
C–O bond hydrogenolysis vs. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C group hydrogenation of furfuryl alcohol: towards sustainable synthesis of 1,2-pentanediol
C group hydrogenation of furfuryl alcohol: towards sustainable synthesis of 1,2-pentanediol
Dominik
Götz
,
Martin
Lucas
and
Peter
Claus
*
Department of Chemistry, Ernst-Berl-Institute/Chemical Technology II, Technical University of Darmstadt, Alarich-Weiss-Straße 8, D-64287 Darmstadt, Germany. E-mail: claus@tc2.tu-darmstadt.de; Fax: +49 615 116 4788; Tel: +49 615 116 5369
First published on 11th December 2015
Abstract
The conversion of furfuryl alcohol into 1,2-pentanediol and tetrahydrofurfuryl alcohol by aqueous phase hydrogenolysis using highly active supported ruthenium catalysts is a promising sustainable route substituting for petro-based production processes. An on-line analysis of the reaction, performed by ATR-IR spectroscopy, is necessary to show the formation of solid deposits during the reaction.
Nowadays, non-renewable fossil resources are the common basis for the production of chemicals and fuels. Crude oil plays a key role in the production of hydrocarbons which are in turn the platform for the production of important base and fine chemicals. In this context, 1,2-pentanediol (1,2-PeD) is currently produced via a cost-intensive multistep route by selective oxidation of pentene to pentene oxide and subsequent hydrolysis.1,2 This diol is frequently used as a monomer for polyesters, as a key intermediate of low-toxic microbicides1 and as an ingredient of various cosmetic products.3
Biomass will surely play an important role in the modern chemical industry because approximately 170 quadrillion kilograms are produced by photosynthesis every year, but only a small percentage is used.4,5 In the next years, the rising demand for crude oil will lead to an increasing shortage, thus the displacement of exhaustible raw materials through biomass is clearly desirable as a sustainable source of energy and organic carbon. Polysaccharides like cellulose or hemicellulose are the major part of biomass, consisting of C5- or C6-monosaccharides. While the direct selective conversion of cellulose into valuable chemicals is still a challenging research topic,6,7 various bulk chemicals like furfuryl alcohol (FA) are already produced from biomass via established production routes.8 Furfuryl alcohol can be produced via hydrogenation of furfural or 5-hydroxymethylfurfural (5-HMF) and be converted into various valuable alcohols.
Fig. 1 presents a simplified reaction network including the main components in the aqueous phase hydrogenolysis of FA. One well-known route is its conversion into tetrahydrofurfuryl alcohol (THFA) which can be done very selectively using ruthenium at low temperatures.9 Its subsequent conversion into 1,5-pentanediol is also receiving increasing interest.10 In the past years, only little attention was paid to its conversion into 1,2-pentanediol. So far, most of the catalysts reported for the hydrogenolysis of FA have disadvantages with regard to their toxicity (e.g. copper chromite11,12) or their need for additives or special solvents (e.g. Adams' catalyst13). This is clearly not desirable for green and sustainable chemistry, as summed up in the popular “12 principles of green chemistry”.14 In contrast to the aforementioned catalysts, supported ruthenium catalysts are non-toxic and are stable against water under hydrothermal conditions. Zhang et al. have recently reported the formation of 1,2-PeD using ruthenium supported on manganese oxide.15 However, the industrial availability and applicability of these catalyst supports are limited. Hence, the use of stable and available supports is obviously needed. This work presents the performance of ruthenium supported on alumina, silica as well as carbon (Table 1). Additionally, because the formation of carbon in biomass-based reactions is often overlooked, we applied on-line ATR-IR spectroscopy during FA hydrogenolysis to get insight into the role of such species during the course of the reaction.
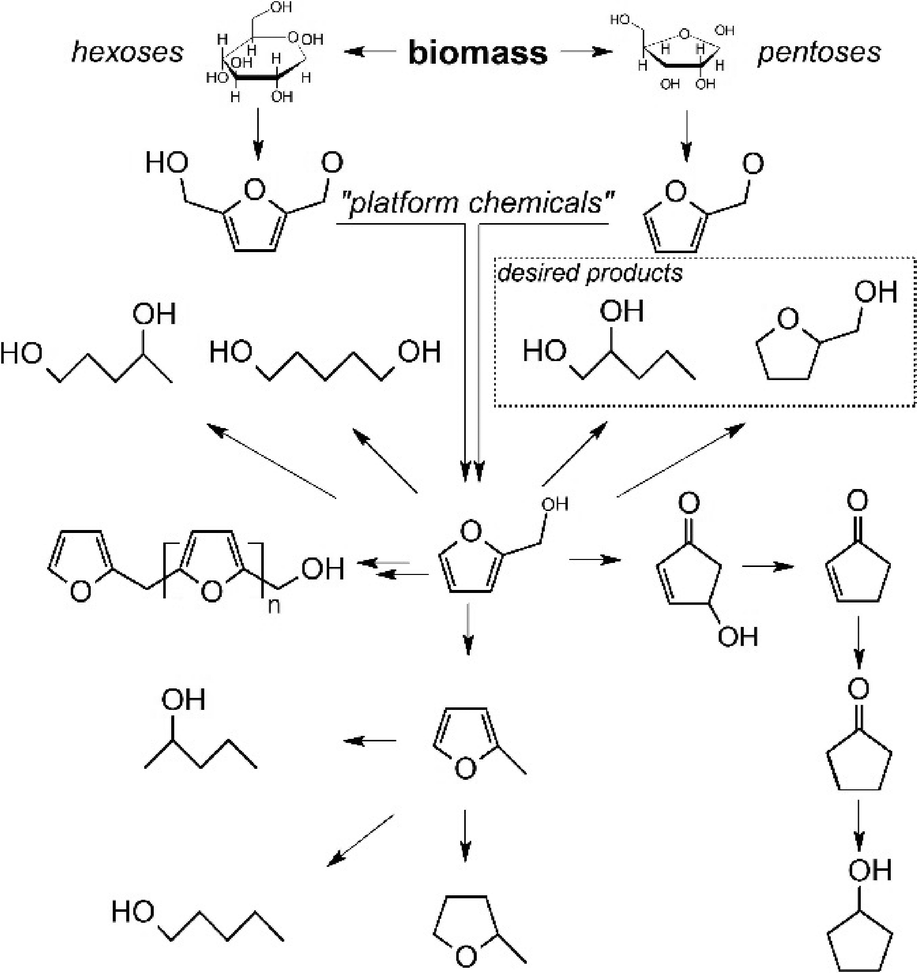 | ||
| Fig. 1 Simplified reaction network of the main components during the aqueous phase hydrogenolysis of furfuryl alcohol. | ||
| Catalyst | Selectivity | t/min | ||||
|---|---|---|---|---|---|---|
| Disp./% | 1,2-PeD/% (TOF/s−1) | THFA/% (TOF/s−1) | Others/% | Sum/% | ||
| Reaction conditions: T = 473 K, pH2 = 100 bar, agitation speed = 1000 rpm, m(cat) = 500 mg.a c(FA) = 7.46 g per 100 ml.b c(FA) = 40 g per 100 ml.c m(Na2CO3) = 100 mg per 100 ml.d m(Na2CO3) = 300 mg per 100 ml. | ||||||
| 5Ru/Ca | 35 | 20 (0.07) | 49 (0.16) | 11 | 80 | 45 |
| 10Ru/Ca | 17 | 21 (0.07) | 60 (0.20) | 16 | 97 | 45 |
| 10Ru/Al2O3a | 22 | 26 (0.10) | 53 (0.21) | 11 | 90 | 30 |
| 10Ru/Al2O3b,c | 22 | 32 (0.34) | 57 (0.60) | 11 | 100 | 60 |
| 10Ru/SiO2a,d | — | 14 (—) | 32 (—) | 27 | 73 | 200 |
| Adams' catalyst13 | — | 20 (—) | 35 (—) | 19 | 74 | — |
In this context, especially carbon- and alumina-supported catalysts show good selectivity values up to about 30% towards 1,2-pentanediol. Therefore, they are promising candidates with respect to the industrial implementation of this reaction. The other main product is tetrahydrofurfuryl alcohol (THFA) which is also of interest as a valuable product due to its use as a green solvent.16 Besides 1,2-PeD, two isomeric diols, namely 1,4-pentanediol (1,4-PeD) and 1,5-pentanediol (1,5-PeD), are formed and it is interesting to see that especially the latter is formed only in small amounts. In contrast to 1,5-PeD, which can be formed from THFA, experiments using THFA as a substrate indicate that the hydrogenolysis to the diols seems to be a parallel reaction to the hydrogenation to THFA under these conditions. This is also in agreement with the results obtained by Zhang and co-workers,15 who postulate two different active sites responsible for the hydrogenolysis towards 1,2-PeD and 1,5-PeD.
Apart from that, the reaction is often accompanied by the formation of a yellow to red insoluble solid, responsible for the unbiased carbon balance in the liquid phase found by gas chromatography. By using attenuated total reflection infrared spectroscopy (ATR-IR), these solids can be identified as polymers of furfuryl alcohol exhibiting similar characteristic vibrational modes (Fig. 2). In addition to this, various dimers and oligomers are formed during the reaction. Since the self-polymerization of furfuryl alcohol is known to be catalysed by Lewis as well as Brønsted acids,17 it is obvious that the formation of polymers is strongly enhanced by the low pH value of the reaction mixture as well as the heterogeneous acid sites of the supporting material. In this context, NH3-TPD measurements of the alumina and carbon supports show only weak acid sites indicated by an early desorption at about 423 K. The amount of ammonia adsorbed on the carbon supports (5Ru/C: 0.35 mmol g−1, 10Ru/C: 0.21 mmol g−1) is higher compared to that on the alumina support (0.05 mmol g−1), but the dependence on the reaction parameters and catalytic activity seems to be way more pronounced than that on the amount of acid sites of the support.
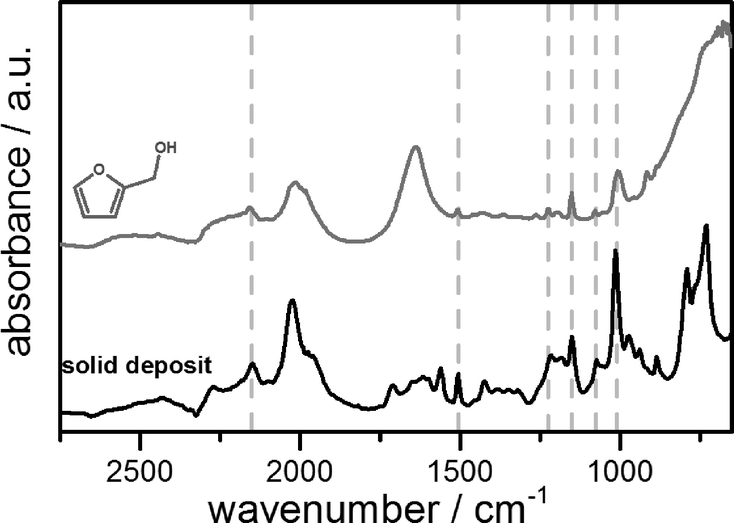 | ||
| Fig. 2 Infrared spectrum (ATR) of the insoluble solid deposit (below) in comparison to furfuryl alcohol (above, dissolved in water). | ||
Although the polymerisation can be reduced by adding a homogeneous basic additive (e.g. Na2CO3), the use of additives is not desirable and should be avoided if possible. Especially at lower hydrogen pressures desirable for industrial realisation, the reaction parameters must be changed to milder conditions in order to avoid the formation of solid deposits. While the FA concentration has a rather small influence on the 1,2-PeD selectivity, higher FA concentrations would favour the polymerisation, increasing the risk of forming solid deposits. Thus, a rather low FA concentration of 5 wt% was chosen in the subsequent experiments. In contrast to this, the reaction temperature strongly influences the selectivity to the desired diol (1,2-PeD) as shown in Table 2.
| Temperature/K | Selectivity at full conversion | t/min | |||
|---|---|---|---|---|---|
| 1,2-PeD/% (TOF/s−1) | THFA/% (TOF/s−1) | Others/% | Sum/% | ||
| Reaction conditions: p(H2) = 30 bar, agitation speed = 1000 rpm, c(FA) = 5 g per 100 ml, m(cat) = 125 mg, PTFE reactor inlet. Selectivity sums greater than 100% are due to the estimated calibration factors of the side products. | |||||
| 403 | 12 (0.08) | 74 (0.49) | 10 | 96 | 60 |
| 423 | 17 (0.15) | 70 (0.62) | 14 | 102 | 45 |
| 443 | 17 (0.15) | 52 (0.46) | 18 | 87 | 45 |
| 463 | 4 (0.04) | 9 (0.08) | 37 | 50 | 45 |
Since the C–O bond hydrogenolysis is enhanced at higher reaction temperatures in comparison to the hydrogenation of the double bonds, lower temperatures favour the formation of THFA. While higher temperatures lead to a higher 1,2-PeD selectivity, they also promote side reactions that do not depend on the catalyst and result in a decrease in 1,2-PeD selectivity at higher temperatures. Besides the self-polymerization, the isomerization to 4-hydroxycyclopentanone (4-HCP) is the dominant reaction in this context. 4-HCP is the precursor compound for cyclopentanone which is hydrogenated to cyclopentanol.18,19 The hydrogenation of cyclopentanone to cyclopentanol is known to be inhibited by the furfuryl alcohol polymers, thus the intermediate product reaches yields of about 15%.20 These cyclopentanol rings are currently of increasing interest as another valuable product in this reaction chain. But regarding the use of high purity 1,2-PeD for cosmetics, the formation of side products is disadvantageous. An elaborate purification process would be necessary, so intermediate reaction temperatures in the range of 130 to 150 °C are favourable due to the low formation of side products, although the selectivity might not be at its maximum.
In order to compete with the catalysts' independent side reactions, high reaction rates are needed. Hydrogen consumption measured during the reaction shows a nearly linear decay until full conversion during the reaction. This is presumably due to the strong adsorption of furfuryl alcohol over ruthenium, leading to a highly covered metal surface. In order to study these fast reactions, a reliable and fast online process analysis method is required to monitor the reaction progress with a high temporal resolution. In this context, the aforementioned ATR-IR spectroscopy is a promising tool due to its fast measuring frequency without the need for taking and analysing offline samples. Although the strong IR absorption of water is challenging in aqueous systems, the experiments show that ATR-IR spectroscopy is able to detect and also quantify the main components of the reaction mixture, when compared to the GC samples, in a reliable way (Fig. 3).
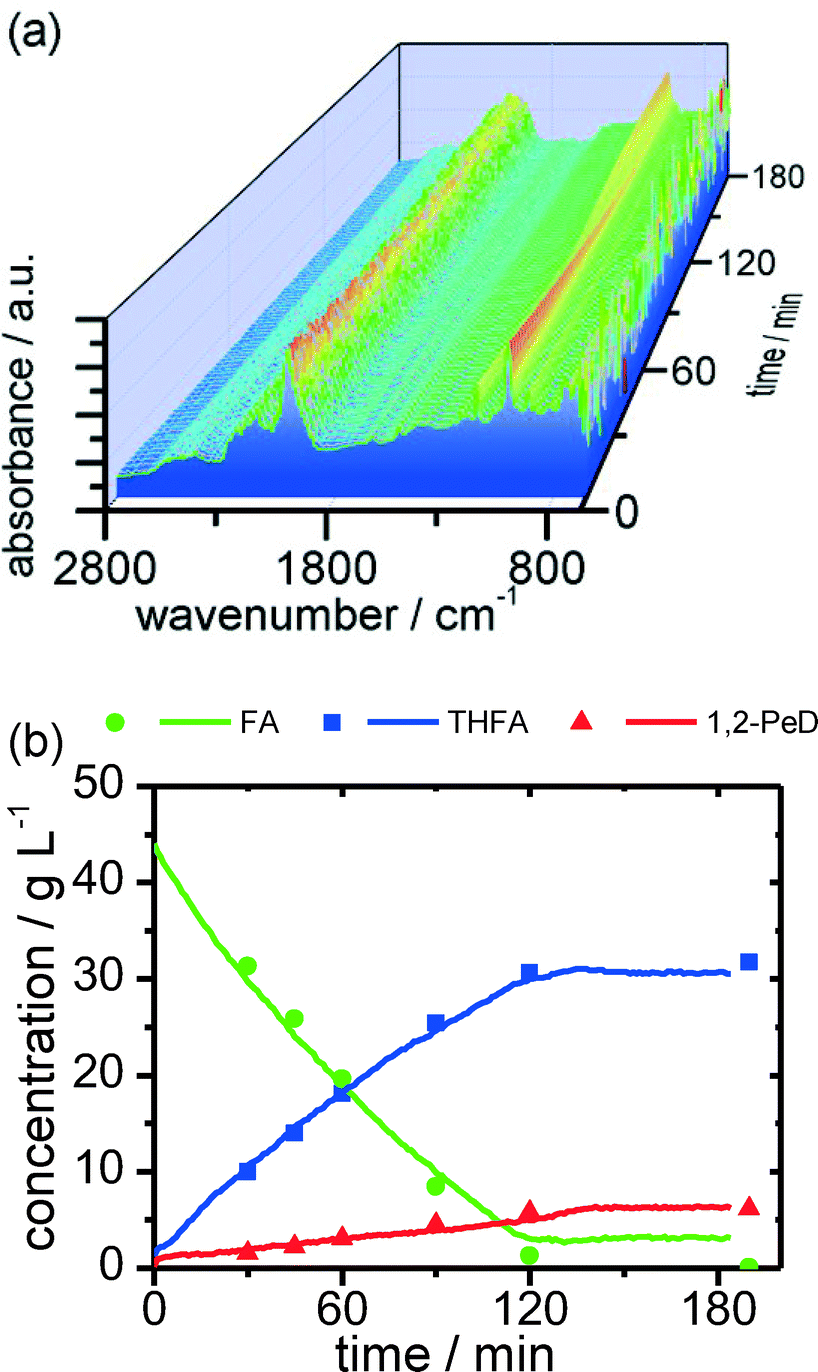 | ||
| Fig. 3 (a) Temporal change of the ATR spectra (after subtracting the IR spectrum of the catalyst slurry) during the reaction. (b) Comparison of concentrations measured by ATR-IR (lines) and GC (symbols) during the reaction. Reaction conditions: 5% Ru/Al2O3, T = 403 K, c(FA) = 50 g L−1, m(cat) = 0.125 g, p(H2) = 30 bar. | ||
The ATR spectroscopy confirms that an almost constant decay of FA is reached shortly before full conversion. On the verge of full conversion of furfuryl alcohol, the reaction rate decreases significantly.
A full conversion of FA is desirable due to the formation of FA–water azeotropes, because the subsequent separation of the product mixture is more complicated in the presence of FA.21 It is remarkable that besides the detection of soluble hydrocarbons, the ATR probe head is also very sensitive to deposition of the insoluble solid polymers formed by the self-polymerization as shown in Fig. 2. This means that the ATR spectrometer can instantly detect the formation of polymers at an early stage during the process. This is important to ensure a failure-free and safe process with regard to the industrial implementation of this biomass-derived reaction.
Conclusions
The aqueous phase hydrogenolysis of furfuryl alcohol represents a promising sustainable alternative for the production of 1,2-pentanediol. The hydrogenolysis can be successfully carried out by using highly active ruthenium catalysts supported on common materials like alumina or carbon. ATR spectroscopy is able to show the formation of poly(furfuryl alcohol) deposits during the reaction. Although the undesired polymerisation can be reduced by base additives, this can also be done by adjusting the reaction conditions. Additionally, for the detection of solid deposits, ATR-IR spectroscopy is useful as an in-time process monitoring tool, which is helpful to the improvement of the efficiency and safety of furfuryl alcohol hydrogenolysis.Materials and methods
Catalytic experiments are carried out using a 300 mL stainless steel batch reactor (Parr Instruments). The reactor is equipped with an external storage for the subsequent introduction of furfuryl alcohol (98%, Sigma Aldrich) into the catalyst–solvent slurry after being heated up. The catalysts are prepared by incipient wetness impregnation with a ruthenium precursor or supplied by Evonik Industries or Alfa Aesar. Dispersity is determined by CO pulse-chemisorption at 273 K (TPD/R/O 1100, Thermo Fisher Scientific). Acid sites are characterized by temperature-programmed desorption of ammonia. During the reaction, liquid phase samples are taken and analysed by gas chromatography (GC as well as GC/MS). Additionally, the reactor can be equipped with a high pressure attenuated total reflection infrared spectrometer (Mettler Toledo ic45m, DiComp) for in situ infrared spectroscopy. Quantitative analysis of the infrared spectra is performed by using the quantification module icQuant in the software package icIR (Mettler Toledo, version 4.0.641.1). The multivariant quantification model was trained using calibration samples at room temperature and also using the spectra recorded under the reaction conditions of the offline samples.Notes and references
- R. Siegmeier, G. Prescher, H. Maurer and G. Hering, EP0183029 A3, Degussa Aktiengesellschaft, 1985.
- F. Jiansong, W. Jiyuan, S. Chao, Z. Dongmei and W. Zhongping, CN103864575 A, China Petroleum & Chemical, Sinopec Shanghai Petrochemical, 2014.
- I. Agostini and S. Cupferman, US6296858 B1, L'oreal, 2001.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- H. Röper, Starch – Stärke, 2002, 54, 89–99 CrossRef.
- K. Fabičovicová, M. Lucas and P. Claus, Green Chem., 2015, 17, 3075–3083 RSC.
- K. Fabičovicová, O. Malter, M. Lucas and P. Claus, Green Chem., 2014, 16, 3580–3588 RSC.
- B. A. Tokay, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a04_099.
- F.-A. Khan, A. Vallat and G. Süss-Fink, Catal. Commun., 2011, 12, 1428–1431 CrossRef CAS.
- S. Koso, I. Furikado, A. Shimao, T. Miyazawa, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 2035–2037, 10.1039/b822942b.
- H. Adkins and R. Connor, J. Am. Chem. Soc., 1931, 53, 1091–1095 CrossRef CAS.
- R. Connor and H. Adkins, J. Am. Chem. Soc., 1932, 54, 4678–4690 CrossRef CAS.
- W. E. Kaufmann and R. Adams, J. Am. Chem. Soc., 1923, 45, 3029–3044 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30 Search PubMed.
- B. Zhang, Y. Zhu, G. Ding, H. Zheng and Y. Li, Green Chem., 2012, 14, 3402 RSC.
- M. A. Tike and V. V. Mahajani, Ind. Eng. Chem. Res., 2007, 46, 3275–3282 CrossRef CAS.
- M. Choura, N. M. Belgacem and A. Gandini, Macromolecules, 1996, 29, 3839–3850 CrossRef CAS.
- M. Hronec, K. Fulajtárová and T. Soták, J. Ind. Eng. Chem., 2014, 20, 650–655 CrossRef CAS.
- M. Hronec, K. Fulajtárova and T. Soták, Appl. Catal., B, 2014, 154–155, 294–300 CrossRef CAS.
- M. Hronec, K. Fulajtárova and M. Mičušik, Appl. Catal., A, 2013, 468, 426–431 CrossRef CAS.
- H. E. Hoydonckx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, in Ullmann's Encyclopedia of Industrial Chemistry, 2007, DOI:10.1002/14356007.a12_119.pub2.
| This journal is © The Royal Society of Chemistry 2016 |