
Biobased poly(ethylene furanoate-co-ethylene succinate) copolyesters: solid state structure, melting point depression and biodegradability
Zoe Terzopouloua,
Vasilios Tsanaktsisa,
Dimitrios N. Bikiaris*a,
Stylianos Exarhopoulosbc,
Dimitrios G. Papageorgioud and
George Z. Papageorgiou*b
aLaboratory of Polymer Chemistry and Technology, Department of Chemistry, Aristotle University of Thessaloniki, Thessaloniki, Macedonia GR-541 24, Greece. E-mail: dbic@chem.auth.gr
bChemistry Department, University of Ioannina, P.O. Box 1186, 45110 Ioannina, Greece. E-mail: gzpap@cc.uoi.gr
cDepartment of Food Technology, Technological Educational Institute of Thessaloniki, PO Box 141, GR-57400 Thessaloniki, Greece
dSchool of Materials and National Graphene Institute, University of Manchester, Oxford Road, Manchester M13 9PL, UK
First published on 31st August 2016
Abstract
Poly(ethylene furanoate) (PEF) is a fully bio-based polyester with unique gas barrier properties, considered an alternative to poly(ethylene terephthalate) (PET) in food packaging applications. However, it is not biodegradable. For this reason, copolymerization with an aliphatic succinic acid monomer was investigated. The respective poly(ethylene furanoate-co-ethylene succinate) (PEFSu) copolymers were prepared via melt polycondensation from 2,5-dimethylfuran-dicarboxylate, succinic acid and ethylene glycol at different ratios. 1HNMR spectroscopy showed the copolymers are random. The crystallization and melting of the copolymers were thoroughly evaluated. Isodimorphic cocrystallization was concluded from both the WAXD patterns and the minimum in the plots of melting temperature versus composition. The pseudo-eutectic melting point corresponded to an ethylene succinate content of about 30 mol%. The enzymatic hydrolysis tests using Rhizopus delemar and Pseudomonas cepacia lipase revealed that the copolymers with up to 50 mol% ES units show measurable weight loss rates. For higher ES content, the copolymers showed fast hydrolysis.
1. Introduction
The global ecological problems which include limited fossil fuels and the disadvantages that come with the widespread use of them (limited recyclability, relatively high cost and environmental impact) have led governments, academia and (to a lesser extent) industry to look for eco-friendly, alternative solutions. Bioplastics represent one of the most ambitious projects, since a lot of products have been developed over the last years and are focused on the use of renewable resources. Furanoate-based polyesters based on 2,5-furandicarboxylic acid are a viable candidate for the replacement of their naphthalate and terephthalate counterparts, since they originate from monomers derived straight from renewable resources like furfural and hydroxymethylfurfural. For this reason, several works have been dedicated on the detailed study of their production and properties for the use in a wide variety of applications.1–22 Of special importance are the unique barrier properties of PEF and furanoates in general which enable uses as green food packaging materials.11,12,19Poly(ethylene succinate) (PESu) is a semi-crystalline aliphatic polyester, which has been attracting attention since it exhibits excellent biodegrability due to its hydrolysable ester bonds, stability and favorable mechanical properties.23–26 It can be synthesized by either ring-opening polymerization of succinic anhydride or by polycondensation of succinic acid and ethylene glycol.27 PESu has been successfully blended with a number of polymers in an attempt to improve its physical properties and increase its biodegradation rates, such as poly(ε-caprolactone),28 poly(butylene succinate),29,30 poly(octamethylene succinate),31 poly(decamethylene succinate),32,33 poly(ethylene oxide),34 poly(ethylene terephthalate),35 poly(L-lactide),26 poly(diethylene glycol succinate),36 poly(ethylene adipate)37 and others. The majority of those materials exhibit good compatibility in blends and improved biodegradation rates. That is the main reason why the specific polyester was carefully chosen in our attempts to prepare copolymers with PEF. In a similar work by Dubois and coworkers38 involving a member of the furanoates family that was copolymerized, the authors successfully prepared poly(butylene succinate-co-butylene furandicarboxylate) (PBSF) copolyesters with high thermal stability and adequate mechanical properties, displaying a potential for replacement of commodity thermoplastics or elastomers.
Basic research on the relationship between structure, morphology, and properties as well efforts to understand the biodegradation mechanism will enable us to design and synthesize a great variety of biodegradable polymers to fulfill the demands in practical applications.39 Biodegradation rate is undoubtedly one of the most important properties of aliphatic polyesters, since the widespread use of those materials in packaging applications (amongst others) has already caused various environmental problems. The enzymatic degradation of polyesters is sensitive to their chemical structure, the hydrophilic/hydrophobic balance within the main chain, molecular weight, the specific solid-state morphology, crystallinity, and so forth. As for crystalline structure and morphology the spherulite size and the lamellar structure can greatly influence the rate of biodegradation.39
Higher degradation rates can be achieved by copolymers compared to the homopolymers, and this is basically attributed to the limited crystallinity.40 In most of the copolymers where both components are crystallizable, the degree of crystallinity decreases as the minor component content increases, due to incompatibility in crystal lattices of the two components.41–43
On the contrary, if the two crystallizable units are compatible in each crystal lattice, cocrystallization can take place. Two cases of cocrystallization behavior have been reported.41 For components with similar chemical structure isomorphism may occur meaning that only one crystalline phase containing both comonomer units is observed at all compositions.44–46 In isodimorphism two crystalline phases and pseudo-eutectic behavior are observed. Isodimorphism has been observed in most cases of random copolymers.47–49
In terms of the thermodynamic parameters and the cocrystallization of copolymers, various models have been proposed, among them those of Flory,50,51 Sanchez–Eby,52 Baur53 as well as that of Wendling and Suter.54,55 Several of them have been applied to the experimental results of this work.
Based on the above mentioned facts, a series of copolymers based on poly(ethylene furanoate) (PEF) and poly(ethylene succinate) (PESu) have been prepared by the melt polycondensation method. The studies on the crystalline structure and crystallization kinetics of the copolymers are very important on a practical viewpoint since they determine the performance and biodegrability of end products. For this reason a variety of techniques and measurement methods were applied on the produced materials, in order to export solid conclusions and evaluate the performance of those materials on packaging applications.
2. Experimental
2.1. Materials
2,5-Furan dicarboxylic acid (2,5-FDCA, purum 97%), succinic acid (purum 99%), ethylene glycol and tetrabutyl titanate (TBT) catalyst of analytical grade were purchased from Aldrich Co. 2,5-Dimethylfuran-dicarboxylate (DMFD) was synthesized from 2,5-FDCA and methanol as described in our previous work.56 All other materials and solvents used were of analytical grade.2.2. Copolymer synthesis
Neat PEF and PESu polyesters were prepared by the two-stage melt polycondensation method (esterification and polycondensation) in a glass batch reactor as described in our previous works.57 The copolymers under study have been synthesized with the same procedure using different DMFD/succinic acid feeding ratios (Table 1) and ethylene glycol from the beginning of esterification procedure. After the polycondensation reaction was completed, the polyesters were easily removed, milled and washed with methanol.| Sample | IV (dL g−1) | PEFSu feed ratios | PEFSu 1HNMR | R | LnS | LnF |
|---|---|---|---|---|---|---|
| 1 | 0.38 | 5/95 | 2/98 | 0.99 | 50 | 1.02 |
| 2 | 0.39 | 10/90 | 11/89 | 1.00 | 9.09 | 1.12 |
| 3 | 0.36 | 15/85 | 17/83 | 1.02 | 5.88 | 1.20 |
| 4 | 0.41 | 30/70 | 35/65 | 0.99 | 2.85 | 1.53 |
| 5 | 0.38 | 40/60 | 37/63 | 1.01 | 2.70 | 1.58 |
| 6 | 0.34 | 50/50 | 49/51 | 1.02 | 2.04 | 1.96 |
| 7 | 0.36 | 60/40 | 61/39 | 0.99 | 1.63 | 2.56 |
| 8 | 0.42 | 80/20 | 81/19 | 0.99 | 1.23 | 5.26 |
| 9 | 0.40 | 90/10 | 87/13 | 1.00 | 1.14 | 7.69 |
| 10 | 0.36 | 95/5 | 92/8 | 1.01 | 1.08 | 12.5 |
2.3. Polyester characterization
Isothermal crystallization experiments of the polymers at various temperatures below the melting point were performed after self-nucleation of the polyester sample. Self-nucleation measurements were performed in analogy to the procedure described by Fillon et al.58 The protocol used is a modification of that described by Müller et al.59 and can be summarized as follows: (a) melting of the sample at 40 °C above the observed melting point for 5 min to erase any previous thermal history; (b) cooling at 20 °C min−1 to room temperature and crystallization; (c) cold-crystallization to create a ‘‘standard’’ thermal history and partial melting by heating at 5 °C min−1 up to a “self-nucleation temperature”, Ts which was 224 °C for PEF and properly decreased for the rest polymers; (d) thermal conditioning at Ts for 1 min. Depending on Ts, the crystalline polyester will be completely molten, only self-nucleated or self-nucleated and annealed. If Ts is sufficiently high, no self-nuclei or crystal fragments can remain (Ts domain I – complete melting domain). At intermediate Ts values, the sample is almost completely molten, but some small crystal fragments or crystal memory effects remain, which can act as self-nuclei during a subsequent cooling from Ts, (Ts domain II-self – nucleation domain). Finally, if Ts is too low, the crystals will only be partially molten, and the remaining crystals will undergo annealing during the 5 min at Ts, while the molten crystals will be self-nucleated during the later cooling, (Ts domain III – self-nucleation and annealing domain); (e) cooling scan from Ts at 20 °C min−1 to the crystallization temperature (Tc), where the effects of the previous thermal treatment will be reflected on isothermal crystallization; (f) heating scan at 20 °C min−1 for standard DSC scans, or heating at an underlying rate 5 °C min−1 in case of TMDSC studies, to 40 °C above the melting point, where the effects of the thermal history will be apparent on the melting signals. Experiments were performed to check that the sample did not crystallize during the cooling to Tc and that a full crystallization exothermic peak was recorded at Tc. In case that some other method was applied, this will be discussed in the corresponding part.
3. Results and discussion
3.1. Synthesis and molecular characterization of the PEFS copolymers
The PEF and PESu homopolymers along with the PEFSu copolymers were synthesized following the two-step melt polycondensation method. The feeding ratios between PEF/PESu monomers can be seen in Table 1 with the measured intrinsic viscosity values. All synthesized copolymers exhibit very similar molecular weights since their IVs differ very slightly between each other. The chemical structures of PEF, PESu and the copolymers are shown in Fig. 1.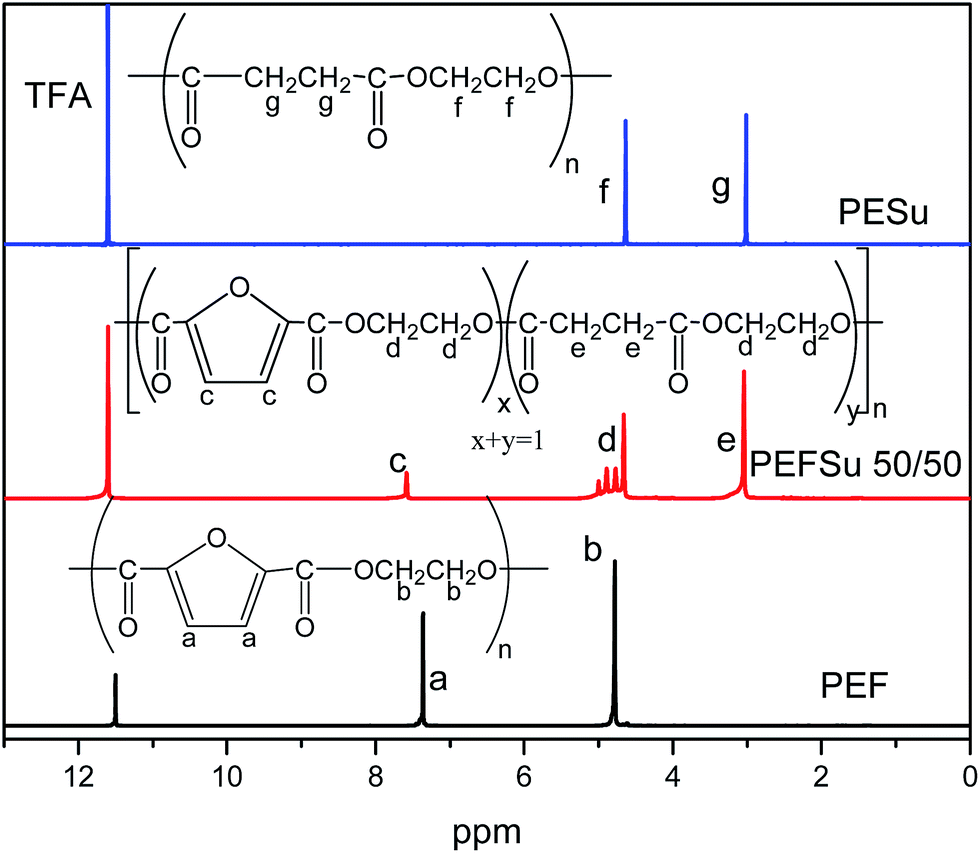 | ||
| Fig. 1 Chemical structures and 1H NMR spectra of PEF, PESu and the PEFSu 50/50 copolymer. | ||
The 1HNMR spectra of the copolymers were used to elucidate their structure (Fig. 1). The protons of the furanoate ring are the most deprotected in the macromolecules due to the carbonyl groups and the π electron system of the ring and they appear at about 7.45 ppm, (the a and c protons of PEF and PEFSu 50/50, respectively). On the other hand, the protons of the succinic moiety appear at 3.02 ppm (e and g protons), i.e. at lower values compared to the protons of the ethylene part which appear at 4.7 ppm giving the characteristic pattern which was used to calculate the real sequence of each comonomer in the macromolecular chain (d and f protons).
The degree of randomness (R) in the PEFSu copolyesters was calculated using the resonance peaks of the ethylene units' aliphatic protons (a). The degree of randomness is defined as:60
| R = PFS + PSF | (1) |
 | (2) |
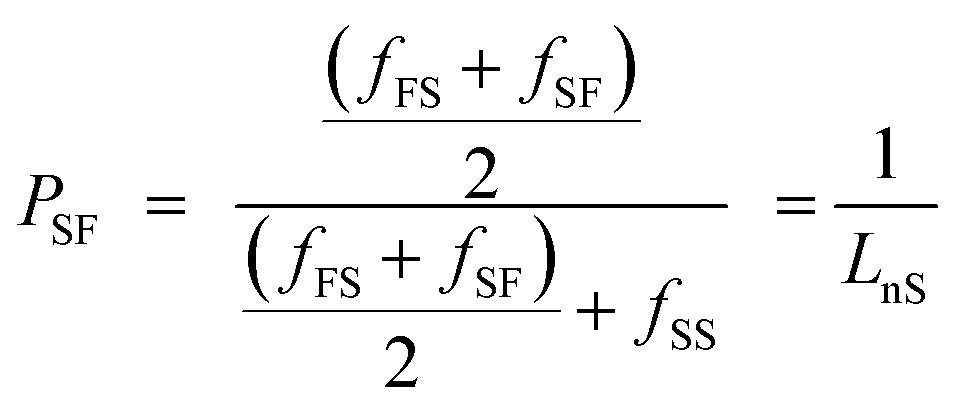 | (3) |
The number average sequence length or block length for furanoate (LnF) and succinate (LnS) units was calculated using eqn (2) and (3) respectively, according to Yamadera and Murano.62 Table 1 summarizes the corresponding values and it can be seen that by increasing the EF ratio in copolymers, the corresponding block length is also increased and the same appears for ES blocks when the ES ratio was higher.
Fig. 2a and b show the DSC traces for the as received samples of ES rich copolymers and EF rich copolymers, respectively. Most of the copolymers crystallized after the solvent treatment. The copolymers with intermediate composition showed broad or multiple melting peaks on heating scans. However, the traces for PEFSu 35/65 and 37/63 samples were characterized by complete absence of melting, indicating their amorphous nature. The phase behavior is highly dependent on the comonomer content and the intense melting peaks decrease with increasing comonomer unit content as was expected. The samples with high EF content (>81 mol%) display peaks which represent the melting of PEF crystals from 185–220 °C, while the peaks of respective ones with high ES content (>83 mol%) reflect the melting of the PESu crystals at 90–107 °C.
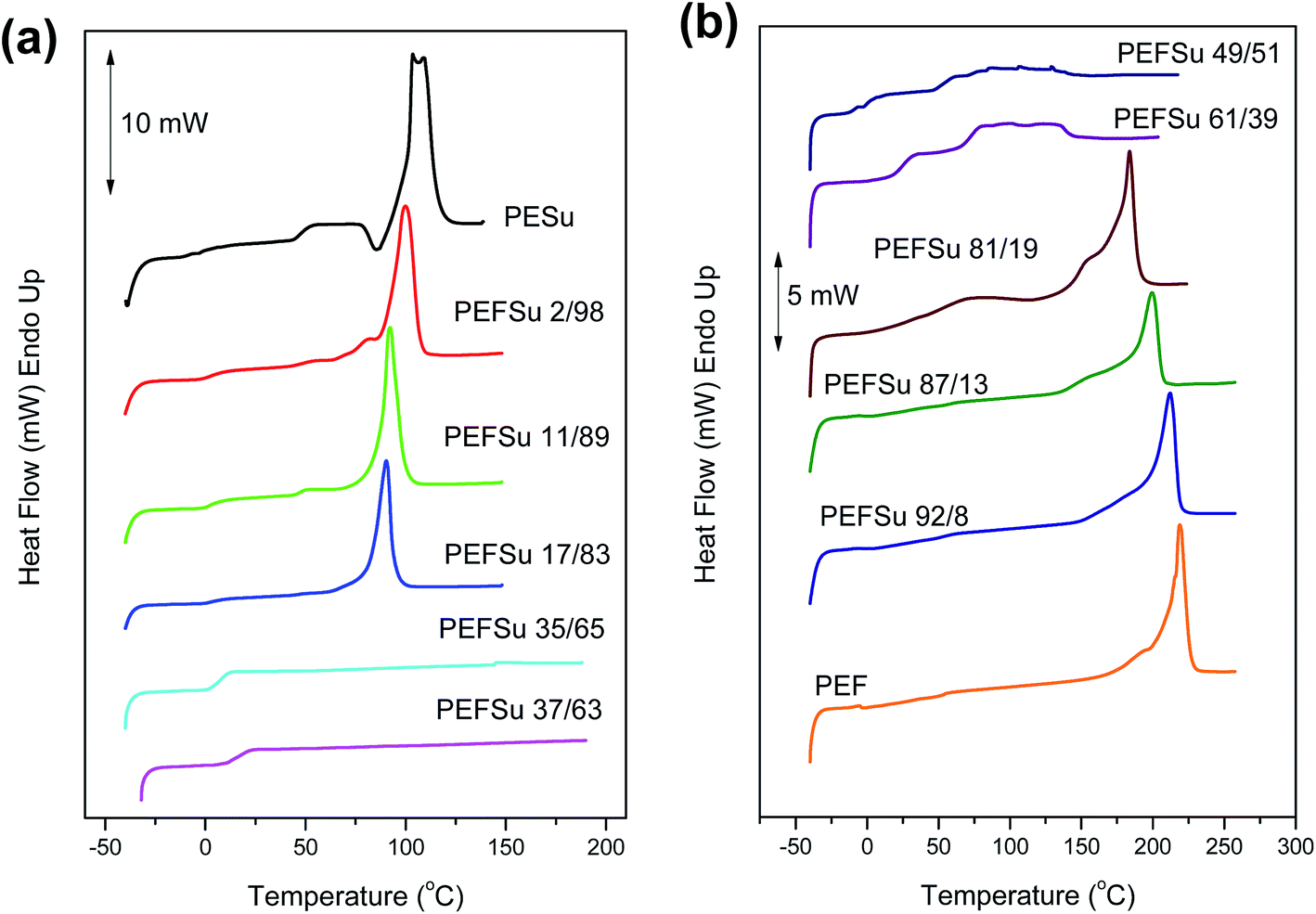 | ||
| Fig. 2 DSC traces of semicrystalline (a) PESu and ES rich copolymers and (b) PEF and EF rich copolymers. | ||
Fig. 3 shows the DSC traces for the quenched copolymer samples. In these curves a single glass transition can be seen for the copolymers. The presence of a single glass transition is another indication of random copolymers, which is in agreement with the results from 1HNMR. However, a single Tg can also be caused by miscible polymers and can be an indication of the state of miscibility of the blend ratios. The PESu sample and the 2/98 copolymer displayed cold crystallization during the heating scan at 20 °C min−1. The rest of the samples could not crystallize upon heating at that rate, because of their relatively slow crystallization rates.
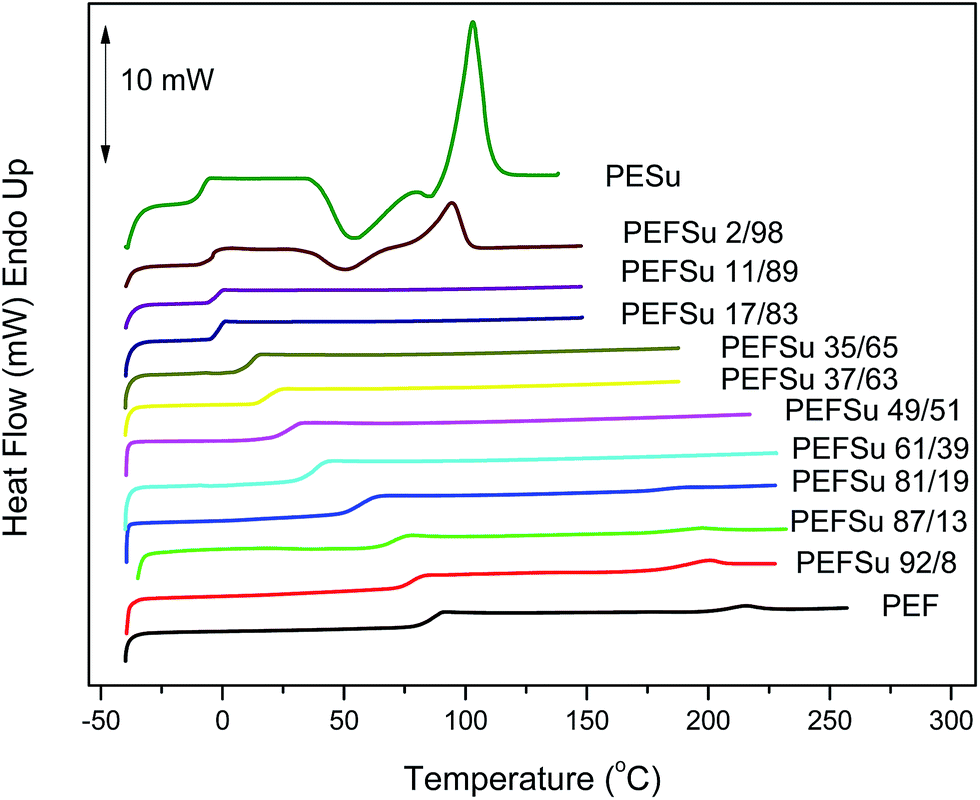 | ||
| Fig. 3 DSC traces of quenched PEF, PESu and PEFSu copolymers. | ||
Melting temperature (Tm) and glass transition temperature (Tg) vs. composition plots were constructed (Fig. 4). A minimum was observed in the plot of the melting temperature vs. composition showing that the copolymers followed a pseudo-eutectic behavior at a EF/ES molar ratio of 35/65. On the one side of the pseudo-eutectic point the crystalline lattice is the one of EF, while on the other side it is the one of ES. Therefore, the crystalline system under study is isodimorphic, implying cocrystallization as will be also discussed in the WAXD study section.
 | ||
| Fig. 4 Variation of the melting temperature (Tm) and glass transition temperature (Tg) of the PEFSu copolymers with composition. | ||
Glass transition temperatures (Tg) of the quenched samples decreased monotonically with ES content between the two values for the neat polymers, which were found to be 87 °C for PEF and −8 °C for PESu. As expected, the EF-rich copolymers present higher rigidity, as a result of their higher glass transition temperatures. Tg is usually a monotonic function of composition in amorphous random copolymers. The most common relationship for the prediction of the Tg as a function of comonomer content is the equation of Fox:63
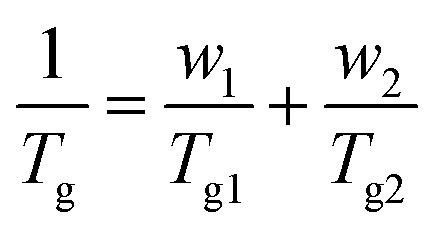 | (4) |
The Couchman–Karasz equation was also tested for the case of the Tg variation for the PEFSu copolymers. The Couchman–Karasz (C–K) equation is given by:64
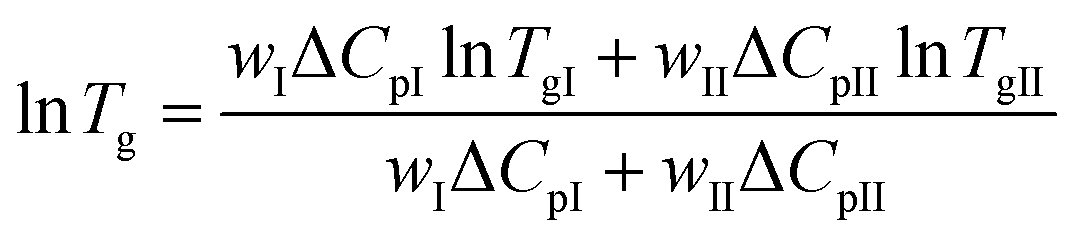 | (5) |
Finally, the Gordon-Taylor equation was elaborated:65
 | (6) |
In general, PESu and especially PEF proved to be slowly crystallizing. The low crystallization rates of PEF originate from the nonlinear character of FDCA and the increased structural rigidity which inhibits the crystallization procedure and leads to slow rates.10 As was expected, the related copolymers showed even slower crystallization rates. Therefore, a self-nucleation scheme was applied prior to accelerate the phenomena and test the isothermal crystallization of the copolymers within a reasonable experimental time-scale. Self-nucleated crystallization of several polymers has been studied recently. In fact, Müller and co-workers, as well as others, proved that measuring isothermal crystallization rates after self-nucleation with DSC, is a reliable method for evaluating crystallization rates even for the application of the Lauritzen–Hoffman analysis.67
Fig. 5a shows the crystallization half-times for PEF and EF rich copolymers. The corresponding plots for PESu and ES rich copolymers are shown in Fig. 5b. From the corresponding plots it is obvious that there is an increase of the crystallization half-times with increasing comonomer unit content, showing retardation in the phenomenon. Also, the temperature window for crystallization shifts to lower temperatures and it becomes narrower. This is caused by two factors. On the one hand, the melting temperatures of the copolymers were reduced with increasing content so, for given crystallization temperature, the degree of supercooling was reduced. On the other hand, incorporation of comonomer units in the macromolecular chain reduces symmetry which in turn causes restrictions in crystallizability.
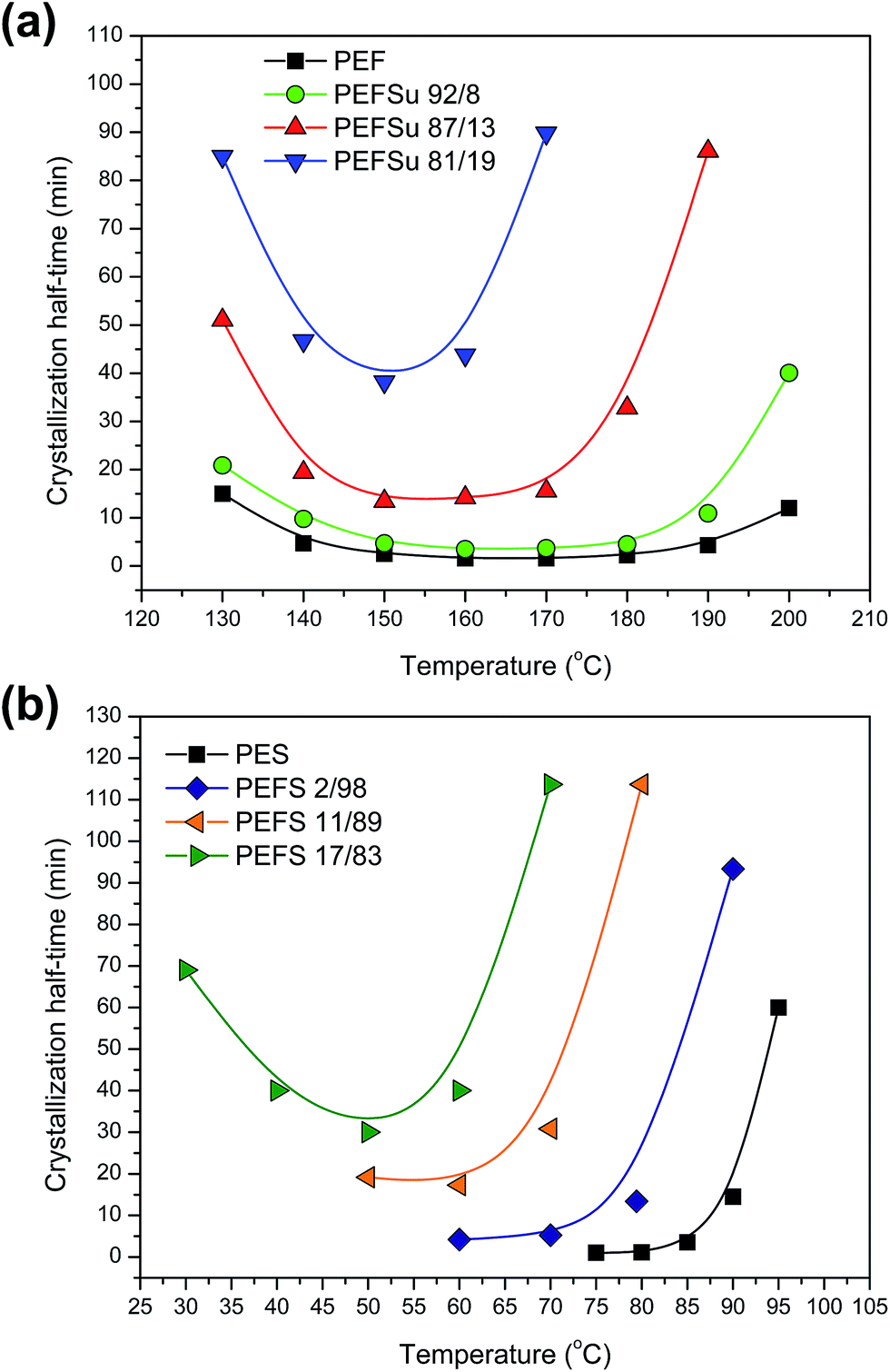 | ||
| Fig. 5 Isothermal crystallization half-times as a function of temperature: (a) for PEF and EF rich copolymers and (b) for PESu and ES rich copolymers. | ||
3.2. Multiple melting behavior
Polyesters most commonly show multiple melting in subsequent heating scans after isothermal crystallization. For PEF it was found that the appearance of multiple melting depends on the crystallization temperature.56,68,69 Sbirrazzuoli and coworkers have reported that the multiple melting of PEF occurs due to recrystallization during heating and not due to the different distributions of lamellar thickness of the material.70 Fig. 6a shows the DSC heating scans of PEFSu 92/8 samples after isothermal crystallizations at different temperatures. The specific copolymer showed mainly double melting. Samples crystallized at lower temperatures (150–165 °C) exhibited also a recrystallization peak in between the two melting peaks. In the traces of the samples crystallized at in the high Tcs region, above 180 °C, a continuous shift of the melting temperature towards higher temperatures could be observed with increasing Tc. This should be attributed to the higher degree of perfection of the generated crystals and to lamellar thickening. Moreover, in the DSC traces shown in Fig. 6b, the melting behavior of the PEFSu 17/83 copolymer after isothermal crystallization at various temperatures can be also seen. Two things should be pointed out in these curves. Firstly, the melting temperature is substantially reduced compared to the neat PESu (Fig. 6c). Secondly, there was a more pronounced multiple melting behavior in the case of the copolymer, but unlike neat PESu, the ultimate melting temperature was not dependent on the crystallization temperature. This should be associated with a much more pronounced crystal perfection and thickening in case of the neat PESu homopolymer. The presence of the comonomer units along the macromolecular chains seemed to restrict not only the crystallization rates but also the crystal stability. | ||
| Fig. 6 DSC traces of (a) PEFSu 92/8, (b) PEFSu 17/83 and (c) neat PESu samples crystallized at different temperatures. | ||
3.3. Cocrystallization behavior
As was reported before, in copolyesters where the two crystallizable components A and B, are also compatible in each crystal lattice, cocrystallization can be observed, which can be classified as isomorphism or isodimorphism.41 Isomorphism is met when the two components occupy about the same volume, so the chain conformations of both corresponding homopolymers are compatible with either crystal lattice and only one crystalline phase which contains both comonomer units is observed at all compositions. The phenomenon is evidenced by a clear melting temperature, or by the appearance of crystallinity over the entire copolymer composition.In isodimorphism two crystalline phases are observed.71 Two sub cases are met in isodimorphism: (a) each crystalline phase contains comonomer units; that is incorporation of B units in A crystal lattice can occur and vice versa and in (b) case the A units can cocrystallize with the incorporation of the B comonomeric units, whereas B crystallizes with the complete rejection of the A units. Isodimorphism is associated with a minimum melting temperature in the plot of melting temperature versus copolymer composition (pseudo-eutectic behavior) and also with lowering in the degree of crystallinity.
For PEF, the crystal structure was estimated in an early study by Kazaryan and Medvedeva.72 According to them the PEF α crystal modification has a triclinic unit cell, with dimensions a = 0.575 nm, b = 0.535 nm, c = 2.010 nm, α = 133.3°, β = 90° and γ = 112°, comprising two repeating units and crystal density of 1.565 g cm−3. The density of the amorphous phase is 1.4299 g cm−3.10 In a previous work it was shown that when PEF crystallizes from solution or after solvent treatment it shows a different crystal modification, called β modification.69
The crystal structure of PESu was first studied by Fuller and Erickson.73 They proposed a monoclinic unit cell with dimensions a = 0.905 nm, b = 1.109 nm and c = 0.832 nm (c being the fiber axis), and β = 102.8°. Later, Ueda et al. determined the crystal and molecular structures of PESu and they proposed slight modification of the crystallographic data.74 Thus, PESu was found to crystallize as a Pbnb-D2b10 space group with unit cell parameters a = 0.760 nm, b = 1.075 nm and c = 0.833 nm (c being the fiber axis), and four macromolecular chains pass through the unit cell. Ichikawa et al. reported the crystal modification of strained PESu fiber.75 This modification was recognized as β form having a 0.95 nm fiber repeat period. In our case, neat PESu showed two strong peaks at 20.1° and 23.3° which correspond to reflections from (120) and (200) planes, respectively.74
The crystalline structure of the PEFSu copolymers was studied by means of WAXD. The comonomer concentration in crystal lattice of the copolymers is strongly dependent on the copolymer composition in both bulk and crystallization conditions, however crystalline reflection peaks were observed for most compositions. The reflections for the EF rich copolymers (EF content higher than 83 mol%) are associated with the PEF β-crystalline form, which is usually observed after solution crystallization. Moreover, for the EF-rich copolymers, the peaks became broader with increasing EF-content, indicating the presence of smaller crystalline size and decreasing crystallinity. In addition, a shift of the peaks towards higher angles can be observed, as a result of the distortion of the a-form PEF unit cell, caused by the presence of the ES co-unit. The PEFSu 35/65 and 37/63 copolymers did not crystallize even from solution since they exhibited the amorphous halo on their diffractograms while crystalline peaks were not clearly evidenced. The results are in agreement with DSC study, where the pseudo-eutectic point was observed on the melting temperatures of the specific samples. On the other hand, the copolymers with ES content higher than 81 mol% preferentially crystallize in the PESu a-crystal structure. The peak positions did not seem to be affected by the presence of the EF co-unit. Interestingly, the PEFSu 49/51 and 61/39 copolymers showed weak crystal reflection peaks at positions consistent with those of both PEF and PESu. In intermediate compositions short comonomer block lengths are expected to lead in comonomer inclusion. This is also associated with lower degree of crystallinity and defective crystal structures. It should be noticed here that these two copolymers showed a broad melting temperature range, with the ultimate melting temperature to be higher than that of pure PESu.
The interplanar spacing was calculated for all copolymers based on the main reflections of the diffractograms presented in Fig. 7a. As it can be seen in Fig. 7b, an increase in the d-spacing and subsequently to the cell unit dimensions, is recorded for most reflection angles with decreasing EF content, as a result of the increasing ES comonomer, which is bulkier than the one of EF. The specific behavior is associated with isodimorphic cocrystallization, as was expected from the study of the melting point and the appearance of a pseudo-eutectic behavior.
 | ||
| Fig. 7 (a) WAXD patterns of the PEFSu copolymers and the PEF and PESu homopolymers, (b) variation of the interplanar distances (d-spacing) with the composition of PEFSu copolymers. | ||
3.4. Thermodynamics of melting point depression
The melting point depression can be used for the thermodynamic analysis of cocrystallization in random copolymers. Several theories have been introduced for the study of the copolymer crystallization. There is distinction between those which assume comonomer exclusion like those of Flory50,51 or Baur53 and those which assume comonomer inclusion in the crystal such as those of Inoue,76 Helfand–Lauritzen77 and Sanchez–Eby.52In the equation of Flory:50
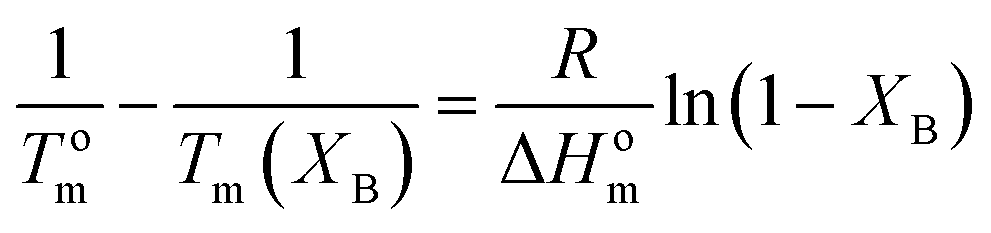 | (7) |
The Sanchez–Eby model assumes that B comonomer units are included into the crystals of A forming defects. The corresponding equation is:52
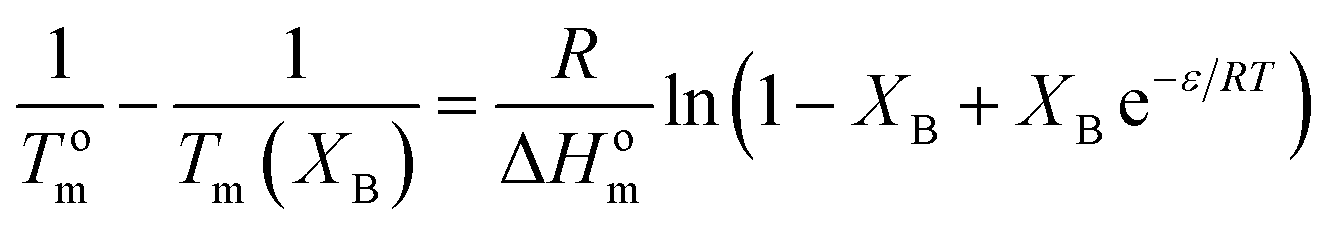 | (8) |
The basic concept in the theory of Baur, is that homopolymer sequences of length ξ may be included into crystals of lamellar thickness corresponding to that length:53
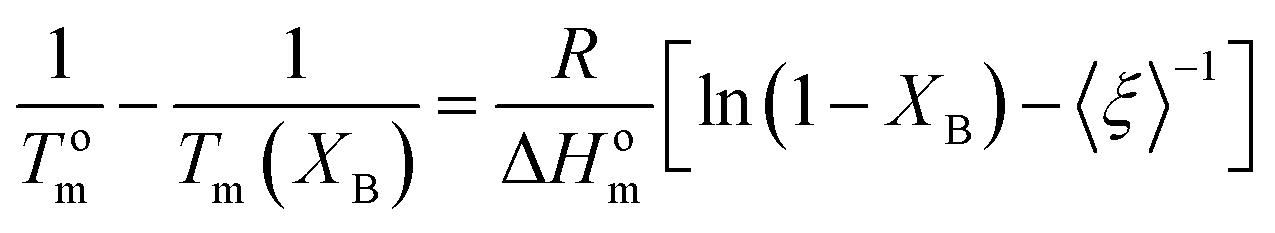 | (9) |
| 〈ξ〉 = [2XB(1 − XB)]−1 | (10) |
Wendling and Suter combined both inclusion and exclusion models to arrive to the following equation:54,78,79
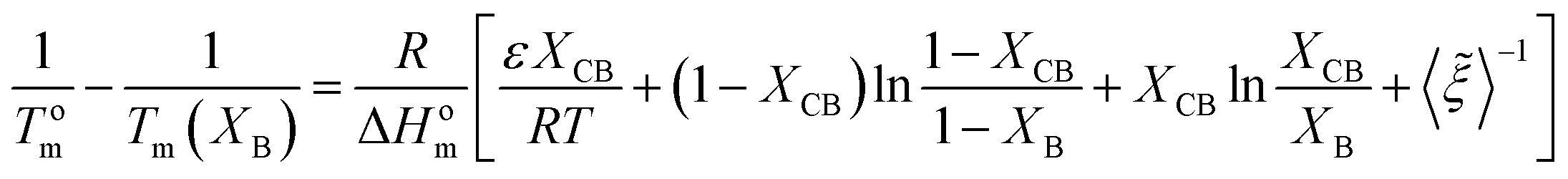 | (11) |
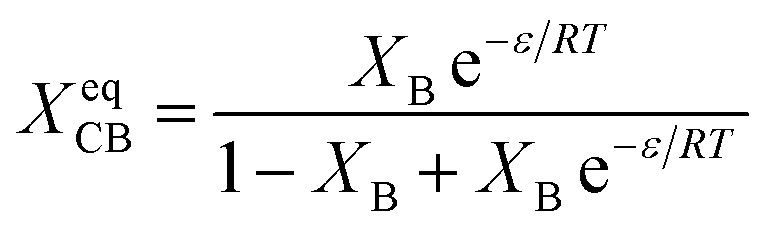 | (12) |
Substitution of XCB in eqn (8) by (12), gives a simplified equation following equilibrium inclusion model:
 | (13) |
〈![[small xi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e119.gif) 〉−1 = 2(XB − XB 〉−1 = 2(XB − XB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−ε/RT)(1 − XB + XBe−ε/RT) e−ε/RT)(1 − XB + XBe−ε/RT)
| (14) |
Uniform inclusion is reached if XCB = XB while for XCB = 0 eqn (8) is reduced to the exclusion model.
Various thermodynamic models were tested for the melting point depression of PEFSu. The temperature corresponding to the end of the melting peak was used, as an approach of the equilibrium melting temperature. Also, in our calculations, the equilibrium melting enthalpy was taken to be 25 kJ mol−1 for PEF56 and 26 kJ mol−1 for PESu.57
As can be seen in Fig. 8 the Wendling–Suter model showed the best fit to the experimental data in the range of low comonomer content, since the regression coefficient was R2 > 0.99. The value of the function ε/RT is determined as an adjustable parameter. A constant ε/RT value is given by the model regardless of the comonomer composition. For PEF best fit was found for ε/RT = 1.8 which results in a value ε = 7.6 kJ mol−1 for the average defect free energy in case of incorporation of ES unit into the PEF crystal, in the limiting case of XES = 0. In the opposite case of incorporation of EF units in the PESu crystal a value ε/RT = 2.5 was needed for best fit, giving ε = 8.1 kJ mol−1 for the average defect free energy. The ε in this case is slightly higher than in case of incorporation of ES units in the PEF crystal.
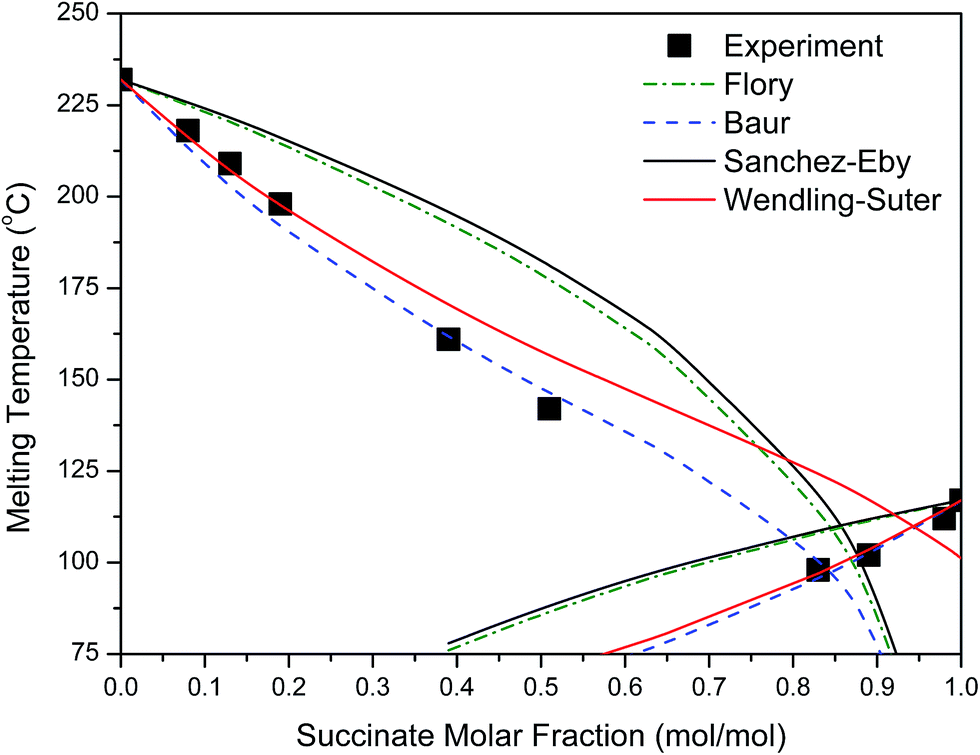 | ||
| Fig. 8 Experimental and predicted melting point as a function of the succinate molar fraction. | ||
3.5. PLOM observations during melt crystallization
Polarized light optical microscopy was utilized for the observation of the spherulitic structure of the copolymers after isothermal crystallization at various temperatures. As it can be seen in Fig. 9a–d, PEF exhibited the characteristic small spherulites with a very high density at all crystallization temperatures.56 On the other hand, PESu displayed significantly larger spherulites with the characteristic maltese cross, while there is a gradual increase in the size with increasing crystallization temperature (Fig. 10a–d). The presence of PESu even at a small percentage on the PEFSu 92/8 sample, increased slightly the sizes of the spherulites, while the nucleation density of the materials remained very high (Fig. 9e and f). On the contrary, as it was expected, the presence of a small amount of PEF on the ES rich samples, affected marginally the spherulitic morphology of the materials, since the nucleation density and the size of the spherulites decreased.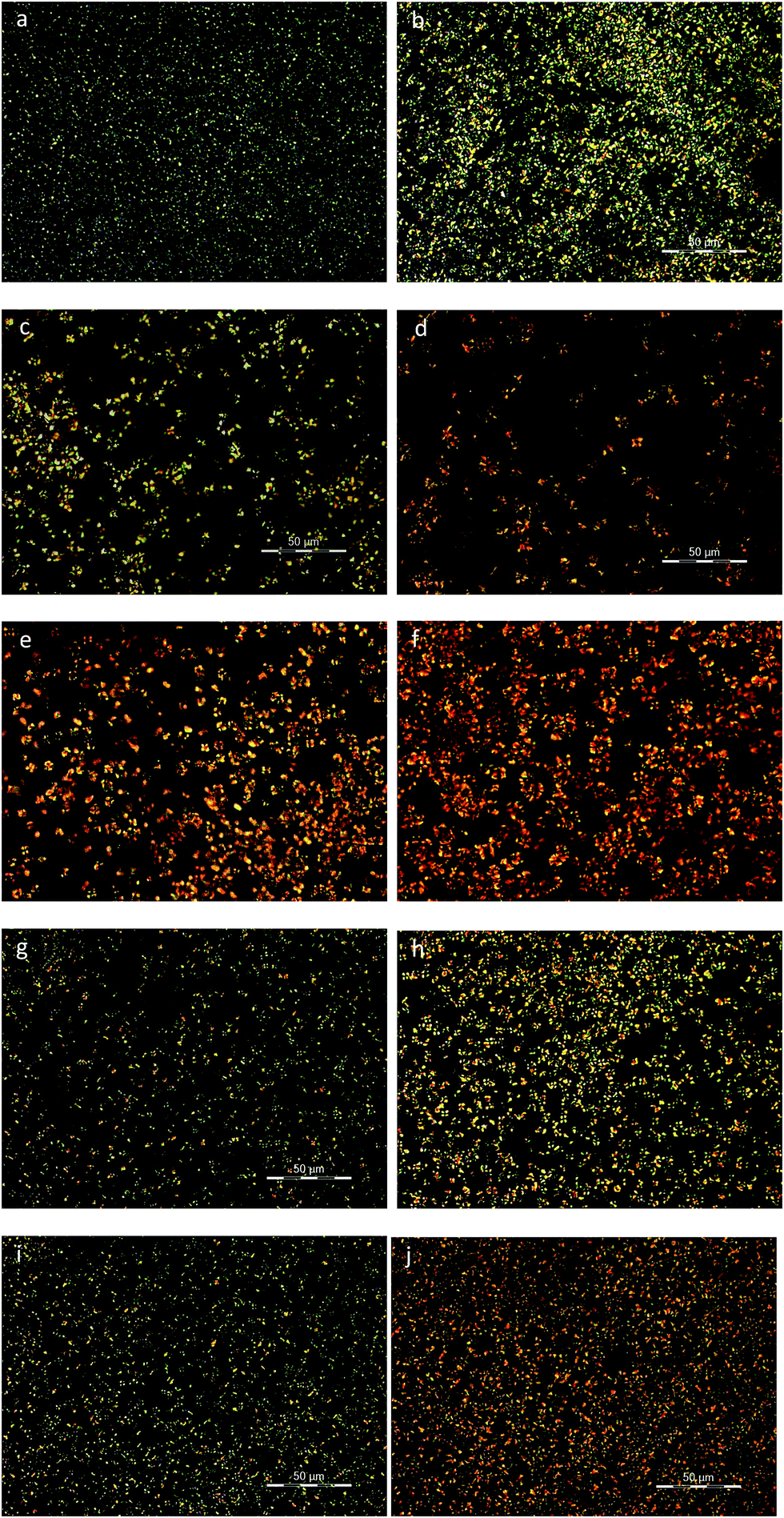 | ||
| Fig. 9 PLOM photos of (a) PEF at 160 °C, 10 min, (b) PEF at 180 °C, 15 min, (c) PEF at 200 °C, 20 min, (d) PEF at 210 °C, 30 min, (e) PEFSu 92/8 at 150 °C, 20 min, (f) PEFSu 92/8 at 160 °C, 20 min, (g) PEFSu 87/13 at 140 °C, 20 min, (h) PEFSu 87/13 at 160 °C, 20 min, (i) PEFSu 81/19 at 130 °C, 20 min and (j) PEFSu 81/19 at 140 °C, 20 min. | ||
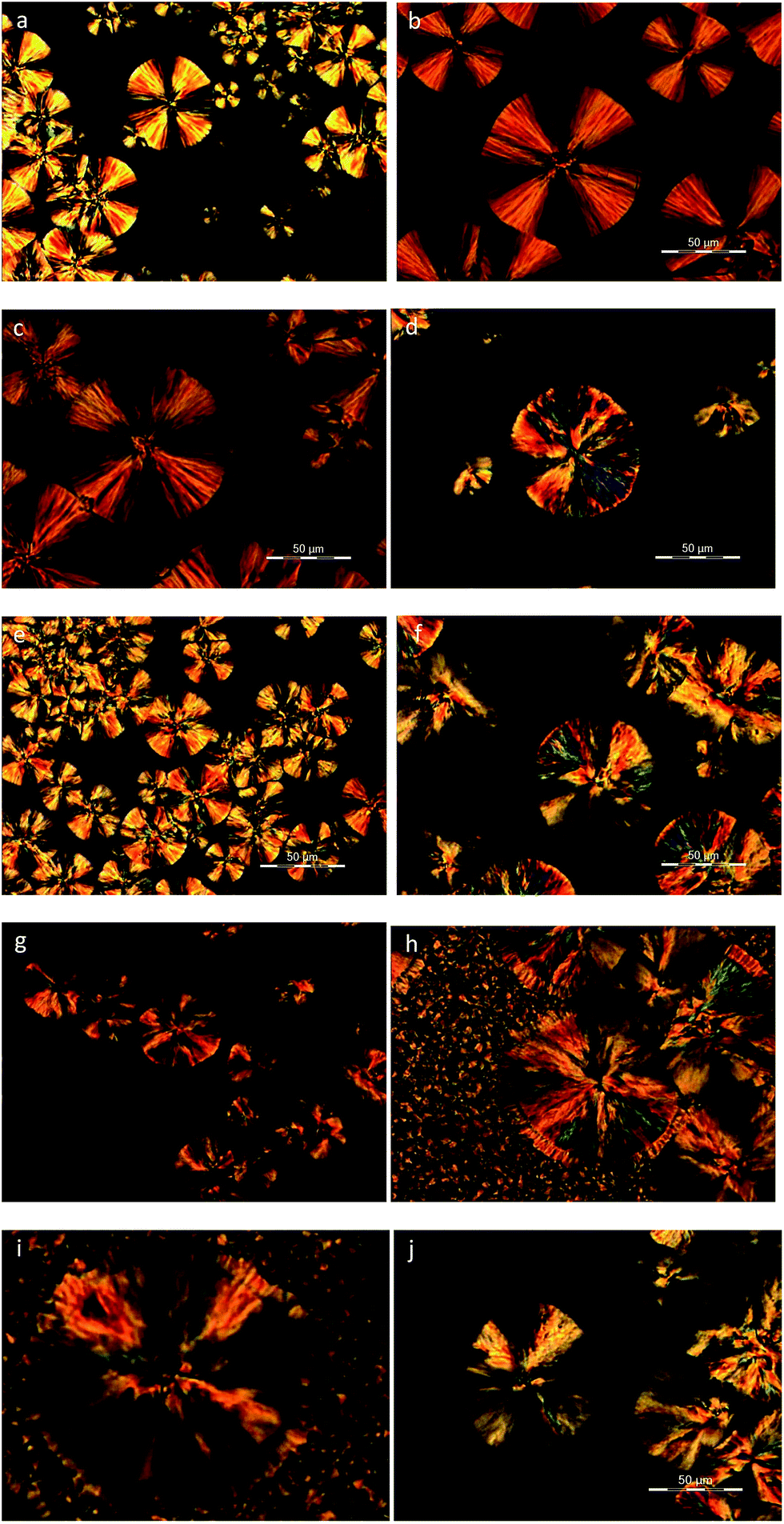 | ||
| Fig. 10 PLOM photos of (a) PESu at 50 °C, 5 min, (b) PESu at 60 °C, 5 min, (c) PESu at 70 °C, 5 min, (d) PESu at 80 °C, 5 min, (e) PEFSu 2/98 at 50 °C, 5 min, (f) PEFSu 2/98 at 60 °C, 7 min (g) PEFSu 11/89 at 60 °C, 7 min, (h) PEFSu 11/89 cooled to room temperature after crystallization at 60 °C for 7 min, (i) PEFSu 11/89 cooled to room temperature after crystallization at 60 °C for 7 min and (j) PEFSu 17/83 at 50 °C, 10 min. | ||
3.6. Enzymatic hydrolysis
As the amount of plastic waste continually grows, the use of biodegradable polymers is becoming more and more essential. 2,5-FDCA-based polyesters can be prepared from monomers derived from renewable resources but these polyesters are not biodegradable materials. However, from soil-burial tests along with enzymatic degradation experiments, it was proposed that these polyesters could be potentially biodegradable.80 In a recent study, PEF powders of various molecular weights (6, 10 and 40 kDa) were synthesized and their susceptibility to enzymatic hydrolysis was investigated.81 According to LC/TOF-MS analysis, it was found that cutinase 1 from Thermobifida cellulosilytica liberated both 2,5-furandicarboxylic acid and oligomers of up to DP4. The enzyme preferentially hydrolyzed PEF with higher molecular weights but was active on all tested substrates. However, this is not a proof the PEF is a biodegradable polyester. On the other hand, while aliphatic polyesters such as PESu are easily hydrolysed, their mechanical properties are poor. Therefore copolymerizing it with aromatic moieties can result in both better properties and acceptable enzymatic hydrolysis rates.82 Commercial aliphatic–aromatic copolyesters are available in the market, for example Ecoflex® which is a product of BASF. Aromatic polyesters such as PET or PEF are regarded as non-biodegradable, so the copolymerization of FDCA with aliphatic acids is expected to induce biodegradability. Enzymatic hydrolysis rate is known to depend on the hydrophilicity, the chemical structure, the mobility of the amorphous phase and also the crystallinity.83,84The weight loss% of the copolyesters during enzymatic hydrolysis over a course of 30 days is presented in Fig. 11. While PESu lost 30% of its initial weight after a month, copolymers containing furanoate units showed negligible hydrolysis rates. As the content in succinate increases, the enzymatic hydrolysis rate is also increased. In succinate contents up to 50%, weight loss is relatively small (maximum 6%) but measureable within the first month. Significant hydrolysis can be induced it higher contents. PEFSu 17/3 and PEFSu 11/89 lost 15% and 12.5% of their initial weights in 1 month. The furan ring makes the access of enzymes to the ester group more difficult, because it causes the macromolecular chain of PEF to be rigid since ring-flipping is hindered by the nonlinear axis of furan ring rotation coupled with the ring polarity. Additionally, melting temperature and glass transition temperature of the copolymers increase with increasing the furanoate content, so the mobility of the macromolecular chains is decreased, further reducing the hydrolysis rates. Similar results were obtained in our previous work with aliphatic–aromatic copolymers,61 as also by other researchers.82,85 In the case of furanoate copolymers it was found that copolymerization of PEF with lactic acid enhanced its hydrolysis rate,86,87 as also the presence of butylene adipate in poly(butylene furanoate) polyester resulted in accelerated hydrolysis by lipase.88 This behavior is attributed to the higher hydrophobicity of the furanoate units and the increased rigidity they provide, therefore less mobility of the macromolecular chains.
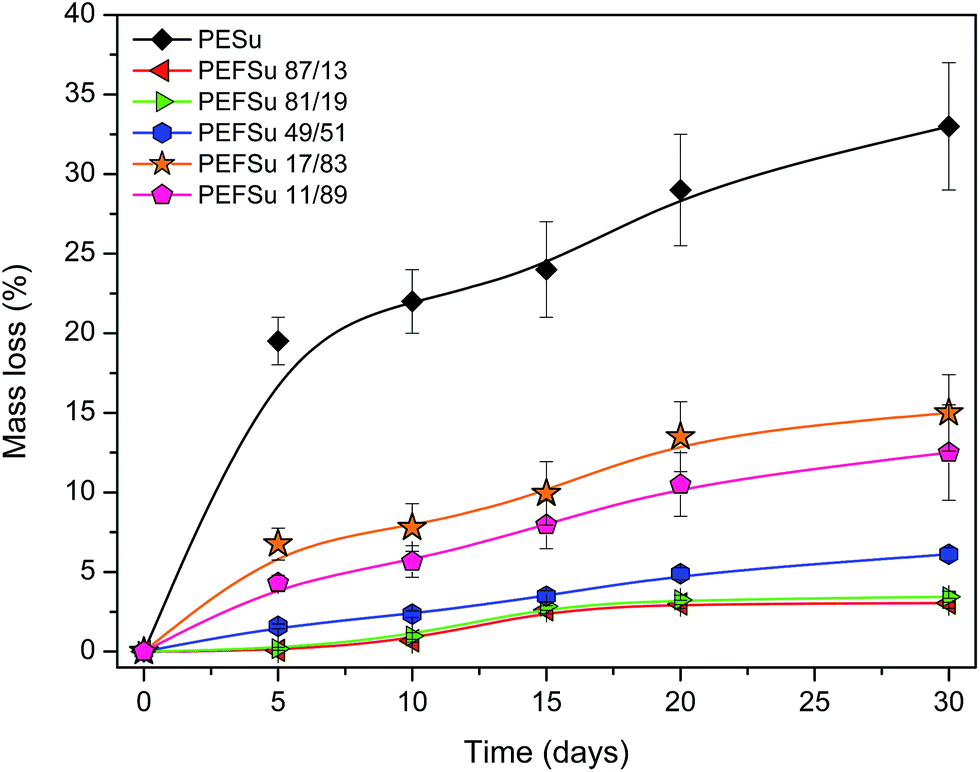 | ||
| Fig. 11 Weight loss plots during enzymatic hydrolysis. | ||
Besides weight loss measurements, SEM micrographs were also obtained after enzymatic hydrolysis of the polyesters for 15 and 30 days, and are presented in Fig. 12. They are in agreement with mass loss, as more abnormalities, gaps and holes are observed in copolyesters with high succinate contents, while surfaces of the polyesters with more furanoate units are not altered significantly. The roughness of surfaces becomes more prominent over the course of enzymatic hydrolysis time.
 | ||
| Fig. 12 SEM micrographs of the copolyesters after enzymatic hydrolysis for 15 and 30 days. | ||
4. Conclusions
Random poly(ethylene furanoate-co-ethylene succinate) copolymers with several fuaranoate/succinate molar ratios have been successfully prepared by melt polycondansation procedure. The influence of the increasing EF (or ES) content on the melting and crystallization behavior, crystalline structure and enzymatic hydrolysis were studied in detail. Most of the produced copolymers are semicrystalline materials with thermal properties depending directly on the composition. The melting and glass transition temperatures were found to decrease with increasing ES content, while increase in the EF content caused a slight decrease in the cell dimensions of the copolymers. Moreover, from the study of the melting point of the copolymers, a pseudo-eutectic behavior was observed at ED/ES fraction of 30/70, which was further confirmed by WAXD. The specific behavior is associated with isodimorphic cocrystallization. From the enzymatic hydrolysis study, the copolymers with high succinic acid amounts are hydrolysable materials with the help of Rhizopus delemar lipase and Pseudomonas cepacia lipase. However, up to a 50 mol% PEF content, measurable losses in weight were shown. Thus, these copolymers are excellent biodegradable materials, which along with the fact that they can be prepared directly from monomers derived from renewable resources, they can be considered quite attractive for various applications and uses.References
- E. de Jong, M. Dam, L. Sipos and G. Gruter, Biobased Monomers, Polymers and Materials, 2012, vol. 1105, pp. 1–13 Search PubMed.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. Coelho, C. S. Freire and A. J. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. Freire, G.-J. M. Gruter, J. F. Coelho and A. J. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- A. Gandini, Polym. Chem., 2010, 1, 245–251 RSC.
- A. Gandini, A. J. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295–298 CrossRef CAS.
- M. Gomes, A. Gandini, A. J. Silvestre and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS.
- J. Ma, X. Yu, J. Xu and Y. Pang, Polymer, 2012, 53, 4145–4151 CrossRef CAS.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1026–1036 CrossRef CAS.
- V. Tsanaktsis, G. Z. Papageorgiou and D. N. Bikiaris, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2617–2632 CrossRef CAS.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- S. K. Burgess, O. Karvan, J. Johnson, R. M. Kriegel and W. J. Koros, Polymer, 2014, 55, 4748–4756 CrossRef CAS.
- S. K. Burgess, R. M. Kriegel and W. J. Koros, Macromolecules, 2015, 48, 2184–2193 CrossRef CAS.
- G. Z. Papageorgiou, V. Tsanaktsis, D. G. Papageorgiou, S. Exarhopoulos, M. Papageorgiou and D. N. Bikiaris, Polymer, 2014, 55, 3846–3858 CrossRef CAS.
- G. Z. Papageorgiou, D. G. Papageorgiou, V. Tsanaktsis and D. N. Bikiaris, Polymer, 2015, 62, 28–38 CrossRef CAS.
- G. Z. Papageorgiou, V. Tsanaktsis, D. G. Papageorgiou, K. Chrissafis, S. Exarhopoulos and D. N. Bikiaris, Eur. Polym. J., 2015, 67, 383–396 CrossRef CAS.
- G. Z. Papageorgiou, N. Guigo, V. Tsanaktsis, D. G. Papageorgiou, S. Exarhopoulos, N. Sbirrazzuoli and D. N. Bikiaris, Eur. Polym. J., 2015, 68, 115–127 CrossRef CAS.
- V. Tsanaktsis, D. N. Bikiaris, N. Guigo, S. Exarhopoulos, D. G. Papageorgiou, N. Sbirrazzuoli and G. Z. Papageorgiou, RSC Adv., 2015, 5, 74592–74604 RSC.
- V. Tsanaktsis, Z. Terzopoulou, S. Exarhopoulos, D. N. Bikiaris, D. S. Achilias, D. G. Papageorgiou and G. Z. Papageorgiou, Polym. Chem., 2015, 6, 8284–8296 RSC.
- M. Vannini, P. Marchese, A. Celli and C. Lorenzetti, Green Chem., 2015, 17, 4162–4166 RSC.
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Green Chem., 2014, 16, 1957–1966 RSC.
- A. F. Sousa, A. C. Fonseca, A. C. Serra, C. S. R. Freire, A. J. D. Silvestre and J. F. J. Coelho, Polym. Chem., 2016, 7, 1049–1058 RSC.
- K. Chrissafis, K. Paraskevopoulos and D. Bikiaris, Thermochim. Acta, 2005, 435, 142–150 CrossRef CAS.
- Z. Gan, H. Abe and Y. Doi, Biomacromolecules, 2000, 1, 704–712 CrossRef CAS PubMed.
- Z. Qiu, M. Komura, T. Ikehara and T. Nishi, Polymer, 2003, 44, 7781–7785 CrossRef CAS.
- J. Lu, Z. Qiu and W. Yang, Polymer, 2007, 48, 4196–4204 CrossRef CAS.
- Y. Tokiwa, B. Calabia, C. Ugwu and S. Aiba, Int. J. Mol. Sci., 2009, 10, 3722 CrossRef CAS PubMed.
- G. Seretoudi, D. Bikiaris and C. Panayiotou, Polymer, 2002, 43, 5405–5415 CrossRef CAS.
- D. P. Kint, A. Alla, E. Deloret, J. L. Campos and S. Muñoz-Guerra, Polymer, 2003, 44, 1321–1330 CrossRef CAS.
- M. Mochizuki, K. Mukai, K. Yamada, N. Ichise, S. Murase and Y. Iwaya, Macromolecules, 1997, 30, 7403–7407 CrossRef CAS.
- X. Li and Z. Qiu, Polym. Test., 2015, 48, 125–132 CrossRef CAS.
- X. Li and Z. Qiu, Ind. Eng. Chem. Res., 2016, 55, 3797–3803 CrossRef CAS.
- X. Li and Z. Qiu, RSC Adv., 2015, 5, 103713–103721 RSC.
- Z. Qiu, T. Ikehara and T. Nishi, Macromolecules, 2002, 35, 8251–8254 CrossRef CAS.
- F. Kondratowicz and R. Ukielski, Polym. Degrad. Stab., 2009, 94, 375–382 CrossRef CAS.
- P. Xue and Z. Qiu, Thermochim. Acta, 2015, 606, 45–52 CrossRef CAS.
- H. Wu and Z. Qiu, CrystEngComm, 2012, 14, 3586–3595 RSC.
- L. Wu, R. Mincheva, Y. Xu, J.-M. Raquez and P. Dubois, Biomacromolecules, 2012, 13, 2973–2981 CrossRef CAS PubMed.
- K. Kuwabara, Z. Gan, T. Nakamura, H. Abe and Y. Doi, Biomacromolecules, 2002, 3, 390–396 CrossRef CAS PubMed.
- G. Z. Papageorgiou and D. N. Bikiaris, Biomacromolecules, 2007, 8, 2437–2449 CrossRef CAS PubMed.
- G. Allegra and I. W. Bassi, in Fortschritte der Hochpolymeren-Forschung, Springer, Berlin, Heidelberg, 1969, pp. 549–574 Search PubMed.
- C. Lavilla, E. Gubbels, A. Alla, A. Martinez de Ilarduya, B. A. J. Noordover, C. E. Koning and S. Munoz-Guerra, Green Chem., 2014, 16, 1789–1798 RSC.
- C. Lavilla and S. Munoz-Guerra, Green Chem., 2013, 15, 144–151 RSC.
- G. Ceccorulli, M. Scandola, A. Kumar, B. Kalra and R. A. Gross, Biomacromolecules, 2005, 6, 902–907 CrossRef CAS PubMed.
- Z. Liang, P. Pan, B. Zhu and Y. Inoue, Polymer, 2011, 52, 2667–2676 CrossRef CAS.
- G. Z. Papageorgiou, A. A. Vassiliou, V. D. Karavelidis, A. Koumbis and D. N. Bikiaris, Macromolecules, 2008, 41, 1675–1684 CrossRef CAS.
- W. J. Orts, R. H. Marchessault and T. L. Bluhm, Macromolecules, 1991, 24, 6435–6438 CrossRef CAS.
- G. Z. Papageorgiou and D. N. Bikiaris, Macromol. Chem. Phys., 2009, 210, 1408–1421 CrossRef CAS.
- G. Z. Papageorgiou, V. Tsanaktsis and D. N. Bikiaris, CrystEngComm, 2014, 16, 7963–7978 RSC.
- P. J. Flory, J. Chem. Phys., 1949, 17, 223–240 CrossRef CAS.
- P. J. Flory, Trans. Faraday Soc., 1955, 51, 848–857 RSC.
- I. C. Sanchez and R. K. Eby, Macromolecules, 1975, 8, 638–641 CrossRef CAS.
- V. H. Baur, Die Makromolekulare Chemie, 1966, 98, 297–301 CrossRef.
- J. Wendling, A. A. Gusev and U. W. Suter, Macromolecules, 1998, 31, 2509–2515 CrossRef CAS.
- J. P. Penning and R. S. John Manley, Macromolecules, 1996, 29, 77–83 CrossRef CAS.
- G. Z. Papageorgiou, V. Tsanaktsis and D. N. Bikiaris, Phys. Chem. Chem. Phys., 2014, 16, 7946–7958 RSC.
- D. N. Bikiaris, G. Z. Papageorgiou and D. S. Achilias, Polym. Degrad. Stab., 2006, 91, 31–43 CrossRef CAS.
- B. Fillon, J. Wittmann, B. Lotz and A. Thierry, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1383–1393 CrossRef CAS.
- D. Cavallo, L. Gardella, G. Portale, A. J. Müller and G. C. Alfonso, Polymer, 2014, 55, 137–142 CrossRef CAS.
- M. Soccio, L. Finelli, N. Lotti, M. Gazzano and A. Munari, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 310–321 CrossRef CAS.
- G. Z. Papageorgiou, A. A. Vassiliou, V. D. Karavelidis, A. Koumbis and D. N. Bikiaris, Macromolecules, 2008, 41, 1675–1684 CrossRef CAS.
- R. Yamadera and M. Murano, J. Polym. Sci., Part A-1: Polym. Chem., 1967, 5, 2259–2268 CrossRef CAS.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123–135 CAS.
- P. Couchman and F. Karasz, Macromolecules, 1978, 11, 117–119 CrossRef CAS.
- M. Gordon and J. S. Taylor, J. Appl. Chem., 1952, 2, 493–500 CrossRef CAS.
- R. E. Prud'homme, Polym. Eng. Sci., 1982, 22, 90–95 Search PubMed.
- A. J. Müller, R. M. Michell, R. A. Pérez and A. T. Lorenzo, Eur. Polym. J., 2015, 65, 132–154 CrossRef.
- G. Stoclet, G. G. du Sart, B. Yeniad, S. de Vos and J. Lefebvre, Polymer, 2015, 72, 165–176 CrossRef CAS.
- V. Tsanaktsis, D. G. Papageorgiou, S. Exarhopoulos, D. N. Bikiaris and G. Z. Papageorgiou, Cryst. Growth Des., 2015, 15, 5505–5512 CAS.
- L. Martino, N. Guigo, J. G. van Berkel, J. J. Kolstad and N. Sbirrazzuoli, Macromol. Mater. Eng., 2016, 301, 586–596 CrossRef CAS.
- G. P. Karayannidis, N. Papachristos, D. N. Bikiaris and G. Z. Papageorgiou, Polymer, 2003, 44, 7801–7808 CrossRef CAS.
- L. G. Kazaryan and F. M. Medvedeva, Vysokomol. Soedin., Ser. B, 1968, 305–306 CAS.
- C. Fuller and C. Erickson, J. Am. Chem. Soc., 1937, 59, 344–351 CrossRef CAS.
- A. S. Ueda, Y. Chatani and H. Tadokoro, Polym. J., 1971, 2, 387–397 CrossRef CAS.
- Y. Ichikawa, K. Noguchi, K. Okuyama and J. Washiyama, Polymer, 2001, 42, 3703–3708 CrossRef CAS.
- N. Kamiya, M. Sakurai, Y. Inoue and R. Chujo, Macromolecules, 1991, 24, 3888–3892 CrossRef CAS.
- E. Helfand and J. I. Lauritzen, Macromolecules, 1973, 6, 631–638 CrossRef CAS.
- J. Wendling, A. A. Gusev, U. W. Suter, A. Braam, L. Leemans, R. J. Meier, J. Aerts, J. v. d. Heuvel and M. Hottenhuis, Macromolecules, 1999, 32, 7866–7878 CrossRef CAS.
- J. Wendling and U. W. Suter, Macromolecules, 1998, 31, 2516–2520 CrossRef CAS.
- M. Okada, K. Tachikawa and K. Aoi, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2729–2737 CrossRef CAS.
- A. Pellis, K. Haernvall, C. M. Pichler, G. Ghazaryan, R. Breinbauer and G. M. Guebitz, J. Biotechnol., DOI:10.1016/j.jbiotec.2016.02.006.
- E. Marten, R.-J. Müller and W.-D. Deckwer, Polym. Degrad. Stab., 2003, 80, 485–501 CrossRef CAS.
- D. N. Bikiaris, N. P. Nianias, E. G. Karagiannidou and A. Docoslis, Polym. Degrad. Stab., 2012, 97, 2077–2089 CrossRef CAS.
- S. Mizuno, T. Maeda, C. Kanemura and A. Hotta, Polym. Degrad. Stab., 2015, 117, 58–65 CrossRef CAS.
- K. Herzog, R.-J. Müller and W.-D. Deckwer, Polym. Degrad. Stab., 2006, 91, 2486–2498 CrossRef CAS.
- M. Matos, A. F. Sousa, A. C. Fonseca, C. S. Freire, J. F. Coelho and A. J. Silvestre, Macromol. Chem. Phys., 2014, 215, 2175–2184 CrossRef CAS.
- H. Wu, B. Wen, H. Zhou, J. Zhou, Z. Yu, L. Cui, T. Huang and F. Cao, Polym. Degrad. Stab., 2015, 121, 100–104 CrossRef CAS.
- W. Zhou, X. Wang, B. Yang, Y. Xu, W. Zhang, Y. Zhang and J. Ji, Polym. Degrad. Stab., 2013, 98, 2177–2183 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |