
Highly efficient and scalable chemoenzymatic syntheses of (R)- and (S)-lactaldehydes†
M. A. K.
Vogel
a,
H.
Burger
a,
N.
Schläger
a,
R.
Meier
a,
B.
Schönenberger
a,
T.
Bisschops
b and
R.
Wohlgemuth
*a
aSigma-Aldrich, Industriestrasse 25, CH-9470 Buchs, Switzerland. E-mail: roland.wohlgemuth@sial.com; Fax: +41 81 7552840; Tel: +41 81 7552640
bSigma-Aldrich, Riedstrasse 2, 89555 Steinheim, Germany
First published on 6th November 2015
Abstract
Biocatalytic asymmetric reductions have been key steps in the enantioselective reduction of 1,1-dimethoxy-2-propanone, catalyzed by suitable ketoreductases, to enantiomerically pure (S)- and (R)-1,1-dimethoxy-2-propanols, obtained in ≥99.9% ee and excellent yield. Removal of the protecting group gave the (S)- and (R)-lactaldehydes in high yield and excellent enantiomeric and chemical purity.
Lactaldehyde has been known since the 19th century and its two enantiomers (R)- and (S)-lactaldehydes (1a and 1b) occur in a variety of metabolic pathways (Fig. 1). (S)-Lactaldehyde 1b is a metabolite of the fructose and mannose metabolism, pyruvate metabolism, propanoate metabolism and microbial metabolism in diverse environments. (R)-Lactaldehyde 1a is a substrate for aldose reductase and glyoxylate reductase and a metabolite of the pyruvate metabolism.
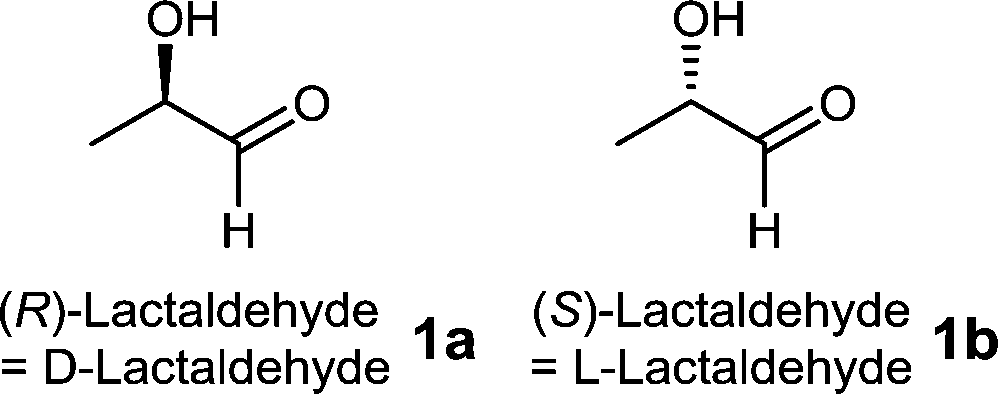 | ||
| Fig. 1 (R)- and (S)-lactaldehydes 1a and 1b. | ||
Although the lactaldehyde metabolism was investigated in yeast cells 80 years ago,1 it was only much later that lactaldehyde dehydrogenases were purified and characterized2 and the crystal structure of lactaldehyde dehydrogenase from E. coli has been determined.3 Both (R)- and (S)-lactaldehydes 1a and b have also been of interest in metabolic engineering of 1,2-propanediol pathways.4
Although the first preparations of racemic5 and later on enantiomerically enriched or pure lactaldehydes6 date back in part more than a century, their isolation in a well-defined and characterized form still remains challenging. Before its first description, even the non-existence of lactaldehyde has been postulated.7 This phenomenon stems from its inherent tendency to dimerise as a neat compound and in solution, as well as from the fact that the dimer can form diastereoisomers.8 Optical rotation values for instance have rarely been published and vary with respect to their absolute values and signs of rotation, which depend on the measurement conditions.9 Therefore, lactaldehydes have often been isolated and characterized as derivatives, such as hydrazones,10 or in their O-protected forms.11 The non-derivatized lactaldehyde enantiomers have been analyzed by enzymatic assays,12 but neither by direct enantiomer separation nor spectroscopic characterization. Alternatively, lactaldehydes have not been isolated but have been used in situ for a subsequent synthetic step.13
Although enantiomerically pure or enriched lactaldehydes have been prepared in several ways, none of the described procedures was found useful for efficient and scalable industrial syntheses. Rosenmund reduction of the acid chloride of L-lactic acid gave (S)-lactaldehyde 1b in a rather low yield due to considerable losses suffered during the tedious work-up.14 (R)-Lactaldehyde 1a is hardly accessible by this pathway owing to the high price of D-lactic acid. Oxidative cleavage of L- or D-threonine followed by a quite lengthy work-up procedure provided both lactaldehydes 1a and 1b in presumably high enantiopurities6 but in a yield below 50% and the disadvantage of the high price of D-threonine. Yeast or horse liver alcohol dehydrogenase as well as a commercially unavailable carbonyl reductase from Candida parapsilosis were applied in the reduction of the dimethylacetal of methylglyoxal (2) to the corresponding (S)-alcohol (3b) in its enantiopure form.15 However, the conversion data given remained somewhat contradictory, and neither the isolation of 3b nor unambiguous ee values were reported. The (R)-alcohol (3a) could be obtained with low enantiomeric excess by application of an ADH from Thermoanaerobium brockii. Alternatively, starting from ethyl L-lactate, L-lactaldehyde 1b was synthesized in a three-step procedure.16 Since the product was never isolated and immediately processed further, the yield stated is of limited significance and the removal of the silyl protecting group using aqueous HF in the last step did not work satisfactorily in our hands. In addition, several enantioselective hydrogenations have been described.17 However, all hydrogenation methods described lack the required high enantiomeric excess and rely on more elaborate techniques, e.g. preparation of commercially unavailable catalysts or handling of hydrogen gas. Enantioselective reduction of ketone 2 to 3b is reported with chiral borohydrides, with an ee of 87% for the (S)-isomer.18 The use of chiral oxazaborolidines in our lab gave compounds 3a and 3b with an ee of only 37%.19 However, all of the methods described above are not industrially viable, because they are neither scalable nor provide the required high chemical and enantiomeric purity.
Thus, our envisaged route had to meet several demanding goals which were not achieved beforehand. Both (R)- and (S)-lactaldehydes, 1a and b, should be accessible by a single methodology, with purity and ee values of >99.0%. Furthermore, the compounds should be available in variable amounts, by an environmentally benign and robust to scale methodology. Over the past two decades, an increasing number of chemoenzymatic methods have been published, benefitting from more efficient enzymatic transformations and allowing straightforward access to complex chiral organic compounds which are otherwise difficult to obtain.20 Consequently, we decided on a two-step chemoenzymatic procedure starting with 1,1-dimethoxy-2-propanone (2) as a cheap and readily available precursor (Scheme 1). The proposed route offers the additional advantage of making also valuable building blocks (R)- and (S)-1,1-dimethoxy-propanols 3a and b available to us.
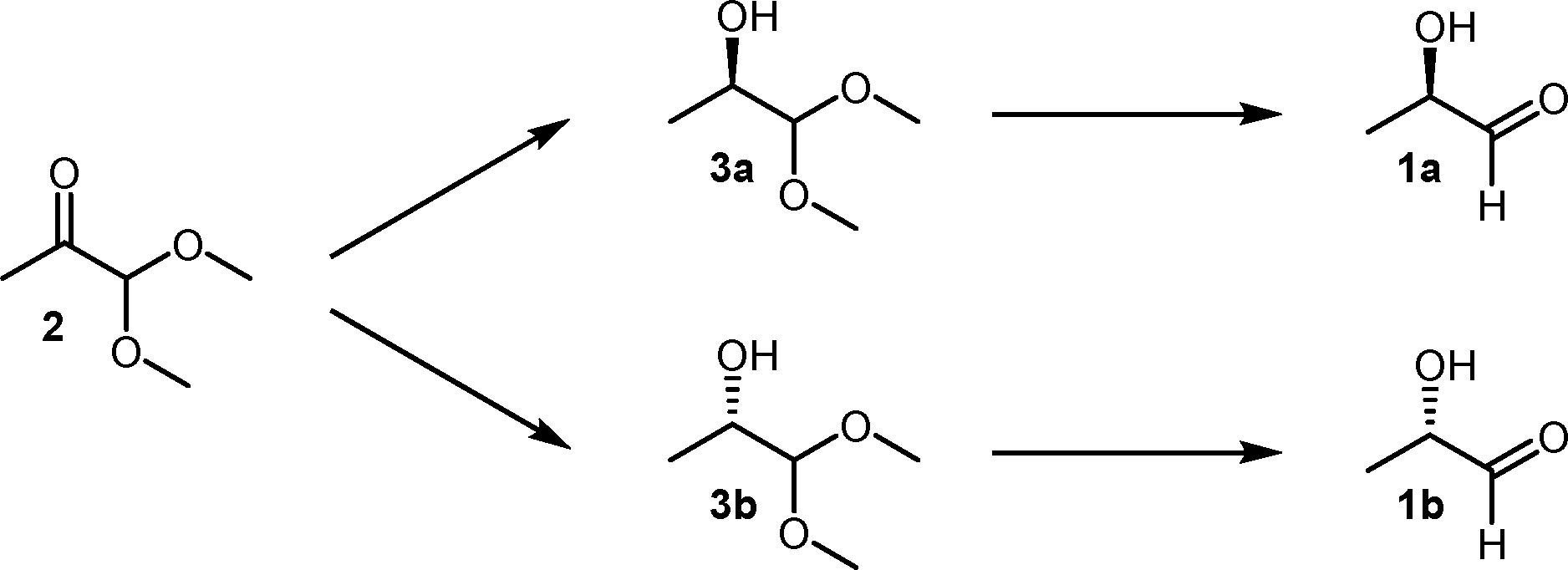 | ||
| Scheme 1 Envisaged route to enantiomerically pure lactaldehydes 1a and 1b. | ||
The enzymatic reduction of a prochiral carbonyl moiety by carbonyl reductases (ketoreductase, KRED) has been proven to yield chiral alcohols efficiently in both (R)- and (S)-configurations with high enantiomeric or diastereomeric excess.21 Therefore, we decided to identify suitably evolved KREDs for ketone 2 reduction. As a cofactor, either NADH or NADPH is required and throughout the catalytic cycle NAD+ or NADP+ is formed. Recycling of the cofactor is a prerequisite and can be performed by various methods.22 Common recycling systems are either by oxidation of isopropanol by the same enzyme or by GDH per conversion of glucose into gluconic acid (Scheme 2).
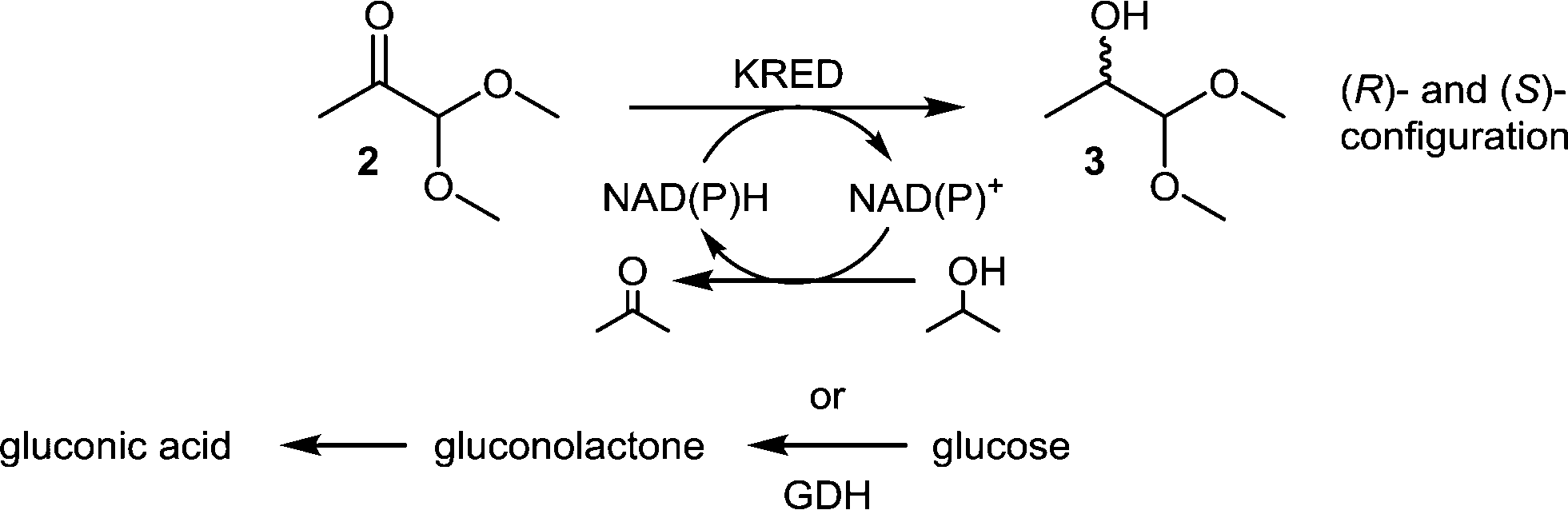 | ||
| Scheme 2 General scheme of a KRED-catalysed reduction with 2 common recycling systems. | ||
Suitable biocatalysts for this reaction have been identified by an enzyme screening using 24 ketoreductases (Codex® KRED Screening Kit, for details see the Experimental section). All screening reactions were conveniently performed with 10 mL of the reaction solution in 40 mL glass vials, equipped with cross-shaped stirrer bars and all reaction components were pipetted sequentially from freshly prepared stock solutions, allowing a fast but accurate handling. From this experiment, a total of 10 enzymes were identified, providing practically full conversion and ee values of >99.0%. 8 enzymes were subjected subsequently to a stepwise increase of substrate loading to identify potential obstacles such as enzyme inhibition and the lack of stability towards the substrate or the product. In total, the substrate load was increased to 150 g L−1 (≈1.25 mol L−1), with all of the hit enzymes performing practically identically. No equilibrium issues were encountered throughout this systematical investigation, and for simplicity reasons, an IPA recycling system was preferred over an enzyme coupled recycling system (see Scheme 2). In a second experiment, the NADP loading was decreased stepwise. In a 3 vial screening, 0.8 g L−1, 0.4 g L−1 and 0.1 g L−1 NADPH were tested. All three reactions showed a similar rate and practically full conversion. Interestingly, stirring of the reaction solution was not even required. Both a sample of a non-agitated mixture and the reaction solution stirred at 75 rpm reached 99.8% conversion after 25 h, indicating a diffusion-controlled reaction.
The results above were taken into a first up-scaling of the reaction. In a 3 L batch, the first multigram amounts of products 3a and b were produced (see Table 1 & Fig. 2). However, foreseeing room for further improvement, a systematic optimization of the catalytic system using DoE (Design of Experiment) was initiated. In total, 4 critical parameters were identified and investigated in a 2 level factorial experiment. The temperature was varied between 30 °C and 40 °C, the NADPH loading from 0.01 g L−1 to 0.1 g L−1, the substrate loading from 150 g L−1 to 250 g L−1 and the phosphate buffer concentration from 0.04 molar to 0.15 molar, targeting a conversion of >99.0% within 30 hours. Thorough evaluation of the data set provided us the optimum process conditions, allowing us to convert 250 g L−1 substrate (2.12 mol L−1) at 40 °C and 0.15 molar buffer concentration. The NADP loading was simultaneously reduced to 0.05 g L−1, corresponding to a NADP loading of 0.003 mol% with respect to the starting material. The performance of the optimized parameter set was verified in a 1 L production run (Fig. 2). Lower NADPH loadings resulted in a reduced rate of conversion. The reaction proceeded in a very clean fashion and reached 99.2% conversion after 28 h, with an ee of >99.9%.
| Parameter | Screening | Target | First phase developmenta | Second phase development (DoE)b |
|---|---|---|---|---|
| a 3 L reaction volume, T = 30 °C. b 1 L reaction volume, T = 40 °C. c With IPA as the major impurity. | ||||
| Substrate | 6 g L−1 | 150 g L−1 | 150 g L−1 | 250 g L−1 |
| KRED | 1 g L−1 | 1 g L−1 | 1 g L−1 | 1 g L−1 |
| NADP | 0.8 g L−1 | 0.1 g L−1 | 0.1 g L−1 | 0.05 g L−1 |
| Ee (%) | >99.0% | ≥99.9% | ≥99.9% | |
| Conv. (%) | >99.0% | 99.6% (29 h) | 99.2% (28 h) | |
| Purity (%) | >99.0% | ≥99.5%c | ≥99.3%c | |
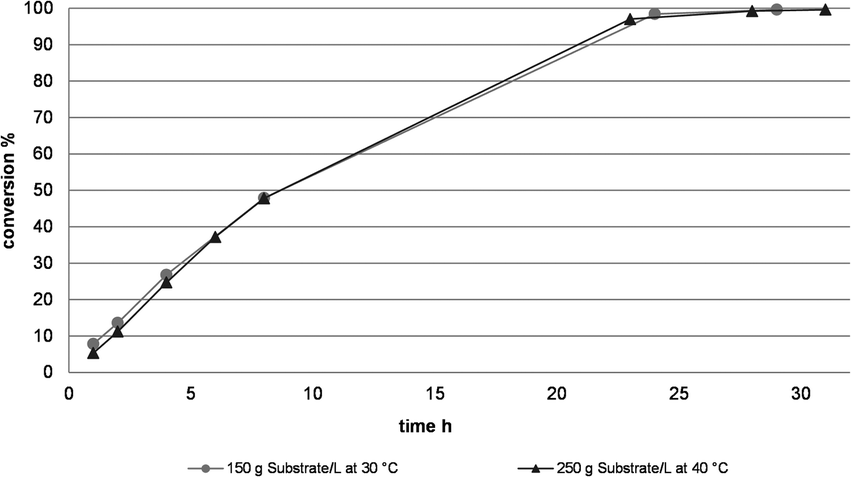 | ||
| Fig. 2 Conversion profiles for the enzymatic transformation of 150 g L−1 and 250 g L−1 substrate loadings. Conditions: 1.) 3 L reaction volume, 150 g L−1 substrate, 30 °C, 1 g L−1 KRED-P2-G03, 0.1 g L−1 NADPH, 15 vol% IPA, in 0.1 molar phosphate buffer; 2.) 1 L reaction volume, 250 g L−1 substrate, 40 °C, 1 g L−1 KRED-P2-G03, 0.05 g L−1 NADPH, 23 vol% IPA (1.44 equivalents with respect to the starting material), in 0.15 molar phosphate buffer. | ||
Owing to the high water solubility of the product, full extraction of 3a and b into MTBE proved to be ineffective, but indispensable to obtain satisfying yields. To avoid the extensive use of organic solvents, the extraction from the reaction buffer solution vs. a NaCl-saturated buffer solution was compared. In contrast to the reaction solution, a fully NaCl saturated solution allowed almost quantitative removal of the product after 3 extractions (Fig. 3).
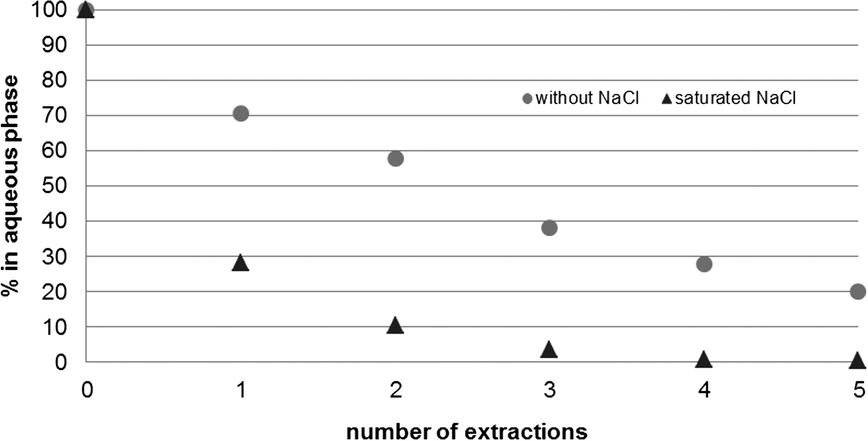 | ||
| Fig. 3 Full extraction of 1,1-dimethoxy-2-propanols 3a and b was achieved by saturating the aqueous reaction solution with NaCl. | ||
On-scale extractions worked well. Depending on the substrate load, a polish filtration to remove enzyme debris might be required. After evaporation of MTBE, products 3a and b were obtained in 98% purity, while the main impurities IPA and MTBE summed up to ca. 2%. Despite careful evaporation of the solvent, slight losses of the products were observed. Therefore, evaporation to just an extent of 6–10 vol% IPA was applied. Final distillation of the product provided us compounds 3a and b in a yield of ≥81% and in excellent purities and stereoselectivity, as highlighted in Table 1.
The homochiral lactaldehydes could easily be prepared by Dowex-H+-catalysed hydrolysis of the corresponding dimethylacetals 3a and 3b, respectively12 (Scheme 3). On prolonged storage, the racemic as well as the homochiral lactaldehydes in aqueous solution were less prone to form complicated NMR spectra than the solvent-free compounds (see the ESI†). Therefore, 1 M aqueous solutions of the lactaldehydes were chosen as preferred forms for long-term stability. The enantiomeric purities of the lactaldehydes have been determined by HPLC after derivatisation to their corresponding 2,4-dinitro-phenylhydrazones using a Chiralcel OD column.23 The absolute configuration of the lactaldehydes and their acetals was confirmed by oxidation (iodine, NaOH) to the corresponding lactic acid enantiomers and comparison of their measured optical rotations with literature values which have been assigned in numerous papers.24
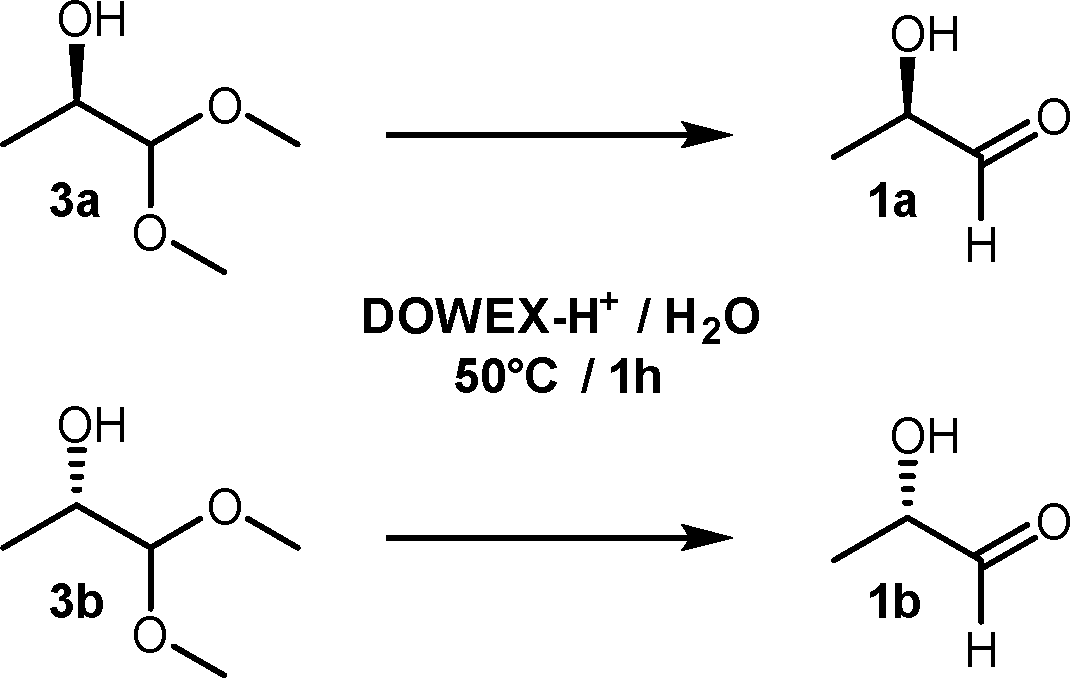 | ||
| Scheme 3 Hydrolysis of lactaldehyde acetals. | ||
Conclusions
The synthesis of (R)- and (S)-1,1-dimethoxy-propanols 3a and b and stable (R)- and (S)-lactaldehydes 1a and b demonstrates the usefulness of modern biocatalysis for fast and efficient development of both highly productive and green chemical processes. This performance has been enabled by a thorough retrosynthetic analysis, taking specifically into account the availability of the evolved enzymes and the use of DoE. In addition, the development of efficient analytical methods turned out to be crucial for purity and stability investigations of the lactaldehyde enantiomers 1a and b. As a consequence, this scalable procedure provides all compounds in any amount desired, from the gram to kilogram scale. The high isolated yields and excellent chemical and enantiomeric purities of each single compound have not been reported so far. In addition, an unambiguous characterization of both lactaldehydes 1a and b has been realized. The inherent simplicity of this approach allowed the development of this process in only a few weeks' time and is not restricted to structures 1a and b, but opens new opportunities to be seriously considered in the retrosynthetic analysis of other chiral 2-hydroxyaldehyde substructures, and in particular the synthesis of metabolites.25Furthermore, the access to larger amounts of well characterised, homochiral, isolated lactaldehydes now simplifies the application of these valuable synthetic building blocks in aldolase26 and transketolase27 mediated aldol condensations as well as classical chemical reactions.28
Experimental section
Preparation of (R)-1,1-dimethoxy-2-propanol
477 g of 0.15 M KH2PO4 buffer solution was placed in a 2.5 L reaction flask equipped with overhead stirring. 250 g of (2.12 mol) methylglyoxal-1,1-dimethylacetal dissolved in 183 g of IPA (3.04 mol) and 50 mg (0.065 mmol) of NADP+ dissolved in 5 g of 0.15 M KH2PO4 buffer solution were subsequently added to the flask. A solution of 1.02 g of KRED-P2-G03 from Codexis in 50 g of 0.15 M KH2PO4 buffer solution was added to start the reaction, applying gentle stirring at ≤100 rpm. With completed addition, the reaction mixture was heated to 40 °C and stirred for 45 h.‡ To the turbid solution, 1.11 kg of MTBE was added. The organic layer was saturated with 170 g of sodium chloride and stirred for 30 min at 20–30 °C. The reaction mixture was filtered over Celite® and the Celite® washed with 0.15 kg of MTBE. The solution was transferred into a separating funnel. After extraction, the layers were separated and the aqueous layer was further extracted with 3 × 1.11 kg of MTBE. The combined organic layers were dried over 80 g of MgSO4, then the solid was filtered off and washed twice with 74 g of MTBE. The solvents were evaporated carefully under reduced pressure. The crude product was distilled using a 30 cm Vigreux column at 80 °C and 70 mbar to give 212 g of (1.76 mol, 84%) (R)-1,1-dimethoxy-2-propanol as a colorless liquid.Analytical data: GC: 99.3%, ee: 99.9% (measured by chiral stationary phase GC), 1H-NMR (d6-DMSO): δ 4.55 (bs, 1H), 3.98 (d, J = 5.8 Hz, 1H), 3.56 (dq, J = 6.4, 5.8 Hz, 1H), 3.30 (s, 6H), 1.00 (d, J = 6.4 Hz, 3H).
Preparation of D- and L-lactaldehydes from (R)- and (S)-1,1-dimethoxy-2-propanols, respectively
To 45 g of Dowex 50WX8 (H+ form) in 900 ml of deionized H2O, 44.3 g (0.37 mol) of enantiomerically pure 1,1-dimethoxy-2-propanol was added at r.t. with stirring. The mixture was heated to 50 °C, kept for 1 h and then cooled to r.t. After filtration and washing of the ion exchanger on the funnel, the filtrate was concentrated on a rotary evaporator (Tbath 35°, ≥10 mbar, the product is somewhat volatile!) to give 25.9 g (0.34 mol, 92%) of a slightly yellowish oil which was immediately taken up in deionized H2O to give a ~1 M solution.
1H-NMR (600 MHz, D2O): 1.10 (d, J = 6.5 Hz, 3H, H3C(3)), 3.59 (m, 1H, H–C(2)), (H–C(1) covered by H2O peak); 13C-NMR (151 MHz, D2O): 16.47 (H3C(3)), 69.79 (H–C(2)), 92.56 (H–C(1)) (see the ESI†); TLC (silica gel 60, toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dioxane:AcOH = 90:25:4, KMnO4): single spot.
:dioxane:AcOH = 90:25:4, KMnO4): single spot.
ee determination of lactaldehydes
To 500 μl of a ~0.4 M 2,4-dinitrophenylhydrazine phosphoric acid solution, 100 μl of ~1 M lactaldehyde solution was added at room temperature with stirring. After 1 h, 1 ml of deionized H2O was added to the resulting suspension to achieve complete precipitation of the formed hydrazone. After filtration, washing with ~1 ml of deionized H2O and drying at high vacuum/room temperature, the yellow powder was analysed on an HPLC (Daicel Chiralcel OD with 2-propanol/hexane 15:85 as eluent, UV detection at 254/350 nm and racemic lactaldehyde–hydrazone as reference) to show 100% ee (see the ESI†).
Absolute configuration
To 5 mL of a 0.1 M solution of each of the homochiral lactaldehydes, 2 mL of 0.5 M aqueous iodine was added and then 3 mL of 1 M NaOH solution. This yellowish, strongly alkaline solution was stirred for 1 h at room temperature until the almost colorless reaction mixture showed no more lactaldehyde on TLC. Then 1 mL of 1 M H2SO4 was added, followed by 1 mL of aqueous 1 M sodium thiosulfate, resulting in a colorless solution. After 15 minutes of stirring at room temperature, the solution was made alkaline by the addition of 1.5 mL of 1 M NaOH. An optical rotation of −0.046° and +0.051° respectively was measured for this solution, translating into a specific rotation of ~−17° and ~+17°. This meets the literature values for D- and L-lactic acids measured in aqueous NaOH: positive values for D-lactic acid with an R configuration and vice versa.Acknowledgements
We thank Patrick Stocker and Mikael Berthet for analytical method development and GC measurements and Hansjörg Tinner for HPLC analyses. We also thank Fabian Luther for his contribution to the experimental DoE work.Notes and references
- C. Neuberg and A. Vercellone, Biochem. Z., 1935, 279, 140 CAS.
- S. Sridhara and T. T. Wu, J. Biol. Chem., 1969, 244, 5233 Search PubMed; Y. Inoue, K. Watanabe, M. Shimosaka, T. Saikusa, Y. Fukuda, K. Murata and A. Kimura, Eur. J. Biochem., 1985, 153, 243 CrossRef CAS PubMed.
- L. Di Costanzo, G. A. Gomez and D. W. Christianson, J. Mol. Biol., 2007, 366, 481 CrossRef CAS PubMed.
- D. C. Cameron, N. E. Altaras, M. L. Hoffman and A. J. Shaw, Biotechnol. Prog., 1998, 14, 116 CrossRef CAS PubMed.
- A. Wohl, Ber. Dtsch. Chem. Ges., 1908, 41, 3599 CrossRef CAS; R. Dworzak and W. Prodinger, Monatsh. Chem., 1928, 50, 459 CrossRef; H. O. L. Fischer and E. Baer, Helv. Chim. Acta, 1935, 18, 514 CrossRef.
- E. Huff and H. Rudney, J. Biol. Chem., 1959, 234, 1060 CAS; B. Zagalak, P. A. Frey, G. L. Karabatsos and R. H. Abeles, J. Biol. Chem., 1966, 241, 3028 Search PubMed.
- J. U. Nef, Justus Liebigs Ann. Chem., 1904, 335, 247 CrossRef CAS.
- H. Takahashi, Y. Kobayashi and N. Kaneko, Spectrochim. Acta, Part A, 1983, 39, 569 CrossRef.
- C. Zioudrou and P. Chrysochou, Tetrahedron, 1977, 33, 2103 CrossRef CAS.
- L. Hough and J. K. N. Jones, J. Chem. Soc., 1952, 4052 RSC; F. Weygand, H. J. Bestmann, H. Ziemann and E. Klieger, Chem. Ber., 1958, 91, 1043 CrossRef CAS.
- S. K. Massad, L. D. Hawkins and D. C. Baker, J. Org. Chem., 1983, 48, 5180 CrossRef CAS.
- C.-H. Wong, D. G. Drueckhammer and H. M. Sweers, J. Am. Chem. Soc., 1985, 107, 4028 CrossRef CAS.
- C.-H. Wong, R. Alajarin, F. Moris-Varas, O. Blanco and E. Garcia-Junceda, J. Org. Chem., 1995, 60, 7360 CrossRef CAS.
- R. Dworzak and H. Zellner, Monatsh. Chem., 1949, 80, 406 CrossRef CAS.
- J. Peters, H.-P. Brockamp, T. Minuth, M. Grotus, A. Steigel, M.-R. Kula and L. Elling, Tetrahedron: Asymmetry, 1993, 4, 1173 CrossRef CAS; J. Peters, T. Zelinski, T. Minuth and M.-R. Kula, Tetrahedron: Asymmetry, 1993, 4, 1683 CrossRef.
- F. N. Palmer and D. K. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 9, 1323 RSC.
- M. Studer, S. Burkhardt and H.-U. Blaser, Chem. Commun., 1999, 1727 RSC; C. Exner, A. Pfaltz, M. Studer and H.-U. Blaser, Adv. Synth. Catal., 2003, 345, 45 CrossRef CAS; G. Szollosi, Z. Makra, M. Fekete, F. Fülöp and M. Bartok, Catal. Lett., 2012, 142, 7–889 CrossRef; H. Ooka, N. Arai, K. Azuma, N. Kurono and T. Ohkuma, J. Org. Chem., 2008, 73, 9084 CrossRef PubMed.
- B. T. Cho and Y. S. Chun, Tetrahedron: Asymmetry, 1994, 5, 1147 CrossRef CAS.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551 CrossRef CAS; E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925 CrossRef.
- Selected publications: F. Hahn and S. Friedrich, Tetrahedron, 2015, 71, 1473 CrossRef CAS; H. Sato, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2015, 54, 4488 CrossRef PubMed; H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171 CrossRef PubMed; M. Müller, Angew. Chem., Int. Ed., 2005, 44, 362 CrossRef PubMed; S. Sivanathan, F. Körber, J. A. Tent, S. Werner and J. Scherkenbeck, J. Org. Chem., 2015, 80, 2554 CrossRef PubMed; S. Sonoike, T. Itakura, M. Kitamura and S. Aoki, Chem. - Asian J., 2012, 7, 64 CrossRef PubMed.
- R. Wohlgemuth, in Synthetic Methods for Biologically Active Molecules: Exploring the Potential of Bioreductions, ed. E. Brenna, Wiley-VCH, Weinheim, 2014, pp. 1–25 RSC; F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285 RSC; I. A. Kaluzna, J. D. Rozzell and S. Kambourakis, Tetrahedron: Asymmetry, 2005, 16, 3682 CrossRef CAS; J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. N. Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193 CrossRef; I. N. Houpis, R. Liu, L. Liu, Y. Wang, N. Dong, X. Zhao, Y. Zhang, T. Xiao, Y. Wang, D. Depre, U. Nettekoven, M. Vogel, R. Wilson and S. Collier, Adv. Synth. Catal., 2013, 355, 1829 CrossRef; N. Richter, M. Neumann, A. Liese, R. Wohlgemuth, A. Weckbecker, T. Eggert and W. Hummel, Biotechnol. Bioeng., 2010, 106, 541 CrossRef PubMed; N. Richter, M. Neumann, A. Liese, R. Wohlgemuth, T. Eggert and W. Hummel, ChemBioChem, 2009, 10, 1888 CrossRef PubMed.
- R. Wichmann and D. Vasic-Racki, Adv. Biochem. Eng./Biotechnol., 2005, 92, 225 CrossRef CAS PubMed; A. Liese, Adv. Biochem. Eng./Biotechnol., 2005, 92, 197 CrossRef PubMed; J. C. Moore, D. J. Pollard, B. Kosjek and P. N. Devine, Acc. Chem. Res., 2007, 40, 1412 CrossRef PubMed; W. Liu and P. Wang, Biotechnol. Adv., 2007, 25, 369 CrossRef PubMed.
- S. Bornemann, D. H. G. Crout, H. Dalton, V. Kren, M. Lobell, G. Dean, N. Thomson and M. M. Turner, J. Chem. Soc., Perkin Trans. 1, 1996, 425 RSC.
- J. Kiegiel, A. Papis and J. Jurczak, Tetrahedron: Asymmetry, 1999, 10, 535 CrossRef CAS.
- R. Wohlgemuth, Biotechnol. J., 2009, 4, 1253 CrossRef CAS PubMed.
- J. R. Durrwachter, H. M. Sweers, K. Nozaki and C.-H. Wong, Tetrahedron Lett., 1986, 27, 1261 CrossRef CAS; D. G. Drueckhammer, J. R. Durrwachter, R. L. Pederson, D. C. Crans, L. Daniels and C.-H. Wong, J. Org. Chem., 1989, 54, 70 CrossRef; A. Ozaki, E. J. Toone, C. H. von der Osten, A. J. Sinskey and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 4970 CrossRef; B. Imperiali and J. Zimmermann, Tetrahedron Lett., 1990, 31, 6485 CrossRef; K. K. C. Liu, T. Kajimoto, L. Chen, Z. Zhong, Y. Ichikawa and C.-H. Wong, J. Org. Chem., 1991, 56, 6280 CrossRef; S. T. Allen, G. R. Heintzelman and E. J. Toone, J. Org. Chem., 1992, 57, 426 CrossRef; I. Henderson, E. Garcia-Junceda, K. K.-C. Liu, Y.-L. Chen, G.-J. Shen and C.-H. Wong, Bioorg. Med. Chem., 1994, 2, 837 CrossRef PubMed; T. D. Machajewski and C.-H. Wong, Synthesis, 1999, 1469 CrossRef; I. Sánchez-Moreno, J. F. García-García, A. Bastida and E. García-Junceda, Chem. Commun., 2004, 1634 RSC; X. Garrabou, J. A. Castillo, C. Guérard-Hélaine, T. Parella, J. Joglar, M. Lemaire and P. Clapés, Angew. Chem., Int. Ed., 2009, 48, 5521 CrossRef PubMed; P. Clapes and X. Garrabou, Adv. Synth. Catal., 2011, 353, 89 CrossRef; M. Rale and S. Schneider, Chem. – Eur. J., 2011, 17, 2623 CrossRef PubMed.
- G. Simon, T. Eljezi, B. Legeret, F. Charmantray, J. A. Castillo, C. Guérard-Hélaine, M. Lemaire, M. Bouzon, P. Marliére, V. Hélaine and L. Hecquet, ChemCatChem, 2013, 5, 784 CrossRef CAS.
- E. W. Baxter and A. B. Reitz, Org. React., 2002, 59 Search PubMed; F. N. Palmer and D. K. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 1323 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5re00009b |
| ‡ The reaction was monitored by HPLC and reached 99.2% conversion after 28 h. To enable convenient work-up of the mixture, the reaction was additionally stirred for 17 h prior to work-up. |
| This journal is © The Royal Society of Chemistry 2016 |