
Functionalised diimidazolium salts: the anion effect on the catalytic ability†
Carla Rizzo,
Francesca D'Anna* and
Renato Noto
Università degli Studi di Palermo, Dipartimento STEBICEF, Sezione di Chimica, Viale delle Scienze, Parco d'Orleans II, 90128 Palermo, Italy. E-mail: francesca.danna@unipa.it; Fax: +39-091596825; Tel: +39-09123897540
First published on 13th June 2016
Abstract
The catalytic ability of some functionalised diimidazolium ionic liquids was tested using the Michael addition of malononitrile to t-chalcone as probe reaction. Diimidazolium salts characterized by the presence of 1-(1-imidazolylmethyl)-3,5-di-[1-(3-octylimidazolylmethyl)]benzene cation and differing in the anion structure were used. Both mono- and dianions were employed and among these some chiral anions generally used as organocatalysts were taken into account. Data collected were analysed both as function of ionic liquids structure and basicity, evaluated using the Hammett basicity function. Although the use of chiral anions did not allow performing a stereochemical control of the reaction, data collected demonstrate the high catalytic ability of the functionalised salts. Indeed, high conversions and yields were obtained under mild conditions and in significantly shorter reaction times with respect to the ones so far reported for this reaction.
Introduction
The choice of solvent and catalyst is crucial for tuning up new synthetic procedures or in the attempt to improve reaction conditions. As far as solvents are concerned, in the last fifteen years, ionic liquids (ILs) have played a significant role.1,2 Thanks to their low vapour pressure and flammability, they have been considered as efficient options to conventional organic solvents (COSs). After their first use as solvent media, also the possibility of incorporating functional groups into the cation or anion has been taken into account. This allows obtaining functionalised ILs generally named “task specific ILs” (TSILs). They were firstly defined by Davis in 2004 as organic salts able to behave not only as reaction media, but also to exert specific action thanks to the presence of a functional group tethered to the cation and/or anion.3 TSILs have been also designed to remove chemically toxic and radiotoxic metal ions from aqueous media.4 They have been frequently considered “rivals” of solid supported catalysts, as they increase the kinetic mobility and allow a large operational surface area. Furthermore, the incorporation of functional groups in the IL structure minimizes the leakage of the catalyst and can increase the recyclability of the solvent–catalyst media for several catalytic cycles. Literature about TSILs has been comprehensively reviewed by Chiappe et al.5 Different kinds of TSILs have been considered. As for imidazolium-based ILs, nitrile-functionalised ILs have been exploited to coordinate metals active in catalysis like palladium. These TSILs have been used both as solvents and stabilizers in palladium catalysed C–C cross-coupling reactions.6,7 On the other hand, a primary amino-functionalised imidazolium IL has been used in [bmim][PF6] as ligand and base for the Heck reaction.8As for TSILs bearing the active function on the anion structure, some years ago we prepared the 1-butyl-3-methylimidazolium azide and we used it as a source of nucleophile, in IL solution, to promote aryl azide formation from corresponding aryl halides.9,10
In the last decade, the further evolution of ILs class has been represented by di- and polycationic ones.11 With respect to monocationic ILs, they show higher thermal stability and lower miscibility with conventional organic solvents (COSs). Recently, the opportunity to anchor a catalytic functionality on the cation or anion structure to obtain functionalised di- or polycationic ILs has been evaluated (TSDILs and TSPILs, respectively). So, acidic TSDILs have been used for biomass transformation in products of industrial value.12,13 On the other hand, a radical functionality has been also tethered on some diimidazolium ILs and used in the oxidation of primary and secondary alcohols.14
In this context, as a development of our work aimed to study ILs properties and applications,15,16 two years ago we synthesized some TSDILs characterized by the presence of 1-(1-imidazolylmethyl)-3,5-di-[1-(3-octylimidazolylmethyl)]benzene ([2C8bti2+]) cation and having different mono- and dianions.17 The presence of a neutral imidazole unit on the cation structure made these TSDILs very efficient reaction media for the base catalysed mononuclear rearrangement of heterocycles (MHR) reaction. They acted both as solvents and catalysts allowing the obtainment of triazoles with high yields and the recycle of the solvent–catalyst system.18 Among anions used, adipate ([ad]) and suberate ([sub]) were the ones able to award the highest catalytic ability to solvent systems.
In the light of the above results, we thought it could be interesting to test some of the above TSDILs and some new diimidazolium salts (from now on they will be referred as functionalised dicationic organic salts, TSDILs) for the study of Michael addition of malononitrile to t-chalcone (Scheme 1). As probe reaction, it could be of a certain interest as, otherwise the MHR reaction, this is a bimolecular reaction and could allow shedding light on different factors affecting catalytic ability of the used TSDILs. Furthermore, giving rise to a chiral product, it could allow evaluating also the “chiral induction solvent”. This base catalysed reaction is of great significance as it represents a useful synthetic strategy for the obtainment of intermediate nitriles able to give 4-oxobutanamides, which are found in a lot of natural and unnatural compounds displaying both pharmacological and biological properties.19 The Michael addition has been investigated in COSs, both as a function of solvent and base.20,21 Furthermore, in the presence of chiral catalysts such as Cinchona alkaloids and quinidine derivatives, good chemical yields with high levels of diastereo- and enantioselectivities have been detected.22,23
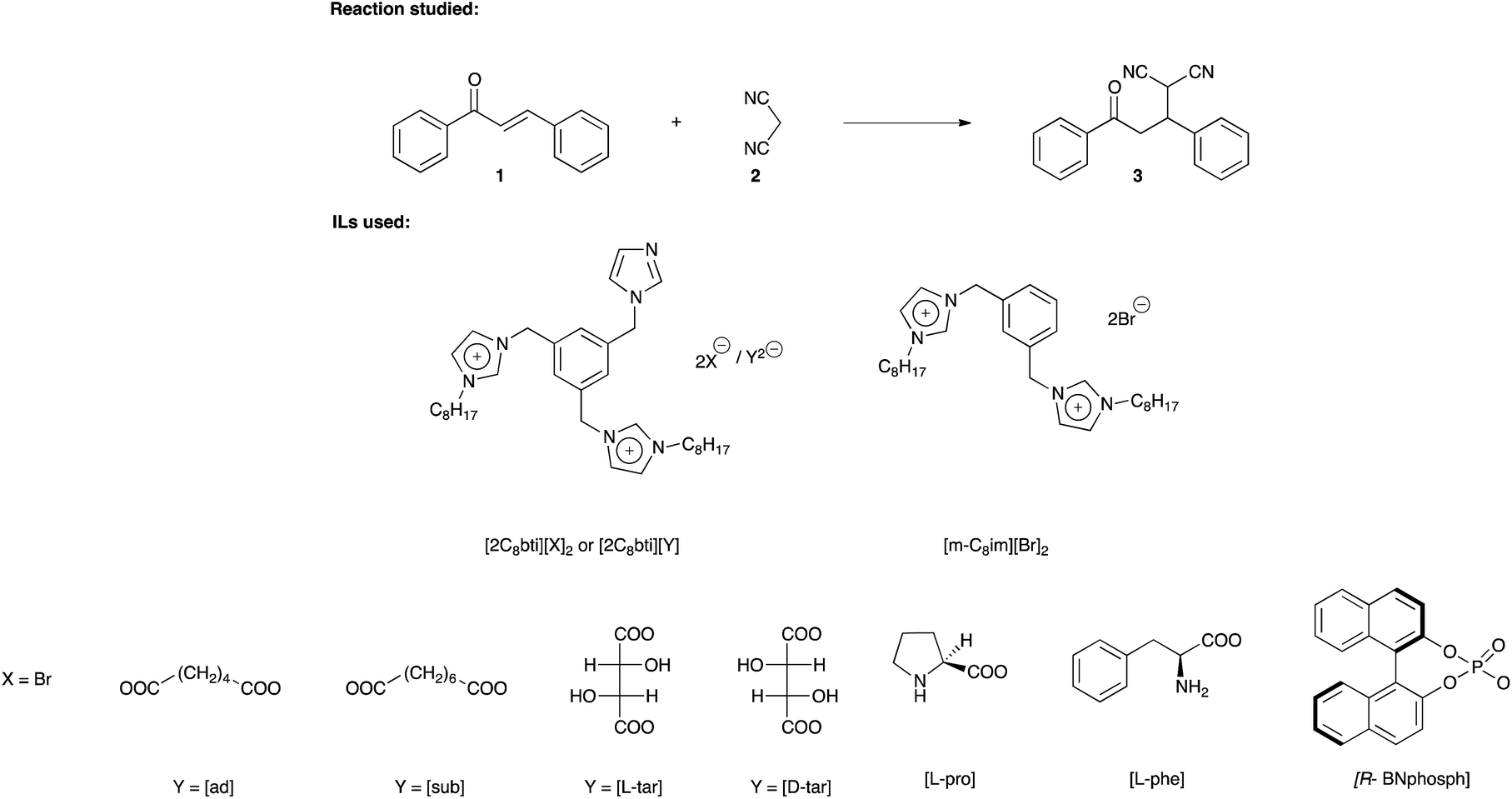 | ||
| Scheme 1 Representation of the reaction studied and TSDILs used. | ||
Michael addition has been also studied in monocationic ILs solution either in the presence or without catalyst.24–27 However, to the best of our knowledge, no data have been so far reported about the study of this reaction in DILs or better in the presence of TSDILs. DILs are frequently claimed for their ability to induce effects markedly different from the ones observed in simple monocationic ILs.28–30 Consequently, this has raised some questions: (i) can the use of TSDILs be advantageous in terms of reaction time? (ii) What's the role played by the catalyst tethered to the ionic structure? (iii) Is the anion an “inert” viewer or does it play a role? (iv) Can the combination of a base functionalised cation with chiral anions affect the stereochemical pathway?
To answer these questions, we synthesized some TSDILs based on [2C8bti2+] cation endowed with both mono- ([Br−]) and dianions (Scheme 1). Chiral carboxylates and phosphate anions, such as L-prolinate ([L-pro]), L-phenylalaninate ([L-phe]) and (R)-(−)-1,1′-binaphthyl-2,2′-diyl phosphate ([R-BNphosph]) were used. In particular, [L-pro] and [L-phe] were used as the corresponding aminoacids are among the most widely employed organocatalysts.31–36 On the other hand, in the last few years, different papers have claimed the chiral induction of binaphthyl phosphates.22,37 As for dianions, together with [ad] and [sub] used for good performances showed in MHR reaction, also chiral dicarboxylates, such as L- and D-tartrate ([tar]), were considered.
Finally, to evaluate the relevance of the base catalyst on the dication structure, the Michael addition was also performed using the 3,3′-di-N-octyl-1,1′-(1,3-phenylenedimethylene)diimidazolium dibromide ([m-C8im][Br]2) as solvent system and 1-methylimidazole as basic catalyst.
Results and discussion
In order to apply the best reaction conditions, we first performed the Michael addition in a conventional solvent, tetrahydrofuran (THF) as this is one of the solvents most commonly used.38 Indeed, the reaction is not favoured in highly polar solvents. It has been claimed that solvents, like DMF and DMSO, under basic conditions, favour the conversion of dicyanide (3) in the monocyanide product (4) through an aerobic oxidation and a subsequent elimination reaction (Chart 1).20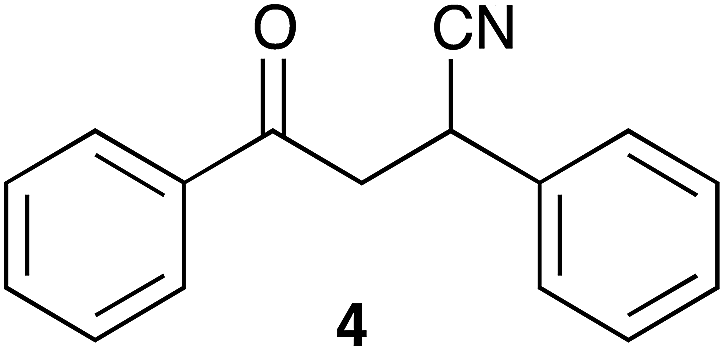 | ||
| Chart 1 Monocyanide deriving from dicyanide (3).20 | ||
On the other hand, it is well known that inorganic bases, like K2CO3 or KOH, catalyse the reaction.21
We took in consideration a weaker organic base such as 1-methylimidazole, as it represents the basic functionality of the TSDILs used.
Obviously, other important experimental parameters, like the ratio between t-chalcone and malononitrile, the temperature and time of reaction were evaluated (Table 1). For a useful comparison, data previously collected in THF using K2CO3 as base are also reported.20 In all cases, we used the catalyst in stoichiometric amount with respect to t-chalcone and the final product was isolated by flash chromatography.
| Entry | Mole ratio 1/2 | Catalyst | T (°C) | Time (h) | Yieldb (3) (%) |
|---|---|---|---|---|---|
| a Reactions were performed using 1 μL of THF.b Yields were reproducible within 4%. Product isolated by flash chromatography.c From ref. 20. | |||||
| 1 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 :1.1 |
K2CO3 | 25 | 12 | 94c |
| 2 | 1:1.3 |
1-Methylimidazole | 25 | 16 | 0 |
| 3 | 1:1.3 |
1-Methylimidazole | 25 & US | 1 | 12 |
| 4 | 1:1.3 |
1-Methylimidazole | 40 | 1 | 41 |
| 5 | 1:10 |
1-Methylimidazole | 25 | 22 | 43 |
Data collected show that, in our experimental conditions, 1-methylimidazole was not able to catalyse the reaction at 25 °C (Table 1, entry 2). On the other hand, keeping constant t-chalcone/malononitrile ratio to 1/1.3, it was necessary to use ultrasound irradiation or a temperature of 40 °C to obtain a low or moderate yield in a short reaction time (1 h) (Table 1, entries 3 and 4). Even though only 12% and 41% yields were respectively obtained, further increasing the temperature caused the formation of some by-products. On the other hand, maintaining the temperature at 25 °C, the increase of t-chalcone/malononitrile mole ratio from 1:1.3 to 1:10 brought to a higher yield but with a long reaction time (22 h) and using less green reaction conditions (Table 1, entry 5). The lower catalytic efficiency of 1-methylimidazole, with respect to K2CO3, is perfectly ascribable to its lower basicity (Table 1, entries 1 and 2). On the grounds of these results, we subsequently studied TSDIL effects performing reactions at 25 °C and using a small excess of the methylene active compound (t-chalcone/malononitrile 1:1.3; Tables 2, 3 and 5).
| Entry | [2C8bti][ad] | ||
|---|---|---|---|
| MeOH (μL) | Reaction times | Conversiona,b | |
| a Conversion obtained at 25 °C. Conversions were reproducible within 4%. Product isolated by flash chromatography.b Conversions and yields were almost coincident.c Reaction performed at room temperature assisted by ultrasound irradiation.d Formation of by-products. | |||
| 1 | 0 | 3.25 hc | 72% |
| 2 | 20 | 0.5 h | 85% |
| 3 | 40 | 0.5 h | 83% |
| 4 | 60 | 0.5 h | >95% |
| Entry | TSDIL | Reaction timeb (h) | Yieldb% (3) | Yieldb% (4) | Conversionb% |
|---|---|---|---|---|---|
| a Reactions were performed using a 1/1/1 substrate/TSDIL/MeOH molar ratio.b Yields and conversions were reproducible within 4%. Products were isolated by flash chromatography. | |||||
| 1 | [m-C8im][Br]2 | 24 | 74 | 0 | 78 |
| 2 | [2C8bti][Br]2 | 5.5 | 93 | 0 | >95 |
| 3 | [2C8bti][ad] | 0.5 | >95 | 0 | >95 |
| 4 | [2C8bti][sub] | 0.5 | 74 | 0 | 78 |
| 5 | [2C8bti][D-tar] | 24 | 47 | 20 | 67 |
| 6 | [2C8bti][L-tar] | 24 | 47 | 24 | 71 |
| 7 | [2C8bti][L-pro]2 | 0.15 | 63 | 25 | 92 |
| 8 | [2C8bti][L-phe]2 | 0.5 | 56 | 35 | 91 |
| 9 | [2C8bti][R-BNphosph]2 | 24 | 78 | 0 | 83 |
It is worth of remembering that some of our TSDILs at 25 °C were waxy solid and their consistency made difficult the evaluation of volume. Consequently, we performed our investigation keeping constant the TSDIL/substrate molar ratio (1; ≈0.15 mmol), and this was smaller than the one used for the MHR reaction (4.5).18 Furthermore, among salts used, only [2C8bti][R-BNphosph]2 had a melting point higher than 100 °C (see Experimental) and cannot be exactly defined a TSDIL. However, we decided to use it to test its “chiral induction ability” on the probe reaction.
Having in mind the results obtained in the MHR reaction,18 we firstly used TSDILs presenting the best catalytic ability, i.e. [2C8bti][sub] and [2C8bti][ad]. Table 2 summarizes results obtained in terms of percentage conversion of t-chalcone as function of amount and nature of cosolvent.
The [ad]-based TSDIL was employed as reference solvent, and we performed the first investigation at 25 °C. However, as above stated, at room temperature our TSDILs were solid like or waxy solids. Consequently, the reaction required long reaction time and the use of ultrasound irradiation to give 72% yield (Table 2, entry 1).
In order to improve the reaction conditions and lower the energetic demand of the process, we performed the reaction in the presence of a cosolvent. This should decrease the system viscosity favouring the contact between the reagents. At a first sight, MeOH appeared to be a suitable choice. Indeed, our TSDILs were really soluble in this solvent and its use could avoid the presence of different microdomains in the solvent system, exerting different effects on the outcome of the reaction. Then, we searched for the smallest cosolvent volume and, fixing the reaction time at 30 minutes, we performed investigation gradually increasing the MeOH volume (Table 2, entries 2–4).
In the presence of a really low MeOH volume (60 μL – molar ratio substrate/TSDIL/MeOH = 1/1/1), we obtained an almost quantitative substrate conversion and this definitely induced us to avoid the use of energy source in neat TSDIL as it did not induce improvements both in terms of yield and reaction time. It is noteworthy that, differently from data collected in THF, in the presence of [2C8bti][ad], we detected a significant conversion of t-chalcone in spite of the short reaction time (30 min). Clearly, this points out a positive effect of the used TSDIL. The result obtained could be ascribed to a higher acidity of methylene compound in this medium, as well as to a significant increase in basicity of neutral imidazole unit borne on the [2C8bti2+] cation. The first hypothesis well agrees with the one previously reported by Toma et al. studying the Michael addition in monocationic ILs.27 On the other hand, some years ago, performing base catalysed reactions in monocationic ILs, we also evidenced a higher catalytic ability of aliphatic amines with respect to the one detected in COSs and we ascribed the effect observed to the less extensive solvation of reagents operating in IL solution.39,40 These effects could operate also in this case giving rise to results collected.
Then, we tested the reaction in the presence of [2C8bti][sub], using the same conditions (25 °C, 30 min, 60 μL of MeOH) and we obtained a 74% conversion of substrate (Table 2, entry 10). As second stage, we became interested whether it is feasible to analyse the effect of cosolvent nature. Indeed, some years ago performing the MHR reaction in IL/COS binary mixtures, we demonstrated that the rate of the above reaction changed as a function of cosolvent nature and most significant effects could be detected when small volumes of COS were added to IL.41 To pursue the above aim, we decided to use [2C8bti][sub] as TSDIL because conversion of t-chalcone was not quantitative and better than [2C8bti][ad] it could allow evaluating the role played by the cosolvent nature (Table 2).
We always used 60 μL of cosolvent and conversions ranged from 73% in the presence of 1,4-dioxane to 95% in the presence of toluene or DMF. The conversion of the substrate is high using cosolvents of low or medium polarity such as toluene, chloroform, THF and 1-propanol. However, in these cases, it is comprehensive of the presence of some by-products, which could not be easily separated from the product desired and identified. Differently, 1,4-dioxane, MeOH, DMF and ACN only give the desired product.
Considering the above results, we decided to use MeOH as cosolvent as it gave rise to a good conversion allowing investigating TSDILs effects (see later) better than DMF or ACN that presented the almost total conversion of the substrate in the desired product.
The further step of our investigation was the study of the influence of TSDILs anions on the Michael addition, determining reaction times and percentage yields (Table 3).
Firstly, to evaluate the relevance of the use of TSDILs, we carried out the reaction using the unfunctionalised DIL, [m-C8im][Br]2, in the presence of 1-methylimidazole in equimolar amount to DIL and t-chalcone. After 24 h, we obtained a 74% yield (Table 3, entry 1). On going from [m-C8im][Br]2 to [2C8bti][Br]2, we observed a significant decrease in reaction time and a good improvement in yield (Table 3, entries 1 and 2), evidencing the advantage of using TSDILs and supporting the importance of the catalyst directly linked to the IL cation.
Then, we performed the reaction in the presence of all TSDILs. Reaction times range from 10 min up to 24 h. In general, they represent a great challenge with respect to the ones previously reported in COSs as well as in IL solution. Indeed, in the first case, at room temperature, reaction times ranging from 12 up to 100 h are reported.42,43 On the other hand, as above stated, the Michael addition has been performed in monocationic ILs. Although reaction times going from 30 s up to 3 h have been reported for thiols addition to t-chalcone, when malononitrile was used as nucleophile, reaction times of 2 or 22 h were detected as piperidine or L-proline were the basic catalysts, respectively. Furthermore, the before-mentioned reactions, were performed using higher IL/substrate mole ratio than the one used by us (∼5 and 1 for monocationic26,27 and TSDILs, respectively). This further sheds light on the catalytic ability of salts used in this work.
Among TSDILs, the Michael addition selectively proceeded in the presence of [2C8bti][Br]2, [2C8bti][R-BNphosph]2, [2C8bti][ad] and [2C8bti][sub]. The latter two are the best reaction media for the target reaction, perfectly recalling what we previously observed studying the MHR reaction.18
In all the other cases ([2C8bti][tar], [2C8bti][L-pro]2 and [2C8bti][L-phe]2), we detected the presence of two products in the reaction mixture. After separation by chromatography and characterization by means of NMR and GC-MS investigation, we identified both di-(3) and monocyanide (4). However, while in the case of [2C8bti][tar], moderate yields were obtained in a long reaction time (Table 3, entries 5 and 6), aminoacids-based TSDILs were able to give fastly moderate yields in the product desired.
Taking into account the base catalysis operating on the reaction, and to have a further support to explain the above experimental trend, we determined the basicity function (H−), for the new chiral TSDILs in MeOH solution, using the Hammett method. This colorimetric method has been applied by us and others, to assess the basicity of functionalised dicationic ILs.18,44 We are aware of the fact that situation detected in MeOH solution could be different from the one occurring in the reaction mixtures. However, the evaluation of H− function could give general information about TSDILs basicity.
Basicity showed significant changes as a function of the nature of the anion. This is the reason why both bromocresol green and p-nitrophenol were used as solvatochromic probes. Data collected are reported in Table 4; whereas UV-vis spectra are displayed in Fig. 1.
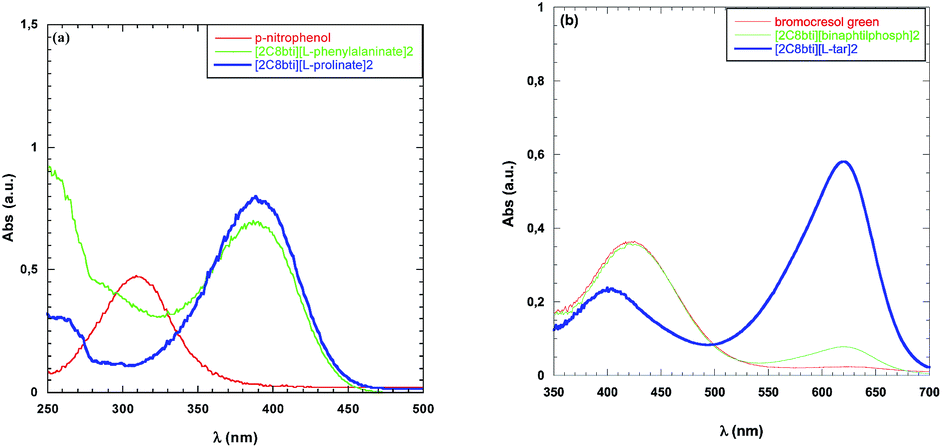 | ||
| Fig. 1 UV-vis spectra of (a) p-nitrophenol and (b) bromocresol green collected in MeOH and at a fixed concentration of TSDIL. | ||
Among TSDILs used, the lowest basicity was detected for [2C8bti][R-BNphosph]2 whereas the highest was the one corresponding to [2C8bti][L-pro]2. Obviously, in the latter case (as well as in the case of [2C8bti][L-phe]2), a contribution to basicity can also be due to the presence of amino group in the anion. The above two TSDILs were the ones in which the highest and lowest reaction times were detected, respectively. On the grounds of H− values, basicity of TSDILs increases along the series: [2C8bti][R-BNphosph]2 < [2C8bti][Br]2 < [2C8bti][tar] < [2C8bti][sub] < [2C8bti][ad] < [2C8bti][L-phe]2 < [2C8bti][L-pro]2. The above trend can not explain differences in reaction times. Indeed, it does not justify the ones between [tar]- and [Br]-based TSDILs. Furthermore, H− values do not allow justifying the almost quantitative conversion detected in the presence of both [2C8bti][ad] and [2C8bti][sub].
In general, the increase in basicity favours the substrate conversion. However, higher basicity ([L-phe] and [L-pro] anions) or longer reaction time ([tar] anions) speed up the further transformation of dicyanide (3) in monocyanide (4) inducing a drop in the selectivity of the reaction.
Taking in consideration the above results, we fixed the reaction time at 30 min to have a better comparison among catalytic ability of TSDILs (Table 5).
| Entry | TSDIL | Yieldb% (3) | Yieldb% (4) | Conversionb% |
|---|---|---|---|---|
| a Reactions were performed using a 1/1/1 substrate/TSDIL/MeOH molar ratio.b Yields and conversions were reproducible within 4%. Products were isolated by flash chromatography. | ||||
| 1 | [2C8bti][Br]2 | 58 | 0 | 63 |
| 2 | [2C8bti][ad] | >95 | 0 | >95 |
| 3 | [2C8bti][sub] | 74 | 0 | 78 |
| 4 | [2C8bti][D-tar] | 51 | 0 | 57 |
| 5 | [2C8bti][L-tar] | 52 | 0 | 56 |
| 6 | [2C8bti][L-pro]2 | 32 | 39 | 76 |
| 7 | [2C8bti][L-phe]2 | 56 | 35 | 91 |
| 8 | [2C8bti][R-BNphosph]2 | 40 | 0 | 46 |
Considering product (3), the percentage yields obtained were higher in non-chiral TSDILs, the highest values were detected in [2C8bti][ad] and [2C8bti][sub] TSDILs. The trend observed as function of different TSDILs anions is the following one: [2C8bti][L-pro]2 < [2C8bti][R-BNphosph]2 < [2C8bti][D-tar] ≈ [2C8bti][L-tar] < [2C8bti][L-phe]2 ≈ [2C8bti][Br]2 < [2C8bti][sub] < [2C8bti][ad].
According to data collected for the MHR reaction,18 also in this case the anion flexibility plays a role in determining the outcome of the reaction as accounted for by high yields obtained in [ad]- and [sub]-based TSDILs (Table 5, entries 2 and 3). Differently, as previously observed for some naphthalenedisulfonate-based TSDILs,18 the extended π-surface of [R-BNphosph] anion induces a drop in yield value. Probably, this anion can easily interact with the cation through π–π stacking, lowering the cation conformational freedom and making it less available to interact with the substrate (Table 5, entry 8).
On the other hand, the lower catalytic ability of [tar]-based TSDILs (Table 5, entries 4 and 5), with respect to [ad]- and [sub]-based TSDILs, could be ascribed to a loss of catalytic efficiency as a consequence of ability of hydroxyl groups to coordinate the nitrogen of the neutral imidazole unit through hydrogen bond formation. This could induce a decrease in its propensity to act as a base towards malononitrile.
A different analysis has to support data obtained in [L-pro]- and [L-phe]-based TSDILs (Table 5, entries 6 and 7). Indeed, conversion values of t-chalcone (76% and 91% respectively) indicate an enhanced catalytic ability of the solvent systems with respect to [tar]-based TSDILs, according to the increase in solvent basicity. This however brings to a decrease in the selectivity of the reaction with the formation of products (3) and (4) in comparable amount.
Then, the whole of data collected seems to indicate that the outcome of the reaction is probably determined by a concomitant action of basicity and structure of TSDILs.
As above stated, the use of chiral dianions in the preparation of TSDILs was aimed to control the reaction from a stereochemical point of view. This is the reason why, after purification by flash chromatography, products obtained in chiral TSDILs were analysed by HPLC using a chiral pack column to assess probable enantiomeric excess. Unfortunately, our results were not promising to this aim. Indeed, we obtained a low enantiomeric excess only in the presence of [2C8bti][R-BNphosph]2 (ee = 10%).
In the attempt to improve the above result, we performed the reaction at 4 °C and using toluene as cosolvent. Indeed, it is well known that the decrease in temperature and the low polarity of solvent disfavour racemization processes. On this subject, some years ago Deng et al., performing Michael addition of malononitrile to α,β-unsaturated ketones, in the presence of Cinchona alkaloids as catalysts, obtained high enantiomeric excesses only in low polar solvents.22
In our experimental conditions, we obtained a 55% yield in dicyanide (3) at 5.50 h, but with a significant increase in enantiomeric excess (ee = 35%). The value obtained is higher than the one reported by Bao et al., studying the enantioselective addition of diethylmalonate to 1,3-diphenyl-prop-2-ene-1-one in imidazolium based chiral ILs (ee = 24%).45
With the only exception above discussed, we did not observed the so-called “chiral induction solvent”. This result is not anomalous. Indeed, in general, good or high enantiomeric excesses have been reported only in COSs and in the presence of catalysts having proved chiral induction ability like Cinchona alkaloids. Toma et al. observed a very different behaviour testing organocatalysts in monocationic ILs.27 According to the above report, the low “chiral induction ability” of our TSDILs could be ascribed to the fact that the direct addition of nucleophile to the ketone is faster with respect to the chiral adduct formation that should improve the stereochemical control of the target reaction.
Conclusions
The search for synthetic processes of low environmental impact induced us to test some functionalised DILs in the Michael addition. Results collected shed light on the advantage of bounding a basic catalyst on an ionic tag as testified by the significant decrease in reaction time detected on going from the use of the catalyst in DIL solution to the use of functionalised DILs.All functionalised DILs are able to give high conversions and yields under mild conditions. They allow employing a catalyst/substrate/solvent stoichiometric ratio and promote the reaction in significantly shorter reaction times with respect to the ones so far reported for the same reaction in COSs and, in some cases, also in IL solution. This certainly represents an advantage from an environmental point of view also considering the fact that these TSDILs allow easily obtaining of useful organic intermediates like nitrile functionalised products.
Data collected point out that the catalytic efficiency of the TSDILs is the result of a combined action of their basicity and anion structure. As for basicity, although it favours t-chalcone conversion, it also induces a drop in the selectivity of the reaction. On the other hand, taking into account anion structure, information obtained indicates that best performances are detected in the presence of anions able to warrant the conformational freedom of the cation and its ability to act both as solvent and catalyst.
However, the high catalytic ability brings a loss in the stereochemical control, as testified by the fact that moderate enantiomeric excess was obtained only in the presence of the most rigid binaphthyl phosphate anion, at low temperature.
Experimental section
Materials and measurements
All TSDILs and [m-C8im][Br]2 were prepared as previously reported.15,17 α,α′-1,3-Dibromoxylene, 1,3,5-tris(bromomethyl)benzene, 1-bromooctane, imidazole, sodium hydroxide, Amberlite IRA 400 resin, 1,4-butanedicarboxylic acid, 1,6-hexanedicarboxylic acid, D-tartaric acid, L-tartaric acid, L-proline, L-phenylalanine, (R)-(−)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate, t-chalcone, malononitrile, potassium carbonate, 1-methylimidazole, p-nitrophenol, bromocresol green, 1-propanol, dimethylformamide, tetrahydrofuran, acetonitrile, dietyl ether, toluene and 1,4-dioxane were analytical reagents purchased from commercial sources and used as received. Chloroform and methanol were distilled before use.1H-NMR and 13C-NMR spectra were recorded using 300 MHz nuclear magnetic resonance spectrometer. Chemical shifts were reported relative to SiMe4.
Polarimetric measurements
Solutions of chiral TSDILs were prepared in volumetric flasks (C = 1 × 10−2 M). Optical rotations were measured using a glass polarimetric cell (light path 1 dm) and obtained as the average value over 50 readings. Measurements were performed at 25 °C and 589 nm.Base strength
A suitable amount of TSDIL was added to a methanol solution of the indicator, observing a color change. The resulting solution was analyzed by means of UV-vis spectroscopy, using a quartz cuvette with light path of 0.2 cm. The indicators used, the relative concentrations, and the TSDIL concentration were as follows: p-nitrophenol (pKHI = 11.30; [HI] = 2 × 10−4 M; [TSDIL] = 2 × 10−3 M); bromocresol green (pKHI = 9.8; [HI] = 1 × 10−4 M; [TSDIL] = 5 × 10−4 M). Basic strength was expressed by a basicity function (H−)44 defined by the following eqn (1).| H− = pKHI + log[I−]/[HI] | (1) |
Reaction conditions
The catalytic reactions were performed using a TSDIL/t-chalcone mole ratio of 1 and a malononitrile/t-chalcone mole ratio of 1.3. In a typical experiment TSDIL (≈100 mg, 0.154 mmol), t-chalcone (30 mg, 0.154 mmol) and malononitrile (≈12 mg, 0.178 mmol) were weighed into a 4 mL screw capped vial, then an appropriate amount of cosolvent was added. The reaction was performed under magnetic stirring at room temperature or at 4 °C and was followed via TLC (eluent: petroleum ether/ethyl acetate 5/1). Unfortunately, the low solubility of the reaction products in diethyl ether did not allow their extraction from TSDILs. So, MeOH was added to the reaction mixture in order to separate the products with silica flash chromatography (eluent, petroleum ether/ethyl acetate 10/1 and 5/1). The organic phase was concentrated in vacuum and solubilized in 2-propanol to determine enantiomeric excess by HPLC.HPLC analysis
HPLC was performed using a Chiralpak AS-H column with eluent 2-propanol/hexane = 30/70, flow rate 0.5 mL min−1 and λ = 234 nm: tmonocyanide = 15.2 min, tminor dicyanide = 19 min, tmajor dicyanide = 22 min.Acknowledgements
We thank MIUR (FIRB 2010RBFR10BF5V) for financial support.References
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- J. J. H. Davis, Chem. Lett., 2004, 33, 1072–1077 CrossRef CAS.
- H. Li, B. Wang, L. Zhang and L. Shen, J. Radioanal. Nucl. Chem., 2015, 303, 433–440 CrossRef CAS.
- C. Chiappe and C. S. Pomelli, Eur. J. Org. Chem., 2014, 6120–6139 CrossRef CAS.
- Z. Fei, D. Zhao, D. Pieraccini, W. H. Ang, T. J. Geldbach, R. Scopelliti, C. Chiappe and P. J. Dyson, Organometallics, 2007, 26, 1588–1598 CrossRef CAS.
- D. Zhao, Z. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 15876–15882 CrossRef CAS PubMed.
- J. Liu, H. Liu and L. Wang, Appl. Organomet. Chem., 2010, 24, 386–391 CAS.
- F. D'Anna, S. Marullo and R. Noto, J. Org. Chem., 2008, 73, 6224–6228 CrossRef PubMed.
- F. D'Anna, S. Marullo and R. Noto, J. Org. Chem., 2010, 75, 767–771 CrossRef PubMed.
- F. D'Anna and R. Noto, Eur. J. Org. Chem., 2014, 4201–4223 CrossRef.
- D. Fang, J. Yang and C. Jiao, ACS Catal., 2011, 1, 42–47 CrossRef CAS.
- H.-F. Liu, F.-X. Zeng, L. Deng, B. Liao, H. Pang and Q.-X. Guo, Green Chem., 2013, 15, 81–84 RSC.
- H. A. Beejapur, V. Campisciano, F. Giacalone and M. Gruttadauria, Adv. Synth. Catal., 2015, 357, 51–58 CrossRef CAS.
- F. D'Anna, F. Ferrante and R. Noto, Chem.–Eur. J., 2009, 15, 13059–13068 CrossRef PubMed.
- F. D'Anna, P. Vitale and R. Noto, J. Org. Chem., 2009, 74, 6224–6230 CrossRef PubMed.
- F. D'Anna, H. Q. Nimal Gunaratne, G. Lazzara, R. Noto, C. Rizzo and K. R. Seddon, Org. Biomol. Chem., 2013, 11, 5836–5846 Search PubMed.
- C. Rizzo, F. D'Anna, S. Marullo and R. Noto, J. Org. Chem., 2014, 79, 8678–8683 CrossRef CAS PubMed.
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed.
- S. Lin, Y. Wei and F. Liang, Chem. Commun., 2012, 48, 9879–9881 RSC.
- E. Wei, B. Liu, S. Lin and F. Liang, Org. Biomol. Chem., 2014, 12, 6389–6392 CAS.
- X. Li, L. Cun, C. Lian, L. Zhong, Y. Chen, J. Liao, J. Zhu and J. Deng, Org. Biomol. Chem., 2008, 6, 349–353 CAS.
- A. Russo, A. Perfetto and A. Lattanzi, Adv. Synth. Catal., 2009, 351, 3067–3071 CrossRef CAS.
- H. Hagiwara, T. Okabe, T. Hoshi and T. Suzuki, J. Mol. Catal. A: Chem., 2004, 214, 167–174 CrossRef CAS.
- P. Kotrusz, S. Toma, H.-G. Schmalz and A. Adler, Eur. J. Org. Chem., 2004, 1577–1583 CrossRef CAS.
- M. Mečiarová and Š. Toma, Chem.–Eur. J., 2007, 13, 1268–1272 CrossRef PubMed.
- M. Meciarova, S. Toma and P. Kotrusz, Org. Biomol. Chem., 2006, 4, 1420–1424 CAS.
- A. Chinnappan and H. Kim, Chem. Eng. J., 2012, 187, 283–288 CrossRef CAS.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, Eur. J. Org. Chem., 2011, 5681–5689 CrossRef.
- X. Han and D. W. Armstrong, Org. Lett., 2005, 7, 4205–4208 CrossRef CAS PubMed.
- A. Bottoni, M. Lombardo, G. P. Miscione, E. Montroni, A. Quintavalla and C. Trombini, ChemCatChem, 2013, 5, 2913–2924 CrossRef CAS.
- M. Gruttadauria, F. Giacalone, P. Lo Meo, A. Mossuto Marculescu, S. Riela and R. Noto, Eur. J. Org. Chem., 2008, 1589–1596 CrossRef CAS.
- M. Lombardo, E. Montroni, A. Quintavalla and C. Trombini, Adv. Synth. Catal., 2012, 354, 3428–3434 CrossRef CAS.
- E. Machuca, G. Granados, B. Hinojosa and E. Juaristi, Tetrahedron Lett., 2015, 56, 6047–6051 CrossRef CAS.
- A. Psarra, C. G. Kokotos and P. Moutevelis-Minakakis, Tetrahedron, 2014, 70, 608–615 CrossRef CAS.
- M. Yoshida, H. Ukigai, K. Shibatomi and S. Hara, Tetrahedron Lett., 2015, 56, 3890–3893 CrossRef CAS.
- C. Bonnot, J.-C. Chambron, E. Espinosa, K. Bernauer, U. Scholten and R. Graff, J. Org. Chem., 2008, 73, 7871–7881 CrossRef CAS PubMed.
- L. Yue, W. Du, Y.-K. Liu and Y.-C. Chen, Tetrahedron Lett., 2008, 49, 3881–3884 CrossRef CAS.
- F. D'Anna, V. Frenna, R. Noto, V. Pace and D. Spinelli, J. Org. Chem., 2005, 70, 2828–2831 CrossRef PubMed.
- F. D'Anna, V. Frenna, V. Pace and R. Noto, Tetrahedron, 2006, 62, 1690–1698 CrossRef.
- F. D'Anna, V. Frenna, S. La Marca, R. Noto, V. Pace and D. Spinelli, Tetrahedron, 2008, 64, 672–680 CrossRef.
- J. A. Castro-Osma, J. W. Comerford, S. Heath, O. Jones, M. Morcillo and M. North, RSC Adv., 2015, 5, 3678–3685 RSC.
- W. Yang, Y. Jia and D.-M. Du, Org. Biomol. Chem., 2012, 10, 332–338 CAS.
- M. Fan, J. Yang, P. Jiang, P. Zhang and S. Li, RSC Adv., 2013, 3, 752–756 RSC.
- Z. Wang, Q. Wang, Y. Zhang and W. Bao, Tetrahedron Lett., 2005, 46, 4657–4660 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR and 13C-NMR spectra of TSDILs. See DOI: 10.1039/c6ra12037g |
| This journal is © The Royal Society of Chemistry 2016 |