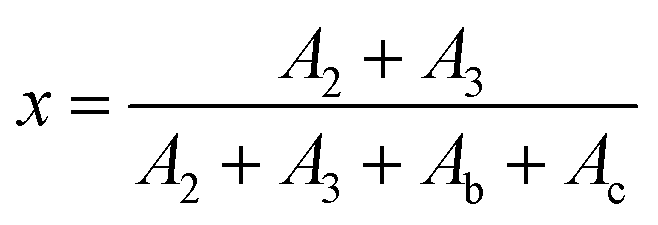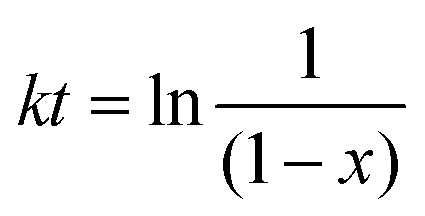DOI:
10.1039/C6RA14659G
(Paper)
RSC Adv., 2016,
6, 73140-73147
Thermally self-healing silicone-based networks with potential application in recycling adhesives†
Received
6th June 2016
, Accepted 28th July 2016
First published on 29th July 2016
Abstract
A simpler approach is developed to obtain recyclable self-healing polysiloxane networks based on the thiol-ene reaction and Diels–Alder (DA) reaction. Tetramethyltetravinylcyclotetrasiloxane (D4Vi) was first modified with furan via a thiol-ene reaction, giving furan-functionalized cyclotetrasiloxane (D1). D1 then reacted with bismaleimide to form thermally dynamic reversible crosslinking linkages via a Diels–Alder (DA) reaction. Thus, crosslinked networks (CNs) were synthesized and their depolymerization through the retro-DA reactions was thoroughly studied. The kinetics of the DA reaction was studied by 1H-NMR spectroscopic analysis of the model compounds. Results indicated that the DA reaction performs better at 60 °C and follows second-order kinetics. The CNs show strong photoluminescence when irradiated by UV light and exhibited potential application in recyclable adhesives for glass.
Introduction
Polysiloxane-based materials are commonly used materials that possess interesting properties owing to their unique structures and characteristics of the Si–O–Si bond.1,2 Highly crosslinked macromolecules possessing excellent mechanical properties and solvent resistance have been widely used in paints, adhesives, dental restorative materials, and elastomers.3–5 However, traditional crosslinking of siloxane networks could provide a remediable ability because of the absence of recombination. Given that polysiloxane materials are commonly utilized under severe conditions for long-term service, integration with superior self-healing properties is valuable. Three kinds of self-healing systems, including vascular, capsule-based, and intrinsic healing systems, have been developed in recent years.6–9 In particular, intrinsic healing systems have attracted most attention for their wide application and their facile post-functionalization.10 However, traditional intrinsic self-healing polymers are usually constructed by using tedious methods, which restrict their applications. Among many potential improvements for the self-healing systems is to create new materials via simpler reaction routes, and thus widen the range of applications for self-healing materials.
Inspired by the self-healing property in biological systems, recycling materials prepared by highly efficient chemical methods, which are often called “click” chemistry, have attracted increasing attention.11–13 The Diels–Alder (DA) reaction is a widely used and important reaction in chemical industries and organic synthesis.14 In addition, the DA reaction and retro-DA (rDA) analogue represent a highly promising route to introducing self-healing properties to macromolecular systems. It is found that the self-healing ability provided excellent recyclability, which is usually seen to be difficult for recycling polymers. DA reaction has been employed as a polymerization process between difurfuryl and bis(maleimide) monomers to achieve novel polymeric structures, such as optically active materials or resins.15 DA reaction has also been used to crosslink between multifunctional furan-functionalized polymers and bis(maleimide)s.16 Among these contributions, studies on polysiloxane materials are rare.17 The most notable finding is that the preparation of furan-functionalized polymers is usually tedious and costly. Therefore, to develop a simpler approach to obtain recyclable self-healing polysiloxane networks is of great significance. Thiol-ene “click” reaction, an efficient, modular, and stereospecific reaction, has been widely used in polymer functionalization.18–20 The benefits of thiol-ene click reaction like high atom economy, room temperature reaction, simple synthetic and handle procedures, and minimum waste generation has been mentioned in previous work.21,22 Selecting thiol-ene reaction to obtain furan modified siloxane matrix also meets the concept of “green chemistry”.23 This work aims to construct a reversible crosslinked silicone network by using a simple and easy-to-handle synthesis route.
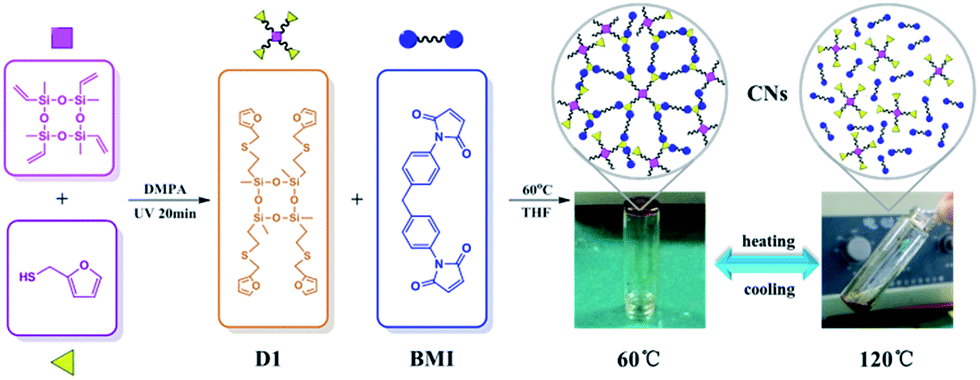
Here, we report the synthesis of a novel reversible crosslinked networks (CNs) via DA reaction with varied molar ratios between N,N′-4,4′-diphenylmethane-bismaleimide (BMI) and functionalized cyclotetrasiloxane (D1), which is initially synthesized through tetramethyltetravinylcyclotetrasiloxane (D4Vi) modified by furan via facile photomediated thiol-ene reaction (Scheme 1). Meanwhile, to investigate the kinetics of the DA reaction, we designed the model compound (M1) (Scheme 2) obtained through furan-functionalized disiloxane (MMF), which was synthesized from divinyltetramethyldisiloxane (MMVi) via thiol-ene reaction. The results show that the DA reaction plays a dominant role at 60 °C and follow the second-order kinetics. However, de-crosslinking of the cured material through the rDA reaction took place at 120 °C. The furfuryl-maleimide DA adduct displayed a relatively low temperature for de-crosslinking via its rDA reaction, which further opens the way to wide applications for recycling materials. CNs were found to possess potential applications in recyclable adhesives especially for glass bonding. The concise manner of the synthesis route, the wide range of commercially available polysiloxane precursors, and the excellent recyclability of this material could lead to a deeper scientific understanding of recyclable materials and would bring about a concept for recyclable composites.
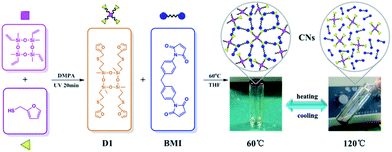 |
| | Scheme 1 Synthesis route and the self-healing behavior of CNs. | |
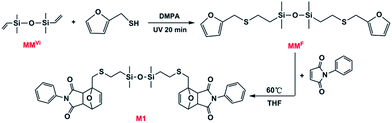 |
| | Scheme 2 Synthetic route toward the model compound (M1) using MMVi as starting molecule. | |
Experimental
Materials
2,2-dimethoxy-2-phenylacetophenone (DMPA) and furfuryl mercaptan (FM) are purchased from Aladdin Co. (China) and used without further purification. Tetramethyltetravinylcyclotetrasiloxane (D4Vi) and divinyltetramethyldisiloxane (MMVi) were obtained as commercial products and used directly. N,N′-4,4′-Diphenylmethane-bismaleimide (BMI) was from Sigma-Aldrich and used as received. Tetrahydrofuran (THF) was purified using routine procedure and distilled over sodium before use.
Characterization and measurements
The thiol-ene reaction was irradiated by UV on a Spectroline Model SB-100P/FA lamp (365 nm, 100 w). UV intensity is 4500 μW cm−2 at a distance of 38 cm.1H NMR spectra were recorded on a Bruker AVANCE 400 spectrometer at 25 °C using CDCl3 as solvent and without tetramethylsilane (TMS) as an interior label. Contact angle was recorded on a Data physics OCA-20 contact angle analyzer with distilled water as the test liquid. Scanning electronic microscopy (SEM) images were obtained with Hitachi S-4800 (7 kV). The samples were cut and coated with a thin layer of gold before the investigation. DSC measurements were studied using SDTQ 600 of TA Instruments. The films were loaded in aluminum pans, heated from 25 to 200 °C. The heating and cooling temperature ramping rates were 10 °C min−1. Atomic Force Microscope (AFM) was measured by using a NanoScoPe IIIA multimode atomic force microscope under ambient condition in tapping mode. The luminescence (excitation and emission) spectra of the samples were determined with a Hitachi F-4500 fluorescence spectrophotometer using a monochromated Xe lamp as an excitation source. Excitation and emission slits measured 5 nm and 5 nm, respectively. Ultraviolet absorption (UV) spectra in THF solution were detected using a Beijing TU-1901 double beam UV-vis spectrophotometer.
Synthesis of MMF and D1
MMF and D1 were synthesized by using the classic thiol-ene reaction procedure.24 The synthesis route of MMF was shown in Scheme 2. 0.93 g (5.0 mmol) MMVi, 1.14 g (10.0 mmol) furfuryl mercaptan, 2 wt% of DMPA, and THF were placed in a glass vessel and then exposed to a UV light source (365 nm, 100 W) with stirring for 20 min at room temperature. MMF was obtained after evaporation of the solvents with a yield of 98%.
Data of MMF. 1H-NMR (400 MHz, CDCl3, ppm): δ = 0.01–0.30 (12H, m, –SiCH3), 0.83–0.91 (4H, m, –SiCH2CH2S–), 2.61–2.69 (4H, m, –SiCH2CH2SCH2–), 3.75–3.80 (4H, m, –SCH2–CH(CH2)O–), 6.20 (2H, s, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH2)–), 6.30 (2H, s, –CHCHC(CH2)–), 7.30 (2H, s, CHCH–O–).13C-NMR (400 MHz, CDCl3, ppm): 151.99; 141.95; 110.77; 110.36; 28.09; 20.98; 17.55; −0.82. HR-MS (FAB) found: C18H30O3S2Si2 (M + H+): 415.13, calcd for C18H30O3S2Si2: 414.12.
C(CH2)–), 6.30 (2H, s, –CHCHC(CH2)–), 7.30 (2H, s, CHCH–O–).13C-NMR (400 MHz, CDCl3, ppm): 151.99; 141.95; 110.77; 110.36; 28.09; 20.98; 17.55; −0.82. HR-MS (FAB) found: C18H30O3S2Si2 (M + H+): 415.13, calcd for C18H30O3S2Si2: 414.12.D1 was synthesized through the same route to MMF with a yield of 96%, as illustrated in Scheme 1.
Data of D1. 1H-NMR (400 MHz, CDCl3, ppm): δ = 0.09–0.30 (12H, m, –SiCH3), 0.80–0.92 (8H, m, –SiCH2CH2S–), 2.52–2.70 (8H, m, –SiCH2CH2SCH2–), 3.70–3.80 (8H, m, –SCH2–CH(CH2)O–), 6.20 (4H, s, –CHC(CH2)–), 6.30 (4H, s, –CHCHC(CH2)–), 7.31 (4H, s, –CHCH–O–).13C-NMR (400 MHz, CDCl3, ppm): 152.02; 141.95; 110.50; 107.14; 28.04; 20.98; 17.52; −0.58. HR-MS (FAB) calcd for C32H48O8S4Si4 (M): 800.13, found (M + H+): 801.13, (M + Na+): 823.11, (M + K+): 839.11.
Preparation of cross-linked networks (CNs)
A series of networks (see Table 1) were prepared following the same procedure. Taking C5 as an example, at first, D1 (2.00 g, 2.5 mmol) and BMI (1.79 g, 5.0 mmol) were dissolved in 50 mL anhydrous THF as solvent in a 100 mL round bottom flask. Then, most solvent were evaporated off. The flask was put into a oil bath at 60 °C for 5 h. Cross-linked network was obtained after evaporation of THF as a yellow block solid (C5).
Table 1 Formula of samples (C1 to C5)
| Sample |
C1 |
C2 |
C3 |
C4 |
C5 |
Molar ratio (furan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :vinyl) :vinyl) |
1:0.2 |
1:0.4 |
1:0.6 |
1:0.8 |
1:1 |
| D1 (g) |
2 |
2 |
2 |
2 |
2 |
| BMI (g) |
0.36 |
0.72 |
1.07 |
1.43 |
1.79 |
Testing of gel fraction and swelling ratio
According to the classic method, the swelling ratio and the gel fraction of the samples (CNs) were measured by soaking them (W1) in toluene for 72 h at room temperature to ensure swelling equilibrium.25,26 After that, the swollen samples were taken out from the solvent to weigh (W2), then the insoluble network was dried in a vacuum oven at 60 °C to the constant weight (W3). Therefore, the gel fraction and the swelling ratio were calculated by the following equations.
| Gel fraction = W3/W1 × 100% |
| Swelling ratio = W2/W1 × 100% |
where W1, W2 and W3 are the original weight, the swollen weight, and the dried weight after swelling of the samples, respectively.
Results and discussion
Model compounds for DA reaction
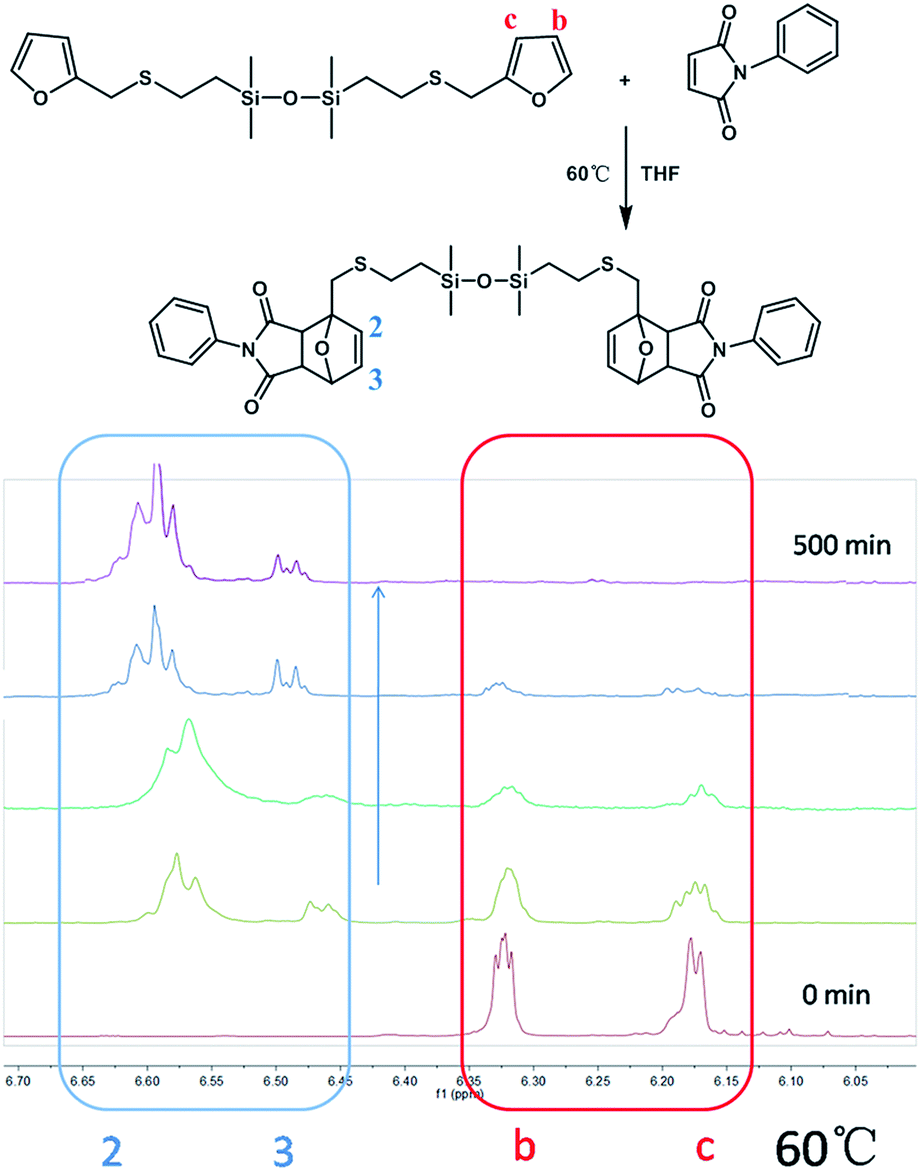
Although the kinetics of DA reaction has been thoroughly studied, reports on DA reaction of silicone-containing molecules are rare. To evaluate the effect of incorporating silicone atoms between furfuryl and maleimide groups in the DA reaction, as illustrated in Scheme 2, a model reaction was designed and started from divinyltetramethyldisiloxane (MMVi). Furan-functionalized disiloxane (MMF) was obtained via thiol-ene reaction, and then the model compounds were synthesized by DA reaction between MMF and N-phenylmaleimide (N-PMI).
The UV absorption change in the Fig. 1 shows the reaction of the model compound. Compared with N-PMI, the decrease of absorption peak for M1 at 320 nm indicated the loss of conjugated double bonds in the maleimide OC–CC–CO group upon the formation of the adduct. It is worth noting that all of the spectra show a single peak at about 250 nm, indicating that no side-reaction occurred when the DA reaction proceeded.27
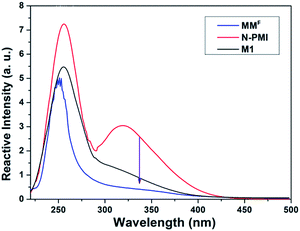 |
| | Fig. 1 UV spectra of MMF, N-PMI, and M1. | |
The DA reaction between MMF and N-PMI was further investigated by 1H-NMR.28,29 An equivalent molar mixture of MMF and N-PMI was dissolved in THF. Then, the samples were taken out at given times to monitor the DA reaction at different temperatures. As shown in Fig. 2, no signals at 6.4 and 6.6 ppm were exhibited corresponding to the DA adducts until after 30 min, wherein peaks appeared. In addition, the intensity of those peaks becomes more apparent with the reaction. The peaks at about 6.1 and 6.3 ppm that corresponded to the signal of –CHCH–CHCH– disappeared gradually and faded away completely after reaction for 500 min. The accessibility of DA reaction between MMF and N-PMI is thus proven.
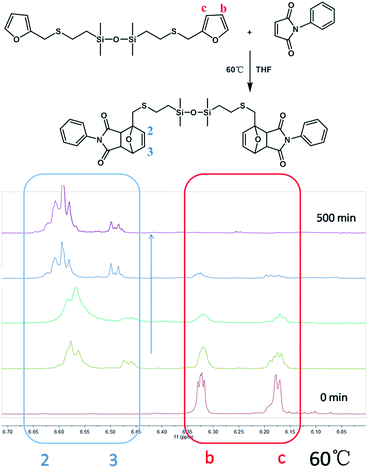 |
| | Fig. 2 Model DA reaction between MMF and N-PMI, and the 1H-NMR spectra of model DA reaction at 60 °C versus time (reaction for 500 min). | |
As shown in Fig. 2, the peaks at 6.1 and 6.3 ppm should be assigned to the b and c protons located in the unreacted furan group, meanwhile, peaks at 6.4 and 6.6 ppm are attributed to the 2 and 3 protons of the DA adduct; thus, the quantification of the DA addition products and unreacted furfuryl group could be achieved from the integrals of b and c peaks, as well as 2 and 3 peaks, respectively. Therefore, the conversion (x) of DA reaction could be calculated by the following eqn (1):
| |
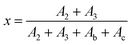 | (1) |
where
A2 and
A3 are the integrals of peaks of 2 and 3 protons, while
Ab and
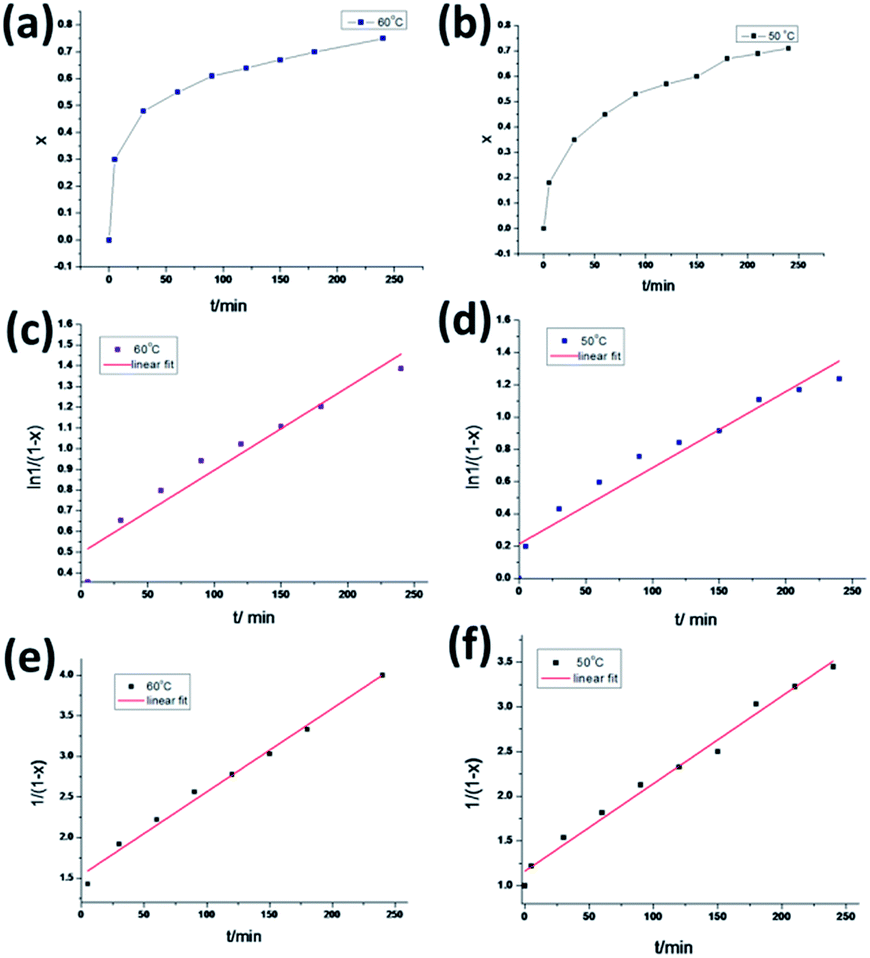
Ac are the integrals of peaks b and c, respectively. The function between conversion and the reaction time are displayed in
Fig. 3, showing that the conversion increases rapidly within the first 100 min and slows down after 100 min. The first-order kinetics of the DA reaction could be obtained from
eqn (2),
| |
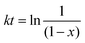 | (2) |
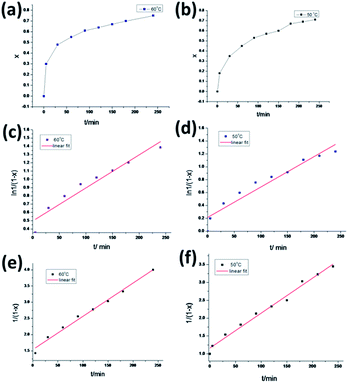 |
| | Fig. 3 Conversion rate (x) versus reaction time for the model reaction from 1H-NMR spectra obtained at 60 °C (a) and 50 °C (b). First-order reaction kinetics of the model DA reaction studied by 1H-NMR spectroscopy at 60 °C (c) and 50 °C (d). Second-order reaction kinetics of the model DA reaction studied by 1H-NMR spectroscopy at 60 °C (e) and 50 °C (f). | |
While the second-order kinetic could be obtained from eqn (3),
where
k is the rate constant. First- and second-order reaction kinetics of the model DA reaction calculated from the
1H NMR data are illustrated in
Fig. 3. At 50 °C, a linear fit of the data for the first-order kinetics obtained a rate constant of
K1 = 0.00472 min
−1 with a confidence coefficient square (
R2) of 0.931. For the second-order kinetics,
K2 = 0.00979 mol L
−1 min with a
R2 of 0.986, which indicates that the DA reaction of the model compound conformed to the second-order kinetics. Unexpectedly, the reaction at 60 °C also has a bigger confidence coefficient square for the second-order kinetics of 0.987 than the first-order. Therefore, this reaction was calculated to follow a second-order kinetic.
Using the known rate constant (K2) accorded with the second-order kinetics at different temperatures, the activation energy (E) obtained from the Arrhenius equation (eqn (4)) is 41.06 kJ mol−1. The kinetic results were similar to the previous reports about DA reaction, which indicated that the incorporation of Si–O–Si structure units have little effect on the DA reaction itself.
| | |
lnk = A − E/RT
| (4) |
Synthesis of D1 (furan-functionalized D4)
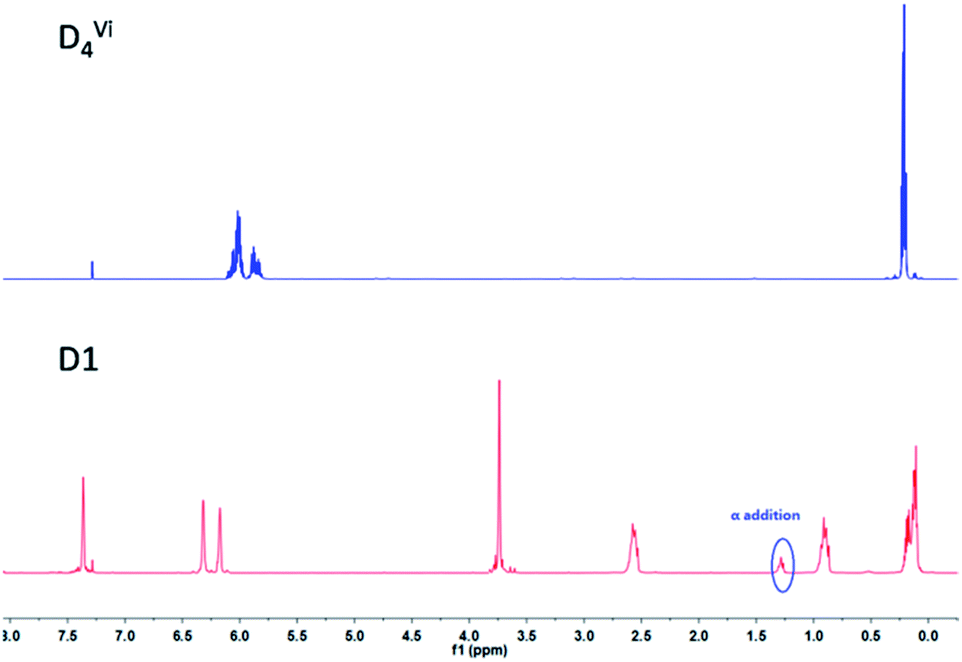
As shown in Scheme 1, D4Vi was functionalized by furfuryl mercaptan (FM) via a facile thiol-ene “click” reaction. Then the furfuryl-functionalized D4 (D1) obtained very high yields within 20 min, as confirmed by the 1H-NMR data. As illustrated in Fig. 4, a vinyl peak (–Si–CHCH2) at 5.70 ppm to 6.22 ppm diminished while a peak at 2.61 to 2.70 ppm assigned to S–CH2 emerged. It is worth noting that a peak existed at about 1.3 ppm, which should be ascribed to the Markovnikov adduct. However, a Markovnikov adduct could not have affected the integral structure of the final crosslinked network because a α-addition product only accounts for a small proportion that is about 5% of the peak. 1H-NMR data indicates that the thiol-ene “click” reaction is an efficient approach to bring furfuryl groups into cyclosiloxanes and that it is possible to accurately control the functionalization densities by simply mixing the cyclosiloxanes with furfuryl in a controlled molar ratio. The IR spectra of MMF and D1 were shown in Fig. S2.† MMF and D1 both exhibited characteristic absorption peak of furan rings at 1250 and 1148 cm−1. The results of the C-NMR, IR, and MS analyses all indicated the successful modification of furan groups to both MMF and D1.
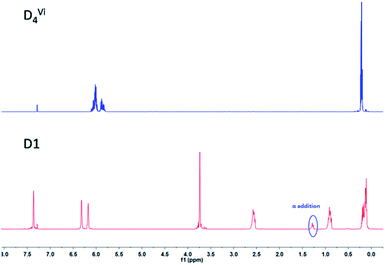 |
| | Fig. 4 1H-NMR spectra of D4Vi and D1. | |
Construction of crosslinked network via DA reaction
The purpose of this contribution is to fabricate a thermally reversible crosslinking siloxane network through incorporating thermally reversible groups into the CNs. The kinetic study above indicated that the DA reaction proceeds better at 60 °C; thus, the crosslinking reactions between D1 with BMI with varied molar ratios were carried out at 60 °C for 24 h. The CNs (C1 to C5) were then fabricated and bear two kinds of crosslinked covalent bonds: (1) a permanent covalent bond from the thiol-ene reaction, and (2) a reversible bond from the DA reaction between the maleic and furan groups. Possessing these structural features, C1 to C5 were expected to have self-healing properties.
Solvent resistance and swelling property of CNs
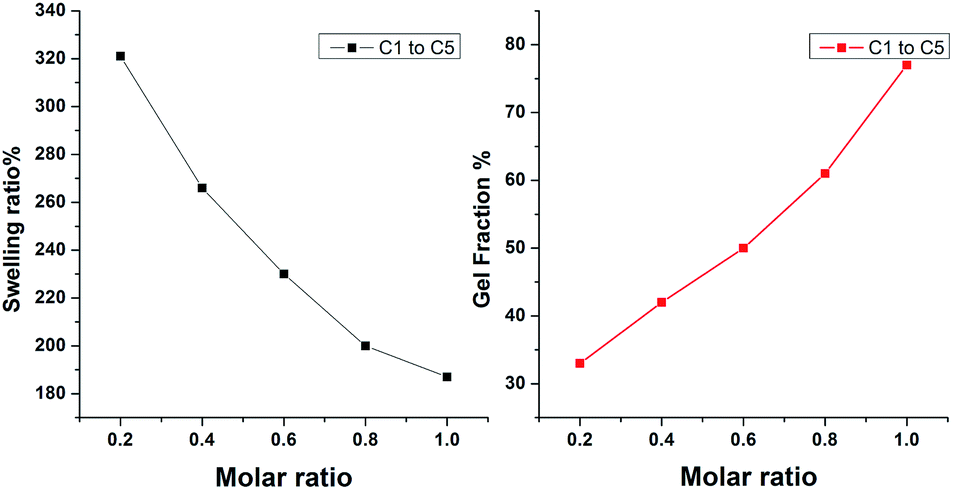
Sol–gel method was used to prove full curing of the networks. At room temperature, the solid CNs were put into THF, toluene, and CHCl3 solutions, respectively, and it was observed that the sample only swells but do not dissolve in these common solvents, which reflected the existence of CNs in the sample. The insoluble fraction for C1 to C5 complexes are shown in Fig. 5. We found that the dynamic crosslinked CNs could swell in toluene which is a good solvent for D1 and BMI. In addition, the crosslinking extent of CNs networks could be illustrated by their swelling ratio as well as gel fraction. As shown in Fig. 5(a), the swelling ratio for C1 is 321% and decreases gradually with the increase of BMI and finally reaches a minimum of 187% of C5. However, when it comes to the gel fraction, as shown in Fig. 5(b), the numerical change presented a reverse trend. The gel fraction for C1 is about 33% and increased to 78% for C5. We can conclude that increasing the content of BMI could increase the gel fraction but cause a decrease in the swelling ratio. The swelling ratio and the gel fraction are both indexes that reflect the crosslinking degree for CNs networks. As a result, the crosslinking degree could be easily regulated by just altering the molar ratio of D1 and BMI, which was facilitated for us to adjust the properties of the CNs.
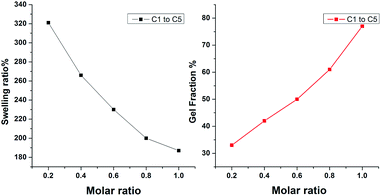 |
| | Fig. 5 Swelling ratio and gel fraction of CNs were measured in toluene. | |
Remolding behavior of the crosslinked networks
To investigate the remolding behavior of the CNs, thermal treatment was adopted. As illustrated in Fig. 6, the network (C1) was crushed down into powder and then remolded at 120 °C for 20 min. re-DA reaction is dominant and as a result, the powder fused into viscous liquid at an evaluated temperature of 120 °C. When cooled down to approximately 60 °C, the DA reaction played a main role. Afterwards, a solid polymeric bulk network was re-formed and re-shaped after cooling down to room temperature. The heating-cooling process could be repeated for several times; thus, it could be deduced that the furfuryl functionalized cyclotetrasiloxane D1 could be de-crosslinked and re-crosslinked by using BMI repeatedly in their solid state. The excellent remolding properties of the CNs could endow them broad applications in recyclable materials field. To investigate the r-DA reaction performed in the remolding process, FT-IR analysis was applied. The de-forming sample of C1 mixture was obtained by dissolving C1 into toluene at 120 °C for 20 min, and then quickly cooling down to room temperature. As shown in Fig. S3,† we could found that the shoulder peak at about 1770 cm−1 disappeared, which should be attributed to the DA adducts of maleimides. The IR result indicated that the r-DA reaction occurred upon heating.
 |
| | Fig. 6 Digital images of C1 to illustrate the remolding process. | |
Photoluminescence properties of the networks
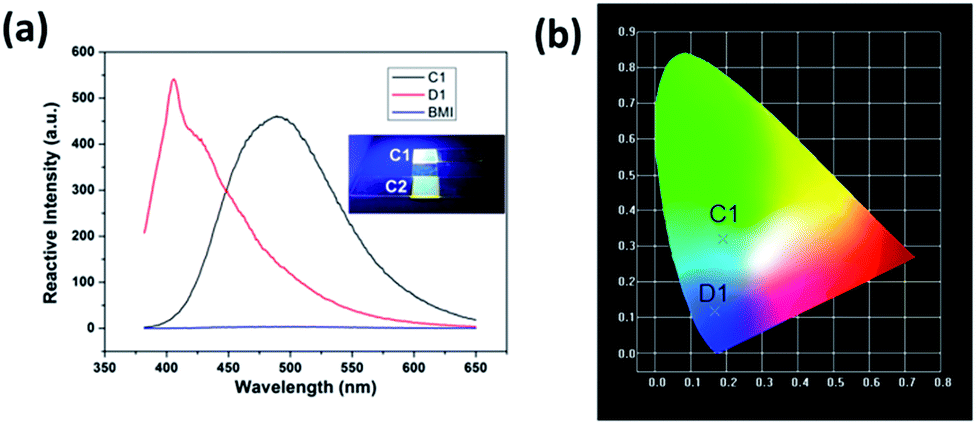
CNs show strong photoluminescence under a 365 nm UV light; the luminescence of C1 and C2 can be seen in the inserted images in Fig. 7(a). The luminescent properties of D1 and BMI were tested in THF solution excited by a wavelength of 330 nm. The photoluminescent properties of CNs in their solid states were also investigated at room temperature, using the luminescence spectra of C1 and D1 as examples, shown in Fig. 7(a). D1 exhibited a strong emission at about 400 nm, while the emission shifted to 470 nm (C1) after the curing process. Since the BMI showed a fairly weak emission that was negligible under the same excited condition, the luminescence of the CNs should be derived from D1. Thus, the luminescence is generated from the Si → S coordination bond which was easy to form in D1 after the thiol-ene addition. The five degenerated 3d orbits of Si atom split due to the effect of the Si → S coordination bond and therefore an electron transition in the split orbits occurred after absorbing the energy from the UV light,23,30 known as “d–d transition”. As shown in Fig. 7(b), based on the CIE-1931 RGB colorimetric program, the bright blue luminescence of the C1 and D1 could be easily seen upon excitation at 330 nm, as shown in Fig. 7(a). The fine luminescent performance of CNs show potential applications in luminescent devices.
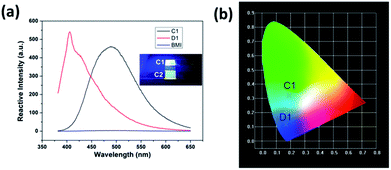 |
| | Fig. 7 (a) Fluorescence emission spectra of C1, D1, and BMI (excitation at 330 nm), the inserted: digital image of C1 and C2 under UV light (365 nm), (b) emission colors of C1 and D1 in CIE 1931 chromaticity diagram; CIE color coordinates: C1 (0.187, 0.322) and D1 (0.175, 0.124). | |
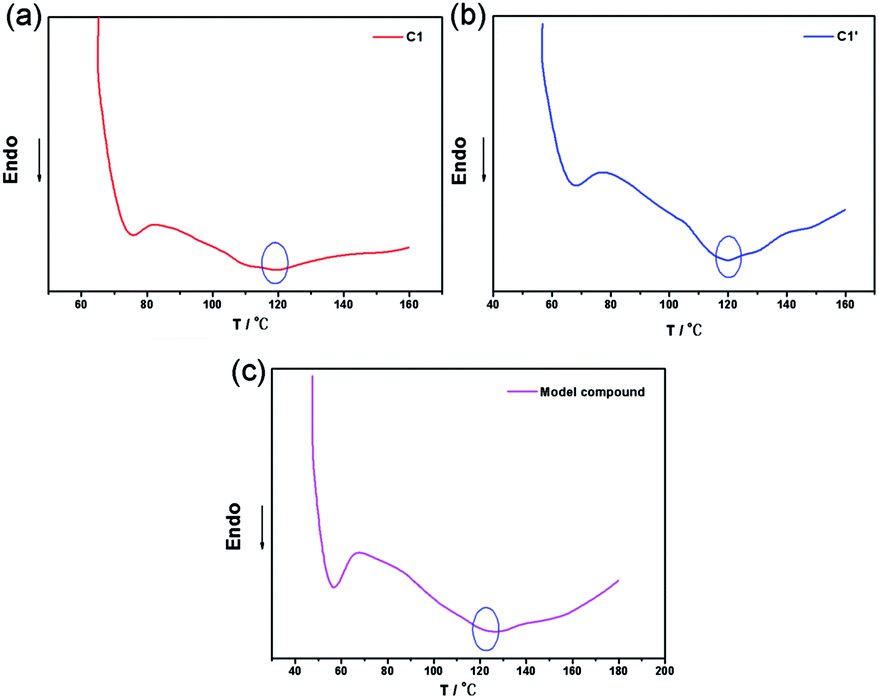
Differential scanning calorimetry (DSC) analysis
To further investigate the thermal reversibility of the CNs, DSC analysis was utilized. Typical DSC curves of C1, C1′, and M1 are presented in Fig. 8 wherein C1 exhibited an endothermic peak at about 120 °C (Fig. 8(a)) which corresponds to the rDA reaction. C1′ is the sample of C1 which was conducted with a heating and cooling process. DSC curve of C1′ (Fig. 8(b)) also exhibited an endothermic peak at 120 °C. The results illustrate that furan and maleic imide functional groups can regenerate DA covalent bond. M1 show analogous DSC curves because of the similarity of their inherent chemical structures. The DSC results confirmed the existence of rDA reaction in the CNs, and further highlights the capability of the crosslinked network as self-healing materials.26 The cooling curves of the samples were shown in Fig. S1 (ESI†) to show the re-construction process of the networks. C1 and C2 were selected for all the structures of the network were analogous. Heating and cooling down rate was 10 °C min−1. Upon cooling in a DSC cell, C1 and C2 both performed a DA reaction at about with an exothermal peak at 60 to 70 °C.
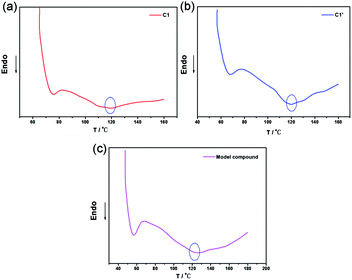 |
| | Fig. 8 DSC curves of C1 (a), C1′ (b), and M1 (c). | |
Contact angle analysis
To get a deeper understanding of the surface properties of the fabricated networks, contact angle analysis was performed to study the effect of the introduction of BMI on hydrophilic surfaces. Purified water was used as the testing liquid. The contact angles were calculated from the shape of the drop with an accuracy of 0.1° using an image analysis program. Polysiloxane-based materials are usually used as hydrophobic surfaces because they bear a water contract angle about 109°.31 Contact angles of C1 to C5 are depicted in Fig. 9, which shows that C1 bears a lower contact angle than that of pure PDMS (i.e., 98.5°), caused by the incorporation of S atoms which are hydrophilic and thereby decreasing hydrophobic properties of the surface. Meanwhile, the contact angles decreased gradually with the increase of the BMI to a lowest value of 76.7° for C5, demonstrating the hydrophilic property of BMI. The surface contact angle of the CNs could be easily tuned by merely altering the molar ratio between D1 and BMI.
 |
| | Fig. 9 Contact angle images of C1 to C5. | |
Morphology analysis
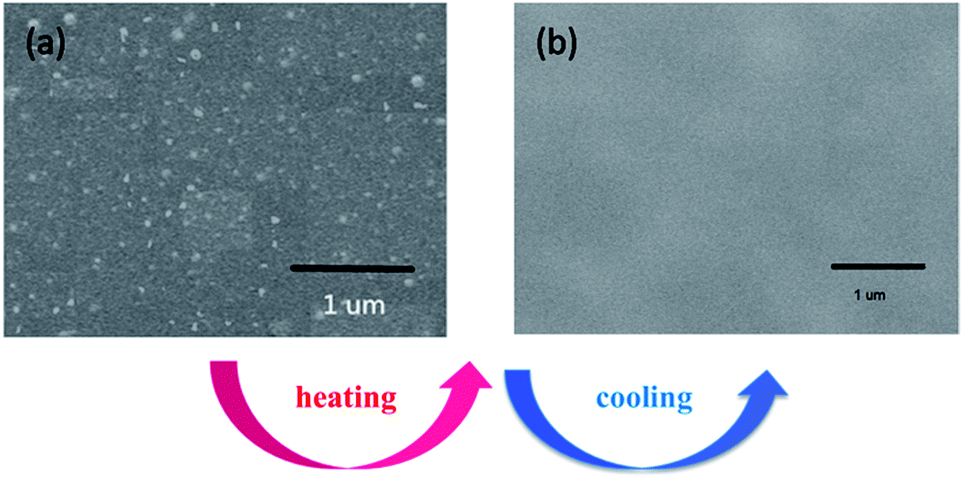
AFM (Atomic Force Microscope) is usually selected to investigate the micro-phase separation behavior reflected by various hardness of materials.32 Taking C1 as an example, AFM micrographs of the upper surface of C1 are depicted in Fig. 10. The probe reacting on the surface with a different hardness lost energy and produced an altered phase contrast. The bright regions stand for the hard phase (BMI) and the dark regions represent the soft phase (D1) in the image. Fig. 10(a) presents the initial surface morphology of C1 before the heating and cooling process. Microphase separation was found in Fig. 10(a). Crosslinking process led to the emergence of the hard phase for the crosslinking structure which is attributed to the hard phase of BMI. After a cycle of heating and cooling down, a homogeneous surface was observed (Fig. 10(c) and (d)) where hard and soft phases were difficult to differentiate. The results demonstrated that the heating and cooling procedure gave the CNs a more uniform surface. SEM testing of the upper surface of the original C1 and after heating and cooling down process are depicted in Fig. 11. The results indicate that the networks show microphase separation before heating and cooling down (Fig. 11(a)) while presenting a homogeneous single phase after being heated and cooled down (Fig. 11(b)). The SEM results were consistent with the previous AFM consequences. The heating and cooling down process causes C1 to become a continuous phase structure.
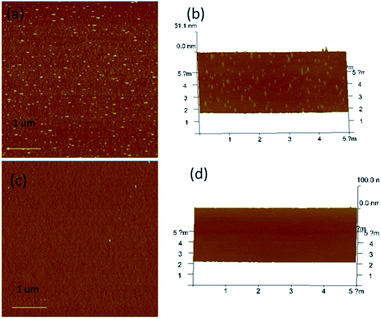 |
| | Fig. 10 (a) AFM micrograph of C1 before heating and cooling process, (b) 3D AFM micrograph of C1 before heating and cooling process, (c) AFM image of C1 after healing and cooling process, (d) 3D AFM micrograph of C1 after healing and cooling process. | |
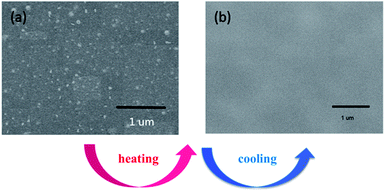 |
| | Fig. 11 SEM micrograph of C1 before (a) and after (b) heating and cooling down process. | |
Application as a glass adhesive
We found that the samples were hard to remove from the bottle walls in synthetic experiments. To investigate the potential applications of CNs in adhesives, a simple adhesive test was carried out to verify their adhesive ability on glass. Two glass slides were cleaned using CHCl3, acetone, ethanol in sequence by ultrasonic waves. Granular C5 was spread onto the slide and heated to 120 °C. The second slide was clung to another slide using molten C5. As shown in Fig. 12(a) and (c), coherent slide was fabricated after cooling down. The coherent slide could hold a weight of 2.330 kg without any damage to the structure (Fig. 12(b)). The results indicated the good performance of CNs in adhesion. C5 was selected to investigate the recyclability. At first, granular C5 was spread onto the slide and heated to 120 °C. The second slide was clung to another slide using molten C5. A coherent slide was fabricated after cooling down. This heating and cooling process was repeated for 5 times. After each time the coherent slide was break and the weight was tested and listed in Table 2. We could found from the table that the force at break did not change prominently after the heating and cooling process. The result illustrated the good recyclability of the polymers as adhesives. Furthermore, due to the self-healing and remolding abilities, these novel materials could be used as recyclable adhesives.
 |
| | Fig. 12 Digital images of the coherent glass slide to show the adhesive ability of CNs. | |
Table 2 Weight at break of the coherent slide with heating and cooling time varied
| Repeat times |
1 |
2 |
3 |
4 |
5 |
| Weight at break (kg) |
2.75 |
2.68 |
2.71 |
2.77 |
2.59 |
Conclusions
In summary, we demonstrated a novel molecular design method and synthesized a cyclosiloxane that contains furan moieties via facile thiol-ene “click” reaction. The functionalized D4 can be effectively used in a simple one-pot preparation of high-performance and recyclable polysiloxane CNs. The crosslinked silicone network also exhibits excellent luminescent properties. The thermally reversible ability of the networks can be achieved through DA and rDA reactions between furan and maleic moiety. Therefore, these networks can be easily fixed and reshaped. The kinetics of DA reaction was elucidated by in situ 1H-NMR experiments. Moreover, the silicone networks are environmentally friendly and show potential for use as recyclable adhesives, especially in glass. The proposed method may be further used in preparation of other self-healing materials, shape-memory networks, and thermo-responsive materials.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21274080, and 21502105), Special Fund for Shandong Independent Innovation and Achievements transformation (No. 2014ZZCX01101), and the National Science Foundation of Shandong Province (ZR2015BQ008).
Notes and references
- F. Abbasi, H. Mirzadeh and A. A. Katbab, Polym. Int., 2001, 50, 1279–1287 CrossRef CAS
 .
. - İ. Yilgör and J. E. McGrath, in Polysiloxane Copolymers/Anionic Polymerization, Springer, 1988, pp. 1–86 Search PubMed .
- A. M. Peterson, R. E. Jensen and G. R. Palmese, ACS Appl. Mater. Interfaces, 2009, 1, 992–995 CAS .
- S. Chen, F. Wang, Y. Peng, T. Chen, Q. Wu and P. Sun, Macromol. Rapid Commun., 2015, 36, 1687–1692 CrossRef CAS PubMed .
- M. Behl, M. Y. Razzaq and A. Lendlein, Adv. Mater., 2010, 22, 3388–3410 CrossRef CAS PubMed .
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2015, 48, 1329–1334 CrossRef CAS .
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS .
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed .
- I. L. Hia, V. Vahedi and P. Pasbakhsh, Polym. Rev., 2016, 56, 225–261 CrossRef CAS .
- K. Guo, D. L. Zhang, X. M. Zhang, J. Zhang, L. S. Ding, B. J. Li and S. Zhang, Angew. Chem., 2015, 127, 12295–12301 CrossRef .
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS .
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS PubMed .
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed .
- R. Gheneim, C. Perez-Berumen and A. Gandini, Macromolecules, 2002, 35, 7246–7253 CrossRef CAS .
- Y. L. Liu and C. Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905–913 CrossRef CAS .
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS .
- J. R. Mcelhanon, E. M. Russick, D. R. Wheeler, D. A. Loy and J. H. Aubert, J. Appl. Polym. Sci., 2002, 85, 1496–1502 CrossRef CAS .
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed .
- L. M. Campos, I. Meinel, R. G. Guino, M. Schierhorn, N. Gupta, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3728–3733 CrossRef CAS .
- Y. Zuo, J. Cao and S. Feng, Adv. Funct. Mater., 2015, 25, 2754–2762 CrossRef CAS .
- W. Zhou, H. Zheng, Y. Li, H. Liu and Y. Li, Org. Lett., 2010, 12, 4078–4081 CrossRef CAS PubMed .
- R. Kumar, Saima, A. Shard, N. H. Andhare, Richa and A. K. Sinha, Angew. Chem., Int. Ed. Engl., 2015, 54, 828–832 CrossRef CAS PubMed .
- Y. Zuo, D. Wang, J. Zhang and S. Feng, RSC Adv., 2014, 4, 62827–62834 RSC .
- L. Xue, D. Wang, Z. Yang, Y. Liang, J. Zhang and S. Feng, Eur. Polym. J., 2013, 49, 1050–1056 CrossRef CAS .
- Y. Zuo, H. Lu, L. Xue, X. Wang, L. Ning and S. Feng, J. Mater. Chem. C, 2014, 2, 2724–2734 RSC .
- J. Bai, H. Li, Z. Shi, M. Tian and J. Yin, RSC Adv., 2015, 5, 45376–45383 RSC .
- A. Gandini, D. Coelho and A. J. Silvestre, Eur. Polym. J., 2008, 44, 4029–4036 CrossRef CAS .
- Q. Tian, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2009, 19, 1289–1296 RSC .
- X. Liu, P. Du, L. Liu, Z. Zheng, X. Wang, T. Joncheray and Y. Zhang, Polym. Bull., 2013, 70, 2319–2335 CrossRef CAS .
- Y. Zuo, Z. Gou, J. Cao, Z. Yang, H. Lu and S. Feng, Chem. - Eur. J., 2015, 21, 10972–10977 CrossRef CAS PubMed .
- J. T. Han, D. H. Lee, C. Y. Ryu and K. Cho, J. Am. Chem. Soc., 2004, 126, 4796–4797 CrossRef CAS PubMed .
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra14659g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.