
A facile one-pot method to synthesise 2-alkylated indole and 2,2′-bis(indolyl)methane derivatives using ketones as electrophiles and their anion sensing ability†
Sinan
Bayindir
ab and
Nurullah
Saracoglu
 *a
*a
aDepartment of Chemistry, Faculty of Sciences, Atatürk University, 25240, Turkey. E-mail: nsarac@atauni.edu.tr
bDepartment of Chemistry, Faculty of Sciences and Arts, Bingöl University, 12000, Turkey
First published on 27th July 2016
Abstract
Indole derivatives are of great importance because of their biological activity and application in technology. This study explores the synthesis of 2-alkylated indoles derivatives and 2,2′-bis(indolyl)methanes, and their application in anion sensing. The synthesis of a wide range of 2-alkylated indoles and some 2,2′-bis(indolyl)methanes, which cannot be synthesized by previously reported methods, was for the first time accomplished employing dipole exchange of the indole ring towards electrophilic substitution. Some of the indole derivatives exhibited selective recognition and sensing ability towards F− and HSO4− anions through naked-eye detectable color changes. The sensing details of the indole derivatives were also evaluated using UV-Vis spectroscopy and 1H NMR titration techniques.
Introduction
Indole and its derivatives are frequently present in the structure of pharmaceutics, agricultural chemicals, functional substances and natural products.1–3 Therefore, many studies have reported the effective derivatization of indole, and the development of new synthetic methods and applications of new indole derivatives have been explored extensively since indole was discovered in 1869.4–6 Bis(indolyl)methane alkaloids and substituted indole derivatives were previously discovered, and because of their importance in biology and technology applications, there has been an increasing interest to develop cheap, effective, and facile methods for synthesis of new derivatives.7–12 But, the directly synthesis of 2-substituted indoles from indole are extremely difficult and limited. Consequently, the most frequently used methods to access 2-substituted indoles are often based on the construction of the heterocycles via cyclization reactions (Scheme 1a).13,14 Despite its importance and widespread use, asymmetric bis(3-indolyl) methanes and symmetric/asymmetric bis(2-indolyl)methanes remain difficult to synthesize effectively, except for synthesis of symmetric bis(3-indolyl)methanes. In the literature, the studies concerning the synthesis of bis(2-indolyl)methanes are very restricted and these indoles are currently substituted at the 3-position (Scheme 1b).15 | ||
| Scheme 1 Strategies for the synthesis of 2-alkyl indoles and 2,2′-bis(indolyl)methanes. | ||
In our previous studies, we developed an efficient protocol for the preparation of 2-substituted indoles through Michael-type addition of 4,7-dihydroindole (1) using Michael acceptors as electrophile followed by an oxidation step (Scheme 1c).16,17 Our strategy also provided the most important alternative option for accessing chiral 2-substituted indoles.18 Herein, we aim to develop a highly efficient, facile and atom economical protocol to prepare 2-alkylated indoles and bis(2-indolyl)methanes (Scheme 1d). The synthesis of 2-alkylindoles are previsualized from Friedel–Crafts reaction between 4,7-dihydroindol (1) and ketones such as cyclic/acyclic aliphatic ketones, bisaryl ketones and aryl–alkyl ketones via redox isomerization including the dehydration and hydrogen shift as in situ. The preparation of bis(2-indolyl)methanes is predicted via two consecutive Friedel–Crafts alkylation of two equivalent of 4,7-dihydroindole (1) with one equivalent of a ketone followed by an oxidation step.
Molecules such as indole, carbazole, bisindole and indolocarbazole form an import class of hydrogen bond donors. In addition to their biological properties, bis(indolyl)methane and alkyl indoles are very attractive compounds for chemosensing.19–25 Because of their sensitivity and selectivity, these compounds have served in the determination of ions hazardous to the environment and health. Within this context, there is just an increasing interest in the synthesis of colorimetric sensors for detection of anions. Anions play an important role in chemical and biological systems, and fluoride, acetate and bisulphate anions sometimes play a role in environmental systems. For example, fluoride ion has an important application in the food and toxicity industry. Among the various anions, bisulphate ion is of particular interest because of its essential role in biological and industrial areas. Upon decomposition of bisulphate ion at high pH, toxic sulfate ion (SO42−) is released, which can cause respiratory paralysis.11 For these reasons, we are concerned with determining whether new indole derivatives have specific chemoselectivity for ions by using spectroscopic analysis methods, including UV-vis and NMR titration as well as the color change observed by the naked eye.
Results and discussions
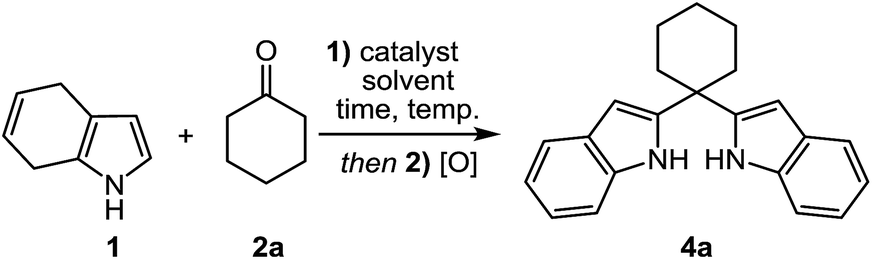
At first, 4,7-dihydro-1H-indole (1) as staring material was obtained by Birch reduction of indole (12) with Li in liquid ammonia.16 Our effort was initially focused on the alkylation of 4,7-dihydro-1H-indole (1) with cyclohexanone (2a) as a model reaction. As shown in Table 1, after treatment of 1 (1.0 equiv.) with 2a (1.0 equiv.) in the presence of Bi(NO3)3·5H2O (10% mol) in methylene chloride at room temperature, the desired alkylation product was not observed (Table 1, entry 10). When the reaction was carried out using TFA (trifluoroacetic acid), AlCl3 and PhCOOH as catalysts at room temperature, a complex product mixture was obtained. Therefore, optimization study was performed by varying the parameters such as reaction time, catalyst, temperature and solvent (Table 1). Notably, when the reaction was carried out at 80 °C in acetonitrile in the presence of the Bi(NO3)3·5H2O, the desired 2-alkylated indole 3a was isolated in 97% yield (Table 1, entry 11).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Time | Yieldb (%) |
| a Conditions: 4,7-dihydro-1H-indole (1, 1 equiv.), cyclohexanone (2a, 1 equiv.), catalyst (10% mmol) and solvent (10 mL). b Isolated yields of 2-cyclohexyl-1H-indole (3a). c Complex reaction mixture. d Under N2. | |||||
| 1 | TFA | CH2Cl2 | 25 | 30 min | 0c,d |
| 2 | AlCl3 | CH2Cl2 | 25 | 30 min | 5c,d |
| 3 | ZrCl4 | CH2Cl2 | 25 | 5 h | 25 |
| 4 | ZrCl4 | MeCN | 80 | 30 min | 15c,d |
| 5 | PhCOOH | MeCN | 80 | 30 min | 17c,d |
| 6 | Cu(OTf)2 | MeCN | 80 | 5 h | 80 |
| 7 | InCl3 | MeCN | 80 | 5 h | 75 |
| 8 | BiCl3 | MeCN | 80 | 5 h | 78 |
| 9 | Zn(OTf)2 | MeCN | 80 | 5 h | 88 |
| 10 | Bi(NO3)3·5H2O | CH2Cl2 | 25 | 12 h | 0 |
| 11 | Bi(NO3)3·5H2O | MeCN | 80 | 5 h | 97 |

With the optimized reaction conditions in hand, we next explored the scope and limitations of the reaction by using 1 (1.0 equiv.) and various ketones (1.0 equiv.) and the desired products (3a–l, 3o–p) were obtained in high yields (Table 2). In contrast, the reaction of 2-acetylpyrrole (2m) failed to produce the desired product 3m under the optimized conditions and only lead to the recovery of the starting material (Table 2). We assume that the possible resonance contribution for 2m could decrease the reactivity of carbonyl carbon toward the nucleophilic attack of dihydroindole 1. When N-Boc-2-acetylpyrrole (2n) reacted with 1, the formation of a trace amount product 3n was estimated by 1H NMR spectra (Table 2).
|
|
|---|
| a Conditions: 1 (1.0 mmol), 2 (1.0 mmol), Bi(NO3)3·5H2O (10% mmol), MeCN (10 mL), 80 °C, under air; isolated yields are shown. |

|
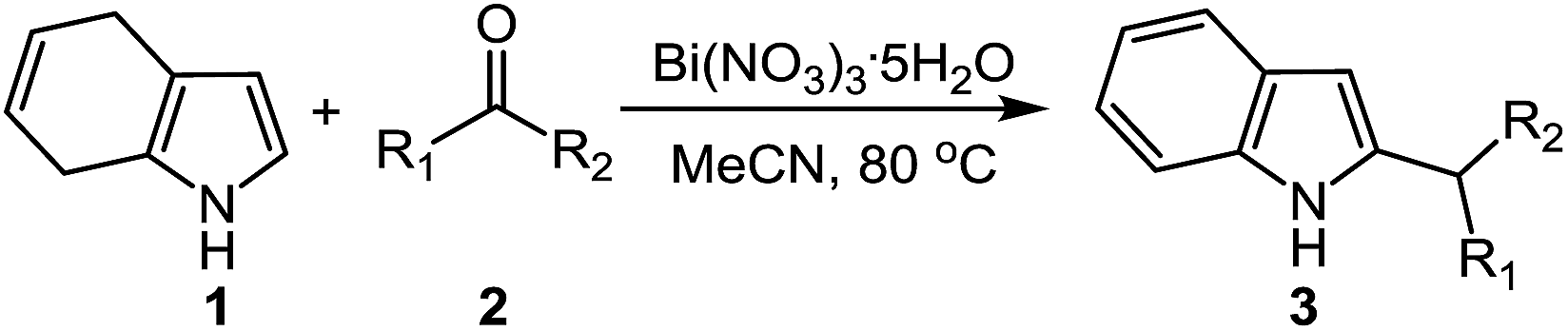
We next examined the reaction between 1 (2.0 equiv.) and ketones (1.0 equiv.) to yield bis(2-indolyl)methanes. The reaction of 1 (2.0 equiv.) and 2a (1.0 equiv.) was selected as model reaction, and carried out by changing the parameters such as reaction time, catalyst, temperature and solvent. Although the formation of the expected 2,2′-(cyclohexane-1,1-diyl)bis(4,7-dihydro-1H-indole) was confirmed by 1H NMR spectra, the purification of the crude product by column chromatography on the silica gel failed. Therefore, we attempted to oxidize the crude product and to isolate the corresponding bis(2-indolyl)methane. Among the tested parameters as shown results in Table 3, the best result was obtained in refluxing acetonitrile with the catalyst of bismuth nitrate for 5 h to give 4a in a yield of 74% followed by the usual work-up and the oxidation of the crude product with p-benzoquinone (PBQ) in methylene chloride (entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Time | Yieldb (%) |
| a Conditions: (1) 4,7-dihydro-1H-indole (1, 2 equiv.), cyclohexanone (2a, 1 equiv.), catalyst (10% mmol) and solvent (10 mL) and then (2) PBQ (p-benzoquinone, 2.5 equiv.) and CH2Cl2 (25 mL). b Isolated yields of 2,2′-(cyclohexane-1,1-diyl)bis(1H-indole) (4a). c Complex reaction mixture. d 2-Cyclohexyl indole (3a) occurred. | |||||
| 1 | TFA | CH2Cl2 | 25 | 30 min | 0c |
| 2 | AlCl3 | CH2Cl2 | 25 | 30 min | 10c,d |
| 3 | ZrCl4 | CH2Cl2 | 25 | 5 h | 12d |
| 4 | ZrCl4 | CH3CN | 80 | 30 min | 34c,d |
| 5 | PhCOOH | MeCN | 80 | 30 min | 27c,d |
| 6 | Cu(OTf)2 | MeCN | 80 | 5 h | 25 |
| 7 | InCl3 | MeCN | 80 | 5 h | 55 |
| 8 | BiCl3 | MeCN | 80 | 5 h | 71 |
| 9 | Zn(OTf)2 | MeCN | 80 | 5 h | 70 |
| 10 | Bi(NO3)3·5H2O | MeCN | 80 | 5 h | 74 |
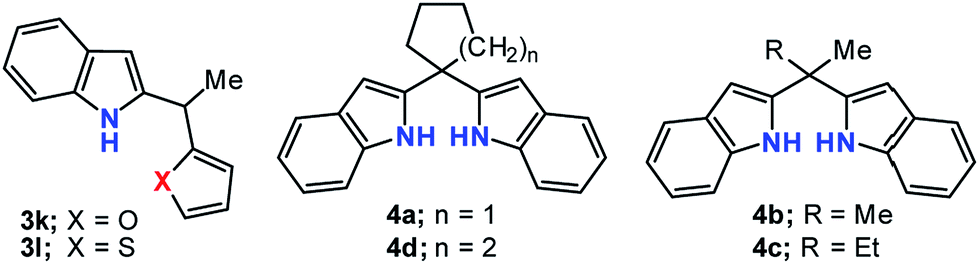
Furthermore, although the reactions were performed over a wide range of ketones, the desired 2,2′-(bis-1H-indolyl)methanes 4a–d were only able to be obtained from with cyclic or acyclic aliphatic ketones 2a–d (Table 4, entry 1–4). In the case of the other ketones 2e–o, while the reactions only gave the 2-alkylated indole derivatives 3e–o, the excess of dihydroindole 1 was recovered (Table 4, entry 5–10). Similar results were likewise obtained from with excess equivalents, regardless of solvent and temperature conditions.
|
|
|---|
| a Conditions: (1) 1 (2.0 mmol), 2 (1.0 mmol), Bi(NO3)3·5H2O (10% mmol), MeCN (10 mL), 80 °C, under air; (2) PBQ (2.5 equiv.) and CH2Cl2 (25 mL), isolated yields are shown. Ketones 2e–o provided 2-alkylated indoles. |

|

A possible mechanism for the formation of the 2-alkylated indole 2a and bisindolylmethane 4a was formulated in Scheme 2. The formation of the 2-alkylated indole 2a may proceed via an intermolecular Friedel–Crafts product 5 between pyrrole 1 and electrophile 2a in the presence of the catalyst followed by a redox isomerization involving hydration and proton shift steps. In the presence of a Lewis acid, the intermediate 5 may be captured by a second equivalent of 1 as a nucleophile to afford bis-pyrrolyl-methane 7, which readily oxidised to 4a with PBQ. As mentioned above, the aryl–aryl or aryl–alkyl ketones under same conditions do not yield the expected bisindolylmethane derivatives in each case, but rather give the corresponding 2-alkylated indoles. We assume that the carbocation intermediate formed in the first step of the Friedel–Crafts reaction has a key role, in that it directly influences the progress of the reaction. Some representatives of the carbocation intermediates can be seen in Fig. 1. We predict that the intermediate 6a possess a coplanar structure, which can tolerate both reaction pathway. In case of the intermediate 6h or 6p, we propose that these carbocations are to be propeller-shaped due to steric crowding of the ortho hydrogens. Thus, these intermediates are twisted out of coplanarity, and their resonance stabilisation is decreased. With this closed geometry as a result of the steric hindrance, the empty p orbital of these carbocations is shielded from the approaching the second dihydroindole 1, and the aromatization to second Friedel–Crafts reaction is preferred.
 | ||
| Scheme 2 Proposed mechanism for the formation of 3a and 4a. | ||
 | ||
| Fig. 1 Representative carbocation intermediates. | ||
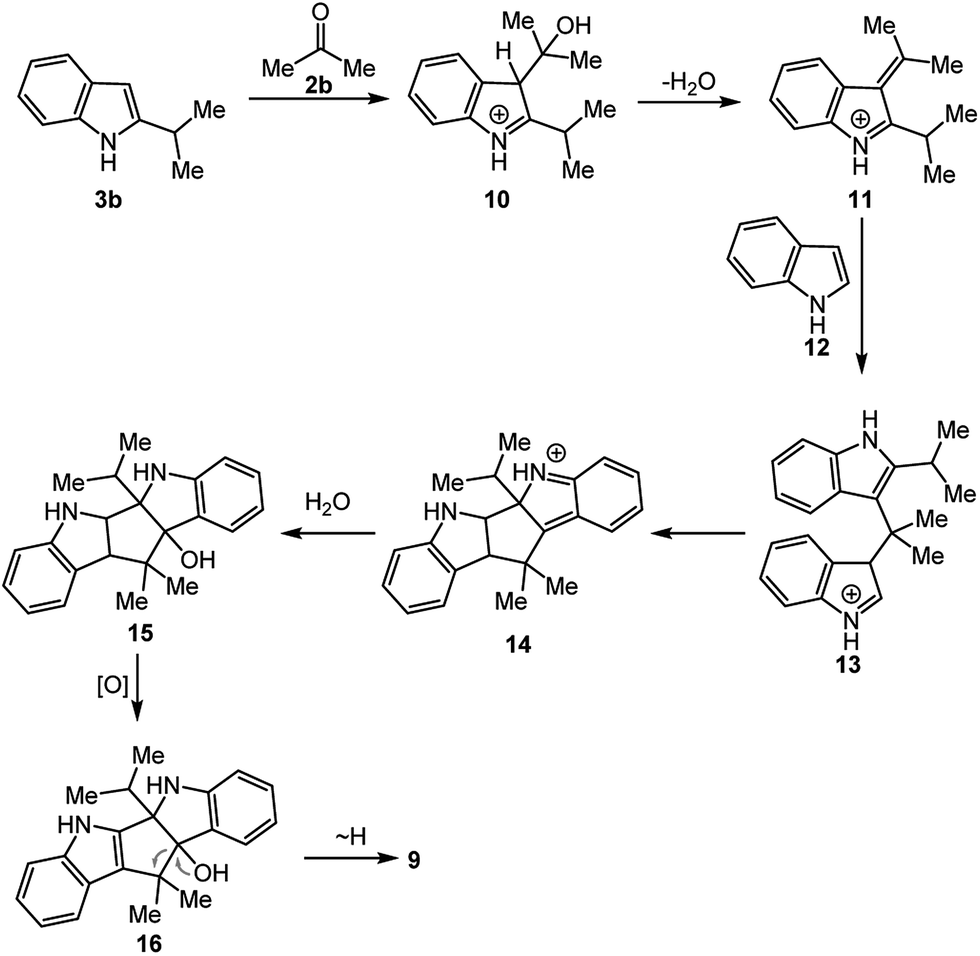
In order to synthesize 2,2′-(propane-2,2-diyl)bis(1H-indole) (4b), the reaction of 4,7-dihydro-1H-indole (1) with acetone (2b) was carried out (Scheme 3). The 1H NMR spectrum of the crude reaction mixture shows a mixture of 2,2′-(propane-2,2-diyl)bis(4,7-dihydro-1H-indole) (8b) as main product and 2-alkylated indole 3b as the byproduct along with unreacted 1. During the separation over silica gel column, while 3b isolated in 12% yield, the main product 8a could not be obtained. Then, the crude mixture was submitted to the oxidation with p-benzoquinone in acetonitrile. After the purification, 2-isopropyl-2-(2-isopropyl-1H-indol-3-yl)indolin-3-one (9) as a novel indole derivative was isolated, as well as the targeted bisindolylmethane 4b. The structure of 9 was established by NMR spectroscopy. The 1H NMR spectrum of the indole derivative 9 showed signals at 7.95 and 5.08 ppm for the NH groups of indole and oxindole rings. The resonance signals for eight aromatic protons were observed at 6.77–8.02 ppm. Additionally, the resonances of the two isopropyl methine protons were present as a multiplet at 3.97–3.93 ppm and 3.15–3.12 ppm. Because of both the chirality resulting from possible blocked rotation of the bond between the indole and oxindole rings by two isopropyl groups and the quaternary stereogenic carbon center in oxindole ring, the methyl protons were resonated as four doublets at 1.32, 1.22, 0.95 and 0.88 ppm. The carbon resonance signal observed at 203.4 ppm on APT 13C NMR of indole–oxindole derivative 9 confirms the existence of a ketone group in the molecule and the fourteen carbon resonance signals at the olefinic area support the proposed structure. Furthermore, the aliphatic quaternary carbon and methine carbon signals in 9 were observed at 75.0, 34.7 and 26.7 ppm, respectively, whereas the methyl carbons were resonated as four signals at 23.7–16.4 ppm due to chirality.
 | ||
| Scheme 3 Reaction of acetone (2b, 1 equiv.) with 1 (2 equiv.). | ||
The probable mechanism for the formation of the unexpected product 9 is shown in Scheme 4. We believe that this product occurred through a series of reactions between 2-isopropyl-1H-indole (3b), acetone (2b) and indole (12). The mechanism involves bismuth nitrate-catalyzed nucleophilic addition of 2-alkylated indole 3b to acetone to afford the intermediate 10. The subsequent dehydration of 10 is likely to proceed via the formation of an azafulvenium salt which in turn undergoes further addition with indole (12) derived from the oxidation of dihydroindole 1 by PBQ as in situ leading to the formation of iminium intermediate 13. The proposed mechanism completes a subsequent intramolecular nucleophilic addition of 13 followed by the hydration, oxidation and oxidative C–C bond cleavage steps to afford 9.
 | ||
| Scheme 4 Proposed mechanism for the formation of 2-isopropyl-2-(2-isopropyl-1H-indol-3-yl)indolin-3-one (9). | ||
In connection with studies on the synthesis of bisindolylmethanes, the reaction of 4,7-dihydro-1H-indole (1; 2.0 equiv.) and 2-acetylfuran (2k; 1.0 equiv.) gave a mixture resulting in the formation of two unexpected products 17 and 12 along with 2-alkylated indole 3k without the oxidation reaction (Scheme 5). This surprising result may be due to a hydride transfer from 1 to the initially formed intermediate 6k to yield 17 and 12 via an intermolecular redox-type reaction (Scheme 5).
 | ||
| Scheme 5 Reaction of 1 (2 equiv.) with 2k (1 equiv.) and proposed mechanism for the products. | ||
Next, the sensing abilities of some of appropriate H-donor receptors depicted in Fig. 2 were monitored by UV-vis, 1H NMR spectroscopic methods and naked-eye observation. As an initial test, we checked the colour changes of receptors in various solvent systems (MeCN, MeCN/H2O, DMSO, THF, THF/H2O etc.) upon the addition of tetrabutylammonium (TBA) salts of F−, Cl−, Br−, I−, HSO4−, AcO−, CN− and SCN−. As shown in Fig. 2, a noticeable color changes by the naked eye could be detected from the interaction of 3l with F−, 4b and 4c with HSO4−. The interactions between other anions and receptors did not result in selective color changes (Fig. 3).
 | ||
| Fig. 2 Structures of the receptors. | ||
 | ||
Fig. 3 Colour changes observed upon addition of 5 × 10−5 M anions to MeCN (a and b) or MeCN/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) (c–f) of 1 × 10−5 M ligands [3k (a), 3l (b), 4a (c), 4b (d), 4c (e) and 4d (f)]. :1, v/v) (c–f) of 1 × 10−5 M ligands [3k (a), 3l (b), 4a (c), 4b (d), 4c (e) and 4d (f)]. | ||
Anion recognition properties of receptors with the aforementioned anions in the various solvent systems were further evaluated by UV-vis absorption spectroscopy (Fig. 4). The free receptor 3l displayed two sharp absorption peaks at 226 nm and 273 nm. Interaction of 3l (1 × 10−5 M) with anions caused changes to appear in this region of the spectrum. Upon addition of F−, the sharp peaks at 226/273 nm red shifted to 270/320 nm, which was attributed to the color change resulting from the interaction of 3l with F− ions that can be detected by the naked eye. Furthermore, a new absorption peak formed at 573 nm (Fig. 4a). The UV-vis spectrum of 4a in CH3CN/H2O (4:1, v/v) showed two sharp peaks at 250 and 290 nm. Upon addition of SCN−, F−, HSO4−, and CN−, a new peaks for 4a were observed at 515, 525, 530, and 540 nm, respectively (Fig. 4b). Although a red-shift occurred as a result of interaction of 4a with anions, 4a was not selective. Different solvents and pH systems were explored, but in each case, there was no selectivity for a specific anion. It was determined that the two sharp characteristic peaks of 4b shifted from 246/293 nm to 248/294 nm (shift to red) resulted from the interaction of 4b with HSO4− ion, and a new interaction absorption peak formed at 520 nm (Fig. 4c). It was noted that 4c showed similar behaviors in CH3CN/H2O (4:1, v/v), and the interaction with HSO4− led to a decrease in sharp bands at 245/289 nm and a shift to 237/272 nm (blue-shift). A further peak formed at 505 nm due to the interaction (Fig. 4d). A series of control studies with the naked eye and UV-vis showed that there was no color change observed for anions in solutions containing only MeCN.
 | ||
| Fig. 4 Change in the UV-vis absorption spectrum of ligands [3l (a), 4a (b), 4b (c) and 4c (d); (1 × 10−5 M)] in MeCN (a) or MeCN/H2O (4:1, v/v) (b–d) solution upon additions of anions (F−, Cl−, Br−, I−, HSO4−, AcO−, CN− and SCN−; tetrabutyl ammonium salts, 5 × 10−5 M). | ||
Following these results, UV-vis and 1H NMR spectrophotometric titrations were conducted in order to understand the binding phenomena of receptors with F− and HSO4−. First, interaction of 3l and F− ions was carried out in a concentration of [Bu4N]F from 0 to 100 equivalents. It was observed that the sharp peaks at 226/273 nm of the receptor decreased upon interaction with 2 equivalent of F−, and they shifted to 270/320 nm upon increasing [Bu4N]F concentration. Furthermore, a new band formed at 573 nm, and its intensity increased as the [Bu4N]F concentration increased. A new peak at 573 nm and a very radical color change were attributed to the exchange of N–H proton of indole unit, which was caused by the interaction with F− (Fig. 5a and b). The titrations of bis(2-indolyl)methanes 4b (1 × 10−5 M) and 4c (1 × 10−5 M) with HSO4− were realized with increasing concentration of [Bu4N]HSO4 from 0 to 100 equivalent. The intensity of the sharp peaks at 246/293 nm of 4b increased upon the addition of 1 equivalent of [Bu4N]HSO4, and a new red-shifted peak was observed at 520 nm (Fig. 6a and b). The interaction of 4c with 1 equivalent of HSO4− anion resulted in a blue-shift and the formation of an additional new band at 505 nm (Fig. 7a and b). It should be noted that fluorescence studies on the receptor–anion interactions did not show any fluorescence emission.
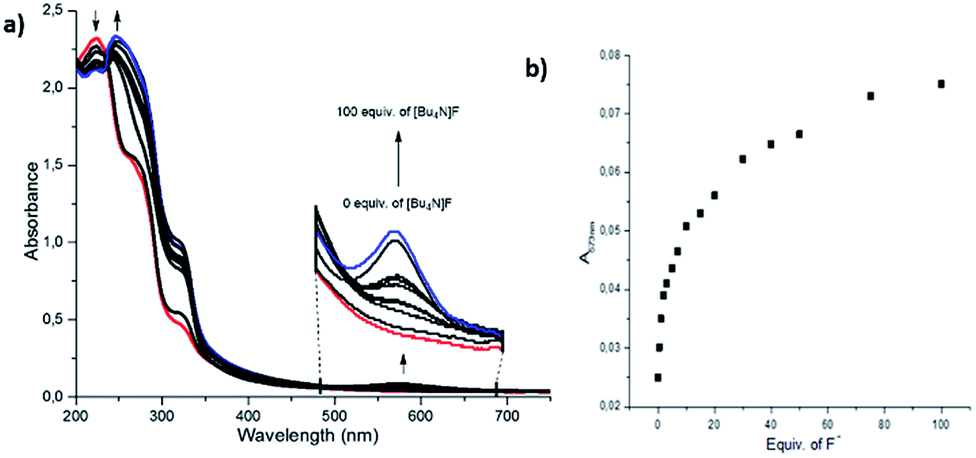 | ||
| Fig. 5 (a) UV-vis titration of the sensor 3l (1 × 10−5 M) in CH3CN solution with standard solution of [Bu4N]F, (b) absorption of sensor 3l at 573 nm vs. equivalent of fluoride anion. | ||
 | ||
| Fig. 6 (a) UV-vis titration of the sensor 4b (1 × 10−5 M) in CH3CN solution with standard solution of [Bu4N]HSO4, (b) absorption of sensor 4b at 520 nm vs. equivalent of bisulfate anion. | ||
 | ||
| Fig. 7 (a) UV-vis titration of the sensor 4c (1 × 10−5 M) in CH3CN solution with standard solution of [Bu4N]HSO4, (b) absorption of sensor 4c at 505 nm vs. equivalent of bisulfate anion. | ||
To obtain insight into the binding ability of the receptors with F− and HSO4−, 1H NMR titration experiments were carried out in CD3CN at 298 K. It is clear that the broad signal of NH proton of the indole moiety disappeared when 2.0 equivalent of [Bu4N]F was added into a solution of 3l. This indicates the formation of strong hydrogen bond between fluoride anion and active NH group (Fig. S21, ESI†). With increasing additional equivalents of F−, the signals of Ha, Hb, Hd (indole) and Hf (thiophene) of 3l showed upfield shifts, whereas the signal of He (indole) shifted toward downfield. To elucidate the binding mode of the bis(2-indolyl)methanes 4b and 4c with increasing HSO4− (from 0 to 10 equivalent), 1H NMR titration spectra were undertaken, which illustrated the characteristic structural changes that occurred upon interaction with [Bu4N]HSO4 in CD3CN at 298 K. While the signal of NH proton of indole of 4b continuously shifted downfield, the signals of Ha, Hb, Hd protons exhibited upfield shifts, and the He proton shifted slightly downfield (Fig. 8). With increasing equivalent of HSO4−, a downfield shift of the NH proton from 9.04 to 9.48 ppm of 4c was especially observed (Fig. S22, ESI†). The evidence above can be ascribed to a hydrogen bonds are responsible for observed chemical shifts upon NH-fluoride ion and NH-bisulfate ion interactions (Fig. 9). Thus, these interactions, which induce polarization of N–H bond, where the partial positive charge creates a downfield shift. Consequently, the increasing electron density on the indole ring promotes an upfield shift of the C–H protons and especially C3–H proton in pyrrole moiety.
 | ||
| Fig. 8 1H NMR (400 MHz) spectra in CD3CN of the sensor 4b (1 × 10−2 M) with presence of [Bu4N]HSO4; (a) 0 equiv. of [Bu4N]HSO4, (b) 1 equiv. of [Bu4N]HSO4, (c) 2 equiv. of [Bu4N]HSO4, (d) 4 equiv. of [Bu4N]HSO4, (e) 6 equiv. of [Bu4N]HSO4, (f) 8 equiv. of [Bu4N]HSO4 and (g) 10 equiv. of [Bu4N]HSO4. | ||
 | ||
| Fig. 9 Plausible intermediates from the interaction between receptors with fluoride and bisulphate anions. | ||
Conclusions
In conclusion, the synthesis studies to create 2-alkylated indoles and bis(2-indolyl)methanes are very limited. Therefore, we have designed and reported an effective and inexpensive method for the synthesis of new 2-alkylated indoles and bis(2-indolyl)methanes from 4,7-dihydroindole (1) using various ketones as the electrophile source of the alkylation and the formation mechanisms are also discussed. Furthermore, studies concerning the chemosensor properties of these 2-alkylated indoles and bis(2-indolyl)methanes were performed, and some of these derivatives show sensing of F− and HSO4− anions via naked eye detection of color changes as well as of absorption signals. The interactions of the receptor and anions were further studied using 1H NMR titrations. Reactions using aldehydes are currently underway.Experimental section
General procedure (GP1) for the synthesis of 2-alkylated indoles
To a solution of 4,7-dihydro-1H-indole (1; 1.0 equiv.) in MeCN (5 mL) was added ketone (2a–p; 1.0 equiv.) and Bi(NO3)3·5H2O (0.1 mmol). The reaction mixture was stirred magnetically in a flask at 80 °C. The reaction was monitored by TLC. After the completion of the reaction, the mixture was diluted with ethyl acetate (30 mL) and washed with water (2 × 50 mL). The organic phase was collected, dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel column chromatograph and isolated compounds were given according to the elution sequence (EtOAc/hexane) in general.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, 1H), 7.30 (d, J = 7.7 Hz, CH, 1H), 7.13–7.04 (m, CH, 2H), 6.23 (s, CH, 1H), 2.75–2.69 (m, CH, 1H), 2.10–2.07 (m, CH2, 2H), 1.87–1.74 (m, CH2, 2H), 1.54–1.29 (m, CH2, 6H); 13C NMR (100 MHz, CDCl3): δ 145.3, 135.7, 128.8, 121.1, 120.1, 119.8, 110.5, 97.7, 37.5, 29.9, 26.5, 26.3; IR (KBr, cm−1): 3335, 2921, 2871, 1624, 1452, 1431, 1397, 1356, 1163, 1122, 908, 858, 811; anal. calcd for C14H17N: C, 84.37; H, 8.60; N, 7.03; found: C, 84.32; H, 8.10; N, 7.27; TLC: Rf = 0.72 (EtOAc/hexane (5%), 254 nm).
CH, 1H), 7.27–7.26 (m, CH, 1H), 7.25 (d, J = 1.1 Hz, CH, 1H), 7.15–7.06 (m, CH, 2H), 6.98–6.96 (m, CH, 1H), 6.91–6.90 (m, CH, 1H), 6.41 (s, CH, 1H), 4.59–4.54 (m, CH, 1H), 1.80 (d, J = 7.0 Hz, CH3, 3H); 13C NMR (100 MHz, CDCl3): δ 148.4, 142.5, 136.2, 128.7, 127.0, 124.4, 124.3, 121.8, 120.5, 120.0, 110.8, 99.4, 34.8, 22.5; IR (KBr, cm−1): 3082, 3028, 2972, 2839, 1605, 1479, 1452, 1384, 1332, 1309, 1256, 1232, 1184, 1023, 882, 824; anal. calcd for C14H13NS: C, 73.97; H, 5.76; N, 6.16; S, 14.11, found: C, 73.88; H, 5.72; N, 6.24; S, 14.13; TLC: Rf = 0.52 (EtOAc/hexane (15%), 254 nm).
CH, 1H), 7.30 (d, J = 7.7 Hz, CH, 1H), 7.13–7.04 (m, CH, 2H), 6.23 (s, CH, 1H), 2.75–2.69 (m, CH, 1H), 2.10–2.07 (m, CH2, 2H), 1.87–1.74 (m, CH2, 2H), 1.54–1.29 (m, CH2, 6H); 13C NMR (100 MHz, CDCl3): δ 145.3, 135.7, 128.8, 121.1, 120.1, 119.8, 110.5, 97.7, 37.5, 29.9, 26.5, 26.3; IR (KBr, cm−1): 3335, 2921, 2871, 1624, 1452, 1431, 1397, 1356, 1163, 1122, 908, 858, 811; anal. calcd for C14H17N: C, 84.37; H, 8.60; N, 7.03; found: C, 84.32; H, 8.10; N, 7.27; TLC: Rf = 0.72 (EtOAc/hexane (5%), 254 nm).
CH, 1H), 7.27–7.26 (m, CH, 1H), 7.25 (d, J = 1.1 Hz, CH, 1H), 7.15–7.06 (m, CH, 2H), 6.98–6.96 (m, CH, 1H), 6.91–6.90 (m, CH, 1H), 6.41 (s, CH, 1H), 4.59–4.54 (m, CH, 1H), 1.80 (d, J = 7.0 Hz, CH3, 3H); 13C NMR (100 MHz, CDCl3): δ 148.4, 142.5, 136.2, 128.7, 127.0, 124.4, 124.3, 121.8, 120.5, 120.0, 110.8, 99.4, 34.8, 22.5; IR (KBr, cm−1): 3082, 3028, 2972, 2839, 1605, 1479, 1452, 1384, 1332, 1309, 1256, 1232, 1184, 1023, 882, 824; anal. calcd for C14H13NS: C, 73.97; H, 5.76; N, 6.16; S, 14.11, found: C, 73.88; H, 5.72; N, 6.24; S, 14.13; TLC: Rf = 0.52 (EtOAc/hexane (15%), 254 nm).
General procedure (GP2) for the synthesis of 2,2′-bis(indolyl)methanes
To a solution of 4,7-dihydro-1H-indole (1; 2.0 equiv.) in MeCN (5 mL) was added ketone (2a–p; 1.0 equiv.) and Bi(NO3)3·5H2O (0.1 mmol). The reaction mixture was stirred magnetically in a flask at 80 °C. The reaction was monitored by TLC. After the completion of the reaction, the mixture was diluted with ethyl acetate (30 mL) and washed with water (2 × 50 mL). The organic phase was collected, dried over Na2SO4, filtered and concentrated. The crude product was dissolved in CH2Cl2 (15 mL) and p-benzoquinone (2.0 equiv.) was added. The mixture was stirred at the room temperature for overnight. After completion of the reaction, the solvent was evaporated and the crude product was dissolved with ethyl acetate (30 mL) and the organic phase was washed with NaOH (2 N, 2 × 30 mL), brine (30 mL), and dried over Na2SO4. The crude product was purified by silica gel column chromatograph and isolated compounds were given according to elution sequence (EtOAc/hexane) in general.CH, 2H), 7.20 (d, J = 7.9 Hz, CH, 2H), 7.13–7.07 (m, CH, 4H), 6.55 (s, CH, 2H), 2.34–2.31 (m, CH2, 4H), 1.72–1.67 (m, CH2, 4H), 1.57–1.55 (m, CH2, 2H); 13C NMR (100 MHz, CDCl3): δ 144.1, 136.1, 128.5, 121.9, 120.4, 120.0, 111.0, 99.5, 41.0, 36.8, 26.2, 23.0; IR (KBr, cm−1): 3039, 2968, 2928, 2109, 1509, 1464, 1451, 1305, 1228, 1163, 1122, 1084, 979, 908, 870, 811; anal. calcd for C22H22N2: C, 84.04; H, 7.05; N, 8.91, found: C, 84.06; H, 7.01; N, 8.87; TLC: Rf = 0.29 (EtOAc/hexane (15%), 254 nm).
Acknowledgements
We are grateful to the Department of Chemistry and Atatürk University and Bingöl University for financial support for this work.Notes and references
- (a) T. Wiggins, S. Kumar, S. R. Markar, S. Antonowicz and G. B. Hanna, Cancer Epidemiol., Biomarkers Prev., 2015, 24, 32–33 CrossRef CAS PubMed; (b) A. Carbone, B. Parrino, G. D. Vita, A. Attanzio, V. Spano, A. Montalbano and G. Cirrincione, Mar. Drugs, 2015, 13, 460–492 CrossRef PubMed; (c) B. D. Horning and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 135, 6442–6445 CrossRef PubMed.
- (a) M. Ruiz, P. L. Alvarado and J. C. Menendez, Eur. J. Org. Chem., 2013, 14, 2802–2812 CrossRef; (b) R. He, B. C. Wang, L. C. Zhu and D. F. Liu, Chem. Res. Chin. Univ., 2013, 34(12), 2772–2777 CAS; (c) R. B. Williams, J. F. Hu, K. M. Olson, V. L. Norman, M. G. Goering, M. O. Johnson and C. M. Starks, J. Nat. Prod., 2010, 73, 1008–1011 CrossRef CAS PubMed; (d) D. Shan, Y. Gao and Y. X. Jia, Angew. Chem., Int. Ed., 2013, 52, 4902–4905 CrossRef CAS PubMed.
- (a) A. D. Martin, A. B. Robinson, A. F. Mason, J. P. Wojciechowski and P. Thordarson, Chem. Commun., 2014, 50, 15541–15544 RSC; (b) P. C. Xue, B. Q. Yao, P. P. Wang, J. B. Sun, Z. Q. Zhang and R. Lu, RSC Adv., 2014, 4, 61548 RSC; (c) L. Maity, H. S. Parmar, D. B. Rasale and A. K. Das, J. Mater. Chem. B, 2014, 2, 5272–5279 RSC; (d) R. Martinez, A. Espinosa, A. Tarraga and P. Molina, Tetrahedron, 2008, 64, 2184–2191 CrossRef CAS.
- (a) M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC; (b) C. L. Zhu, Z. B. Liu, G. Y. Chen, K. Zhang and H. F. Ding, Angew. Chem., Int. Ed., 2015, 54, 879–882 CrossRef CAS PubMed; (c) D. Beukeaw, K. Udomsasporn and S. Yotphan, J. Org. Chem., 2015, 80, 3447–3454 CrossRef CAS PubMed.
- (a) A. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef PubMed; (b) W. Tan, X. Li, Y. X. Gong, M. D. Ge and F. Shi, Chem. Commun., 2014, 50, 15901–15904 RSC; (c) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2009, 110, 2250–2293 CrossRef PubMed; (d) P. Praveen, P. S. Parameswaran and M. S. Majik, Synthesis, 2015, 47, 1827–1837 CrossRef CAS.
- (a) H. Kilic, S. Bayindir, E. Erdogan and N. Saracoglu, Tetrahedron, 2012, 68, 5619–5630 CrossRef CAS; (b) S. Bayindir, E. Erdogan, H. Kilic and N. Saracoglu, Synlett, 2010, 2010, 1455–1458 CrossRef; (c) W. B. Liu, X. Zhang, L. X. Dai and S. L. You, Angew. Chem., Int. Ed., 2012, 51, 5183–5187 CrossRef CAS PubMed; (d) S. Chen, Y. Liao, F. Zhao, H. Qi, S. Liu and G. J. Deng, Org. Lett., 2014, 16, 1618–1621 CrossRef CAS PubMed; (e) G. M. Shelke, V. K. Rao, R. K. Tiwari, B. S. Chhikara, K. Parang and A. Kumar, RSC Adv., 2013, 3, 22346–22352 RSC; (f) J. Tummatorn, M. P. Gleeson, S. Krajangsri, C. Thongsornkleeb and S. Ruchirawat, RSC Adv., 2014, 4, 20048–20052 RSC; (g) C. Y. Liu and F. S. Han, Chem. Commun., 2015, 51, 11844–11847 RSC.
- (a) S. Y. Wang and S. J. Ji, Tetrahedron, 2006, 62, 1527–1535 CrossRef CAS; (b) A. Carbone, B. Parrino, P. Barraja, V. Spano, G. Cirrincione, P. Diana and H. H. Fiebig, Mar. Drugs, 2013, 11, 643–654 CrossRef CAS PubMed; (c) S. Alvarado, B. F. Roberts, A. E. Wright and D. Chakrabarti, Antimicrob. Agents Chemother., 2013, 57, 2362–2364 CrossRef CAS PubMed.
- (a) Y. Zhang, X. Chen, J. Liang and Z. C. Shang, Synth. Commun., 2011, 41, 2446–2454 CrossRef CAS; (b) T. Abe, S. Nakamura, R. Yanada, T. Choshi, S. Hibino and M. Ishikura, Org. Lett., 2013, 15, 3622–3625 CrossRef CAS PubMed; (c) T. P. Pathak, J. G. Osiak, R. M. Vaden, B. E. Welm and M. S. Sigman, Tetrahedron, 2012, 68, 5203–5208 CrossRef CAS PubMed; (d) L. Iglesias, C. Aguilar, D. Bandyopadhyay and B. K. Banik, Synth. Commun., 2010, 40, 3678–3682 CrossRef CAS; (e) H. Veisi, A. Sedrpoushan, M. A. Zolfigol and F. Mohanazadeh, J. Heterocycl. Chem., 2011, 48, 1448–1453 CrossRef CAS; (f) N. Seyedi, K. Saidi and H. Khabazzadeh, Synth. Commun., 2009, 39, 1864–1870 CrossRef CAS.
- D. X. Shu, G. N. W. McPherson, W. Z. Song and W. P. Tang, Org. Lett., 2013, 15, 4162–4165 CrossRef CAS PubMed.
- (a) L. Hong, C. X. Liu, W. S. Sun, L. Wang, K. Wong and R. Wang, Org. Lett., 2009, 11, 2177–2180 CrossRef CAS PubMed; (b) B. Xiao, X. C. Dong and L. Zhou, Org. Biomol. Chem., 2013, 11, 1490–1497 RSC; (c) B. W. Gong, J. J. Shi, X. W. Wang, Y. N. Yan, Q. Li, Y. Q. Meng and W. Yi, Adv. Synth. Catal., 2014, 356, 137–143 CrossRef CAS; (d) K. Dittmann and U. Pindur, Arch. Pharm., 1985, 318, 340–350 CrossRef CAS.
- (a) X. He, S. Hu, K. Liu, Y. Gou, J. Xu and S. Shao, Org. Lett., 2006, 8, 333–336 CrossRef CAS PubMed; (b) L. Wang, X. He, Y. Guo, J. Xu and S. Shao, Org. Biomol. Chem., 2011, 9, 752–757 RSC.
- (a) O. Aydin, H. Kilic, S. Bayindir, E. Erdogan and N. Saracoglu, J. Heterocycl. Chem., 2015, 52, 1540–1553 CrossRef CAS; (b) S. Bayindir, E. Erdogan, H. Kilic, O. Aydin and N. Saracoglu, J. Heterocycl. Chem., 2015, 52, 1589–1594 CrossRef CAS.
- (a) C. S. Cho, J. H. Kim, T. J. Kim and S. C. Shim, J. Chem. Res., 2004, 9, 630–631 Search PubMed; (b) L. Zhou, Y. Shi, Q. Xiao, Y. Z. Liu, F. Ye, Y. Zhang and J. B. Wang, Org. Lett., 2011, 13, 968–971 CrossRef CAS PubMed; (c) T. B. Xiao, X. C. Dong and L. Zhou, Org. Biomol. Chem., 2013, 11, 1490–1497 RSC; (d) Y. Wang, L. Ye and L. Zhang, Chem. Commun., 2011, 47, 7815–7817 RSC; (e) I. Ambrogio, S. Cacchi, G. Fabrizi and A. Prastaro, Tetrahedron, 2009, 65, 8916–8929 CrossRef CAS; (f) M. Alajarin, A. Vidal and M. M. Ortin, Tetrahedron Lett., 2003, 44, 3027–3030 CrossRef CAS; (g) J. Gao, Y. Shao, J. Zhu, J. Zhu, H. Mao, X. Wang and X. Lv, J. Org. Chem., 2014, 79, 9000–9008 CrossRef CAS PubMed; (h) F. Zhao, D. Zhang, Y. Nian, L. Zhang, W. Yang and H. Liu, Org. Lett., 2014, 16, 5124–5127 CrossRef CAS PubMed; (i) B. Z. Lu, H. X. Wei, Y. Zhang, W. Zhao, M. Dufour, G. Li, V. Farina and C. H. Senanayake, J. Org. Chem., 2013, 78, 4558–4562 CrossRef CAS PubMed; (j) A. Gogoi, S. Guin, S. K. Rout and B. K. Patel, Org. Lett., 2013, 15, 1802–1805 CrossRef CAS PubMed; (k) B. Prasad, R. Adepu, S. Sandra, D. Rambabu, G. R. Krishna, C. M. Reddy, G. S. Deora, P. Misraa and M. Pal, Chem. Commun., 2012, 48, 10434–10436 RSC.
- (a) M. McLaughlin, M. Palucki and I. W. Davies, Org. Lett., 2006, 8, 3307–3310 CrossRef CAS PubMed; (b) K. C. Majumdar, S. Samanta and B. Chattopadhyay, Tetrahedron Lett., 2008, 49, 7213–7216 CrossRef CAS; (c) N. C. de Mattos, S. Alatorre-Santamaria, V. Gotor-Fernandez and V. Gotor, Synthesis, 2007, 2007, 2149–2152 CrossRef; (d) N. Sakai, K. Annaka, A. Fujita, A. Sato and T. Konakahara, J. Org. Chem., 2008, 73, 4160–4165 CrossRef CAS PubMed; (e) M. Layek, Y. S. Kumar, A. Islam, R. Karavarapu, A. Sengupta, D. Halder, K. Mukkantib and M. Pal, MedChemComm, 2011, 2, 478–485 RSC; (f) M. Amjad and D. W. Knight, Tetrahedron Lett., 2004, 45, 539–541 CrossRef CAS.
- P. J. Das and J. Das, Tetrahedron Lett., 2012, 53, 4718–4720 CrossRef CAS.
- H. Cavdar and N. Saracoglu, Tetrahedron, 2005, 61, 2401–2405 CrossRef CAS.
- H. Cavdar and N. Saracoglu, J. Org. Chem., 2006, 71, 7793–7799 CrossRef CAS PubMed.
- H. Kilic, S. Bayindir and N. Saracoglu, Curr. Org. Chem., 2014, 11, 167–181 CAS.
- (a) R. M. F. Batista, E. Oliveira, C. Nunez, S. P. G. Costa, C. Lodeiro and M. M. M. Raposo, J. Phys. Org. Chem., 2009, 22, 362–366 CrossRef CAS; (b) P. A. Gale, Chem. Commun., 2008, 4525–4540 RSC.
- (a) P. Bose and P. Ghosh, Chem. Commun., 2010, 46, 2962–2964 RSC; (b) P. Bose, B. N. Ahamed and P. Ghosh, Org. Biomol. Chem., 2011, 9, 1972–1979 RSC; (c) R. Martinez, A. Espinosa, A. Tarraga and P. Molina, Tetrahedron, 2008, 64, 2184–2191 CrossRef CAS; (d) R. M. F. Batista, S. P. G. Costa, R. M. P. Silva, N. E. M. Lima and M. M. M. Raposo, Dyes Pigm., 2014, 102, 293–300 CrossRef CAS.
- P. A. Gale, Chem. Soc. Rev., 2010, 39, 3746–3771 RSC.
- J. R. Hiscock, C. Caltagirone, M. E. Light, M. B. Hursthouse and P. A. Gale, Org. Biomol. Chem., 2009, 7, 1781–1783 CAS.
- P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215–220 RSC.
- D. Makuc, M. Lenarcic, G. W. Bates, P. A. Gale and J. Plavec, Org. Biomol. Chem., 2009, 7, 3505–3511 CAS.
- (a) L. T. Wang, X. M. He, Y. Guo, J. A. Xu and S. J. Shao, Org. Biomol. Chem., 2011, 9, 752–757 RSC; (b) P. A. Gale, S. E. G. Garrido and J. Garric, Chem. Soc. Rev., 2008, 37, 151–190 RSC; (c) K. Ghosh, D. Kar, S. Joardar, A. Samander and A. R. K. Bukhsh, RSC Adv., 2014, 4, 11590–11611 RSC; (d) M. Nishiki, W. Oi and K. J. Ito, J. Inclusion Phenom. Macrocyclic Chem., 2008, 61, 61–69 CrossRef CAS; (e) P. Bose and P. Ghosh, Chem. Commun., 2010, 46, 2962–2964 RSC; (f) M. G. Murali, K. A. Vishnumurthy, S. Seethamraju and P. C. Ramamurthy, RSC Adv., 2014, 4, 20592–20598 RSC; (g) H. Khanmohammadi and K. Rezaeian, RSC Adv., 2014, 4, 1032–1038 RSC; (h) C. F. Wan, S. T. Yang, H. Y. Lin, Y. J. Chang and A. T. Wu, Luminescence, 2014, 29, 500–503 CrossRef CAS PubMed; (i) R. Pegu, R. Mandal, A. K. Guha and S. Pratihar, New J. Chem., 2015, 39, 5984–5990 RSC; (j) W. C. Lin, J. W. Hu and K. Y. Chen, Anal. Chim. Acta, 2015, 893, 91–100 CrossRef CAS PubMed; (k) D. Sain, C. Kumari, A. Kumar and S. Dey, Supramol. Chem., 2016, 28, 239–248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, spectroscopic data, NMR (1H and 13C). See DOI: 10.1039/c6ra16192h |
| This journal is © The Royal Society of Chemistry 2016 |