
Computational insights into CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I)†
Zhengguo Huang*,
Yuan Yuan,
Le Sun,
Xiaohong Wang and
Yuying Li
Tianjin Key Laboratory of Structure and Performance for Functional Molecules, Key Laboratory of Inorganic–Organic Hybrid Functional Materials Chemistry (Tianjin Normal University), Ministry of Education, College of Chemistry, Tianjin Normal University, Tianjin 300387, People's Republic of China. E-mail: hsxyhzg@126.com
First published on 31st August 2016
Abstract
The C–X bond activation products, CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I) formed by the insertion of coinage metal atoms into C–X bonds of methane and halomethanes, were investigated by density functional theory (DFT). Equilibrium geometries, harmonic vibrational frequencies, and energies were calculated. Bader's atoms-in-molecule (AIM), natural population charge (NPA) and fuzzy bond orders (FBO) calculations were performed to investigate the bonding interactions in CH3MX. As X varies from F to I, the thermodynamic stability of CH3MX with respect to CH3X + M increases, and the order of the thermodynamic stability for different coinage metals is CH3CuX > CH3AuX > CH3AgX. Although the CH3MX (M = Cu, Ag and Au; X = Cl, Br and I) were predicted to be more stable thermodynamically than the others (e.g. CH3MH and CH3MF) observed in matrix isolation experiments, they have not been identified experimentally yet, and one of the probably key reasons is that their vibrational fingerprints (νC–M and νM–X) are so low that they are beyond the detection limit of an infrared spectrometer. AIM analyses show that both C–M and M–X bonds in CH3MX exhibit mainly closed-shell interaction character, and partial covalent character contributes to them. The BCP of M–H bond just locates at the boundary between the charge concentration region and the charge depletion region, which lead to the covalent character of M–H bond being overestimated by the AIM topological parameters.
1. Introduction
Selective metal-mediated activation of C–H and C–X (X = F, Cl, Br and I) bonds is an active research field in organometallic chemistry. In recent years, various kinds of small transition-metal complexes were produced by the reactions of laser-ablated transition metals with small alkanes and halomethanes.1–26 These simple metal complexes usually have unique structures and often show interesting photochemical properties.Group 11 metals, the so-called coinage metals, are at the borderline between the main group elements and transition metals, and have high electronegativity and catalytic activity due to their d10 electronic configuration and single valence electron (s1).8,9 Unlike other transition metals, the coinage metals often behave as Lewis acids because of their relatively high electron affinities. CH3MH (M = Cu, Ag and Au) and its negative ion form have been identified in low-temperature matrix isolation experiments by the insertion of coinage metals into C–H bond of methane,6 moreover, the electron affinities of the insertion complexes are higher than the coinage metals, which is believed to be the driving force to form the rare anionic insertion complex.
The investigations of the reactions of transition metal with halomethanes CH3X (X = F, Cl, Br and I) can provide us an insight into C–X bond activation. On one hand, CH3X are interesting substrates for reactions with metals (and their cations) because they constitute a simple gas-phase model that can provide an opportunity for analyzing the competition between C–H and C–X bond activation. On the other hand, the stabilities and reactivities of the products usually have considerable variations by halogen substitution, which mainly due to the general preference for the M–X bond over the M–H bond.1,15,25,27 CH3CuF was first prepared by the reaction of laser-vaporized Cu atoms with methyl fluoride in the matrix ESR spectra,7,28 CH3MF (M = Ag and Au) have been identified in the matrix IR spectra,8 and reactions of Au with CH3Cl and CH3Br yield CH3AuX (X = Cl and Br) as well.9 In addition, M+CH3F complexes for coinage metal cations have been observed using mass spectroscopy.29 In addition, some anionic adducts, [CH3AuI]− (ref. 10) and [CH3MH]− (M = Cu, Ag and Au),6 have been characterized experimentally. To our best knowledge, CH3MI (M = Cu, Ag and Au) and other CH3MX (M = Cu and Ag; X = Cl and Br) have not been prepared and identified experimentally yet. Of what interest is the causes for the failure of the preparation of these species. Are the failures caused by technological reason? Or are these species not stable enough to be detected? Or is it something else? In the light of these questions, a systematic theoretical research on CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I) was performed in this paper. Our aims are to understand the nature of CH3MX and explore the reason for the failure of the preparation of some CH3MX species by studying the thermodynamically stability, IR character and bonding interactions of CH3MX. This article is organized as follows. The next section (Section 2) provides an overview of the theoretical methods used in the calculations. In Section 3, the reliable geometries and the relative energies of CH3MX were calculated, then the bonding interactions in CH3MX were analyzed. A short summary is given in Section 4.
2. Theoretical calculations
For H, C, F and Cl atoms, the augmented Dunning's correlation consistent valence triple-zeta (aug-cc-pVTZ) basis sets30,31 were used. To consider relativistic effect of heavy elements, the aug-cc-pVTZ-PP small-core relativistic effective core potential (RECP) together with their corresponding basis sets32,33 were used for all other atoms, and the RECP retains 25 and 19 explicit electrons for halogen atoms (Br and I) and coinage-metal atoms (Cu, Ag and Au), respectively. The Becke's three-parameter hybrid exchange functional34 with Lee–Yang–Parr correlation functional (B3LYP)35 were used to optimize the structures of the molecules under investigation. The equilibrium structures of CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I) were optimized by synchronous transit-guided quasi-Newton (STQN) method. Harmonic vibrational frequencies calculation using analytic second derivatives at the same level for all species were performed in order to confirm that the structures were minima and evaluate zero-point vibrational energies (ZPVE). Base on the optimized geometries, the single-point CCSD(T)36–38 energies have been carried out to obtain more accurate energies using the same RECP and basis sets.Bader's atoms-in-molecule (AIM),39,40 natural population charge (NPA) and fuzzy bond orders (FBO)41,42 calculations were performed to understand the nature of the bonding interactions in CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I). To avoid the negative effects of diffuse functions, the cc-pVTZ/cc-pVTZ-PP basis sets removing diffuse functions were used to NPA calculations. In order to obtain reasonable results in chemical sense, all-electron basis sets (WTBS basis set43,44 for both I and Xe, and cc-pVTZ for other atoms) rather than ECP basis sets mentioned above were used to generate wave functions for AIM analysis, moreover, the same basis sets were also used for the FBO calculations since FBO with diffuse function augmentation of basis sets may be unreliable.45 The DFT and NPA calculations were carried out using the Gaussian09 program.46 FBO and AIM calculations were performed by the software Multiwfn.47
3. Results and discussions
3.1 Structures
The model of CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I) molecules was illustrated in Fig. 1, and the selected structural parameters were listed in Table 1. Some CH3MX molecules have been synthesized in solid noble-gas matrices. For example, CH3MH and CH3MF (M = Cu, Ag and Au) has been observed in reactions of coinage-metal with methane in solid matrices,6–8 and reactions of Au with CH3Cl and CH3Br yield CH3AuX (X = Cl and Br) as well.9 This is the first report on all other CH3MX molecules (including CH3MI, CH3CuCl, CH3AgCl, CH3CuBr and CH3AgBr) since no concerning experimental or theoretical result is found. | ||
| Fig. 1 The model of CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I) molecules. | ||
| CH3MH | CH3MFc | CH3MCl | CH3MBr | CH3MI | |
|---|---|---|---|---|---|
| a Atomic covalent radii:48 C: 0.750; H: 0.320; F: 0.640; Cl: 0.990; Br: 1.140; I: 1.330; Cu: 1.120; Ag: 1.280; Au: 1.240.b Bond lengths R in Å, bond angle θ in degree.c CH3AgF is only one with a highly symmetry of C3v, and its electronic state is 2A1 rather than 2A′. | |||||
| Electronic state | 2A′ | 2A′ | 2A′ | 2A′ | 2A′ |
| Symmetry | Cs | Cs | Cs | Cs | Cs |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| M = Cu | |||||
| RC–Cu (ΔRC–Cu) | 1.938 (0.068) | 1.903 (0.033) | 1.909 (0.039) | 1.912 (0.042) | 1.917 (0.047) |
| RCu–X (ΔRCu–X) | 1.508 (0.068) | 1.751 (−0.009) | 2.102 (−0.008) | 2.235 (−0.025) | 2.423 (−0.027) |
| θC–Cu–X | 129.0 | 140.0 | 142.4 | 141.9 | 141.5 |
|
|||||
| M = Ag | |||||
| RC–Ag (ΔRC–Ag) | 2.182 (0.152) | 2.098 (0.068) | 2.150 (0.120) | 2.154 (0.124) | 2.159 (0.129) |
| RAg–X (ΔRAg–X) | 1.649 (0.049) | 1.985 (0.065) | 2.318 (0.048) | 2.442 (0.022) | 2.611 (0.001) |
| θC–Ag–X | 130.4 | 180.0 | 146.6 | 143.1 | 141.7 |
|
|||||
| M = Au | |||||
| RC–Au (ΔRC–Au) | 2.120 (0.130) | 2.036 (0.046) | 2.059 (0.069) | 2.063 (0.073) | 2.071 (0.081) |
| RAu–X (ΔRAu–X) | 1.605 (0.045) | 1.965 (0.085) | 2.291 (0.061) | 2.420 (0.040) | 2.593 (0.023) |
| θC–Au–X | 138.3 | 146.2 | 146.1 | 145.7 | 146.0 |
As illustrated in Table 1, all CH3MX molecules except CH3AgF are non-planar with Cs symmetry and belong to 2A′ electronic state. The ground-state 2A1 CH3AgF is only one with a highly symmetry of C3v, and the F, Ag and C atoms are collinear. Generally, the M–X bond is constituted by the sp/sd hybrid orbitals of M atoms and the sp hybrid orbitals (mainly p orbital) of X atoms. However, the Ag–F bond is constituted by the sd hybrid orbital (mainly s orbital) of Ag atoms and the sp hybrid orbitals (mainly p orbital) of F, which is mainly responsible for the C3v structure of CH3AgF. As shown in Table 1, θC–Cu–X of CH3CuH is 129.0° and is smaller than those of CH3CuX (X = F, Cl, Br and I), which fall in the range of 140–142.5°. Similar things happen in CH3AgX and CH3AuX (X = H, F, Cl, Br and I) since θC–M–H of CH3MH (M = Ag and Au) is the smallest, which attribute to that the p orbitals of halogen atoms are involved the M–X bonds. For CH3MX (X = F, Cl, Br and I) with the same coinage metal, the C–M bond is weakened as X varies from F to I due to its elongation, so the smallest RC–M is found in CH3MF, which indicates that the C–M bond in CH3MF is the strongest. Moreover, the C–M bond in CH3MH is the weakest C–M bond since its C–M bond length is the longest. It is noteworthy that information concerning the strength of C–M bond with different coinage metals cannot be given directly by their lengths. Likewise, although the M–H bond in CH3MH is the shortest among M–X bonds in CH3MX (X = H, F, Cl, Br and I) with the same coinage metal, we cannot infer that the M–H bond in CH3MH is the strongest due to the different atomic radius of different X atoms. In this situation, the M–X bond length is also inappropriate to be used to evaluate the strength of M–X bond directly. Therefore, in order to compare M–X (or C–M) bonds involving different M (or X) atoms, we define one structural parameter ΔR which allow us to unify interactions to estimate their strengths even if different pairs of atoms:49,50
| ΔRA–B = RA–B − RA − RB | (1) |
3.2 Fragmentation energies of CH3MX
To test the stability of CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I) molecules, the energies associated with different formation pathways were calculated using the CCSD(T) method with the same RECP and basis sets to B3LYP, in which ZPVE correction was not considered since it is very time-consuming at the CCSD(T) level. Three possible formation paths that might reasonably occur| CH3X + M → CH3MX | (1) |
| ΔE1 = E(CH3MX) − E(CH3X) − E(M) |
| CH3 + MX → CH3MX | (2) |
| ΔE2 = E(CH3MX) − E(CH3) − E(MX) |
| CH3M + X → CH3MX | (3) |
| ΔE3 = E(CH3MX) − E(CH3M) − E(X) |
The formation energies of CH3MX were given in Table 2. The formations of CH3MX (M = Cu, Ag and Au; X = Cl, Br and I) and CH3CuF along path (1) were expected to be exothermic, while the formations of CH3MH (M = Cu, Ag and Au) and CH3MF (M = Ag and Au) along path (1) were expected to be endothermic, so the preparations of CH3MH (M = Cu, Ag and Au) and CH3MF (M = Ag and Au) via CH3X react with coinage metals are unfavorable thermodynamically. However, previously studies showed that the excitation of coinage-metal atoms in laser ablation process and subsequent UV irradiation can provide the required energy for C–X insertion by excited M*. For example, the lowest-lying excited Cu (2D), Ag (2P) and Au (2D) are 134.0, 353.4 and 109.6 kJ mol−1 higher than their ground state (2S),51–53 respectively. Therefore, it is favorable thermodynamically for the C–X insertion of CH3X by excited coinage metals. As shown in Table 2, as X varies from H to I, the decreasing of ΔE1 indicates the thermodynamically stability of CH3MX increases with respect to CH3X + M, and CH3MI is the most thermodynamically stable although it has not been identified experimentally so far. For different coinage metals, the order of the thermodynamically stability is CH3CuX > CH3AuX > CH3AgX (X = F, Cl, Br and I). Indeed, only CH3AgF has been identified experimentally,8 while other CH3AgX (X = H, Cl, Br and I) have not been characterized experimentally yet. Of course, one plausible reason is that it is difficult to excite Ag atom since it requires 3 times more energy than the excitation of Au atom.
| CH3MX | CH3X + M | CH3 + MX | CH3M + X | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cu | Ag | Au | Cu | Ag | Au | Cu | Ag | Au | |
| H | 100.2 | 186.8 | 59.4 | −95.7 | −51.9 | −96.5 | −126.1 | −95.9 | −137.8 |
| F | −78.5 | 58.0 | 7.6 | −145.5 | −82.1 | −180.8 | −311.0 | −230.9 | −195.8 |
| Cl | −121.4 | −8.6 | −74.1 | −120.3 | −64.2 | −155.8 | −237.7 | −181.3 | −161.4 |
| Br | −125.1 | −23.3 | −93.9 | −115.4 | −62.4 | −151.8 | −204.2 | −158.9 | −144.0 |
| I | −122.9 | −37.3 | −115.7 | −106.7 | −60.6 | −144.3 | −160.5 | −131.5 | −124.3 |
Previous matrix isolation experimental researches showed that CH3X can be decomposed to X atom and CH3 radical in solid matrices by laser ablation, and subsequent CH3M and MX are formed conveniently by coinage metals reacting with CH3 radical as well as X atom, respectively.6 Therefore, it is possible that CH3MX is formed along path (2) or path (3) although the amount of CH3M and MX are very small. However, we think that path (1) should be the main pathway of the formation of CH3MX because the amount of CH3X is significantly larger than those of MX or CH3M in solid matrices. The formations of CH3MX along paths (2) and (3) were expected to be exothermic, which indicates that CH3MX is thermodynamically stable with respect to CH3 + MX or CH3M + X, respectively. Moreover, because both CH3 + MX and CH3M + X are less thermodynamically stable than CH3X + M, CH3MX shows more thermodynamically stabilities with respect to CH3 + MX (or CH3M + X) than that with respect to CH3X + M.
3.3 Frequencies
IR spectroscopy is generally viewed as a powerful tool to characterize active species in matrix isolation experiments because the vibrational fingerprints can provide important information for molecular identification and structural analysis. The selected harmonic vibrational frequencies of CH3MX (M = Cu, Ag and Au; X = H, Cl, Br and I) calculated at the B3LYP level of theory were listed in Table 3, and the selected harmonic vibrational frequencies of CH3X (X = H, Cl, Br and I) and CH3M (M = Cu, Ag and Au) calculated at the same level were listed in Table S1 (see ESI†). For most of CH3MX molecules, the most intense peak is the CH3 deform mode, however, one thing to be careful of is that both CH3M and CH3X have similar peaks. The calculated the CH3 deform frequencies (τCH3) of CH3MF (M = Cu, Ag and Au) and CH3AuX (X = Cl and Br) are in line with previous works.8,9 The CH3 deform mode in CH3MX (X = H, F, Cl, Br and I) illustrates larger red shift with respect to that of CH3X, meanwhile it also shows smaller red-shift behavior compared with that of CH3M. Such different shift behaviors combined with isotopic shift can be used to identify these CH3MX molecules in matrix isolation experiments, especially when other vibrational fingerprints (such as the C–M and M–X stretching vibrational modes) are beyond the detection limit of infrared spectrometer.| CH3MH | CH3MF | CH3MCl | CH3MBr | CH3MI | |
|---|---|---|---|---|---|
| a Frequencies are in cm−1 and intensities (in parentheses) are in km mol−1.b Mixes heavily with CH3 rocking vibrational mode.c Mixes with CH3 rocking vibrational mode.d Mixes with Ag–F stretching vibrational mode.e Mixes heavily with C–Ag rocking vibrational mode.f The C–Au stretching mixes with the Au–X stretching vibrational mode. | |||||
| M = Cu | |||||
| νC–Cu, stretch | 491.3 (0) | 522.4 (1)b | 537.9 (17) | 530.0 (10) | 523.4 (8) |
| νCu–X, stretch | 1774.3 (46) | 638.2 (107)c | 376.9 (23) | 276.1 (13) | 230.2 (7) |
| τCH3, deform | 1079.3 (48) | 1138.8 (33) | 1106.0 (96) | 1099.2 (121) | 1090.4 (166) |
| νCH2, sys stretch | 3017.9 (6) | 3010.7 (3) | 3010.0 (3) | 3012.9 (4) | 3015.0 (4) |
| νCH2, asys stretch | 3117.6 (7), 3148.4 (4) | 3110.3 (8), 3147.9 (2) | 3113.3 (7), 3150.4 (2) | 3115.2 (7), 3150.1 (2) | 3116.4 (7), 3149.8 (2) |
|
|||||
| M = Ag | |||||
| νC–Ag, stretch | 356.1 (2) | 395.5 (45)d | 368.5 (8) | 367.9 (19) | 367.9 (23) |
| νAg–X, stretch | 1641.3 (20) | 532.6 (92) | 308.8 (35)e | 221.0 (10) | 176.5 (4) |
| τCH3, deform | 989.6 (101) | 1002.5 (128) | 986.6 (170) | 987.9 (234) | 987.1 (353) |
| νCH2, sys stretch | 3054.8 (2) | 3039.6 (0) | 3051.4 (0) | 3053.1 (0) | 3055.0 (0) |
| νCH2, asys stretch | 3176.2 (2), 3205.6 (1) | 3167.0 (1) | 3181.0 (1), 3211.1 (0) | 3180.9 (1), 3209.9 (0) | 3180.5 (1), 3207.3 (0) |
|
|||||
| M = Au | |||||
| νC–Au, stretch | 424.5 (2) | 530.4 (16)f | 502.7 (0) | 497.6 (0) | 492.0 (0) |
| νAu–X, stretch | 1927.5 (11) | 511.7 (45)f | 324.6 (18) | 216.8 (6) | 172.0 (2) |
| τCH3, deform | 1093.7 (42) | 1165.2 (51) | 1146.5 (113) | 1146.2 (144) | 1142.7 (206) |
| νCH2, sys stretch | 3033.1 (2) | 2992.2 (2) | 3011.4 (1) | 3018.6 (1) | 3025.6 (1) |
| νCH2, asys stretch | 3146.3 (3), 3183.1 (1) | 3121.9 (7), 3170.2 (0) | 3128.8 (8), 3171.9 (1) | 3129.8 (10), 3169.2 (1) | 3130.7 (11), 3165.5 (1) |
Because the C–M and M–X stretching vibrational modes (νC–M and νM–X) cannot be observed in CH3M and CH3X, they are regarded as the vibrational fingerprints of CH3MX although they are usually weaker than the CH3 deform mode. As shown in Table 3, the νM–H of CH3MH and the νM–F of CH3MF are within the ranges of 1640–1930 cm−1 and 510–640 cm−1, respectively, which have been used to identify these molecules in matrix isolation experiments.6,8 It is noticeable that both the Cu–F and Au–F stretching frequencies are considerably underestimated by the DFT calculation, which is due to the mixture of these vibrational modes with the CH3 deformation mode. The νM–X of other CH3MX (M = Cu, Ag and Au; X = Br and I) are lower than 400 cm−1, which are beyond the detection limit of infrared spectrometer. Perhaps this is one of the reasons why some CH3MX molecules with enough thermodynamically stabilities have not been identified by IR spectroscopy in matrix isolation experiments yet, so other techniques besides IR spectroscopy are required to identify such molecules, and ESR might be an appropriate alternative since it has been used to characterized CH3CuF molecules in noble-gas matrix.28 Similarly, although the calculated harmonic νC–M of CH3MX (M = Cu, Ag and Au; X = Br and I) are higher than 490 cm−1, it can hardly play an active role in the characterization of these CH3MX molecules due to the weak intensity. Nevertheless, some useful information on the bonding of CH3MX can probably be learned from these vibrational modes. As shown in Table 3, the decreasing νC–M of CH3MX (X = F, Cl, Br and I) with the same coinage metal indicates that the C–M bond is weakened as X varies from F to I, which is agree with the above-mentioned structural results. Moreover, the νC–M of CH3MH is the smallest than those in CH3MX (X = F, Cl, Br and I), so the C–M bond in CH3MH should be the weakest one. Similar things also happened in M–X stretching vibrational mode since its frequency decreases as X varies from F to I as well, and the νM–H is the largest, which is different from the case of C–M stretching vibrational mode. However, this cannot be the adequate basis for the evaluation on the order of the strength of M–X bond because that different X atoms are involved in M–X stretching vibrational modes, and the decreasing of the νM–X is partly attributed to the increasing of the reduced mass concerning to the M–X stretching modes as X varies from F to I. Likewise, as M varies from Cu to Au, the decreasing of the νM–X in CH3MX (M = Cu, Ag and Au) does not provide the adequate evidence for the evaluation on the order of the strengths of M–X bonds with the same halogen atom as well. However, unlike M–X stretching vibrational mode, the νC–M in CH3AuX with the same halogen atom is the larger than that of CH3AgX, especially the νC–M in CH3AuF is the largest one among those of CH3MF (M = Cu, Ag and Au), so it is reasonable to assume that the C–Au bond seems to stronger than C–Ag bonds even the effects of reduced mass is taken into account.
3.4 Bond analyses
AIM is a useful tools to explore the nature of the bonding interactions and has been used widely. According to AIM, the first descriptor of bond is the existence of the bond critical point (BCP), and the various characters at BCP are used to analyze the nature of interactions, such as the electron density (ρb) as well as its Laplacian (∇2ρb), potential energy density (Vb), kinetics energy density (Gb), total energy density (Hb), ratio of |Vb|/Gb,54 and eta index (ηb). It has been proved that both Hb and ∇2ρb at the BCP can be used to discriminate interaction types:54–57 Hb < 0 and ∇2ρb < 0 means an accumulation of charge density at BCP and therefore a covalent interaction between the interacting atoms; Hb > 0 and ∇2ρb > 0 means a depletion of charge density at BCP and therefore a closed-shell interaction between the interacting atoms; Hb < 0 but ∇2ρb > 0 means partially covalent interactions. The |Vb|/Gb ratio allow the interaction to be characterized: |Vb|/Gb < 1 indicates pure closed-shell interaction; |Vb|/Gb > 2 indicates pure covalent (open-shell) interaction; while 1 < |Vb|/Gb < 2 indicates intermediate interaction.54AIM analyses have also been carried out to explore the nature of bonds in CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I), and the results were presented in Table 4. The contour line diagrams of Laplacian of electron density (∇2ρb) for CH3MX were presented in Fig. 2. As shown in Fig. 2, the presence of dashed isosurfaces around C–H suggest that valence-shell electrons are strongly concentrated on these regions, it is typical pattern of covalent bonding. No shell structure can be found for the hydrogen atom since it just has 1s valence shell. The C atom has a region of charge concentration towards the metal atom, while the metal atom has a region of charge depletion region along the M–C bond line direction. Similar things also happened in the M–X bond in all studied molecules. Therefore, both the M–X and M–C bonds illustrate closed-shell interaction characters, and there is charge transfer (CT) from the metal atom to the X atom and CH3 group. It is noteworthy that the BCP of M–H bond just locates at the boundary between the charge concentration region and the charge depletion region, which must be taken into account for the study of the nature of M–H bond, otherwise, a unreliable information might be obtained just by the topological parameters (e.g. ∇2ρb, Hb and |Vb|/Gb ratio), which will be discussed later.
| M–C | M–X | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ρb | ∇2ρb | |Vb|/Gb | Hb | ηb | ρb | ∇2ρb | |Vb|/Gb | Hb | ηb | |
| CH3CuH | 0.116 | 0.149 | 1.561 | −0.048 | 0.348 | 0.127 | 0.055 | 1.844 | −0.075 | 0.457 |
| CH3CuF | 0.128 | 0.134 | 1.630 | −0.057 | 0.373 | 0.138 | 0.817 | 1.197 | −0.050 | 0.198 |
| CH3CuCl | 0.126 | 0.131 | 1.628 | −0.055 | 0.370 | 0.103 | 0.297 | 1.329 | −0.036 | 0.235 |
| CH3CuBr | 0.124 | 0.132 | 1.622 | −0.054 | 0.367 | 0.089 | 0.202 | 1.368 | −0.029 | 0.248 |
| CH3CuI | 0.122 | 0.132 | 1.615 | −0.053 | 0.363 | 0.071 | 0.103 | 1.480 | −0.024 | 0.269 |
| CH3AgH | 0.079 | 0.184 | 1.333 | −0.023 | 0.247 | 0.111 | 0.118 | 1.677 | −0.062 | 0.382 |
| CH3AgF | 0.099 | 0.175 | 1.454 | −0.036 | 0.293 | 0.100 | 0.656 | 1.087 | −0.016 | 0.150 |
| CH3AgCl | 0.085 | 0.180 | 1.375 | −0.027 | 0.261 | 0.078 | 0.307 | 1.183 | −0.017 | 0.170 |
| CH3AgBr | 0.084 | 0.180 | 1.368 | −0.026 | 0.259 | 0.069 | 0.228 | 1.206 | −0.015 | 0.177 |
| CH3AgI | 0.083 | 0.184 | 1.355 | −0.025 | 0.253 | 0.053 | 0.159 | 1.190 | −0.009 | 0.162 |
| CH3AuH | 0.103 | 0.201 | 1.426 | −0.037 | 0.280 | 0.140 | 0.056 | 1.870 | −0.094 | 0.457 |
| CH3AuF | 0.126 | 0.188 | 1.545 | −0.057 | 0.318 | 0.117 | 0.786 | 1.109 | −0.024 | 0.157 |
| CH3AuCl | 0.119 | 0.191 | 1.514 | −0.050 | 0.308 | 0.093 | 0.340 | 1.226 | −0.025 | 0.185 |
| CH3AuBr | 0.118 | 0.192 | 1.507 | −0.049 | 0.306 | 0.081 | 0.246 | 1.252 | −0.021 | 0.192 |
| CH3AuI | 0.115 | 0.195 | 1.489 | −0.047 | 0.299 | 0.059 | 0.174 | 1.208 | −0.011 | 0.169 |
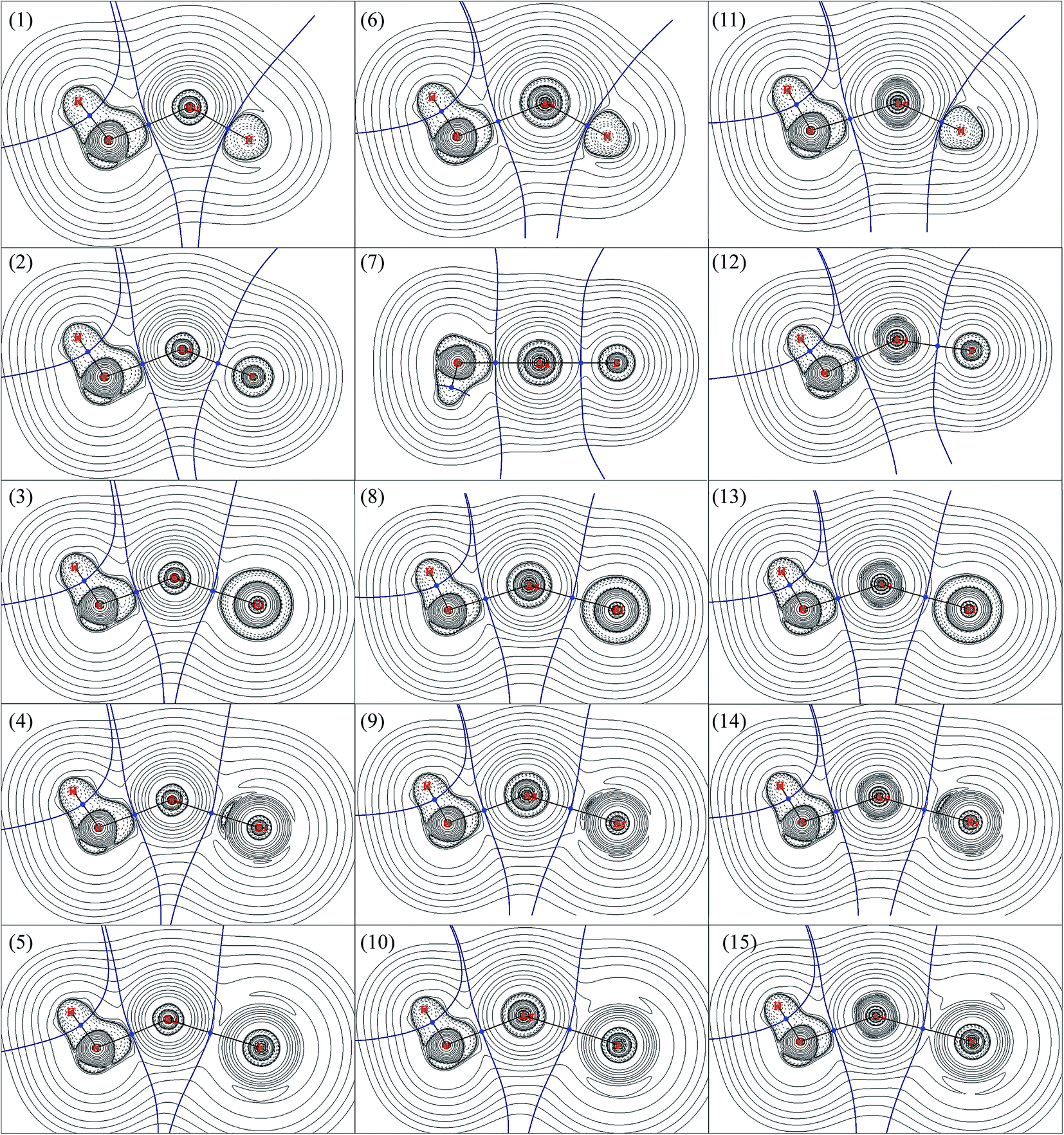 | ||
| Fig. 2 Contour line diagrams of ∇2ρb for CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I), obtained by B3LYP method with all-electron basis sets. (1) CH3CuH; (2) CH3CuF; (3) CH3CuCl; (4) CH3CuBr; (5) CH3CuI; (6) CH3AgH; (7) CH3AgF; (8) CH3AgCl; (9) CH3AgBr; (10) CH3AgI; (11) CH3AuH; (12) CH3AuF; (13) CH3AuCl; (14) CH3AuBr; (15) CH3AuI. Dashed lines indicate areas of charge concentration (∇2ρb < 0), while solid lines show areas of charge depletion (∇2ρb > 0). The bold brown solid lines connecting the atomic nuclei are the bond paths and the solid blue lines separating the atomic nuclei indicate the zero-flux surfaces in the molecular plane. The crossing points of the bond paths and zero-flux surfaces are the bond critical points (BCP). | ||
As shown in Table 4, the ∇2ρb at the BCPs of both M–C and M–X in all studied molecules are positive, and their total energy density (Hb) are negative, which reveals that both M–C and M–X bonds are partially covalent interactions. The point was confirmed by the |Vb|/Gb ratios since all |Vb|/Gb ratios are within the range of 1–2. The BCP of M–C in CH3MH (M = Cu, Ag and Au) has larger Hb > 0 and ∇2ρb than other that of M–C in CH3MX (M = Cu, Ag and Au; X = F, Cl, Br and I), which indicates that the M–C bond in CH3MH is weaker than the one in CH3MX. Of particular note is that the |Vb|/Gb ratio at the BCP of M–H bond, especially Cu–H and Au–H bonds, is obviously larger than those of M–X bonds, so the M–H bond seems to be stronger and exhibits more covalent than the M–X bonds, which is supported by the more negative Hb and smaller ∇2ρb. However, such conclude is inconsistent with the discussions mentioned above, and the covalent character of M–H bond is overestimated because the BCP of M–H bond just locates at the boundary between the charge concentration region and the charge depletion region. Therefore, it is not advisable to study the nature of bonds by AIM descriptors without considering the particular circumstances of each case.
The |Vb|/Gb ratio of M–C BCP in CH3MX decrease as X varies from F to I, which indicates that the M–C bond is weakened simultaneously with the decreasing of the electronegativity of X atom, and such trend can also be learned from the decreasing ∇2ρb and the increasing Hb. The M–X bond shows a reverse trend with respect to the M–C bond, and is strengthened due to the increasing |Vb|/Gb ratio and Hb as well as the decreasing ∇2ρb as X varies from F to I. Compare the descriptors including ∇2ρb, Hb as well as |Vb|/Gb ratios of BCPs of either M–X bonds in CH3MX (M = Cu, Ag, Au) containing the same halogen atom, it is not hard to find that the Cu–X bond is stronger than both Ag–X and Au–X, and the Ag–X bond is the weakest bond. Similarly, the order of the strengths of M–C bond is Cu–C > Au–C > Ag–C. Therefore, CH3CuX is expected to be more stable than CH3AuX and CH3AgX which attribute to the stronger Cu–C and Cu–X bonds, whereas CH3AgX should have the less stability due to the weakest M–C and M–X bonds.
To understand the bonding interactions in CH3MX (M = Cu, Ag and Au; X = Cl, Br and I), the fuzzy bond order (FBO) were calculated using B3LYP method, and cc-pVTZ basis sets removing the diffusion basis functions were used to obtain reliable results. The calculated fuzzy bond orders (FBO) of CH3MX were listed in Table 5. As shown in Table 5, the order of FBOs of C–M bond in CH3MX (M = Cu, Ag and Au) with the same halogen is C–Cu > C–Au > C–Ag, so the C–Cu bond is stronger than the other two, while the C–Ag bond is the weakest one, which is consistent with the discussion above. The FBO of the M–X bond demonstrates a similar order of Cu–X > Au–X > Ag–X. As X varies from F to I, the FBO of C–M in CH3MX decreases, which means that the C–M bond is weakened. However, the M–X illustrates one reverse trend and is strengthened as X varies from F to I due to the increasing FBO.
| C–M | M–X | |||||
|---|---|---|---|---|---|---|
| Cu | Ag | Au | Cu | Ag | Au | |
| CH3MH | 1.188 | 0.954 | 1.063 | 0.992 | 0.949 | 1.029 |
| CH3MF | 1.232 | 1.017 | 1.128 | 1.634 | 1.307 | 1.384 |
| CH3MCl | 1.195 | 0.915 | 1.075 | 1.719 | 1.524 | 1.588 |
| CH3MBr | 1.184 | 0.912 | 1.065 | 1.751 | 1.578 | 1.642 |
| CH3MI | 1.164 | 0.898 | 1.039 | 1.697 | 1.515 | 1.565 |
The calculated NPA charges of CH3MX (M = Cu, Ag and Au; X = Cl, Br and I) were presented in Table 6. As shown in Table 6, the charge of M is positive, which indicates that there is electron transfer occured from metal atom to CH3 group and the halogen atom. The extent of CT decreases as X varies from F to I due to the decreasing charge of M, which is attributed mainly to the influence of halogen atoms. Moreover, the first ionization potential of Au is the highest among coinage-metal atoms, which is in line with that the extent of CT in CH3AuX is the largest. However, although the first ionization potential of Cu is larger than that of Ag, our results show that the extent of CT is CH3CuX > CH3AgX. The charge of halogen atom is more negative than that of methyl group, which reveals that the halogen atom is the main electron acceptor rather than the methyl group. Due to the smaller electronegativity of the H atom, the charge of the H atom in CH3MH is obviously larger than that of the halogen atom in CH3MX, which lead to the smallest CT effect happened in CH3MH.
| CH3CuX | CH3AgX | CH3AuX | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CH3 | Cu | X | CH3 | Ag | X | CH3 | Au | X | |
| CH3MH | −0.315 | 0.585 | −0.270 | −0.171 | 0.400 | −0.229 | −0.165 | 0.272 | −0.107 |
| CH3MF | −0.347 | 0.984 | −0.637 | −0.114 | 0.861 | −0.747 | −0.148 | 0.694 | −0.546 |
| CH3MCl | −0.288 | 0.781 | −0.493 | −0.078 | 0.619 | −0.541 | −0.137 | 0.501 | −0.364 |
| CH3MBr | −0.284 | 0.705 | −0.421 | −0.095 | 0.554 | −0.459 | −0.144 | 0.427 | −0.283 |
| CH3MI | −0.281 | 0.603 | −0.322 | −0.116 | 0.468 | −0.352 | −0.159 | 0.328 | −0.170 |
4. Conclusions
CH3MX (M = Cu, Ag and Au; X = H, F, Cl, Br and I) have been investigated by DFT calculation. Equilibrium geometry, harmonic vibrational frequencies, energies were calculated, and AIM calculations were also performed to understanding the bonding interactions in the studied molecules.(1) The ground states of all molecules are doublet, and all molecules except for CH3AgF are planar structures with Cs symmetry, while CH3AgF has C3v symmetry with a linear H–Ag–C bond angle.
(2) As X varies from F to I, the thermodynamically stability of CH3MX with respect to CH3X + M increases, and the order of the thermodynamically stability for different coinage metals is CH3CuX > CH3AuX > CH3AgX. Such conclusion is supported by the AIM and FBO calculations.
(3) Although CH3MH (M = Cu, Ag and Au) and CH3MF (M = Ag and Au) were predicted to be unstable with respect to CH3X + M, most of them have been prepared and identified in low-temperature matrix isolation experiments. Besides the involvement of the excited coinage-metal atoms in laser ablation process and UV irradiation, the strong M–H (or M–F) stretching vibrational modes make a crucial contribution to the identification of these species.
(4) The most intense peak in CH3MX is the CH3 deform mode, which illustrates larger red shift with respect to that of CH3X, and smaller red shift with respect to that of CH3M. Such different shift behaviors combined with isotopic shift can be used to identify these CH3MX molecules in matrix isolation experiments. Of note is that some CH3MX molecules with enough thermodynamically stabilities have not been identified by IR spectroscopy in matrix isolation experiments yet, and one of probably reasons is that their vibrational fingerprints (C–M and M–X stretching vibrational modes) are weak or too low to be detected by infrared spectrometer.
(5) AIM analyses show that both C–M and M–X bonds exhibit mainly closed-shell interaction character, and partial covalent character contributes to them. The BCP of M–H bond just locates at the boundary between the charge concentration region and the charge depletion region, which lead to that the covalent character of M–H bond is overestimated by the AIM topological parameters.
In brief, although the CH3MX molecules with more stabilities should be more chances to be observed experimentally, one of the challenges is that both νC–M and νM–X are weak and are beyond the detection limit of infrared spectrometer, so how to overcome such challenge should be the next research direction, and this paper might provide useful information for the next research.
Acknowledgements
This work is supported by the Natural Science Foundation of Tianjin (No. 12JCYBJC13400) and the Program for Innovative Research Team in University of Tianjin (TD12-5038).References
- L. Andrews and H. G. Cho, Organometallics, 2006, 25, 4040–4053 CrossRef CAS.
- H.-G. Cho and L. Andrews, Organometallics, 2007, 26, 4098–4101 CrossRef CAS.
- H.-G. Cho and L. Andrews, Organometallics, 2007, 26, 633–643 CrossRef CAS.
- J. T. Lyon, H.-G. Cho and L. Andrews, Organometallics, 2007, 26, 6373–6387 CrossRef CAS.
- H. G. Cho, J. T. Lyon and L. Andrews, Organometallics, 2008, 27, 5241–5251 CrossRef CAS.
- H. G. Cho and L. Andrews, Dalton Trans., 2011, 40, 11115–11124 RSC.
- L. B. Knight, J. T. Petty Jr, S. T. Cobranchi, D. Feller and E. R. Davidson, J. Chem. Phys., 1988, 88, 3441–3450 CrossRef CAS.
- H.-G. Cho and L. Andrews, Inorg. Chem., 2011, 50, 10319–10327 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, Organometallics, 2013, 32, 2753–2759 CrossRef CAS.
- S. Muramatsu, K. Koyasu and T. Tsukuda, J. Phys. Chem. A, 2016, 120, 957–963 CrossRef CAS PubMed.
- L. Andrews and H. G. Cho, J. Phys. Chem. A, 2005, 109, 6796–6798 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, J. Phys. Chem. A, 2007, 111, 2480–2491 CrossRef CAS PubMed.
- J. T. Lyon and L. Andrews, Organometallics, 2007, 26, 2519–2527 CrossRef CAS.
- H.-G. Cho and L. Andrews, J. Phys. Chem. A, 2008, 112, 12293–12295 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, J. Am. Chem. Soc., 2008, 130, 15836–15841 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, Eur. J. Inorg. Chem., 2008, 2537–2549 CrossRef CAS.
- H.-G. Cho and L. Andrews, Organometallics, 2008, 27, 1786–1796 CrossRef CAS.
- H.-G. Cho and L. Andrews, Inorg. Chem., 2008, 47, 1653–1662 CrossRef CAS PubMed.
- H.-G. Cho, J. T. Lyon and L. Andrews, J. Phys. Chem. A, 2008, 112, 6902–6907 CrossRef CAS PubMed.
- J. T. Lyon and L. Andrews, Eur. J. Inorg. Chem., 2008, 1047–1058 CrossRef CAS.
- H.-G. Cho and L. Andrews, Organometallics, 2009, 28, 1358–1368 CrossRef CAS.
- H.-G. Cho, L. Andrews, B. Vlaisavljevich and L. Gagliardi, Organometallics, 2009, 28, 6871–6879 CrossRef CAS.
- H.-G. Cho, L. Andrews, B. Vlaisavljevich and L. Gagliardi, Organometallics, 2009, 28, 5623–5632 CrossRef CAS.
- H.-G. Cho and L. Andrews, J. Phys. Chem. A, 2010, 114, 8056–8068 CrossRef CAS PubMed.
- H.-G. Cho and L. Andrews, Dalton Trans., 2010, 39, 5478–5489 RSC.
- H.-G. Cho and L. Andrews, Organometallics, 2010, 29, 2211–2222 CrossRef CAS.
- H. G. Cho and L. Andrews, Organometallics, 2009, 28, 1358–1368 CrossRef CAS.
- L. G. Knight Jr, S. T. Cobranchi, B. W. Gregory and G. C. Jones Jr, J. Chem. Phys., 1988, 88, 524–532 CrossRef.
- X. Zhao, G. K. Koyanagi and D. K. Bohme, J. Phys. Chem. A, 2006, 110, 10607–10618 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877–13883 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- L. Chengteh, Y. Weitao and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- M. Sprik and M. L. Klein, J. Chem. Phys., 1987, 87, 5987–5999 CrossRef CAS.
- G. E. Scuseria, C. L. Janssen and H. F. Schaefer, J. Chem. Phys., 1988, 89, 7382–7387 CrossRef CAS.
- G. E. Scuseria and H. F. Schaefer, J. Chem. Phys., 1989, 90, 3700–3703 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, U.K., 1990 Search PubMed.
- C. F. Matta and R. J. Boyd, The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007 Search PubMed.
- L. George, A. Kalume, B. J. Esselman, J. Wagner, R. J. McMahon and S. A. Reid, J. Chem. Phys., 2011, 135, 124503 CrossRef PubMed.
- I. Mayer and P. Salvador, Chem. Phys. Lett., 2004, 383, 368–375 CrossRef CAS.
- S. Huzinaga and M. Klobukowski, Chem. Phys. Lett., 1993, 212, 260–264 CrossRef CAS.
- S. Huzinaga and B. Miguel, Chem. Phys. Lett., 1990, 175, 289–291 CrossRef CAS.
- L. Goodman and R. R. Sauers, J. Comput. Chem., 2007, 28, 269–275 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, E.01, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- P. Pyykko and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- H. K. Wang, Z. G. Huang, T. T. Shen and L. F. Guo, Struct. Chem., 2012, 23, 1163–1172 CrossRef CAS.
- Q. Wang, B. Zhang and Z. Huang, Chem. Phys. Lett., 2014, 614, 5–9 CrossRef CAS.
- J. Sugar and A. Musgrove, J. Phys. Chem. Ref. Data, 1990, 19, 527–616 CrossRef CAS.
- T. Badr, M. D. Plimmer, P. Juncar, M. E. Himbert, J. D. Silver and G. D. Rovera, Eur. Phys. J. D, 2004, 31, 3–10 CrossRef CAS.
- J. C. Ehrhardt and S. P. Davis, J. Opt. Soc. Am., 1971, 61, 1342–1349 CrossRef.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835–12841 CrossRef CAS.
- S. Jenkins and I. Morrison, Chem. Phys. Lett., 2000, 317, 97–102 CrossRef CAS.
- S. J. Grabowski, W. A. Sokalski and J. Leszczynski, J. Phys. Chem. A, 2006, 110, 4772–4779 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18033g |
| This journal is © The Royal Society of Chemistry 2016 |