
Multiple pH responsive zwitterionic micelles for stealth delivery of anticancer drugs†
Jin Maa,
Ke Kanga,
Qiangying Yi*a,
Zhirong Zhangb and
Zhongwei Gu*a
aNational Engineering Research Center for Biomaterials, Sichuan University, 29 Wangjiang Road, Chengdu 610064, China. E-mail: zwgu@scu.edu; qyi@scu.edu.cn; Tel: +86-028-85410336
bWest China School of Pharmacy, Sichuan University, China
First published on 29th June 2016
Abstract
In this work, we reported zwitterion-functionalized “stealth” micelles for intelligent delivery of anticancer drugs. The amphiphilic polymer, poly(carboxybetaine methacrylate)-block-poly[2,2-di(acryloyloxy-1-ethoxy)propane-co-4,4-trimethylene dipiperidine [pCBMA-b-p(ADA-TMDP)], was synthesized via ATRP and subsequent Michael addition reaction. The assembled polymeric micelles were demonstrated as nanoscale (69 ± 20 nm) carriers possessing zwitterionic tentacles. Specifically, the hydrophilic zwitterionic tentacles, which were composed of the pCBMA component, could not only significantly resist protein adsorption to realize “stealth” transportation and prolong the circulation time of the drug delivery system in the blood stream, but also reverse its surface charge to facilitate cellular uptake in response to the extracellular pH environment (pH 6.8); the steady drug encapsulation cavities, as assemblies of p(ADA-TMDP) units, endowed the micelles intracellular environmental pH-responsive properties for potential endosomal escape and desired drug release after their accumulation in tumor sites through the EPR effect. The anticancer activity of these zwitterionic micelles carrying doxorubicin (DOX) was systematically studied here. At low pH (e.g., pH 5.0), cleavage of the ketal bonds of the ADA segments and protonation of the TMDP segments both contributed to the swelling and disassembly of the zwitterionic micelles. Moreover, protonation of TMDP segments caused water influx and thus facilitated micelle endosomal escape. The drug release profiles revealed that these zwitterionic micelles could accelerate drug release remarkably more (3.26-fold) at pH 5 than pH 7.4, indicating the great potential to quickly release the vast majority of the drug in tumor tissue. Both in vitro and in vivo evaluations verified the high anticancer efficiency and greatly reduced side effects of DOX-loaded zwitterionic micelles as compared with the free drug DOX·HCl.
1. Introduction
Cancer has been a severe disturbance to public health because of its substantial morbidity and mortality all around the world.1 Chemotherapy, which uses one or more chemical substances as anti-cancer drugs, has been developed as one of the significant therapeutic strategies in cancer treatment.2 Although the administrated chemotherapeutic agents would clinically either reduce symptoms or prolong the life of patients, their side effects such as high toxicity and insignificant selectivity between tumor and normal cells remarkably hinder their applications in clinical practices.3 With fast development of nanoscience and nanotechnology, emerged nanoscale drug delivery systems with good biocompatibility and unique functions allow controlled manners to deliver therapeutic substances for better pharmacokinetics and preferred bio-distribution.4 Micelles, self-assembled from amphiphilic polymers in water, represent a catalog of widely studied colloidal drug delivery systems.5 In particular, polymeric micelles provide stable storage for anti-cancer drugs, especially the poor water soluble ones, without any complicated chemical modification on the drug molecules. Their attractive characteristics such as small size (<100 nm) and large drug encapsulation content have endowed them growing scientific attention and great potential to be employed in clinical areas.6Design of appropriate delivery systems for drug transportation requires careful consideration of many parameters, including possible interactions of the carrier itself and surrounding environments. As for the micelles, one of the most concerned issues should be how to ensure safe transportation of the carrier system in blood stream post administration. Crosslinking or decrease of the critical micelle concentration (CMC) value should be optional approaches to stabilize micelle/carrier formations.7 However, the emphasis here should be focused on micelle scavenging by mononuclear phagocyte system (MPS) due to possible non-specific plasma protein adsorption or cell attachment during their transportation in circulation. On this basis, reducing the biomolecules interactions to micelle surfaces has been a crucial task for the development of polymeric micelles. As reported previously, PEGylation has been the common and most applied methods to keep nonfouling property of the carrier (micelles, nanoparticles, liposomes, microcapsules, etc.) surfaces.8 However, recent research demonstrated that repeated injections of the PEGylated nanomaterials led to rapid clearance from the blood and therefore to lose the expected long circulation in blood stream at the second dose, this effect was also known as the accelerated blood clearance (ABC) phenomenon.9 Ishida and co-workers performed series research works on the ABC phenomenon, and revealed that this phenomenon was mediated by the soluble serum factor anti-PEG immunoglobulin (Ig) M antibody (anti-PEG IgM); they also suggested that the repeating subunits of PEG would be an immunogenic epitopes and binding sites for anti-PEG IgM.10 Inspired by the external surface of the mammalian cell membrane, which rich in phospholipids bearing zwitterion headgroups,11 biocompatible zwitterionic segment poly(carboxybetaine methacrylate) was therefore introduced in this work as promising alternative. Schlenoff has well summarized the zwitterionic functionality and illustrated rational explanations of the antifouling mechanism of zwitterions/zwitterionic coatings:12 hydrated zwitterions contain a lot of water as other nonfouling surfaces; the zwitterionic surface possess low interfacial energy with water, which discourage adsorption driven by interfacial energy change; zwitterions show no disruption to H-bonding of the associated water nearby, thus high energy cost is required to disrupt this balance. Based on this background, utilization of the zwitterions facilitates generation of super low fouling surfaces on various carriers to achieved “stealth” delivery and long circulation in blood stream.13
Particular physiological characteristics of tumors should be another important issue to be addressed for design of drug delivery systems. Vascular pathophysiological change leads to an enhanced vascular permeability, providing opportunities to passive accumulate long-circulating micelles (in the size range of 30–200 nm) at solid tumors through the surrounding leaky vasculature by the EPR (enhanced permeability and retention) effect.14 On the other hand, tumor microenvironments enable design of smart drug delivery systems for cancer therapy. Specific endogenous stimuli factors, such as pH, glutathione concentration, and enzymes are particularly used to trigger release of payloads in desired manners.15 Typically, subtle difference of pH values existing in healthy tissues (∼7.4), extracellular environment of solid tumors (6.5–7.2) and cell organelles (e.g., endolysosomes) (4.0–5.5) could be exploited as triggering stimuli to stimulate functionalization of the micelles studied in this work, likely to help their cellular uptake, endosomal escape and disassembly inside tumor cells. Here, we specially introduce the 2,2-di(acryloyloxy-1-ethoxy)propane (ADA) and 4,4′-trimethylenedipiperidine (TMDP) pH-sensitive units for micelle fabrication. For the former, low pH would cleave its ketal structure.16 For the latter, which is an amine donor, could be protonated in acidic environments to realize sharp transition from hydrophobic to hydrophilic state to evoke water influx.17 Thus, employment of these units for micelle building could help stable storage of hydrophobic therapeutic agents at normal physiological pH (∼7.4), and further to allow drug release at lower pH through pH-induced micelle disassembly in the form of polymer cleavage and water influx.
In this work, we are aiming to fabricate zwitterionic micelles for cancer therapy, and functionalization of micelles in tumor sites is expected to be activated by tumor environmental pH conditions, as shown in Scheme 1. Generally, the multiple pH responsive amphiphilic polymer poly(carboxybetaine methacrylate)-block-poly[2,2-di(acryloyloxy-1-ethoxy)propane-co-4,4-trimethylene dipiperidine[pCBMA-b-p(ADA-TMDP)] was synthesized for building up of the zwitterionic micelles. Specially, wide-spectrum anti-cancer drug doxorubicin (DOX) was studied as the typical example for tumor treatment. At physiological pH (pH 7.4), the hydrophilic zwitterionic pCBMA tentacles would function as superior biointerfaces in the peripheral of the polymeric micelles, to realize stealth transportation and prolong circulation time in blood; the hydrophobic p(ADA-TMDP) units could provide excellent hydrophobic interspace for DOX encapsulation. The multiple pH responses of the zwitterionic micelles would be achieved step by step corresponding to practical pH gradient along their way in transportation: (1) outside the tumor cells (pH 6.8), zeta potential of the zwitterionic-micelles would be changed from slightly negative to positive to facilitate micelle–cell interactions and therefore to help cellular uptake; after endocytosis, acidic endolysosomes (pH 4.0–5.5) would (2) fast protonate TMDP segment to enable efficient micelle escape from endosomes, and (3) also trigger switches for micelles swelling/disassembly through acid cleavage of ketal bonds in ADA segments and consequent drug release. Investigations of the in vitro protein adsorption and pH-induced zeta potential changes would exam feasibility of micelle stealth delivery. Furthermore, bio-safety of the micelle carriers, DOX release profiles, cellular uptake behaviors as well as both in vitro and in vivo antitumor capabilities of the DOX encapsulated micelles would also be carefully studied.
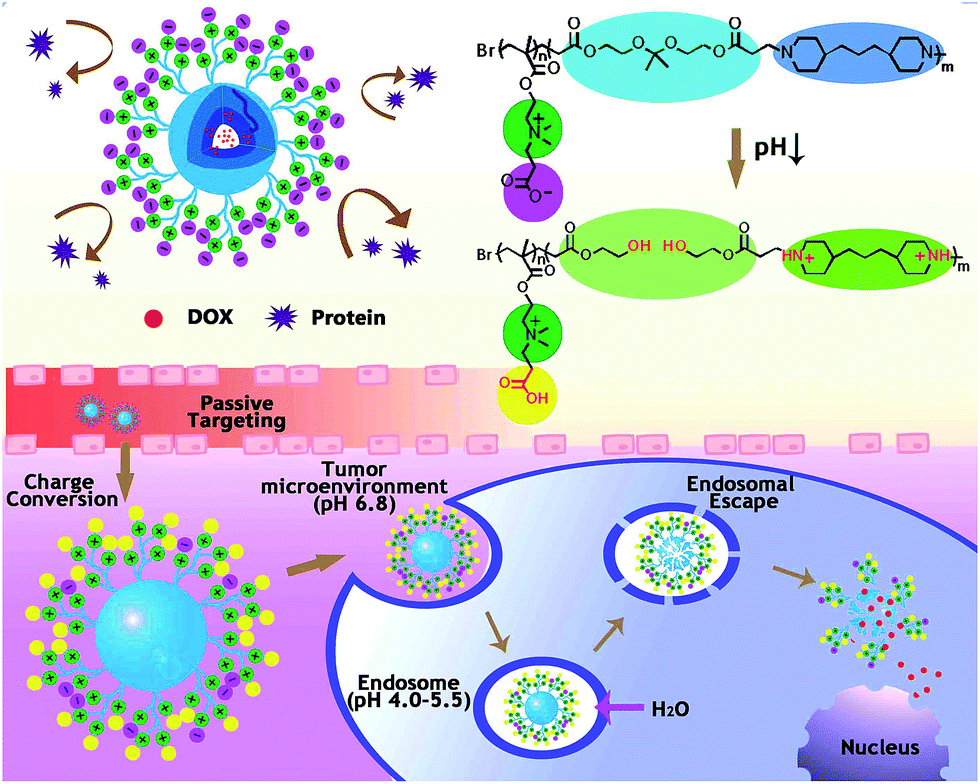 | ||
| Scheme 1 Multiple pH responses of zwitterionic micelles triggered by subtle microenvironmental pH along their way in transportation. | ||
2. Materials and methods
2.1 Materials
Cell counter kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). Dulbecco's minimum essential medium (DMEM) medium, penicillin, streptomycin and fetal bovine serum (FBS) were purchased from Hyclone (USA). HepG2 cell line (Human hepatoma carcinoma cell) and NIH 3T3 fibroblast cell line were purchased from Chinese Academy of Science Cell Bank for Type Culture Collection (Shanghai, China). β-Propiolactone (95%) and 2,2-dimethoxypropane (99%) were purchased from Aladdin (China). Doxorubicin hydrochloride (DOX·HCl) was purchased from Hisun Pharmaceutical (Zhejiang, China). Triethylamine (TEA) was purchased from Asta Tech Pharmaceutical (Chengdu, China). Bovine Serum Albumin (BSA), 2-hydroxyethylacrylate (96%), 2-(N,N′-dimethylamino)ethyl methacrylate (DMAEM, 99%), CuBr (99.99%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%), ethyl α-bromoisobutyrate (EBiB, 98%), 4,4′-trimethylenedipiperidine (TMDP, 97%), 2-(dimethylamino)ethyl methacrylate (DMAEMA, 98%), 4′,6-diamidino-2-phenylindole (DAPI), p-toluenesulfonic acid (p-TSA), methanol (MeOH), dimethyl formamide (DMF) and other solvents were purchased from Sigma-Aldrich. All the chemicals were used as received without further purification.2.2 Chemical synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent, and the product was obtained as a colorless oil.:1 mixture of MeOH–DMF was carefully transferred into the flask using a syringe under protection of nitrogen. Another purge cycles were carried out followed by addition of the catalyst CuBr (0.06 mmol). Then the flask was sealed, transferred to an oil bath at 60 °C. After reacting for 24 h, the resulting solution was dialyzed (MWCO 500–1000) against water to remove small molecules (e.g., free monomers, catalyst and ligand). Finally, the polymer were lyophilized and stored for further use.:1). After three cycles of degassing and nitrogen bubbling, CuBr (0.1 mmol) and 4,4′-trimethylenedipiperidine (1.476 mmol) was added into the reaction system and stirred for 48 h at 70 °C. The resulting solution was dialyzed against water (MWCO 3500) to remove unreacted monomers and other small molecules. Then centrifugation at 1000 rpm was used to remove the insoluble components. Finally, the polymer were lyophilized and stored for further use. The polymer pDMAEM-b-p(ADA-TMDP) was synthesized as the control material, where the DMAEM monomers were used to synthesize the hydrophilic component instead of zwitterionic monomers (CBMA). The reaction process was similar as proposed above.
:1) as the eluent, and the product was obtained as a colorless oil.:1 mixture of MeOH–DMF was carefully transferred into the flask using a syringe under protection of nitrogen. Another purge cycles were carried out followed by addition of the catalyst CuBr (0.06 mmol). Then the flask was sealed, transferred to an oil bath at 60 °C. After reacting for 24 h, the resulting solution was dialyzed (MWCO 500–1000) against water to remove small molecules (e.g., free monomers, catalyst and ligand). Finally, the polymer were lyophilized and stored for further use.:1). After three cycles of degassing and nitrogen bubbling, CuBr (0.1 mmol) and 4,4′-trimethylenedipiperidine (1.476 mmol) was added into the reaction system and stirred for 48 h at 70 °C. The resulting solution was dialyzed against water (MWCO 3500) to remove unreacted monomers and other small molecules. Then centrifugation at 1000 rpm was used to remove the insoluble components. Finally, the polymer were lyophilized and stored for further use. The polymer pDMAEM-b-p(ADA-TMDP) was synthesized as the control material, where the DMAEM monomers were used to synthesize the hydrophilic component instead of zwitterionic monomers (CBMA). The reaction process was similar as proposed above.2.3 Micelle preparation and DOX loading
Diafiltration method was applied to prepare micelles in our work. Briefly, 0.5 mL DMF solution containing 1.0 mg pCBMA-b-p(ADA-TMDP) and 0.2 mg DOX together with 3 equiv. of triethylamine (TEA) was slowly added into a 10 mL aqueous solution under ultrasound exposure. Then the micelle suspension was dialyzed against water (MWCO = 3500) for 24 h in dark to remove free DOX and DMF. Obtained zwitterionic micelles with encapsulated DOX were noted as DOX/Z-micelles. The blank zwitterionic micelles (Z-micelles) without addition of DOX and TEA were prepared by the same procedure. On the other hand, non-zwitterionic micelles were prepared, where the material pDMAEM-b-p(ADA-TMDP) was used as building blocks to fabricate the micelles, corresponding micelles with/without DOX loading were noted as DOX/C-micelles and C-micelles respectively.DOX loading efficiency and content were studied by fluorescence spectrometer (Hitachi F-7000, Japan). Briefly, DOX/micelles were diluted in DMF, fluorescence signals of DOX molecules were then detected (λex = 480 nm, λem = 590 nm). Loaded DOX was therefore quantified by reference to the standard calibration curves obtained from DOX solutions with known concentrations (0–10 μg mL−1).
Drug loading efficiency (DLE) and drug loading content (DLC) were calculated according to the following formula:

where the WDOX was the weight of DOX in micelles, WTotal was the weight of DOX-loaded micelles.

2.4 Characterizations of the micelles
Micelle morphologies were investigated using Transmission Electron Microscope (JEM-100CX, Japan). In general, diluted micelle suspensions were dropped on silicon wafers or carbon-coated copper grids, air dried before observation. Specially, micelles were phosphate–tungstic acid-stained before TEM tests.
2.5 Protein adsorption
Protein adsorption on the micelles was studied, and the bovine serum albumin (BSA, 66 kDa) was used as the model protein in this work. In order to quantify the adsorbed proteins, Z-micelles and C-micelles were incubated with BSA solution in pH 7.4 and pH 6.8 buffer solutions at 37 °C under shaking. The final concentrations of the micelle and BSA suspension/solution were 0.15 mg mL−1 and 0.30 mg mL−1 respectively. After incubation for 1, 2 and 4 h, each mixture was centrifuged at 13000g for 15 min to precipitate the protein-adsorbed micelles. BSA concentration in the supernatant was measured by a BCA Protein Assay Kit (BD, Rockford IL, USA) with bovine serum albumin as a standard. 25 μL samples were mixed with 200 μL BCA working reagent in 96-well plates at 37 °C for 4 h. Absorbance was measured at 562 nm with a microplate reader (Thermo Scientific, VARIOSKAN FLASH, USA).
2.6 In vitro drug release
The release profiles of DOX from DOX/Z-micelles were studied in three buffers with varied pH values: (1) PBS buffer (pH = 7.4, 0.01 M), (2) pH = 6.8 acetate buffer (0.01 M) and pH = 5.0 acetate buffer (0.01 M). In brief, 0.5 mg DOX loaded micelles were re-suspended in 1 mL distilled water and transferred to a dialysis bag (MWCO 500–1000). The dialysis bag was immersed in 25 mL buffer solution, and incubated at 37 °C with shaking. At desired time points, 1 mL of dialyzate containing released DOX molecules was taken out for fluorescence measurement. Equal volume of corresponding buffer solution was added to replenish the incubation buffer. Fluorescence intensity of release DOX was detected using fluorescence spectrophotometer (Hitachi F-7000, Japan) (λex = 480 nm, λem = 590 nm, slit width 5 nm). Standard calibration curve of DOX solutions with known concentrations (0–10 μg mL−1) was used to normalize obtained data. All the measurements were carried out in triplicate.2.7 Cell culture and animal modeling
Hepatoma cells (HepG2) and NIH 3T3 fibroblast cells were cultured in Dulbecco's minimum essential medium (DMEM) medium at 37 °C in a humidity atmosphere with 5% CO2. The medium was supplemented with 10% fetal bovine serum, 100 μg mL−1 streptomycin and 100 IU mL−1 penicillin. Female BALB/c nude mice (3 week old, 18–20 g) were purchased from the Dossy Biological technology Co., Ltd. (Chengdu, China). 90% confluence HepG2 cells were harvested and re-suspended in PBS. 8 × 106 HepG2 cells were subcutaneously injected into the right backs of BALB/c nude mice to establish xenograft tumor models. Tumor length (L) and width (W) were measured and used to calculate the tumor volume (V) according to the formula V = L × W2/2.21 Tumor-bearing mice were injected with testing samples (saline, free DOX and DOX/micelles) via tail vein when the tumor size reached about 150 mm3. All animal experiments were complied with the Experimental Animal Administrative Committee of Sichuan University. All animals were adapted to the animal facility for one week before experiment. All animals were fed under conditions of 25 °C and 55% of humidity and allowed free access to food and sterile water. Nude mice were fed under a germ-free environment.2.8 Cytotoxicity and cancer cell growth inhibition
The cytotoxicity of blank micelles against HepG2 cells or 3T3 cells was evaluated with CCK-8 (cell counting kit-8) assay.22 Cells were seeded onto 96-well plates (5 × 103 cells per well). After culture for 24 h, cells were exposed to fresh medium containing series concentration of micelles (Z-micelles or C-micelles). After incubation for 24 h and 48 h, CCK-8 solution (10 μL) was added into each well and further incubated for 2 h at 37 °C. A microplate reader (Thermo Scientific, VARIOSKAN FLASH, USA) was employed to measure the absorbance at 450 nm, and the cell viability was calculated according to the ratio between sample absorbance and that of negative controls without treatment of micelles.To evaluate in vitro cancer cell inhibition ability of DOX loaded Z-micelles (DOX/Z-micelles), half-maximal inhibitory concentration IC50 was investigated using the CCK-8 assay. At the same time, DOX·HCl and DOX/C-micelles were used as controls. Testing samples containing different DOX contents were incubated with HepG2 or 3T3 cells for certain time, and related cell viabilities were detected. Specially, IC50 values of each group were analyzed with the Graph Pad Prism 5 software.
2.9 Cellular uptake and endosomal escape
Confocal laser scanning microscopy (CLSM, Leica TCP SP5) and flow cytometry were used to investigate the cellular uptake of DOX loaded micelles. For CLSM tests, HepG2 cells were seeded on 35 mm glass bottom cell culture dishes (NEST®, Cat. no. 801002) at a density of 1 × 104 cells per well for 24 h. After removing the culture medium, cells were incubated with the culture medium containing DOX/Z-micelles (DOX concentration 5 μg mL−1). DOX·HCl was used as control. After incubation for certain time (0.5 h, 2 h, 6 h), the culture medium was discarded and cells were rinsed with PBS. The cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) and 300 μL PBS was added to replace the culture medium. CLSM was subsequently used to record fluorescent images of the cells. For flow cytometer tests, HepG2 cells (2 × 105 per well) were seeded in 6-well plates and allowed to adhere for 24 h. DOX/Z-micelles and DOX/C-micelles were diluted in culture medium to reach a final DOX concentration of 2.5 μg mL−1. After incubation for 4 h at 37 °C, culture medium was carefully removed and thrice washed with cold PBS. Then the cells were collected and re-suspended in 0.3 mL PBS buffer after twice centrifugation at 1000 rpm for 5 min. Flow cytometry (BectoDickinson, USA) was consequent applied to detect the micelle uptake rates by HepG2 cells (λex = 480 nm, λem = 590 nm). Each test was repeated in triplicate.For endosomal escape study, LysoTracker® Green DND-26 (100 nM) was incubated with the seeded cells for 1 hour at 37 °C to stain the acidic organelles (e.g., lysosomes and endosomes). After that, these cells were incubated with DOX/Z-micelles (DOX concentration 5 μg mL−1) for certain time intervals, carefully washed and observed under CLSM.
2.10 In vivo antitumor efficacy
When the tumor volume reached about 150 mm3, tumor-bearing BALB/c nude mice were randomly divided into four groups, and administrated with 200 μL testing samples (DOX/Z-micelles, DOX/C-micelles, DOX·HCl, saline) via tail veins. Specifically, DOX containing formulations were injected into mice at a dosage of 2.5 mg DOX per kg body weight. The mice were treated with all the formulations for 7 times (at day 1, 4, 7, 11, 15, 19, 23) in the whole duration. Meanwhile, tumor volume and body weight of each mouse were carefully recorded. The tumor inhibition rate was calculated by the equation:23
2.11 Histological and immunohistochemical analyses
All the mice were sacrificed at 35th day since first time injection. Tumor, liver, heart, spleen, lung and kidney were excised and fixed with 4% formaldehyde. After embedded in paraffin, they were cut into 5 μm thick slides. Hematoxylin and eosin (H&E) staining was performed for histopathological evaluation. Platelet/endothelial cell adhesion molecule-1 (CD31) staining and monoclonal antibody Ki-67 response test were performed to detect tumor cell proliferation activity, expressing potential change in either tumor vasculature or cell proliferation. Tumor apoptosis level was displayed by the terminal deoxynucleotidyl transferase mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay.2.12 Statistical analysis
Student's t-test was used to address statistical significance of the experimental groups. The form of mean ± SD was used to represent obtained data, with p < 0.05 indicating statistical significance.3. Results and discussion
3.1 Synthesis of pCBMA-b-p(ADA-TMDP)
ADA was obtained as a colorless oil according to previous research. Its chemical structure was confirmed by 1H-NMR spectrum (CDCl3, 400 MHz, Bruker), as shown in Fig. S1.† CBMA was obtained as a pale powder after purification. Its chemical structure was confirmed by 1H-NMR spectrum (D2O, 400 MHz, Bruker), as shown in Fig. 1a. Polymerization of the CBMA monomers was achieved by EBiB-initiated ATRP. After completion, the disappearance of the vinyl signals of CBMA at 5.6 and 6.0 ppm indicated the successful synthesis of pCBMA, as shown in Fig. 1b. In order to graft the hydrophobic components onto the zwitterionic units, another ATRP and subsequent Michael addition were carried out. Freeze-dried pCBMA was used here as the initiator for ATRP, and the other two pH sensitive components, ADA and TMDP, were introduced in one pot reaction. The 1H-NMR spectrum (CDCl3, 600 MHz, Bruker) and GPC were studied to confirm the successful obtainment of the materials. Fig. 1c showed characteristic peaks corresponding to pCBMA (OCH2, δ = 4.02–4.07 ppm), ADA (COOCH2, δ = 4.22–4.41 ppm) and the TMDP (CH2, δ = 1.21–1.27 ppm) units in 1H-NMR spectrum of synthesized pCBMA-b-p(ADA-TMDP), verifying successful reactions between the hydrophilic zwitterions and hydrophobic p(ADA-TMDP) components. Similarly, chemical structure of synthesized pDMAEM-block-p(ADA-TMDP) was also confirmed by 1H-NMR measurement, as shown in Fig. 1d. The molecular weights of pCBMA segment and the pCBMA-b-p(ADA-TMDP) were detected by Gel Permeation Chromatography (HLC-8320 GPC, Tosoh Corporation) measurements, under the following chromatographic conditions: column, TSK gel Super AWM-H; column temperature, 40 °C; eluent, DMF; flow rate, 0.4 mL min−1; detector, UV254 nm. The results showed that molecular weights were found to be 3.5 kD (PDI = 1.84) for pCBMA, 3.5 kD (PDI = 1.32) for pDMAEM, 15.3 kD (PDI = 1.24) for pCBMA-b-p(ADA-TMDP) and 16.1 kD (PDI = 1.20) for pDMAEM-b-p(ADA-TMDP) respectively(Table S1†). Besides, it also revealed a ratio of 2:3:3 of the repeating subunits (CBMA:ADA:TMDP). This result was in high accordance with the 1H-NMR analysis, of which the ratio of repeating subunits was estimated by the integrals of characteristic peak of each unit.
 | ||
| Fig. 1 1H-NMR spectra of (a) CBMA (D2O, 400 MHz), (b) pCBMA (D2O, 400 MHz), (c) pCBMA-b-p(ADA-TMDP) (CDCl3, 600 MHz) and (d) pDMAEM-b-p(ADA-TMDP) (CDCl3, 600 MHz). | ||
3.2 Characterization of micelles
Hydrodynamic diameters of formed micelles were detected by DLS measurements, as shown in Fig. 2a. Number average diameter of Z-micelles was found to be 69 ± 20 nm. After DOX encapsulation, their hydrodynamic diameter was increased to be 77 ± 19 nm. This result was highly consistent with TEM results. As shown in Fig. 2b, prepared DOX/Z-micelles were visualized as black particles under TEM observation, and their average diameter was found to be 75 ± 13 nm, according to random measurements of at least 30 micelles per sample of the TEM images by using the software Image-Pro Plus v 6.0. Similar characterizations of C-micelles were also studied, as shown in Fig. S2.† In general, number average diameters by DLS tests were found to be 68 ± 15 nm for C-micelles and 76 ± 19 nm for DOX/C-micelles. Besides, average diameter of the DOX/C-micelles was 78 ± 9 nm as represented in TEM image. | ||
| Fig. 2 Characterization of the Z-micelles and DOX/Z-micelles. (a) Size distribution of Z-micelles (red) and DOX/Z-micelles (blue) in PBS solution. (b) TEM images of DOX/Z-micelles. *Scale bar measures 250 nm. (c) Critical micelle concentration of the Z-micelles. | ||
CMC is one of the most important properties of the micelles, representing the concentration at which the amphiphilic polymers start to form micelles. In our work, the critical micelle concentration (CMC) of the Z-micelles was found to be 0.046 mg mL−1 (Fig. 2c), while CMC of the C-micelles was 0.053 mg mL−1 (Fig. S2c†). The relatively low CMC values could ensure that these micelles keep their aggregated states after intravenously injected into blood stream.7a
 | ||
| Fig. 3 pH sensitivity of the micelles. (a) Dynamic light scattering intensity variations of the Z-micelles after exposed to a series of pH buffer solution for 4 h. (b) Zeta potential changes after 4 h of incubation in different pH buffer solutions. | ||
3.3 Protein adsorption
In theory, non-fouling carrier surfaces would greatly reduce non-specific protein adsorption and thus to expect prolonged carrier circulation time in blood stream. To clarify the antifouling property of the Z-micelles, C-micelles with molecular structures highly similar to that of the Z-micelles except lack of terminal carboxylation with β-propiolactone were used to as controls to research the protein (BSA) adsorption, as shown in the Fig. 4a. Generally, at physiological pH condition (pH 7.4), both of the two micelle systems exhibited low protein adsorption amounts after 4 hour incubation (<18.1%) similar to other zwitterionic polymer functionalized nanocarriers.29 In particular, the Z-micelles exhibited a smaller proportion of protein adsorption than the C-micelles in pH 7.4 buffer solution in the whole incubation duration. Significant difference of protein adsorption percentages between the two micelle systems was obvious after 2 hours incubation, as one could find that 10.0% of proteins were adsorbed on Z-micelles compared with 14.7% of proteins adsorbed on C-micelles. Furthermore, the significant difference was still existed after incubation for 4 h, as 15.6% proteins were adsorbed on Z-micelles and 18.1% on C-micelles. The excellent antifouling property of the Z-micelles was attributed to the well hydrated zwitterionic tentacles with fairly low interfacial energy to adsorb protein.12 On the contrary, when the environmental pH decreased to be 6.8, more proteins were adsorbed on Z-micelles (22.3%) than that of the C-micelles (18.3%), as shown in the Fig. 4b. These phenomena could be explained by the pH-induced zeta potential changes of fabricated micelles. For the Z-micelles, they were slightly negatively charged (−2.37 mV) at pH 7.4, thus possible electrostatic interactions between Z-micelles and negative charged proteins were greatly suppressed. At pH 6.8, surface charge of the Z-micelles became positive (+4.41 mV) and consequently to enhance micelle–protein interaction. Compared with Z-micelles, the C-micelles with tentacles terminated with tertiary amines behaved different zeta potentials in varied pH conditions. As discussed above, C-micelles possessed negative charges on their tentacles at pH 6.8, implying limited ability to interact with BSA macromolecules. | ||
| Fig. 4 Protein adsorption on Z-micelles and C-micelles at (a) pH 7.4 and (b) pH 6.8 for 1 h, 2 h, 4 h (means ± SD, n = 3, *p < 0.05). | ||
3.4 Drug release profiles
The in vitro drug release behaviors of DOX/Z-micelles were investigated under different pH conditions, as shown in Fig. 5. At pH 5.0, a rapid release of the encapsulated DOX from the DOX/Z-micelles was found in the beginning 12 hours. After 12 hours, accumulated release of DOX was found to be 22.7%. Later, the release rate of DOX was slowed slightly. 34.8%, 54.3% and 65.4% of encapsulated DOX molecules were detected in the incubation medium after 24, 48 and 72 hours of incubation. Further increase the incubation time to 96 h, the accumulated release of DOX was remained to be 72.7%. On the other hand, incubating the DOX/Z-micelles in other two buffer systems illustrated remarkable decreased release rates. Only 11.05% of encapsulated DOX was released after 24 hours and 22.3% was further accumulated within 96 h in the pH 7.4 buffer solution. Similarly, 27.6% of accumulated DOX was found over the release study period in the pH 6.8 buffer. These different release rates were attributed to the pH related micelles size/stability changes, as discussed above (Fig. 3a). These results revealed that the micelles possessing zwitterionic tentacles could response to varied external pH and therefore to selectively govern their drug release profiles. Promisingly, the Z-micelles studied in our work could enable safe storage of DOX molecules in their inner cavities at physiological environment (pH 7.4), and then to release their payloads inside tumor cells (pH ∼ 5.0) predominantly. | ||
| Fig. 5 DOX release profiles of the DOX/Z-micelles in pH 7.4, 6.8, and 5.0 at 37 °C (means ± SD, n = 3, **p < 0.001). | ||
3.5 In vitro cytotoxicity study
In this work, we propose the zwitterionic micelles serve as novel nanocarriers for cancer treatment, careful study of their in vitro biosafety was therefore of great importance prior to possible applications in living bodies. As shown in Fig. 6a, c and S3,† the cytotoxicity of the micelle carriers against HepG2 and 3T3 cell lines was evaluated. For the micelles without drug loading, both of the Z-micelles and C-micelles showed good biocompatibility co-cultured with either HepG2 cells or 3T3 cells, as proven by the high cell viability (∼100%) cultured for 24 h and 48 h. IC50 values which demonstrate in vitro cell inhibition effect of micelles after drug loading were also examined, as shown in Fig. 6b, d and e. Generally, growth of the two cell lines showed a DOX concentration-dependent manner, where the higher DOX concentration demonstrated the lower cell viability. After 36 h of incubation, IC50 values of free DOX (DOX·HCl) were detected to be 0.27 μg mL−1 for HepG2 cells and 0.15 μg mL−1 for 3T3 cells. The relatively small IC50 value of 3T3 cells verified the fact that cancer cells possess drug resistance ability.30 Incorporating the biocompatible carriers, a tiny increase of the IC50 values of prepared DOX/micelles was detected. As illustrated in our work, the IC50 values of Z-micelles were 0.76 and 0.52 μg mL−1 for HepG2 and 3T3 cell lines; similarly, IC50 values of C-micelles were found to be 0.98 and 0.77 μg mL−1 for HepG2 and 3T3 cell lines, respectively. Compared with the cell inhibition results of free DOX (DOX·HCl), proper explanation here could be attributed to the steady carrier formations (micelles), which limited burst release of DOX molecules and thus to slow down their diffusion and penetration into nuclei.20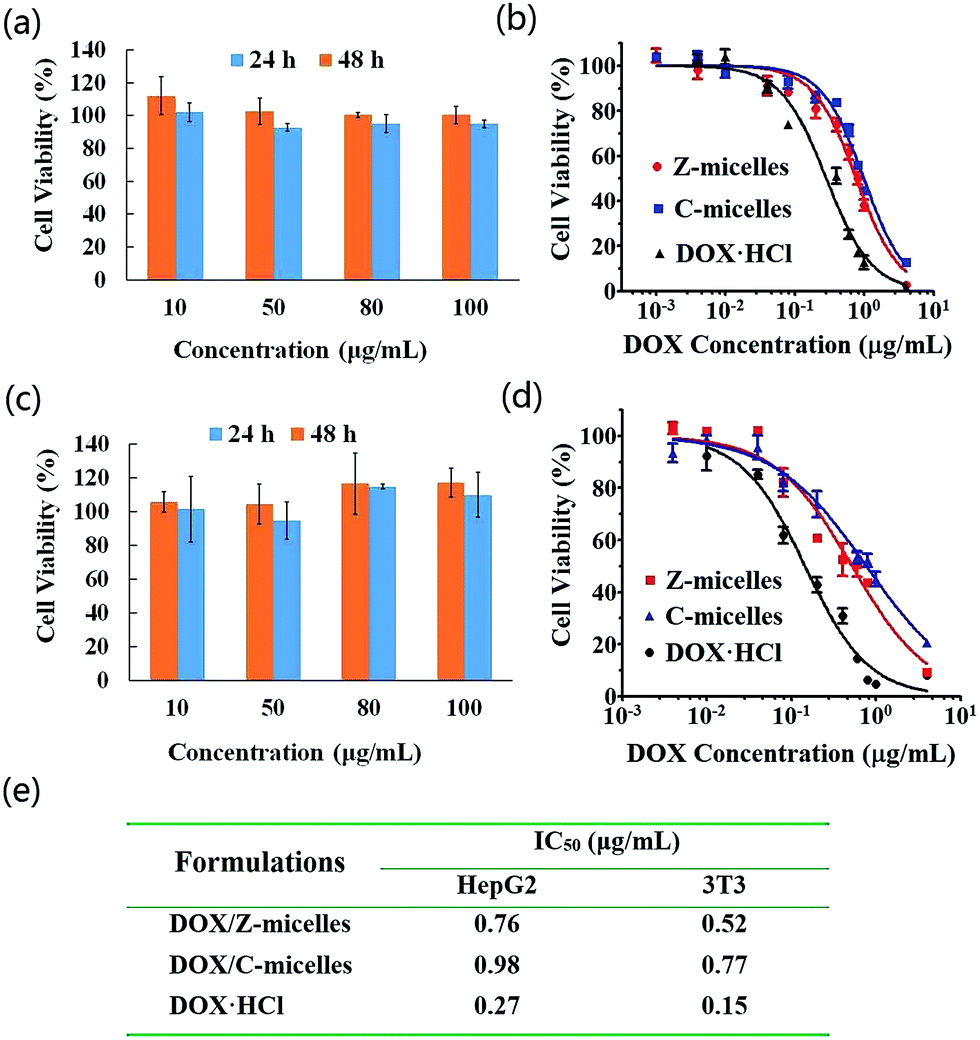 | ||
| Fig. 6 Cytotoxicity of blank Z-micelles to (a) HepG2 cells and (c) 3T3 cells after incubation for 24 h and 48 h. In vitro cytotoxicity of DOX-containing formulations to (b) HepG2 cells and (d) 3T3 cells co-cultivating for 36 h. (e) IC50 values of all the DOX-containing formulations. | ||
3.6 Cellular uptake and endosomal escape
Both flow cytometry and confocal laser scanning microscopy (CLSM) were used to detect the cellular uptake behaviors of the DOX/Z-micelles and DOX/C-micelles. Considering the DOX was loaded in micelles, fluorescent signals of the DOX molecules were recorded directly. First, the flow cytometry measurement was applied to investigate possible difference of the HepG2 cellular uptake of the two micelle systems (DOX/Z-micelles and DOX/C-micelles), as shown in Fig. 7a. In this test, the cancer cells without micelle incubation were used as negative control. The percentage of cells that internalized DOX was quantified into diagram, as shown in Fig. 7b. Percentage of fluorescent cells with internalized DOX/Z-micelles was measured to be 90.4 ± 0.67, in contrast to 62.9 ± 3.6 of the DOX/C-micelles.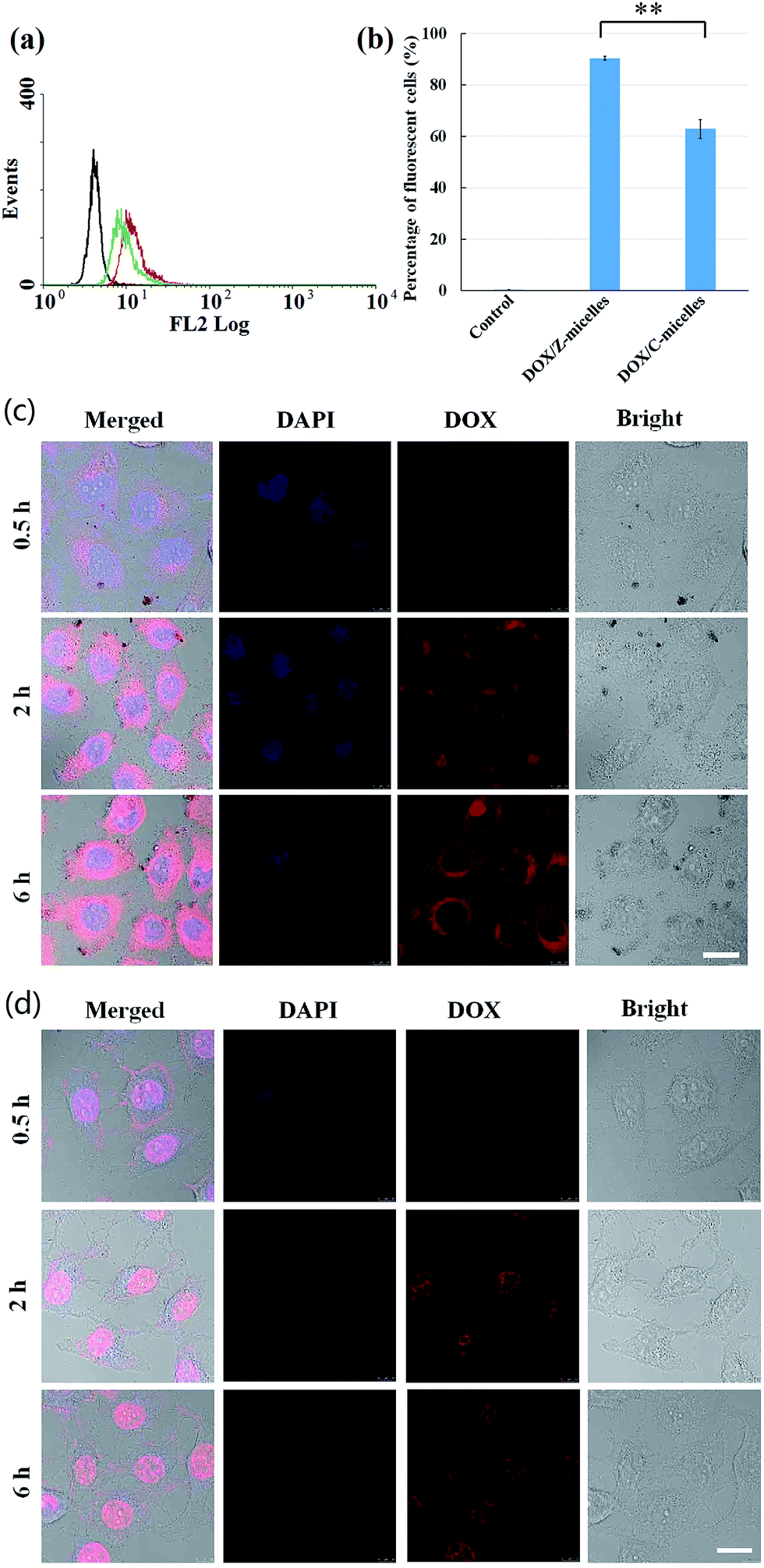 | ||
| Fig. 7 Cellular uptake and intracellular drug delivery to HepG2 tumor cell line of DOX containing formulations: (a) flow cytometry measured the DOX accumulation in HepG2 tumor cells after co-cultured with DOX/Z-micelles, DOX/C-micelles (2.5 μg mL−1 DOX) for 3 h. (b) Percentages of fluorescent cells of each group (means ± SD, n = 3,**p < 0.001). (c) CLSM of DOX/Z-micelles and (d) DOX·HCl (5 μg mL−1 DOX) at 0.5 h, 2 h, 6 h. DOX channel (red), DAPI stained nucleus channel (blue), bright images and overlay of previous images. Scale bar measures 20 μm. | ||
The flow cytometry measurement revealed a 1.44-fold faster uptake of the DOX/Z-micelles than that of the DOX/C-micelles. Consequently, the CLSM study was carried out to visualize cellular internalization of the DOX/Z-micelles over time (Fig. 7c). In this study, the free DOX (DOX·HCl) was used as control (Fig. 7d). In general, DOX·HCl showed a faster cellular uptake rate than the DOX/Z-micelles. At 0.5 h, HepG2 cells treated with DOX·HCl exhibited a stronger fluorescent intensity than these cells treated with DOX/Z-micelles. Moreover, only a small portion of the internalized DOX·HCl was observed in cytoplasm, in comparison with the DOX/Z-micelles which were mostly located in cytoplasm. This rapid accumulation of DOX·HCl in nuclei was because of the passive diffusion of the DOX molecules, as previous study of Gu's group.20 With increase of the incubation time, more DOX molecules, either from free DOX formulation or DOX/Z-micelles, were detected inside cells, as proven by the much brighter red fluorescence signals. Furthermore, for the cells treated with DOX/Z-micelles, the DOX signals were observed both in cytoplasm and nuclei at 6 h. This result could be explained as the fact that the released DOX from Z-micelles was gradually distributed in the cytoplasm and then diffused into the nuclei. In addition, the similar cellular internalization process with DOX/Z-micelles was observed in the DOX/C-micelles study (Fig. S4†). It is worth mentioning, this slow passive release/diffusion of the DOX from micelles was in high accordance with our above cytotoxicity assay, in which the larger IC50 value of encapsulated DOX revealed limitation of the micelle structures (Fig. 6d).
After cellular internalization of the DOX/Z-micelles, the efficient endosomal escape was critical process for the released DOX enter into nuclei. Previous studies have reported the strategies using environmental pH to protonate the poly(β-amino ester) and thus to cause water influx and disruption of endolysosomal membrane.31 In this work, pH-induced protonation of pTMDP segment was proposed to help DOX molecules escape from endolysosomes to cytosol. Here, the time-dependent fluorescent signal changes of specific acidic organelles (green) and DOX/Z-micelles (red) were monitored, as shown in Fig. 8. After incubating for 2 h, the drug was mainly concentrated in the acidic organelles labeled with Lysotracker, as DOX signals (red) were almost overlaid with Lysotracker signals (green). With the increase of incubation time, overlay of the two fluorescent signal channels faded out. From 6 h to 18 h, the DOX molecules were gradually located outside of endolysosomes and entered into cytosol and nuclei, suggesting the efficient drug release and endosomal escape.
 | ||
| Fig. 8 CLSM of DOX/Z-micelles (5 μg mL−1 DOX) at 2 h, 6 h, and 18 h. DOX channel (red), lysotracker green stained acidic organelles channel (green), and overlay of previous images. Scale bar measures 25 μm. | ||
3.7 In vivo inhibition efficiency
In order to study the in vivo tumor inhibition activity of the DOX/Z-micelles, human-derived liver tumor models were established on BALB/c nude mice. The tumor-bearing mice were divided into four groups (n = 5) randomly and administrated with saline, DOX·HCl, DOX/Z-micelles and DOX/C-micelles through the tail veins, respectively. In order to avoid possible side effect of the drug, a general principle of multiple-low-dose treatment was applied. In brief, 7 times of injection (2.5 mg DOX per kg) with the intervals for 3 or 4 days was used in our work. The in vivo antitumor efficacy was evaluated by monitoring tumor size changes over the experimental period. Obviously, the saline group showed fastest tumor growth in comparison with other three groups treated with DOX-containing formulations (Fig. 9a). The average tumor volume of 2186 ± 976 mm3 was measured on the 35th day. On the contrary, DOX/Z-micelles, DOX/C-micelles and DOX·HCl significantly reduced tumor growth, demonstrating less than 30% of tumor volume as compared with that of the saline group. Fig. 9b showed the tumor growth inhibition quantitatively. It was clear that the DOX/Z-micelles achieved a higher tumor inhibition rate (89.3%) than DOX·HCl (69.9%) and DOX/C-micelles (60.2%). It should be noticed that the DOX/Z-micelles achieved a better therapeutic efficacy than that of other two DOX-containing formulations, although the DOX·HCl demonstrated the best cancer cell growth inhibition in vitro. This would be explained as the higher amount of DOX that reached the tumor site via EPR effect.32 As for the different in vivo antitumor efficiencies of the two DOX/micelles systems, reasonable explanation here should be: on one hand, the zwitterionic tentacles of fabricated micelles could benefit safe delivery of anticancer drugs in circulation (Fig. 4) and pH-induced surface charge transition could enable prior uptake of these zwitterionic micelles by cancer cells (Fig. 3b and 7a). On the other hand, these large accumulated DOX/Z-micelles in tumor sites could respond to tumor microenvironment (e.g., pH 5) and thus to realize fast release (Fig. 5) as well as endosomal escape of the anticancer drugs (Fig. 8). In the whole duration of trails, body weight of each mouse was recorded. As shown in Fig. 9c, no significant difference was observed in variations of body weight after treatment, which implied no serious side effect was found after treatment of the DOX-containing formulations over 35 days in our work.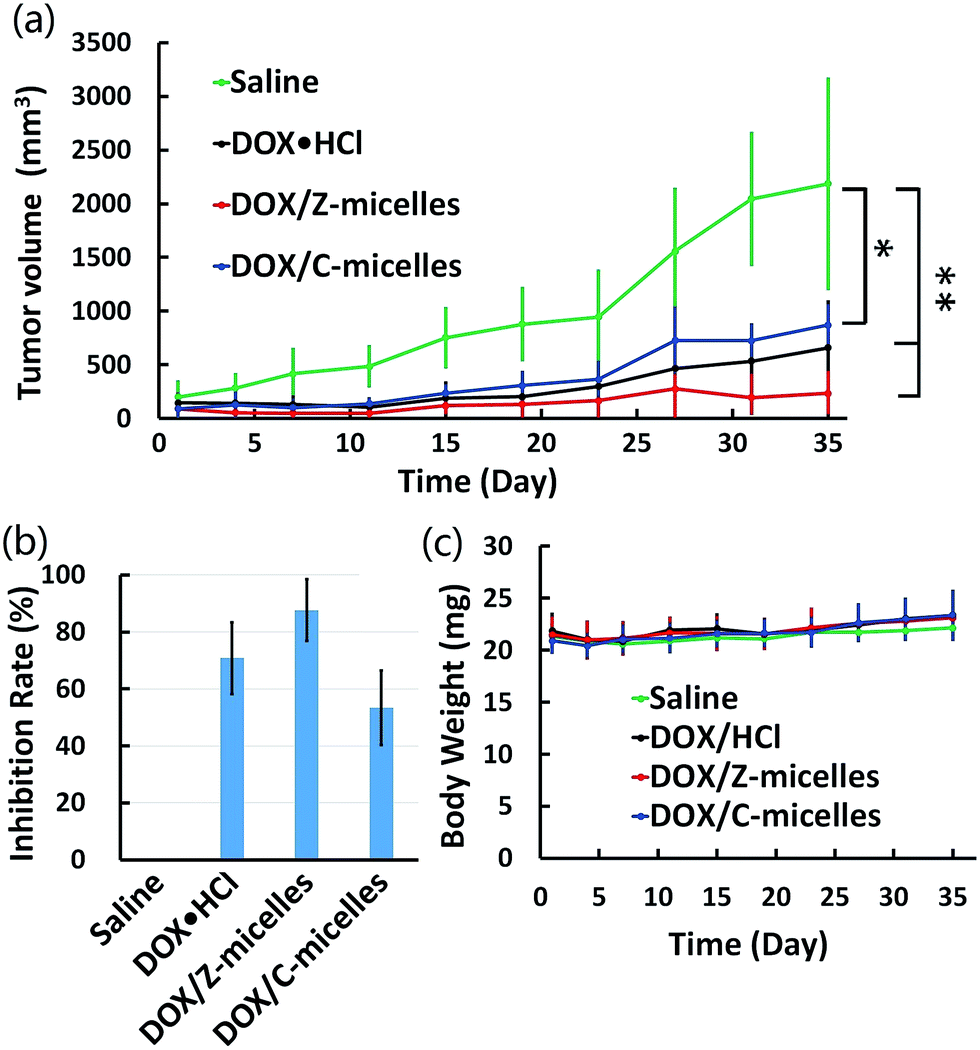 | ||
| Fig. 9 In vivo antitumor efficacy of DOX containing formulations in HepG2 cells tumor-bearing mice. (a) Tumor volume changes due to intravenous injection with saline (control), DOX·HCl, DOX/Z-micelles, DOX/C-micelles (2.5 mg DOX per kg, n = 5,*p < 0.05, **p < 0.001). (b) Tumor growth inhibition rate of each group after administered over 35 days. (c) Body weight changes of tumor-bearing BALB/c nude mice over 35 days. | ||
3.8 Histological and immunohistochemical studies
Besides measurements of the tumor volume changes over drug treatment period, immunohistochemical analysis was carried out to investigate anticancer activities of the DOX/Z-micelles. As shown in Fig. 10a, immunohistochemical studies of excised tumors were analyzed to further illustrate potential antitumor efficacy and tumor suppression mechanism. In addition to H&E staining for basic histopathological evaluation, CD31 (as a marker of endothelium33), Ki-67(cell proliferation34) and TUNEL (in situ cell apoptosis35) stainings were used to investigate specific tumor cell activities. In the H&E assay (Fig. 9a, 1st row), the DOX/Z-micelle group revealed the most apoptosis and necrosis regions of the tumor tissues after treatment with DOX-containing formulations, illustrating large amount of damaged cells with incomplete cellular structures.36 Optical density analysis about CD31 and Ki-67 stainings (Fig. 10a, 2nd and 3rd rows) indicated that the DOX/Z-micelles group showed much less angiogenesis and tumor cell proliferation than DOX·HCl and DOX/C-micelles groups in tumors, which demonstrated higher abilities of tumor inhibition of the DOX/Z-micelles. The tumor microvessel density (MVD) was calculated as the ratio of CD31-positive area (brown) to total tissue area (Fig. 10b). In our work, the MVD of DOX/Z-micelles group was 1.75 ± 0.83, which was obviously smaller than saline (11.3 ± 0.47), DOX·HCl (5.8 ± 0.83) and DOX/C-micelles (8.5 ± 1.11) groups. The results indicated that the stealth zwitterionic micelles could significantly inhibit angiogenesis than other three groups, even the DOX·HCl. Optical density (OD) of Ki-67 positive area (brown) was normalized to total tissue area, as shown in Fig. 10c. Distinct lower density (0.2991 ± 0.00096) was found in DOX/Z-micelles group than saline, DOX·HCl and DOX/C-micelle groups, demonstrating the lowest tumor cell proliferation activity. In addition, the level of tumor cell apoptosis (brown) (Fig. 10a, 4th row, Fig. 10d) caused by DOX/C-micelles and DOX·HCl treatments were 15 ± 1.63% and 29.3 ± 2.86% respectively. By contrast, much higher apoptosis level (64.3 ± 3.30%) was observed in DOX/Z-micelle treated group, indicating the most significant anticancer efficacy of DOX/Z-micelles. Immunohistochemical studies were consistent with the in vivo antitumor efficacy, and these results confirmed that the DOX/Z-micelles could inhibit solid tumor growth by decreasing tumor cell proliferation and inducing tumor cell apoptosis. | ||
| Fig. 10 The H&E, CD31, Ki-67, and TUNEL assays for tumor tissues of various treatment groups (tumor: ×400). (a) The apoptosis and necrosis areas labeled with blue in H&E assay. CD31-positive vessels, Ki-67-positive cells and TUNEL-positive were stained brown. Scale bar measures 100 μm. (b) The ratio of CD31-positive to total area of each group was calculated as tumor microvessel density (MVD). (c) Measurement of Ki-67-positive OD (optical density) of each group. (d) The percentage of TUNEL positive cells of tumor tissues in each group (means ± SD, **p < 0.001, compared to saline). | ||
The H&E examination of main organs (heat, liver, spleen, lung and kidney) was applied to determine possible toxicities caused by administrated DOX formations, as shown in Fig. 11. In our work, the tumor-bearing BALB/c nude mice were treated with DOX containing formulations complying with a multiple-low-dose (2.5 mg DOX per kg, 7 times) principle. Consequently, severe heart damage, which would appear mostly in large dose DOX·HCl (e.g., 5 mg DOX per kg) treated group,37 was avoided in all the four treated groups in our work. However, side effect of DOX·HCl was still found in the kidney after injection of DOX·HCl directly. As shown in Fig. 11 (4th row), glomerular swelling was obvious (pointed out by red arrows), demonstrating the nephrotoxicity induced by DOX·HCl treatment.38 The other groups showed no obvious damages in this organ. Such low side effects to normal tissues/organs of DOX/micelles suggested that encapsulation of the DOX molecules in nanoscale micelles could prevent possible damage to internal organs. The relatively low cytotoxicity of DOX/Z-micelles verified the multiple pH responsive zwitterionic micelles were low toxic and biocompatible. Combining the in vitro and in vivo antitumor activities studied above, one could conclude that the DOX/Z-micelle systems would be promising candidates for anti-cancer drug delivery and cancer treatment.
 | ||
| Fig. 11 Histological analysis for different organs of tumor bearing mice treated with saline, DOX·HCl, DOX/Z-micelles, DOX/C-micelles. (All tissues: ×100). The mice administrated with free DOX·HCl resulted in kidney glomerular swelling (pointed out by red arrows). Scale bar measures 100 μm. | ||
4. Conclusions
Summarily, we have built up nanoscale (69 ± 20 nm) multiple environmental-responsive zwitterionic micelles (Z-micelles) composed of zwitterionic peripheral tentacles (pCBMA) and pH cleavable/sensitive drug encapsulation cavities [p(ADA-TMDP)]. The zwitterionic tentacles exhibited non-fouling feature to address the challenges of non-specific protein adsorption on nanocarriers in circulation. Subtle tumor environmental conditions realized multiple pH responses of micelle carriers and consequently activated specific micelle functionalization. In particular, when the zwitterionic micelles were largely transported to tumor sites by EPR effect, tumor extracellular pH (pH 6.8) turned the micelle surface charge from −2.4 mV to slightly positive(+4.41 mV) to help cellular uptake; acidic conditions of cellular compartments such as endolysosomes accelerated micelle swelling/disassembly through cleavage of pADA and protonation of pTMDP units. Typically, average hydrodynamic diameter of Z-micelles was almost tripled at pH 5.0 with greatly widened size distribution. On this basis, Z-micelles demonstrated a 3.26-fold faster DOX release at pH 5.0 than that of the physiological condition (pH 7.4). Furthermore, answering to the acidic organelles, endosomal escape enabled the DOX/Z-micelles to primarily release their payloads inside tumor cells. In vitro cytotoxicity assay demonstrated that the biocompatible Z-micelles possessed high cancer cell growth inhibition capability after drug encapsulation. In vivo evaluations verified the effective antitumor activity of DOX/Z-micelles. Besides the greatly reduced drug toxicity to major organs, a higher tumor inhibition rate (89.3%) was achieved, as compared with the DOX·HCl (69.9%) and DOX/C-micelles (60.2%).Acknowledgements
Financial supports from the National Natural Science Foundation of China (81471775, 51133004 and 81361140343), the Fundamental Research Funds for the Central Universities (2015SCU04A31), the Joint Sino-German Research Project (No. GZ905) and the Sichuan University Start-Up Research Found (YJ 201405) are greatly appreciated.Notes and references
- P. Boyle and B. Levin, World Cancer Report 2008, International Agency for Research on Cancer (IARC), Lyon, 2008 Search PubMed.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- X. Guo, C. Shi, J. Wang, S. Di and S. Zhou, Biomaterials, 2013, 34, 4544–4554 CrossRef CAS PubMed.
- X. Guo, D. Li, G. Yang, C. Shi, Z. Tang, J. Wang and S. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 8549–8559 CAS.
- (a) J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS PubMed; (b) G. Yang, J. Wang, Y. Wang, L. Li, X. Guo and S. Zhou, ACS Nano, 2015, 9, 1161–1174 CrossRef CAS PubMed; (c) Z. L. Tyrrell, Y. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128–1143 CrossRef CAS.
- W. Wang, D. Cheng, F. Gong, X. Miao and X. Shuai, Adv. Mater., 2012, 24, 115–120 CrossRef CAS PubMed.
- (a) T. Chen, X. Guo, X. Liu, S. Shi, J. Wang, C. Shi, Z. Qian and S. Zhou, Adv. Healthcare Mater., 2012, 1, 214–224 CrossRef CAS PubMed; (b) Q. Yi, D. Wen and G. B. Sukhorukov, Langmuir, 2012, 28, 10822–10829 CrossRef CAS PubMed.
- (a) R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed; (b) J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Adv. Mater., 2013, 25, 3670–3676 CrossRef CAS PubMed; (c) G. H. Gao, M. J. Park, Y. Li, G. H. Im, J.-H. Kim, H. N. Kim, J. W. Lee, P. Jeon, O. Y. Bang and J. H. Lee, Biomaterials, 2012, 33, 9157–9164 CrossRef CAS PubMed.
- (a) T. Ishida, K. Masuda, T. Ichikawa, M. Ichihara, K. Irimura and H. Kiwada, Int. J. Pharm., 2003, 255, 167–174 CrossRef CAS PubMed; (b) E. T. Dams, P. Laverman, W. J. Oyen, G. Storm, G. L. Scherphof, J. W. van der Meer, F. H. Corstens and O. C. Boerman, J. Pharmacol. Exp. Ther., 2000, 292, 1071–1079 CAS.
- A. S. A. Lila, H. Kiwada and T. Ishida, J. Controlled Release, 2013, 172, 38–47 CrossRef PubMed.
- M. S. Bretscher, Nature, 1975, 258, 43–49 CrossRef CAS PubMed.
- J. B. Schlenoff, Langmuir, 2014, 30, 9625–9636 CrossRef CAS PubMed.
- (a) Y. Chang, W.-J. Chang, Y.-J. Shih, T.-C. Wei and G.-H. Hsiue, ACS Appl. Mater. Interfaces, 2011, 3, 1228–1237 CrossRef CAS PubMed; (b) Z. G. Estephan, P. S. Schlenoff and J. B. Schlenoff, Langmuir, 2011, 27, 6794–6800 CrossRef CAS PubMed; (c) R. E. Holmlin, X. Chen, R. G. Chapman, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 2841–2850 CrossRef CAS.
- A. Nori and J. Kopeček, Adv. Drug Delivery Rev., 2005, 57, 609–636 CrossRef CAS PubMed.
- (a) S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed; (b) A. M. Pavlov, G. B. Sukhorukov and D. J. Gould, J. Controlled Release, 2013, 172, 22–29 CrossRef CAS PubMed.
- (a) S. E. Paramonov, E. M. Bachelder, T. T. Beaudette, S. M. Standley, C. C. Lee, J. Dashe and J. M. Fréchet, Bioconjugate Chem., 2008, 19, 911–919 CrossRef CAS PubMed; (b) S. C. Yang, M. Bhide, I. N. Crispe, R. H. Pierce and N. Murthy, Bioconjugate Chem., 2008, 19, 1164–1169 CrossRef CAS PubMed.
- J. Ko, K. Park, Y. S. Kim, M. S. Kim, J. K. Han, K. Kim, R. W. Park, I. S. Kim, H. K. Song, D. S. Lee and I. C. Kwon, J. Controlled Release, 2007, 123, 109–115 CrossRef CAS PubMed.
- W. H. Heath, F. Palmieri, J. R. Adams, B. K. Long, J. Chute, T. W. Holcombe, S. Zieren, M. J. Truitt, J. L. White and C. G. Willson, Macromolecules, 2008, 41, 719–726 CrossRef CAS.
- Z. Zhang, T. Chao, S. Chen and S. Jiang, Langmuir, 2006, 22, 10072–10077 CrossRef CAS PubMed.
- Y. Lai, Y. Lei, X. Xu, Y. Li, B. He and Z. Gu, J. Mater. Chem. B, 2013, 1, 4289 RSC.
- C. Menon, G. M. Polin, I. Prabakaran, A. Hsi, C. Cheung, J. P. Culver, J. F. Pingpank, C. S. Sehgal, A. G. Yodh and D. G. Buerk, Cancer Res., 2003, 63, 7232–7240 CAS.
- Z. Zhang, L. Wang, J. Wang, X. Jiang, X. Li, Z. Hu, Y. Ji, X. Wu and C. Chen, Adv. Mater., 2012, 24, 1418–1423 CrossRef CAS PubMed.
- Y. B. Patil, U. S. Toti, A. Khdair, L. Ma and J. Panyam, Biomaterials, 2009, 30, 859–866 CrossRef CAS PubMed.
- K. Kataoka, T. Matsumoto, M. Yokoyama, T. Okano, Y. Sakurai, S. Fukushima, K. Okamoto and G. S. Kwon, J. Controlled Release, 2000, 64, 143–153 CrossRef CAS PubMed.
- J. Luo, K. Xiao, Y. Li, J. S. Lee, L. Shi, Y.-H. Tan, L. Xing, R. H. Cheng, G.-Y. Liu and K. S. Lam, Bioconjugate Chem., 2010, 21, 1216–1224 CrossRef CAS PubMed.
- M. J. Heffernan and N. Murthy, Bioconjugate Chem., 2005, 16, 1340–1342 CrossRef CAS PubMed.
- H. S. Sundaram, J.-R. Ella-Menye, N. D. Brault, Q. Shao and S. Jiang, Chem. Sci., 2014, 5, 200–205 RSC.
- J. Sankaranarayanan, E. A. Mahmoud, G. Kim, J. M. Morachis and A. Almutairi, ACS Nano, 2010, 4, 5930–5936 CrossRef CAS PubMed.
- Y. Y. Yuan, C. Q. Mao, X. J. Du, J. Z. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476–5480 CrossRef CAS PubMed.
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS PubMed.
- (a) Y. Shen, H. Tang, Y. Zhan, E. A. Van Kirk and W. J. Murdoch, Nanomedicine: Nanotechnology, Biology and Medicine, 2009, 5, 192–201 CrossRef CAS PubMed; (b) A. L. Becker, A. P. Johnston and F. Caruso, Small, 2010, 6, 1836–1852 CAS.
- Y. Li, K. Xiao, J. Luo, W. Xiao, J. S. Lee, A. M. Gonik, L. Kato, T. A. Dong and K. S. Lam, Biomaterials, 2011, 32, 6633–6645 CrossRef CAS PubMed.
- M. I. Koukourakis, A. Giatromanolaki, P. E. Thorpe, R. A. Brekken, E. Sivridis, S. Kakolyris, V. Georgoulias, K. C. Gatter and A. L. Harris, Cancer Res., 2000, 60, 3088–3095 CAS.
- A. Urruticoechea, I. E. Smith and M. Dowsett, J. Clin. Oncol., 2005, 23, 7212–7220 CrossRef CAS PubMed.
- C. Liu, C. Sun, H. Huang, K. Janda and T. Edgington, Cancer Res., 2003, 63, 2957–2964 CAS.
- V. Milacic, S. Banerjee, K. R. Landis-Piwowar, E. H. Sarkar, A. P. Majumdar and Q. P. Dou, Cancer Res., 2008, 68, 7283–7292 CrossRef CAS PubMed.
- N. Li, N. Li, Q. Yi, K. Luo, C. Guo, D. Pan and Z. Gu, Biomaterials, 2014, 35, 9529–9545 CrossRef CAS PubMed.
- F. H. Wapstra, H. van Goor, P. E. de Jong, G. Navis and D. de Zeeuw, J. Pharmacol. Toxicol. Methods, 1999, 41, 69–73 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11645k |
| This journal is © The Royal Society of Chemistry 2016 |