect and making the metal centers least accessible for the nucleophilic attack. Consequently in complex 1 both the metal centers participate in the catalytic process; that is, one metal center accommodates the nucleophile and other binds with the substrate to favor the cooperative action for bond cleavage. The larger Cu(II)–Cu(II) distance of complex 3 in comparison to complex 1, is responsible for lesser catalytic activity of the former due to lack of cooperative action. Complex 2 shows negligible phosphatase-like activity due to lack of appropriate coordinated water or solvent molecules and its stacking layer arrangement.28 The results of our complexes 1 and 3 were compared with already reported complexes for phosphatase-like activities (Table S1, ESI†) and it was observed that the present complexes have promising activities.
Filtration test
The filtration test of the catalyst was performed to confirm the heterogeneous nature of the catalytic process for complex 1.29 During the progress of catalytic process, reaction was stopped after 20 min and catalyst was filtered. The filtrate was allowed to stir at the same reaction conditions and its absorption profile was studied at regular intervals. The filtrate did not afford any significant change in the reaction further, however a very small enhancement in the absorption intensity was observed due to some leaching of metal complex into the solution. Consequently filtrate showed almost negligible change in the absorption intensity of filtrate in comparison to the catalysed reaction and confirmed the heterogeneous catalytic process. The above procedure is repeated with another batch and the catalyst was filtered after 40 min to find the similar results (Fig. 11).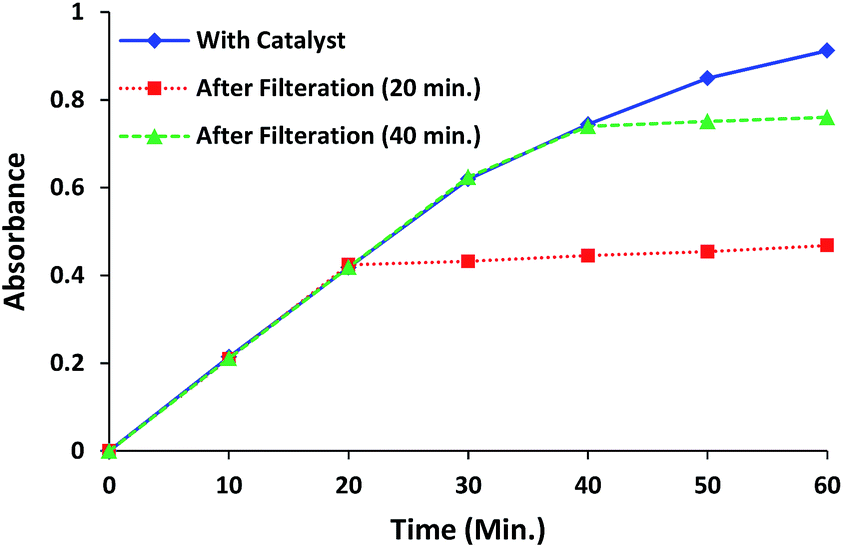 | ||
| Fig. 11 Filtration experiment for complex 1, showing the changes in absorption intensity of filtrate after removing the catalyst (after 20 min and 40 min). | ||
Catalyst reusability
The reusability of catalyst is a significant factor in environmentally friendly heterogeneous catalysis. The reusability of complex 1 and 3 was examined by the repeated HPNP phosphate ester bond cleavage experiments. The catalyst was recovered from the reaction mixture by filtration, washed with HEPES buffer followed by acetonitrile and dried to reuse for further experiments under similar conditions. The stability of the recovered material was confirmed from similar PXRD patterns of complexes before and after third cycle of catalytic experiments (Fig. S5†). However some loss in the intensity of 2θ values was observed.After the first use sample retained its catalytic efficiencies with only 10–12% decrease in the reaction rate. The decrease in the reaction rate is due to leaching of small amount of metal ions into the solution. For further subsequent reuses the reaction showed a very less reduction (<4%) in the rate of reaction (Fig. 12 and S6† for complex 1 and 3, respectively), therefore no further recycling experiments were performed.
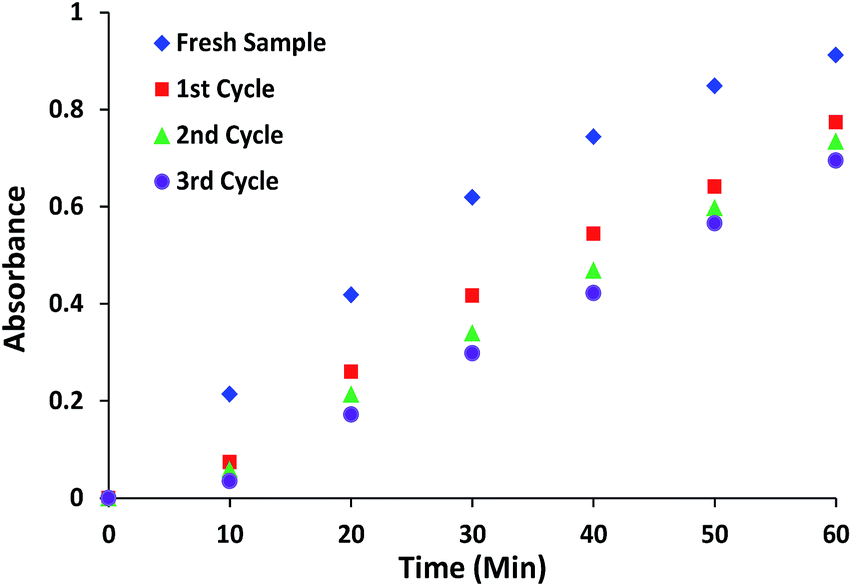 | ||
| Fig. 12 Reusability of complex 1 for repeated HPNP phosphate ester bond cleavage experiments. | ||
Conclusions
Three known one-dimensional Cu(II) coordination polymers {[Cu3(L1)(NO3)2(DMF)(H2O)]·3(DMF)}n (1), [Cu3(L1)(Cl)2(DMF)2]n (2), [Cu3(L2)(NO3)4(H2O)4]n (3) with different coordination environments, were chosen to investigate their potential in heterogeneous catalytic studies for the cleavage of phosphate ester bond. In complex 1, the adjacent trinuclear copper units are connected by bent phenoxo-bridging (μ2-O−), giving rise to sheet like arrangement, whereas in complex 2 and 3 trinuclear copper units are connected through linear bridging of Cl− and NO3− groups, in a μ2-Cl and μ-[O–N–O] mode, respectively resulting in the stacking layer arrangement.A structure activity relationship for all the complexes has been studied. Complexes 1 and 3 were found to be active toward the phosphatase like activity due to presence of coordinated water molecules that act as active nucleophile to catalyse the cleavage of phosphate ester bond. However complex 3 shows lower activity than 1, in spite of the fact that it has more number of coordinated molecules. The highest activity of complex 1 can be attributed to metal coordinated water molecule assisted by favourable metal–metal distance 3.015 Å in comparison to complex 3 with larger metal–metal distance (6.84 Å). Moreover sheet like arrangement of complex 1 is appropriately exposed for the favourable nucleophilic attack in comparison to complex 3 with stacking layer arrangement. Complex 2 showed negligible activity in spite of lesser metal–metal distance than 3, due to lack of appropriate coordinated water/solvent molecules and its stacking layer arrangement. Thus, it can be inferred that although appropriate metal–metal distance is assistive but not the only condition for ester cleavage reactions; however presence of appropriate nucleophile (coordinated water or solvent) along with appropriate metal–metal distance and exposed metal centers facilitate the phosphate ester bond cleavage.
Experimental
Materials and measurements
All solvents were dried by standard methods. Unless otherwise specified, chemicals were purchased from commercial suppliers and used without further purification. Various biological buffers 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (EPPS), tetra-n-butyl ammonium perchlorate (TBAP), 2-(cyclohexylamino)ethanesulfonic acid (CHES) etc. were purchased from Aldrich. 2-Hydroxypropyl-p-nitrophenylphosphate (HPNP) was prepared by literature method.30 The syntheses of the complexes 1–3 was conducted as per our previously reported procedure.16The NMR spectra of the ligands were performed in DMSO-d6 with TMS as an internal reference, on JNM-ECS400 (JEOL) instrument operating at 400 MHz for 1H NMR. The chemical shifts are reported as δ values (ppm) relative to TMS. The absorption spectra were recorded on a Shimadzu UV-1800, UV-vis spectrophotometer. The PANalytical X'PERT PRO diffractometer was used for X-ray diffraction (XRD) with a scan speed of 10° min−1 for 2θ over a range from 10 to 80 (45 kV, 40 mA using Ni-filtered Cu Kα radiations).
Kinetic measurements for phosphate ester bond cleavage
In all the kinetic studies for phosphate ester bond cleavage, 2-hydroxypropyl-p-nitrophenylphosphate (HPNP) was used as substrate against all complexes. All the experiments were performed in buffer solutions. Double distilled deionized water was used for the buffer preparation. The 10 mL solution of each buffer salt (0.1 M) (HEPES, pH < 8.0; EPPS, 8.0 < pH < 8.9; CHES, 8.9 < pH < 11.0) was prepared and mixed with equal volume of 0.2 M, NaNO3 solution (to maintain the ionic strength). These mixtures were titrated with NaOH (1 M) to get the desired pH.Heterogeneous catalytic studies
All the kinetic studies were performed on UV-vis spectrophotometer using following conditions. In a typical reaction employing the heterogeneous catalysis, a suspension of each complex 1–3 (50 μM) in buffer solution was added to 100 μM of the substrate (HPNP), resulting in a MeOH–H2O buffered solution (10%, v/v). The hydrolysis reaction was carried out in a round bottom flask and reaction product present in solution was separated at regular intervals by centrifugation. The increase in absorption band (λmax 400 nm) corresponding to the formation of reaction product (p-nitrophenolate) was recorded after every 5 minutes for one hour. The rates of substrate transesterification in the presence of different complexes were measured by initial rate method. The same procedure was followed for blank experiment using HPNP only. The effect of pH on catalytic activity was studied in the pH range 7.0–10.0 using different buffer solutions. To determine the dependence of the rates on the substrate concentration and various kinetic parameters, 50 μM solutions of different metal complexes were treated with increasing substrate concentration (50–500 μM) in 10% MeOH buffer solution at particular pH. In all the experiments absorption of solutions was recorded directly after fifteen minutes equilibration time at 400 nm. All the measurements were recorded two times and the average values were taken.Acknowledgements
This work is supported by INSPIRE Faculty research grant (IFA-11CH-09) from Department of Science and Technology (DST), Government of India. V.K.B. is also thankful to Indian Institute of Technology, Ropar (I.I.T. Ropar) for research facilities.References
- (a) J. W. Shin, J. M. Bae, C. Kim and K. S. Min, Inorg. Chem., 2013, 52, 2265–2267 CrossRef CAS PubMed; (b) H. Kwak, S. H. Lee, S. H. Kim, Y. M. Lee, E. Y. Lee, B. K. Park, E. Y. Kim, C. Kim, S. J. Kim and Y. Kim, Eur. J. Inorg. Chem., 2008, 408–415 CrossRef CAS; (c) E. Fujita, B. S. Brunschwig, T. Ogata and S. Yanagida, Coord. Chem. Rev., 1994, 132, 195–200 CrossRef CAS; (d) D. Rambabu, C. P. Pradeep and A. Dhir, Inorg. Chem. Front., 2014, 1, 163–166 RSC.
- (a) T. Opstal and F. Verpoort, Angew. Chem., Int. Ed., 2003, 42, 2876–2879 CrossRef CAS PubMed; (b) K. Srinivasan, P. Michaud and J. K. Kochi, J. Am. Chem. Soc., 1986, 108, 2309–2320 CrossRef CAS PubMed; (c) B. Rihter, S. Srihari, S. Hunter and J. Masnovi, J. Am. Chem. Soc., 1993, 115, 3918–3924 CrossRef CAS; (d) A. C. D. Midoes, P. E. Aranha, M. P. dos Sontos, E. Tozzo, S. Romera, R. H. de A. Santos and E. R. Dockal, Polyhedron, 2008, 27, 59–64 CrossRef CAS.
- (a) Y. L. Hou, R. W. Y. Sun, X. P. Zhou, J. H. Wang and D. Li, Chem. Commun., 2014, 50, 2295–2297 RSC; (b) T. Yang, H. Cui, C. Zhang, L. Zhang and C. Y. Su, ChemCatChem, 2013, 5, 3131 CrossRef CAS; (c) Y. Kang, F. Wang, J. Zhang and X. Bu, J. Am. Chem. Soc., 2012, 134, 17881 CrossRef CAS PubMed.
- P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Chem. Soc. Rev., 2000, 29, 75–86 RSC.
- N. Strater, W. N. Lipscomb, T. Klablunde and B. Krebs, Angew. Chem., Int. Ed. Engl., 1996, 35, 2024–2055 CrossRef.
- (a) H. Dugas, Bioorg. Chem., Springer-Verlag, New York, 1988 Search PubMed; (b) J. L. Vanhooke, M. M. Benning, F. M. Raushel and H. M. Holden, Biochemistry, 1996, 35, 6020–6025 CrossRef CAS PubMed.
- (a) E. Y. Tirel and N. H. Williams, Chem.–Eur. J., 2015, 21, 7053–7056 CrossRef CAS PubMed; (b) M. Yashiro, A. Ishikubo and M. Komiyama, J. Chem. Soc., Chem. Commun., 1995, 1793–1794 RSC; (c) P. Molenveld, S. Kapsabelis, J. F. J. Engbersen and D. N. Reinhoudt, J. Am. Chem. Soc., 1997, 119, 2948–2949 CrossRef CAS; (d) K. Yamaguchi, F. Akagi, S. Fujinami, M. Suzuki, M. Shionoya and S. Suzukia, Chem. Commun., 2001, 375–376 RSC; (e) V. Gandin, C. Marzano and A. Erxleben, Eur. J. Inorg. Chem., 2014, 25, 4084–4092 Search PubMed; (f) M. Diez-Castellnou, F. Mancin and P. Scrimin, J. Am. Chem. Soc., 2014, 136, 1158–1161 CrossRef CAS PubMed; (g) J. Adhikary, P. Chakraborty, S. Das, T. Chattopadhyay, A. Bauza, S. K. Chattopadhyay, B. Ghosh, F. A. Mautner, A. Frontera and D. Das, Inorg. Chem., 2013, 52, 13442–13452 CrossRef CAS PubMed.
- (a) S. Anbu, M. Kandawamy and B. Varghese, Dalton Trans., 2010, 39, 3823–3832 RSC; (b) F. Mancin and P. Tecilla, New J. Chem., 2007, 31, 800–817 RSC; (c) A. Guha, T. Chattopadhyay, N. D. Paul, M. Mukherjee, S. Goswami, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2012, 51, 8750–8759 CrossRef CAS PubMed; (d) J. Mukherjee and R. N. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429–438 CrossRef CAS.
- (a) V. K. Bhardwaj and A. Singh, Inorg. Chem., 2014, 53, 10731–10742 CrossRef CAS PubMed; (b) S. Mandal, J. Mukherjee, F. Lloret and R. Mukherjee, Inorg. Chem., 2012, 51, 13148–13161 CrossRef CAS PubMed; (c) V. K. Bhardwaj, M. S. Hundal, M. Corbella, V. Gómez and G. Hundal, Polyhedron, 2012, 38, 224–234 CrossRef CAS.
- (a) S. Bosch, P. Comba, L. R. Gahan and G. Schenk, Inorg. Chem., 2014, 53, 9036–9051 CrossRef CAS PubMed; (b) M. Wall, R. C. Hynes and J. Chin, Angew. Chem., Int. Ed., 1993, 32, 1633–1635 CrossRef.
- (a) S. Naiya, S. Biswas, M. G. B. Drew, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2012, 51, 5332–5341 CrossRef CAS PubMed; (b) P. Kar, R. Biswas, Y. Ida, T. Ishida and A. Ghosh, Cryst. Growth Des., 2011, 11, 5305–5315 CrossRef CAS.
- S. Mandal, V. Balamurugan, F. Lloret and R. Mukherjee, Inorg. Chem., 2009, 48, 7544–7556 CrossRef CAS PubMed.
- (a) D. Desbouis, I. P. Troitsky, M. J. Belousoff, L. Spiccia and B. Graham, Coord. Chem. Rev., 2012, 256, 897–937 CrossRef CAS; (b) F. Mancin, P. Scrimin and P. Tecilla, Chem. Commun., 2012, 48, 5545–5559 RSC.
- (a) B. R. Bodsgard and J. N. Burstyn, Chem. Commun., 2001, 647–648 RSC; (b) A. A. Muxel, A. Neves, M. A. Camargo, A. J. Bortoluzzi, B. Szpoganicz, E. E. Castellano, N. Castilho, T. Bortolotto and H. Terenzi, Inorg. Chem., 2014, 53, 2943–2952 CrossRef CAS PubMed; (c) S. G. Srivastan, M. Parvez and S. Verma, Chem.–Eur. J., 2002, 8, 5184–5191 CrossRef; (d) C. Piovezan, J. M. R. Silva, A. Neves, A. J. Bortoluzzi, W. Haase, Z. Tomkowicz, E. E. Castellano, T. C. S. Hough and L. M. Rossi, Inorg. Chem., 2012, 51, 6104–6115 CrossRef CAS PubMed; (e) B. R. Bodsgard, R. W. Clark, A. W. Ehrbar and J. N. Burstyn, Dalton Trans., 2009, 2365–2373 RSC; (f) G. Zaupa, L. J. Prins and P. Scrimin, Bioorg. Med. Chem. Lett., 2009, 19, 3816–3820 CrossRef CAS PubMed.
- (a) M. K. Sharma, P. P. Singh and P. K. Bharadwaj, J. Mol. Catal. A: Chem., 2011, 343, 6–10 CrossRef; (b) S. K. Yoo, J. Y. Ryu, J. Y. Lee, C. Kim, S. J. Kima and Y. Kim, Dalton Trans., 2003, 1454–1456 RSC; (c) A. Karmakar, G. M. D. M. Rúbio, M. F. C. G. da Silva, S. Hazra and A. J. L. Pombeiro, Cryst. Growth Des., 2015, 15, 4185–4197 CrossRef CAS; (d) T. Maity, S. Bhunia, S. Das and S. Koner, RSC Adv., 2016, 6, 33380–33386 RSC; (e) M. Trivedi, B. A. Kumar, G. Singh, A. Kumar and N. P. Rath, New J. Chem., 2016, 40, 3109–3118 RSC; (f) Y. Chen and S. Ma, Dalton Trans., 2016, 45, 9744–9753 RSC.
- N. Hussain and V. K. Bhardwaj, Dalton Trans., 2016, 45, 7697–7707 RSC.
- V. K. Bhardwaj, Dalton Trans., 2015, 44, 8801–8804 RSC.
- (a) E. Y. Tirel and N. H. Williams, Chem.–Eur. J., 2015, 21, 7053–7056 CrossRef CAS PubMed; (b) M. Yashiro, A. Ishikubo and M. Komiyama, J. Chem. Soc., Chem. Commun., 1995, 1793–1794 RSC; (c) P. Molenveld, S. Kapsabelis, J. F. J. Engbersen and D. N. Reinhoudt, J. Am. Chem. Soc., 1997, 119, 2948–2949 CrossRef CAS; (d) K. Yamaguchi, K. Akagi, S. Fujinami, M. Suzuki, M. Shionoya and S. Suzukia, Chem. Commun., 2001, 375–376 RSC; (e) V. Gandin, C. Marzano and A. Erxleben, Eur. J. Inorg. Chem., 2014, 25, 4084–4092 Search PubMed; (f) M. Diez-Castellnou, F. Mancin and P. Scrimin, J. Am. Chem. Soc., 2014, 136, 1158–1161 CrossRef CAS PubMed; (g) J. Adhikary, P. Chakraborty, S. Das, T. Chattopadhyay, A. Bauza, S. K. Chattopadhyay, B. Ghosh, F. A. Mautner, A. Frontera and D. Das, Inorg. Chem., 2013, 52, 13442–13452 CrossRef CAS PubMed.
- P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, J. Org. Chem., 1999, 64, 6337–6341 CrossRef CAS.
- A. Greatti, M. Scarpellini, R. A. Peralta, A. Casellato, A. J. Bortoluzzi, F. R. Xavier, R. Jovito, M. A. de Brito, B. Szpoganicz, Z. Tomkowicz, M. Rams, W. Haase and A. Neves, Inorg. Chem., 2008, 47, 1107–1119 CrossRef CAS PubMed.
- (a) L. J. Daumann, K. E. Dalle, G. Schenk, R. P. McGeary, P. V. Bernhardt, D. L. Ollis and L. R. Gahan, Dalton Trans., 2012, 41, 1695–1708 RSC; (b) J. W. Chen, X. Y. Wang, Y. G. Zhu, J. Lin, X. L. Yang, Y. Z. Li, Z. Lu and J. Guo, Inorg. Chem., 2005, 44, 3422–3430 CrossRef CAS PubMed; (c) C. Bazzicalupi, A. Bencini, E. Berni, A. Bianchi, V. Fedi, V. Fusi, C. Giorgi, P. Paolettti and B. Valtancoli, Inorg. Chem., 1999, 38, 4115–4122 CrossRef CAS.
- H. Korhonen, T. Koivusalo, S. Toivola and S. Mikkola, Org. Biomol. Chem., 2013, 11, 8324–8339 Search PubMed.
- L. Kugel and M. Halmann, J. Am. Chem. Soc., 1967, 89, 4125–4128 CrossRef CAS.
- K. Selmeczi, C. Michel, A. Milet, I. G. Luneau, C. Philouze, J. L. Pierre, D. Schnieders, A. Rompel and C. Belle, Chem.–Eur. J., 2007, 13, 9093–9106 CrossRef CAS PubMed.
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
- I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814–840 RSC.
- K. D. Karlin, Y. Gultneh, T. Nicholson and J. Zubieta, Inorg. Chem., 1985, 24, 3725–3727 CrossRef CAS.
- (a) T. Koiket and E. Kimura, J. Am. Chem. Soc., 1991, 113, 8935–8941 CrossRef; (b) M. Subat, K. Woinaroschy, C. Gerstl, B. Sarkar, W. Kaim and B. Konig, Inorg. Chem., 2008, 47, 4661–4668 CrossRef CAS PubMed; (c) S. Anbu, S. Kamalraj, B. Varghese, J. Muthumary and M. Kandaswamy, Inorg. Chem., 2012, 51, 5580–5592 CrossRef CAS PubMed; (d) R. Sanyal, A. Guha, T. Ghosh, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 85–92 CrossRef CAS PubMed.
- D. De, T. K. Pal, S. Neogi, S. Senthilkumar, D. Das, S. S. Gupta and P. K. Bharadwaj, Chem.–Eur. J., 2016, 22, 3387–3396 CrossRef CAS PubMed.
- D. M. Brown and D. A. Usher, J. Chem. Soc., 1965, 6558 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Various kinetic plots and table showing comparison of phosphatase like activities between present complexes and other reported complexes. See DOI: 10.1039/c6ra11102e |
| This journal is © The Royal Society of Chemistry 2016 |