
Highly active and selective catalysts for olefin hydrosilylation reactions using metalloporphyrins intercalated in natural clays†
Waheed
Jondi
a,
Ahed
Zyoud
a,
Waseem
Mansour
a,
Ahmad Q.
Hussein
b and
Hikmat S.
Hilal
*a
aSSERL, An-Najah National University, Nablus, Palestine. E-mail: hshilal@najah.edu; hikmathilal@yahoo.com
bDepartment of Chemistry, University of Jordan, Amman, Jordan
First published on 4th November 2015
Abstract
Tetra(4-pyridyl)porphyrinato-manganese(III) (MnTPyP+) ions intercalated inside nano- and micro-size natural clay powders effectively catalyzed the hydrosilylation reaction of 1-octene and tri(ethoxy)silane under mild conditions. The results showed that the homogeneous MnTPyP+ ions catalyzed the reaction where both branched and linear products were observed. Intercalation significantly enhanced both catalyst activity and selectivity. Production of the linear tri(ethoxy)silyl-1-octane as sole product was confirmed by FT-IR and NMR spectra, in the case of the intercalated catalyst. The activity enhancement was more pronounced in the case of the nano-particle supported catalyst system, with turnover frequency (TF) values up to 1200 min−1 and 85% conversion observed in less than 20 min. The intercalated catalyst systems were easy to recover and were reused three times, while retaining up to 80% of their original activity. Effects of different reaction parameters on the initial reaction rate, such as clay particle size, solvent polarity, reactant concentration, catalyst loading and temperature, were investigated. A new plausible mechanism, with evidence and justifications, is proposed.
1. Introduction
Homogeneous catalysts have several advantages, most notably their high activity associated with the accessibility of catalytically active sites to reactants. However, recovery of homogeneous catalysts after reaction cessation cannot be achieved by simple separation methods like filtration or decantation.1–3 Heterogeneous catalysts are thermally stable and easy to recover, but normally have lower activity and demand more forcing conditions than their homogeneous counterparts.4,5 In heterogeneous catalysis, the reactant molecules reach only the surface active sites, while the bulk sites are hidden inside.To combine the advantages of both homogeneous and heterogeneous catalyses, the catalyst species can be supported onto insoluble solids, to yield supported catalysts.6 Supporting the catalysts may be achieved by using different techniques. Ideally speaking, all active sites are accessible to reactant molecules in such catalysts. This increases the activity of the supported catalyst as compared to heterogeneous systems. Moreover, the supported catalysts become easier to isolate and recover after reaction completion.7
The support may also intervene with the reaction process, most notably in zeolites where the cavities exhibit a solvent-like behavior. This may affect the reaction speed and nature of products.8,9 For these reasons, different insoluble materials were attempted to support different types of transition metal complex catalyst systems for different reactions.10–13 In practice, the support may speed up a given reaction, compared to the homogeneous counterpart itself, or it may slow down the reaction. Moreover, the support may enhance the reaction selectivity towards a given product, or may even enhance the selectivity for a given reactant. The effect of the support is thus not limited to make catalyst recovery easier, but the stability, selectivity and activity of the supported catalyst active sites may be modified by the support.2,14,15 Therefore, it is necessary to keep searching for new types of supporting materials for different catalyst systems in different reactions.
Metalloporphyrins, which involve metal cations chemically bonded in the centers of the planar aromatic porphyrinato rings, are a well-known class of stable compounds.16–18 Such compounds were used as homogeneous catalysts for many reactions.19,20 Olefin photo-oxidation,21,22 isomerization,23,24 hydrogenation,25 oxygenation26,27 and other reactions have all been reported. Just like other homogeneous catalysts, metalloporphyrin ions are difficult to isolate after reaction completion, and are therefore being supported on different types of supports.13,28,29
Clays may involve layered structures of silicates and aluminates. Some clay phases are expandable such as montmorillonite and biotite, while others are non-expandable such as kaolinite and illite. Substitution of Si4+ ions with Al3+ ions causes deficiency in total positive charges, and excess in negative charges. Clays, therefore, may function as cationic exchange resins.30 Clays have been described as suitable supports for different types of catalyst systems in many organic reactions.31–34 Like zeolite systems, clay layers may help bring reactants in close proximity to the catalytically active site and may thus affect the reaction.35–37
Clays are naturally abundant materials. In a previous communication, natural clays collected from the Northern areas of the West Bank of the Palestinian Territories were described as hosts for intercalated MnTPYP+ ions. Such clays are safe, low cost, abundant and widely used in ceramics and potteries. They involve expandable phases (such as montmorillonite and biotite) and non-expandable phases (such as kaolinite and illite). Tetra(4-pyridyl)porphyrinato-manganese(III) [MnTPyP]+ ions intercalated readily in micro- and nano-scale clay particles of the expandable phases only, as described in the previous communication.38
This work aims at investigating the catalytic activity of the natural clay-supported MnTPyP+ ions for an industrially important hydrosilylation reaction. Such reactions involve addition of the Si–H to the unsaturated olefin C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group. Olefin hydrosilylation reactions are important to many industries,39 as they have once been described as “the most important application of platinum in homogeneous catalysis”.40 It is imperative to find active and recoverable catalyst systems for such reactions. To our knowledge, metalloporphyrin complexes, in their homogeneous or supported forms, have not been widely described as catalysts for hydrosilylation reactions. In this work, the catalytic activity of MnTPyP+ ions in olefin hydrosilylation reactions will be first tested. The influence of the clay support on the activity and selectivity of the MnTPyP+ ions to produce the saturated product tri(ethoxy)silyl-1-octane will then be investigated in depth.
C group. Olefin hydrosilylation reactions are important to many industries,39 as they have once been described as “the most important application of platinum in homogeneous catalysis”.40 It is imperative to find active and recoverable catalyst systems for such reactions. To our knowledge, metalloporphyrin complexes, in their homogeneous or supported forms, have not been widely described as catalysts for hydrosilylation reactions. In this work, the catalytic activity of MnTPyP+ ions in olefin hydrosilylation reactions will be first tested. The influence of the clay support on the activity and selectivity of the MnTPyP+ ions to produce the saturated product tri(ethoxy)silyl-1-octane will then be investigated in depth.
2. Results and discussion
The hydrosilylation reaction of 1-octene with triethoxysilane, eqn (1), occurred readily with the three types of catalysts examined here, namely the homogeneous MnTPyP+ ions, micro-particle supported catalyst (MnTPyP@Micro-Clay) and nano-particle supported catalyst (MnTPyP@Nano-Clay). The preparation and characterization of the three catalyst systems are described in section 4 below.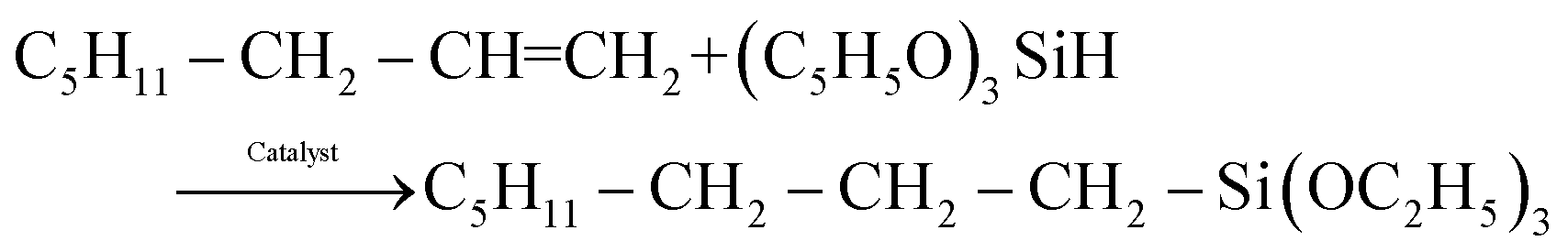 | (1) |
The intensities of the Si–H and CC bands at 2200 and 1641 cm−1, respectively, decreased while the C–Si band at 1253 cm−1 continued to increase. These confirm the addition reaction of Si–H to the CC bond. In the cases of MnTPyP@Nano-Clay and MnTPyP@Micro-Clay catalyst systems, the spectra confirmed the absence of any bands characteristic for tri(ethoxy)silyl-2-octane with silyl group bonded to the internal C atom. Exchange reactions to produce tri(ethoxy)silyl-1-octene did not occur. No olefin isomerization was observed. Under the working conditions, the results indicate that the reaction was selective to olefin hydrosilylation reaction to produce the stated product only. Therefore, in the case of the supported MnTPyP+ catalyst systems, the reaction was totally regioselective to produce the linear hydrosilylation product only. The reaction stereoselectivity will be further discussed based on the proposed mechanism below.
In the case of the homogeneous catalyst system, no olefin isomerization reaction was detected. A small band at 1288 cm−1, the intensity of which continued to increase parallel to the 1253 cm−1 band, occurred due to production of tri(ethoxy)silyl-2-octane as a side product. Therefore, the homogeneous catalyst system is also regioselective but to a lesser extent than the supported catalyst system, as will be further discussed in the proposed mechanism.
Formation of the tri(ethoxy)silyl-1-octane product was further confirmed by NMR spectra. The product was isolated from the supported catalyst reaction mixture by distillation (at 98 °C) under reduced pressure and analyzed by 1H NMR, 13C NMR and DEPT 13C NMR spectra. Considering the product molecular structure shown below, the corresponding NMR signals are shown in Table 1.
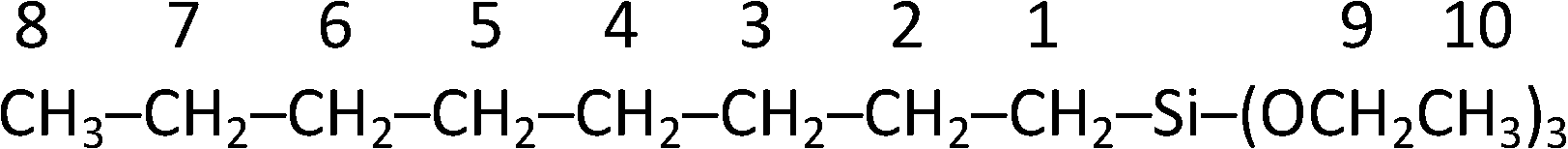
| a Additional product NMR data available. | |
|---|---|
| 1H NMR | δ = 0.628 (2H, H1, t, J = 8.15 Hz); 0.875 (3H, H8, t, J = 7.10 Hz); 1.226 (3H, H10, t, J = 6.25 Hz), 3.819 (2H, H9, q, J = 6.25 Hz); and complex signals from 1.0–1.8 ppm in accordance with 6 (CH2) straight chain. |
| 13C NMR | δ = 10.532 (C1); 14.022 (C8); 18.231 (C10); 22.858 (C2); 22.631 (C7); 22.731 (C3); 29.819 (C6); 31.881 (C4); 33.160 (C5) and 58.220 (C9) ppm. |
| DEPT 135 13C NMR | Only two positive signals for CH3 groups at δ = 14.022 (C8) and 18.231 (C10) ppm. All other signals are inverted which indicates that all the other signals are for CH2 groups, in accordance with the expected structure. |
Different control experiments were performed, as described in Table 2. No reaction was observed in the absence of the MnTPyP+ catalyst. Control experiments with naked clay (entry 1) showed only minimal disappearance of 1-octene and silane (less than 15% each) at the beginning, due to adsorption. Therefore, the actual catalyst was the supported MnTPyP+ ions themselves. Entry 2 shows that silane adsorption (~15%) occurred when it was mixed with the MnTPyP@Nano-Clay system in the absence of 1-octene. Naked clay mixed with free non-intercalated MnTPyP+ ions (entry 3) showed results similar to those observed for the homogeneous system described above (entry 4). This confirms the effect of intercalation on reaction rate enhancement.
| Entry | Description | Loss of silane | Loss of olefin | Observed hydrosilylation product(s) |
|---|---|---|---|---|
| 1 | Naked clay (0.5 g) with added silane (0.72 M) and olefin (1.33 M) | ~15% | ~15% | None |
| 2 | MnTPyP@Nano-Clay (0.5 g) with added silane (0.72 M) | ~15% | — | None |
| 3 | Naked clay (0.5 g) and free MnTPyP+ ions (1.740 × 10−4 M) with added silane (0.72 M) and olefin (1.33 M) | ~17% | ~16% | Linear product <2% and branched product <1% |
| 4 | Free MnTPyP+ ions (1.740 × 10−4 M) with added silane (0.72 M) and olefin (1.33 M) | — | — | Linear product <2% and branched product <1% |
Both supported systems showed higher activity than the homogeneous MnTPyP+ ions. The MnTPyP@Nano-Clay system was more active than the MnTPyP@Micro-Clay system, as shown in Fig. 1. The clay-supported MnTPyP+ ions showed higher catalytic activity than other types of supported catalysts such as Mn2(CO)10 or Ru3(CO)12 reported earlier for hydrosilylation reactions.12,41 This behavior is rationalized by the ability of the clay layers to assess the catalytic reaction. Literature42 showed that clay-supported catalysts may have higher activity than other supported catalysts, due to the ability of the layered clays to host reactant molecules and bring them into close proximity to the catalytically active sites. By this way, the clay layered structure exhibits solvent-like behavior and enhances the rate of the reaction. The perpendicular intercalation of the MnTPyP+ ions gives extra room for the reactant molecules of 1-octene and tri(ethoxy)silane to reach the catalytically active sites.
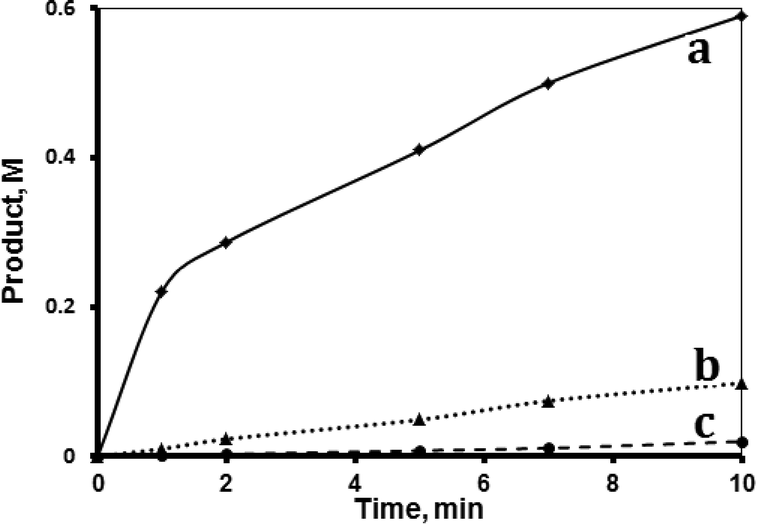 | ||
| Fig. 1 Effect of catalyst type. Hydrosilylation reaction profiles showing product formation with time for the (a) MnTPyP@Nano-Clay, (b) MnTPyP@Micro-Clay and (c) homogeneous MnTPyP+ ions. All reactions were conducted using 1-octene (1.33 M), tri(ethoxy)silane (0.72 M), MnTPyP+ ions (1.740 × 10−4 M) and 1,4-dioxane solvent in a total volume of 15.00 mL at 40 °C. | ||
Fig. 1 shows that the activity for the three catalyst systems varied in the order MnTPyP+ < MnTPyP@Micro-Clay < MnTPyP@Nano-Clay. Table 3 summarizes the values of the turnover number (TN) and the turnover frequency (TF) measured for each catalyst system at different times. The influence of the clay support on MnTPyP+ catalyst activity is rationalized above, as both supported catalysts exhibited higher activity than the homogeneous system. The results are thus consistent with earlier findings reported for some other catalyst systems, as discussed above. On the other hand, the MnTPyP@Nano-Clay catalyst showed higher activity than the MnTPyP@Micro-Clay. This is because the nano-scale system has higher relative surface area than the micro-scale system. Our previous report38 showed that the nano-scale system has a relative surface area of 200 m2 g−1 compared to 130 m2 g−1 for the micro-scale system. With higher relative surface area, the MnTPyP@Nano-Clay catalyst should exhibit higher activity. Higher surface area furnishes higher accessibility for the reactant molecules to reach the catalytically active sites inside the clay particles.
| Catalyst | Values of TN (and TF, min−1) with time | Ref. | ||||
|---|---|---|---|---|---|---|
| 1 min | 5 min | 10 min | 15 min | 30 min | ||
| Homogeneous MnTPyP, at 40 °C | 8 (8.0) | 42 (8.4) | 115 (11.5) | 220 (14.7) | 412 (14.0) | This work |
| MnTPyP@Micro-Clay at 40 °C | 53 (53.0) | 276 (55.2) | 632 (63.2) | — | — | This work |
| MnTPyP@Nano-Clay at 40 °C | 1184 (1184.0) | 2299 (460.0) | 3448 (345.0) | — | — | This work |
| Homogeneous Ru3(CO)12, at 70 °C at 40 °C | — | — | 30 (3.0) 0 | 70 (2.3) 0 | 41 | |
| Polysiloxane/supported Mn2(CO)10 at 40 °C | — | — | 42 (4.2) | 47 (3.1) | — | 5 |
A closer look at Table 3 shows that the ratio between the TF values for the nano- and micro-catalyst systems does not correspond to the ratio between their relative surface areas. At the beginning of the reaction, the TF value ratio was 22 times higher, and after 10 min, the ratio became about 6 times higher. In either case, the ratio was higher than the measured relative surface area ratio, as shown above. This is presumably due to the fact that the molecules of the reactants (especially the silane) used in the reaction process are bulkier than those of acetic acid used in relative surface area determination as described in section 4 below. In the case of the micro-system, the acetic acid molecules are expected to penetrate through different pores with ease, whereas the bulkier silane molecules may not enter the smaller pores of clay particles. On the other hand, nano-particles have more open structures for the silane molecules to go through the particles and reach the catalytically active metalloporphyrin sites. The solvent-like behavior is thus anticipated to be more pronounced in the case of the nano-catalyst system, with further enhancement in catalyst activity.
The results suggest that the nano-catalyst system is more favorable than the homogeneous or micro-catalyst systems. Due to its special activity, the results presented hereinafter describe the MnTPyP@Nano-Clay catalyst only, unless otherwise stated.
The effects of different reaction parameters on the rate of hydrosilylation reaction of tri(ethoxy)silane with 1-octene using the MnTPyP@Nano-clay catalyst system are described here.
2.1 Effect of added solvent type
The hydrosilylation reaction was studied in three aprotic solvents, carbon tetrachloride (CCl4), tetrahydrofuran (THF) and 1,4-dioxane, in addition to the internal standard toluene, at 40 °C. The solvent polarity varies as: CCl4 < dioxane < THF. Fig. 2 shows that the reaction rate varied with solvent as CCl4 < THF < dioxane. The figure shows that, when keeping all other factors the same, the reaction rate was affected by the solvent polarity. CCl4, which is a non-polar solvent (dipole moment 0.0 D), exhibited the slowest reaction, with a TF value up to 490 min−1 within the first minute. The solvent THF, with the highest polarity (dipole moment 1.67 D), exhibited a higher reaction rate with a TF value reaching 870 min−1 within the first minute. The dioxane solvent with intermediate polarity (0.45 D) showed the highest reaction rate, with a TF value more than 1180 min−1 within the first minute. Unless otherwise stated, all reactions described hereinafter were performed using dioxane as added solvent. The effect of solvent polarity on reaction rate will be revisited in the mechanism section.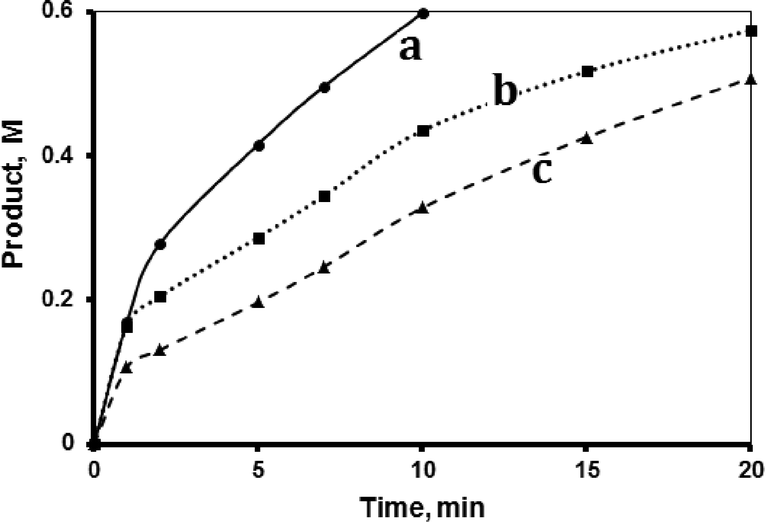 | ||
| Fig. 2 Effect of solvent type. Hydrosilylation reaction profiles showing product formation with time in (a) 1,4-dioxane, (b) THF and (c) CCl4. All reactions were conducted using 1-octene (1.33 M), tri(ethoxy)silane (0.72 M) and fresh MnTPyP@Nano-Clay catalyst (0.500 g supported, net 1.740 × 10−4 M of MnTPyP+) in a total volume of 15.00 mL at 40 °C. | ||
2.2 Effect of temperature
The hydrosilylation reaction was conducted using dioxane as added solvent (in addition to the internal standard, toluene) at different temperatures ranging between 20 and 50 °C. The temperatures are far lower than the boiling points for the used liquids, namely 1,2-dioxane (101 °C); toluene (112); 1-octene (121 °C) and tri(ethoxy)silane (135 °C). The reaction rate increased at higher temperature (Fig. 3). Reactant evaporation should also be ruled out because a condensation column was used to prevent evaporation. The figure shows that the reaction occurred readily with relatively fast rate at relatively ambient temperatures (20–50 °C). At 50 °C, the reaction progressed quickly and was difficult to measure accurately. After reaction cessation, the observed product yield values, based on the silane disappearance, varied with reaction temperature. The measured yield values were 85, 83, 80, 79 and 73% for the samples subjected at 50, 40, 30 and 20 °C, respectively. Unless otherwise stated, all reactions described here were conducted at 40 °C.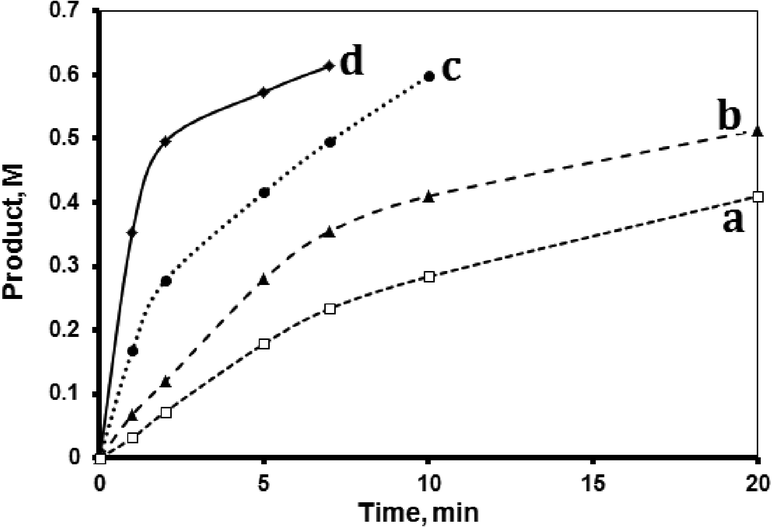 | ||
| Fig. 3 Effect of temperature. Hydrosilylation reaction profiles showing product formation with time at (a) 20 °C, (b) 30 °C, (c) 40 °C, and (d) 50 °C. All reactions were conducted using 1-octene (1.33 M), tri(ethoxy)silane (0.72 M) and fresh MnTPyP@Nano-Clay catalyst (0.500 g supported, net 1.740 × 10−4 M of MnTPyP+) and 1,4-dioxane in a total volume of 15.00 mL. | ||
The value of the activation energy (Eact) was calculated using the Arrhenius equation, by plotting ln(initial rate) vs. 1/T. The value for Eact was ~64 kJ mol−1. This means that the reaction was not merely diffusion controlled, but rather a genuine chemical process with a rate determining step as described below.
2.3 Effect of tri(ethoxy)silane concentration
The hydrosilylation reaction was conducted using different concentrations of tri(ethoxy)silane at 40 °C, while keeping all other parameters the same (Fig. 4). The reaction rate order with respect to the silane was calculated using both the initial rate and the logarithmic methods. | ||
| Fig. 4 Effect of tri(ethoxy)silane concentration. Hydrosilylation reaction profiles showing product formation with time, using different initial concentrations of (a) 1.5, (b) 1.00, (c) 0.72 and (d) 0.36 M. All reactions were conducted using 1-octene (1.33 M) and MnTPyP@Nano-Clay (0.500 g, 1.740 × 10−4 M MnTPYP+ ions) in 1,4-dioxane (enough to make a volume of 15 mL) at 40 °C. | ||
The plot of ln(initial rate) vs. ln[silane] showed the reaction order (n) with respect to the silane being 0.93, which is approximately a first order dependence. The logarithmic relation, assuming a first order reaction with respect to the silane, showed a linear relation, which confirms the first order dependence of the reaction with respect to the silane.
2.4 Effect of 1-octene concentration
The effect of 1-octene concentration on the hydrosilylation reaction was studied at 40 °C by changing the concentration of 1-octene while keeping the other variables constant. Fig. 5 shows the profiles for the reactions using different 1-octene concentrations. Plotting the ln(initial rate) vs. ln[initial 1-octene] showed a slope 0.96, indicating a first order reaction with respect to the octane. The logarithmic method further confirmed the first order behavior.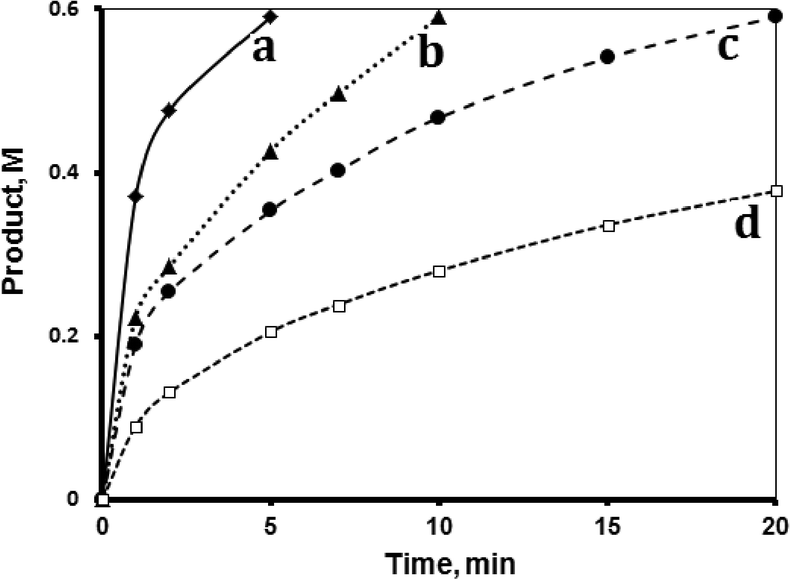 | ||
| Fig. 5 Effect of olefin concentration. Hydrosilylation reaction profiles showing product formation with time using 1-octene concentrations of a) 2.66 M, b) 1.33 M, c) 1.00 M, and d) 0.66 M. All reactions were conducted at 40 °C with tri(ethoxy)silane (0.72 M), fresh MnTPyP@Nano-Clay catalyst (0.500 g supported, net 1.740 × 10−4 M of MnTPyP+) and 1,4-dioxane in a total volume of 15.00 mL. | ||
2.5 Effect of catalyst concentration
The effect of supported MnTPyP+ ion concentration on the reaction rate was studied at 40 °C. Fig. 6 shows that increasing the nominal amount of the catalyst increases the reaction rate. Plotting ln(initial rate) vs. ln[MnTPyP+] showed a slope value of 0.96, which means a first order reaction with respect to the supported MnTPyP+ ions. With more added catalyst, more active sites are available for the reaction to proceed, which increases the initial rate. Such behaviors are common in thermal catalyst system reactions.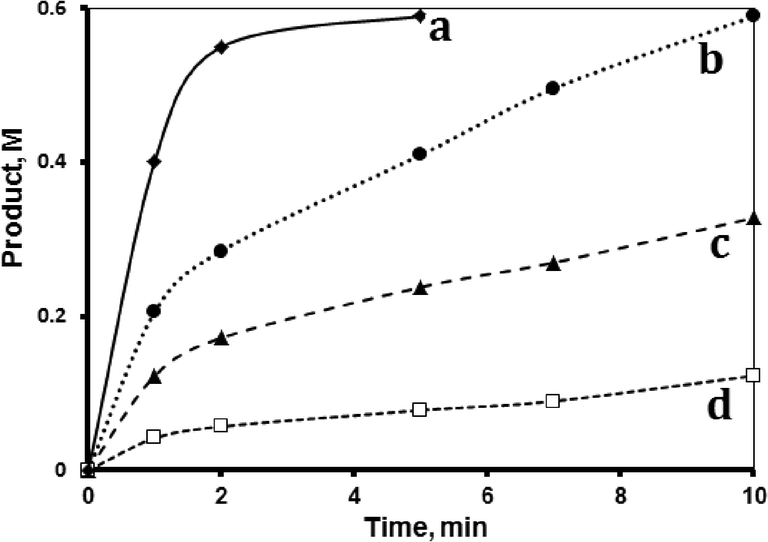 | ||
| Fig. 6 Effect of catalyst concentration. Hydrosilylation reaction profiles showing product formation with time using 1-octene (initial concentration 1.33 M), tri(ethoxy)silane (0.72 M) and different MnTPyP@Nano-Clay amounts making MnTPyP+ ion catalyst concentrations of (a) 3.50 × 10−4 M, (b) 1.75 × 10−4 M, (c) 0.87 × 10−4 M, and (d) 0.35 × 10−4 M. All reactions were conducted in 1,4-dioxane at 40 °C in a total volume of 15 mL. | ||
2.6 Catalyst reproducibility
The MnTPyP@Nano-Clay catalyst was relatively reproducible, when fresh catalyst samples were used in the catalytic experiments under similar conditions. Fig. 7 shows the examples of reproducible reaction profiles measured at 40 °C.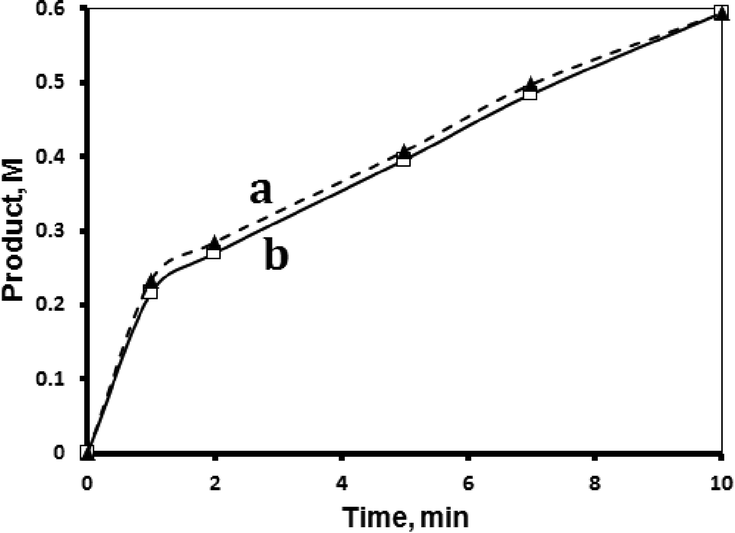 | ||
| Fig. 7 Reproducibility of MnTPyP@Nano-Clay catalyst activity in hydrosilylation reaction. Two different reactions (a) and (b) were conducted using fresh batches of catalyst (0.500 g supported, net 1.740 × 10−4 M of MnTPyP+), tri(ethoxy)silane (0.72 M) and 1-octene (1.33 M) in 1,4-dioxane (total volume of 15 mL) at 40 °C. | ||
2.7 Catalyst recovery and reuse
The MnTPyP@Nano-Clay catalyst was recovered, after cessation of the reaction, and was reused for a fresh reaction mixture. Second time recovery was again performed. Fig. 8 describes the reaction profiles measured for the fresh, first recovered and second recovered catalyst samples. The fresh catalyst showed some loss of activity after the first and second recoveries. Table 4 summarizes the values of TN and TF for the fresh, first recovered and second recovered MnTPyP@Nano-Clay catalyst systems measured after 10 min. The TN value decreased from 3448 (for the fresh sample) to 2814 (for the first recovery) to 2623 (for the second recovery).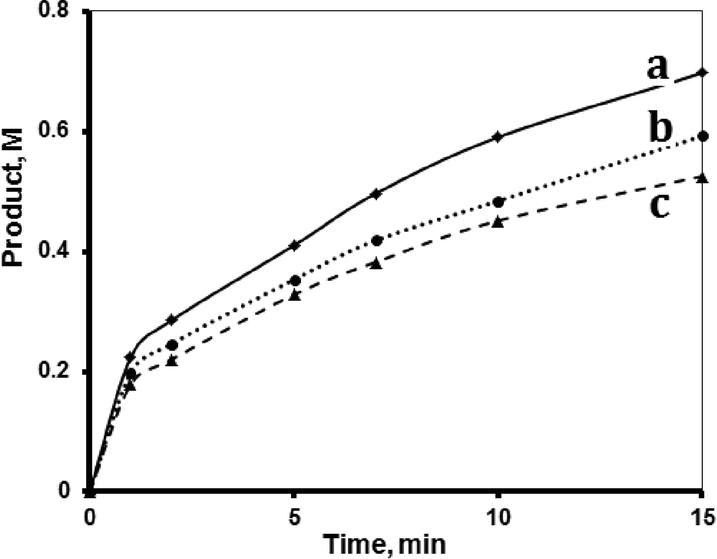 | ||
| Fig. 8 Catalyst recovery and reuse in hydrosilylation reactions. MnTPyP@Nano-Clay (0.5 g, 1.740 × 10−4 M of MnTPyP+) was used as (a) fresh batch, (b) first recovery, and (c) second recovery. All reactions were conducted using fresh batches of tri(ethoxy)silane (0.72 M), 1-octene (1.33 M) in 1,4-dioxane (total volume of 15 mL) at 40 °C. | ||
| Values of TN (and TF, min−1) with time | ||||
|---|---|---|---|---|
| Entry | Catalyst sample | 1 min | 5 min | 10 min |
| 1 | Fresh catalyst | 1184 (1184) | 2299 (460) | 3448 (344.8) |
| 2 | 1st recovery | 1042 (1042) | 1977 (395.4) | 2814 (281.4) |
| 3 | 2nd recovery | 947 (947) | 1816 (263.2) | 2623 (262.3) |
Supported catalysts normally exhibit activity lowering on reuse.2 Such lowering could be due to leaching out of the supported catalyst molecules from the support. This assumption was tested for the supported MnTPyP+ ions present using atomic absorption spectroscopy (AAS). After cessation of the fresh catalytic reaction, the reaction mixture was filtered and the solution phase was analyzed. About 0.13 ppm Mn2+ ion concentration was observed in the solution. By recalculating the amount of MnTPyP+ ions leached from the original MnTPyP@Nano-Clay system, based on this concentration, about 15% of the supported MnTPyP+ ions have leached out. This value is consistent with the activity loss of the fresh catalyst sample on recovery (~18%). Therefore, the loss of activity is not due to any poisoning of the MnTPyP+ ions present. The tendency of supported catalysts to leach out is well-documented in different supported systems. The value of 15% leakage is not high compared to other types of supported catalysts, which may reach up to 40% activity loss on recovery, as reported earlier.43 Work to minimize the tendency of the ions to leach out is underway here.
2.8 A plausible reaction mechanism
To our knowledge, no mechanisms have been proposed for olefin hydrosilylation reactions using metalloporphyrin complex catalysts. A new plausible mechanism for the hydrosilylation reaction of triethoxysilane with 1-octene is thus discussed here, as shown in Scheme 1.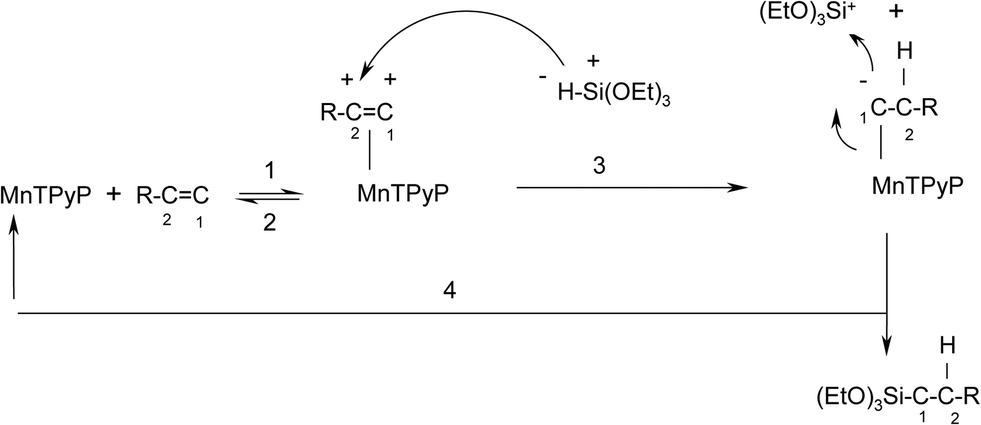 | ||
| Scheme 1 A plausible mechanism for the reaction of tri(ethoxy)silane with 1-octene catalyzed by the MnTPyP+ ion. | ||
The mechanism involves steps that are well-known. Unlike earlier hydrosilylation reaction mechanisms reported for other catalysts,44 all steps here occur at one side of the MnTPyP+ ring only. Steps (1) and (2) are Lewis base association/dissociation processes. Metalloporphyrins are subject to attack by different types of nucleophiles at the central metal ions.45,46 Attack of olefins onto the metal atom of the metalloporphyrin ring in a Lewis base fashion has been proposed earlier.24 Step (3) involves the formation of a single covalent bond between the MnTPyP+ and an alkyl group. Such bond formation is also known.24 Step (3) is a nucleophilic attack by the H atom at the bonded olefin, as discussed below. Step (4) is a nucleophilic attack by the bonded carbon atom at the positively charged Si atom.
The proposed mechanism has evidence from all results discussed. From its kinetics, the reaction is first order with respect to the catalyst, to the olefin and to the silane. The observed rate law can thus be expressed as in eqn (2):
| Rate = kobs[MnTPyP][silane][olefin] | (2) |
| Rate = Rate(3) = k3[MnTPyP–olefin][silane]. | (3) |
| k1[MnTPyP][olefin] = k2[MnTPyP–olefin]. | (4) |
| [MnTPyP–olefin] = k1[MnTPyP][olefin]/k2. | (5) |
| Rate = (k1k3/k2)[MnTPyP][silane][olefin] | (6) |
The mechanism also explains the effect of temperature on the reaction rate. As discussed above, the value of the activation energy (~64 kJ mole−1) shows that the reaction is not merely diffusion controlled. With a rate determining step, it is expected that the overall reaction will be faster by increasing the reaction temperature, as rate(3) should go faster based on the Arrhenius equation.
The proposed mechanism explains the effect of added solvent type on the reaction rate. The results showed that the overall hydrosilylation reaction proceeds faster in solvents that are Lewis bases (DMF and dioxane) than in a non-polar one (CCl4), keeping in mind that the internal standard toluene is not highly polar itself. As step (3) is rate determining, and as it involves dissociation of the silane, solvents with Lewis basic sites should speed up this reaction. However, due to the higher solvent polarity of DMF, the solvent itself competes with the silane to form adducts with the intermediate complex MnTPyP–olefin. In this case, step (3) proceeds slower than expected, and the overall reaction is consequently slowed down. The proposed mechanism predicts faster reactions in the case of solvents having intermediate Lewis basic nature such as dioxane.
Olefin hydrosilylation is normally accompanied by other side reactions such as dehydrosilylation.47,48 In this work, the catalyst showed high selectivity to olefin hydrosilylation. The mechanism explains the regioselectivity of the supported catalyst to yield the linear hydrosilylation product rather than the branched one. Olefin molecules in their free form behave as Lewis bases. The interaction between the free olefin (Lewis base, nucleophile) with the Mn central ion is understandable as discussed above. The bonded olefin molecule readily donates electrons to the electron deficient Mn central ion. The two C atoms (C1 and C2) of the olefin are thus depleted of electrons and carry partial positive charges. Unlike its free counterpart, the bonded olefin molecule behaves as an electrophile. Such inversion in olefin properties is well-documented and is called umpolung behavior.49 Therefore, both C1 and C2 atoms are susceptible to attack by the H atom of the silane molecule, as expected in step (3). This is because that H atom carries a partial negative charge due to the low electronegativity of the Si atom. As a result, one carbon atom bonds to the H atom, while the other becomes bonded to the Mn central atom via a polar covalent bond. As the other carbon atom should now carry a partial negative charge, due to electronegativity difference, it may behave as a nucleophile that can readily interact with the silyl positive ion formed in step (3).
A closer look at step (3) indicates that the H atom may attack at either of the two carbon atoms (C1 and C2). Two factors, the electronic and steric effects, may decide the position of the H atom attack. Based on the electronic factor, the H atom should preferably attack at C1 with higher partial positive charge. This is because the C2 carries two electron donating alkyl groups. As such, the C2 will then be bonded to the silyl group to yield the branched product.
Based on the steric effect, the H atom should attack at C2, leaving C1 bonded to the central Mn ion. Due to geometry around the Mn ion, and due to the size of the R group, such orientation is favored. As such, C1 will be favorably bonded to the silyl group to yield the linear product. The proposed mechanism thus explains the regioselectivity of the hydrosilylation reaction described above.
In the case of the homogeneous MnTPyP+ system, both hydrosilylation reaction products occurred. The regioselectivity was clearly higher in the case of the supported MnTPyP+ catalyst, where the branched product, if any, occurred in immeasurably low concentrations. This is understandable based on the proposed mechanism. In the homogeneous system, the steric effect favors the production of the linear hydrosilylation product, tri(ethoxy)silyl-1-octane, as the major product. However, the steric effect is not strong enough to completely prevent the formation of the branched hydrosilylation product tri(ethoxy)silyl-2-octane, which occurs due to the electronic effect, vide supra.
In the supported MnTPyP+ catalyst system, things are different. The metalloporphyrin ring is encapsulated inside the clay particles. This increases the steric factor. Therefore, the linear tri(ethoxy)silyl-1-octane dominates, and the branched product does not occur in significant concentrations. The proposed mechanism thus successfully explains the special regioselectivity of the supported catalyst.
The mechanism explains the lack of olefin isomerization process, which is well-documented in other catalyst systems.41 Isomerization of 1-octene into other internal olefins has not been observed in this study. For isomerization to occur, the intermediate MnTPyP–olefin should undergo additional steps, one of which is the oxidative addition of the C3–H adduct to the Mn central ion. Such a process is well-known in other catalyst systems and involves π-allyl complex formation.41 Due to the presence of the cyclic ligand around the Mn ion, formation of π-allyl systems must be ruled out as it should involve an intermediate with a coordination number of 7. Therefore, isomerization of 1-octene to internal olefins, such as 2-octene, should not occur in the case of the MnTPyP+ catalyst. As neither homogeneous nor supported MnTPyP+ catalyst systems caused olefin isomerization in this study, the proposed mechanism successfully explains the lack of olefin isomerization products. Involvement of metalloporphyrins in olefin isomerization reactions of different nature, has been reported23,24 but with no need for high coordination numbers or π-allyl intermediates.
The clay-supported MnTPyP+ exhibited higher activity than the homogeneous counterpart. As discussed earlier, the clay support exhibits solvent-like behavior to bring the reactants into close proximity. This enhances the catalyst activity of the MnTPyP+ ions in olefin hydrosilylation reactions. The results show the added value of using layered clay as support for metalloporphyrin catalyst in such reactions.
3. Experimental
Common solvents and starting materials were purchased from Aldrich, Merck or Riedel de Haen, in pure form.An ICE 3000 AA spectrometer from Thermo Scientific was used for atomic absorption spectral (AAS) analysis. A Scientific Model Nicolet IS5 (ASB 1200315) FTIR-spectrophotometer, from Thermo Fisher, was used to record the FT-IR spectra. 1H and 13C NMR spectra were recorded on a Bruker AVANCE-500 (500 MHz) equipment in the laboratories of the University of Jordan, Amman, Jordan. CDCl3 was used as the solvent.
Scanning Electron Microscopy (SEM) was performed on a Jeol Model JSM-6700 F microscope (King Saud University, Riyadh, Saudi Arabia). X-Ray Diffraction (XRD) patterns were recorded using a Philips XRD X'PERT PRO diffractometer, equipped with a Cu Kα (λ1.5418A) source (King Saud University, Riyadh, Saudi Arabia) as described earlier.38
The preparation of tetra(4-pyridyl)porphyrinato-manganese(III) sulfate [MnIII(TPyP)]+ was described in earlier reports.2,7,13,50 5,10,15,20-Tetra(4-pyridyl)21H,23H-porphine (490 mg, 0.792 mmol) and excess MnSO4·H2O (0.900 g, 5.45 mmol) were refluxed in N,N-dimethylformamide (DMF) (180 ml) with magnetic stirring under air for 10 h. The UV-visible spectra confirmed the product formation in DMF showing the characteristic three bands at 569, 512 and 463 nm (Soret). The solvent was evaporated by suction under air to confirm oxidation of manganese. Column chromatography was used, using neutral alumina (Bio-Rad, AGF, 100–200 mesh), to isolate the product. A mixture of methanol/chloroform (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 vol/vol) was used as eluant. The eluate was taken and dried under reduced pressure at room temperature to yield the MnTPyP+ product (480.00 mg, 0.669 mmol).
:85 vol/vol) was used as eluant. The eluate was taken and dried under reduced pressure at room temperature to yield the MnTPyP+ product (480.00 mg, 0.669 mmol).
Pre-calcinated natural clay, collected from the Northern areas of the Palestinian Territories, was ground and sieved to 30–100 mesh, collected and cleaned with excess HCl (50% vol) for 24 h. The cleaned white solid was then filtered and rinsed with excess deionized water until neutral. Mg2+, Fe2+/Fe3+, Ca2+ and other ions were detected in the filtrate acid solution. After drying at 120 °C for 2 h, the cleaned solid was stored in a desiccator.
Details of intercalation of MnTPyP+ inside the clay were also described.38 MnTPyP@Nano-Clay was prepared by refluxing a methanolic solution of MnTPyP+ (100 mL, 4.17 × 10−3 M) and pre-cleaned clay (15.0 g) with vigorous magnetic stirring for 30 hours under dry atmosphere. The solid material was then carefully filtered and rinsed with methanol to remove any remaining free or surface-adsorbed MnTPyP+ ions. MnTPyP@Micro-clay was prepared by the same technique using reflux with vigorous stirring for a shorter time (6 h).
MnTPyP+ ion intercalation was confirmed by different methods, as described earlier.38 The MnTPyP+ FT-IR spectra exhibited shifts in two major bands from 1656 and 1388 cm−1 (for the homogeneous system) to 1650 and 1385 cm−1 (for the intercalated system). Similarly, the electronic absorption spectra exhibited shifts in the Soret band from 462 cm−1 (for the homogeneous system) to 470 and 490 cm−1 (for the intercalated system). The XRD patterns showed expansions in the montmorillonite interplanar distances from 13.4 Å (for the naked clay) to 14.7 Å, 17.3 Å and 23.9 Å for horizontal, diagonal and perpendicular intercalations, respectively.38 In the case of biotite, the interlayer spacing expanded from 8.6 Å to 9.8 Å, which indicates only the horizontal intercalation of the MnTPyP+ ions. Collectively, the characterization results confirmed the intercalation of the MnTPyP+ ions inside montmorillonite and biotite with different geometries.38 The specific surface area values were measured using the acetic acid adsorption method51 for each powder, and the values were 90, 130 and 200 m2 g−1 for the naked clay, MnTPyP@Micro-Clay and MnTPyP@Nano-Clay powders, respectively, as described earlier.38 SEM micrographs showed average particle size values of 625, 316 and 50 nm for the naked clay, MnTPyP@Micro-Clay and MnTPyP@Nano-Clay solids, respectively. The MnTPyP+ ion uptake values for the nano-size and micro-size clay powders were measured by AAS and were 3.8 mg and 2.4 mg of MnTPyP+ per g of solid, respectively.
Catalytic experiment
The hydrosilylation reaction experiment was performed inside a 100 mL three-necked flask (reaction vessel). The flask was equipped with stoppers, a thermostated bath, a cooled refluxing condenser (to prevent evaporation), a magnetic stirrer and a CaCl2 drying tube outlet to prevent contamination with moisture.In a typical catalytic experiment, an internal reference (toluene, 5.00 ml) was mixed with known amounts of 1-octene, tri(ethoxy)silane, catalyst and added solvent (enough to make the total volume 15.0 ml) inside the stoppered reaction vessel. The temperature was controlled as desired using a thermostated bath with stirring. Aliquots were syringed out of the reaction mixture, and immediately filtered to the remove catalyst. The IR spectra were measured, relative to the toluene internal standard, for each aliquot and compared with pre-constructed calibration curves for the 1-octene, the silane and the product.
The reaction progress was followed with time using FT-IR spectral analysis of aliquots syringed out from the reaction mixture with time. The intensity decay of the bands at 2220 cm−1 (characteristic for the Si–H bond) and 1641 cm−1 (for the CC bond) and the building up of the band at 1253 cm−1 (for the terminal C–Si bond), were measured relative to the band at 1604 cm−1 (for the internal reference, toluene).
Production of tri(ethoxy)silyl-1-octene was confirmed by isolating the product in its pure form using distillation under reduced pressure. In addition to the FT-IR analysis, the isolated product was confirmed by NMR spectroscopy as discussed in the Results and Discussion section above.
Control experiments made using no catalyst showed no hydrosilylation reaction. Naked clay powders with no MnTPyP+ ions caused some adsorption of reactants with no hydrosilylation. Other control experiments were performed using naked clay powder mixed with MnTPyP+ ions, with no intercalation, and the reaction progress resembled that of homogeneous MnTPyP+ ions only. Mixing the MnTPyP@Nano-Clay with the silane reactant in the absence of 1-octene showed loss of silane, by adsorption, with no reaction as discussed in section 3 above.
Catalyst recovery was performed by filtering the supported catalyst. The recovered catalyst was reused for fresh batches of reactants.
4. Conclusion
The hydrosilylation reaction of 1-octene with tri(ethoxy)silane can be selectively catalyzed by tetra(4-pyridyl)porphyrinato-manganese(III) ions (MnTPyP+) with no isomerization side product. Catalyst activity and selectivity to produce the linear hydrosilylation product tri(ethoxy)silyl-1-octane have been dramatically enhanced by supporting the MnTPyP+ ions inside layered clay supports. The values of the supported catalyst turnover number and frequency are many times higher than those measured for the homogeneous system. The supported catalyst showed much higher activity than the other catalysts with other supports reported earlier. Both catalyst activity and selectivity are rationalized based on the solvent-like behaviour of the support. Moreover, the supported catalyst was easy to recover after reaction cessation by simple filtration, without high loss of its activity on reuse. The results show the future potential application value of the supported catalyst described here.Acknowledgements
Many results described here are based on W. Mansour's thesis, under the direct supervision of H. S. Hilal and W. Jondi, in the laboratories of An-Najah National University. A. Zyoud performed additional experimental and characterization activities. A. Q. Hussein performed NMR measurements in the laboratories of the University of Jordan. XRD and SEM measurements were conducted in the laboratories of King Saud University, Saudi Arabia. The authors wish to acknowledge the help from the technicians in the Department of Chemistry, ANU. Financial support from the Al-Maqdisi Project and from the Union of Arab Universities is also acknowledged.References
- R. V. Gholap, O. M. Kut and J. R. Bourne, Ind. Eng. Chem. Res., 1992, 31, 2446–2450 CrossRef CAS.
- H. S. Hilal, W. Jondi, S. Khalaf, A. Keilani, M. Suleiman and A. Schreiner, J. Mol. Catal. A: Chem., 1996, 113, 35–44 CrossRef CAS.
- J. Hagen, Industrial catalysis: a practical approach, John Wiley & Sons, 2006 Search PubMed.
- A. Z. Fadhel, P. Pollet, C. L. Liotta and C. A. Eckert, Molecules, 2010, 15, 8400–8424 CrossRef CAS PubMed.
- H. S. Hilal, M. A. Suleiman, W. J. Jondi, S. Khalaf and M. M. Masoud, J. Mol. Catal. A: Chem., 1999, 144, 47–59 CrossRef CAS.
- F. Rao, S. Deng, C. Chen and N. Zhang, Catal. Commun., 2014, 46, 1–5 CrossRef CAS.
- A. Nuzzo and A. Piccolo, J. Mol. Catal. A: Chem., 2013, 371, 8–14 CrossRef CAS.
- J. B. Nicholas, Top. Catal., 1999, 9, 181–189 CrossRef CAS.
- B. C. Gates, Catalytic chemistry, Wiley, New York, 1992 Search PubMed.
- J. Dwyer, H. Hilal and R. Parish, J. Organomet. Chem., 1982, 228, 191–201 CrossRef CAS.
- H. S. Hilal, S. Khalaf, M. Al Nuri and M. Karmi, J. Mol. Catal., 1986, 35, 137–142 CrossRef CAS.
- H. S. Hilal, M. Abu-Eid, M. Al-Subu and S. Khalaf, J. Mol. Catal., 1987, 39, 1–11 CrossRef CAS.
- H. Hilal, C. Kim, M. Sito and A. Schreiner, J. Mol. Catal., 1991, 64, 133–142 CrossRef CAS.
- H. Hilal, M. Sito and A. Schreiner, Inorg. Chim. Acta, 1991, 189, 141–144 CrossRef CAS.
- Y. R. de Miguel, E. Brulé and R. G. Margue, J. Chem. Soc., Perkin Trans. 1, 2001, 3085–3094 RSC.
- K. M. Smith and J. E. Falk, Porphyrins and metalloporphyrins, Elsevier, Amsterdam, 1975 Search PubMed.
- H. R. A. Golf, H.-U. Reissig and A. Wiehe, J. Org. Chem., 2015, 5133–5143 CrossRef CAS PubMed.
- G. Simonneaux, P. Le Maux, D. Carrie and S. Chevance, Asymmetric Oxidation and Carbene Transfer Reactions Catalyzed By Metalloporphyrins in Water, Abstract MA2015-01 966, ECS Meeting Abstracts, Porphyrin Supramolecular Assemblies 2 - May 25 2015, http://ma.ecsdl.org/content/MA2015-01/11/966.abstract Search PubMed.
- K. M. Kadish, L. Frémond, F. Burdet, J.-M. Barbe, C. P. Gros and R. Guilard, J. Inorg. Biochem., 2006, 100, 858–868 CrossRef CAS PubMed.
- J. P. Collman, R. Boulatov, C. J. Sunderland, I. M. Shiryaeva and K. E. Berg, J. Am. Chem. Soc., 2002, 124, 10670–10671 CrossRef CAS PubMed.
- M. Hajimohammadi and N. Safari, J. Porphyrins Phthalocyanines, 2010, 14, 639–645 CrossRef CAS.
- M. L. Merlau, W. J. Grande, S. T. Nguyen and J. T. Hupp, J. Mol. Catal. A: Chem., 2000, 156, 79–84 CrossRef CAS.
- K. Maruyama and H. Tamiaki, J. Inorg. Chem., 1986, 51, 602–606 CAS.
- C.-Y. Zhou, P. W. H. Chan and C.-M. Che, Org. Lett., 2006, 8, 325–328 CrossRef CAS PubMed.
- W. Mori, S. Takamizawa, C. N. Kato, T. Ohmura and T. Sato, Microporous Mesoporous Mater., 2004, 73, 31–46 CrossRef CAS.
- N. W. Kamp and J. R. L. Smith, J. Mol. Catal. A: Chem., 1996, 113, 131–145 CrossRef CAS.
- Y. Murakami and K. Konishi, J. Am. Chem. Soc., 2007, 129, 14401–14407 CrossRef CAS PubMed.
- R. Rahimi, S. Z. Ghoreishi and M. G. Dekamin, Monatshefte für Chemie - Chemical Monthly, 2012, 143(7), 1031–1038 CrossRef CAS.
- E. Zampronio, M. Gotardo, M. Assis and H. Oliveira, Catal. Lett., 2005, 104, 53–56 CrossRef CAS.
- H. Bönnemann, Appl. Organomet. Chem., 2008, 22, 412 CrossRef.
- F. Bedioui, Coord. Chem. Rev., 1995, 144, 39–68 CrossRef CAS.
- H. H. Murray, Applied clay mineralogy: occurrences, processing and applications of kaolins, bentonites, palygorskitesepiolite, and common clays, Elsevier, 2006 Search PubMed.
- F. Uddin, Metall. Mater. Trans. A, 2008, 39, 2804–2814 CrossRef.
- Z. Tong, T. Shichi, G. Zhang and K. Takagi, Res. Chem. Intermed., 2003, 29, 335–341 CrossRef CAS.
- G. Nagendrappa, Appl. Clay Sci., 2011, 53, 106–138 CrossRef CAS.
- J. M. Fraile, J. I. García, J. A. Mayoral and T. Tarnai, Tetrahedron: Asymmetry, 1998, 9, 3997–4008 CrossRef CAS.
- S. Ray, G. Galgali, A. Lele and S. Sivaram, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 304–318 CrossRef CAS.
- A. Zyoud, W. Jondi, W. Mansour, M. A. Majeed Khan and H. S. Hilal, Chem. Cent. J., 2015 Search PubMed , submitted.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- H. Renner, G. Schlamp, I. Kleinwächter, E. Drost, H. M. Lüschow, P. Tews, P. Panster, M. Diehl, J. Lang and T. Kreuzer, Ullmann's Encyclopedia of Industrial Chemistry, 1992 Search PubMed.
- H. S. Hilal, S. Khalaf and W. Jondi, J. Organomet. Chem., 1993, 452, 167–173 CrossRef CAS.
- K. M. Kadish, K. M. Smith and R. Guilard, The porphyrin handbook, Elsevier, 1999 Search PubMed.
- E. S. Bickford, S. Velu and C. Song, Prepr. Pap. - Am. Chem. Soc., Div. Fuel Chem., 2004, 49, 649 CAS.
- B. Marciniec, in Hydrosilylation, Springer, 2009, pp. 3–51 Search PubMed.
- P. Zucca, A. Rescigno, A. C. Rinaldi and E. Sanjust, J. Mol. Catal. A: Chem., 2014, 388, 2–34 CrossRef.
- X. Huang, K. Nakanishi and N. Berova, Chirality, 2000, 12, 237–255 CrossRef CAS PubMed.
- B. Marciniec, A. Kownacka, I. Kownacki, M. Hoffmann and R. Taylor, J. Organomet. Chem., 2015, 791, 58–65 CrossRef CAS.
- H. Dong, Y. Jiang and H. Berke, J. Organomet. Chem., 2014, 750, 17–22 CrossRef CAS.
- A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137(22), 7224–7230 CrossRef CAS PubMed.
- A. Zyoud, R. S. Al-Kerm, R. S. Al-Kerm, W. Mansur, M. H. S. Helal, D. Park, G. Campet, N. Sabli and H. S. Hilal, Electrochim. Acta, 2015, 174, 472–479 CrossRef CAS.
- A. El-Hamouz, H. S. Hilal, N. Nassar and Z. Mardawi, J. Environ. Manage., 2007, 84, 83–92 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5re00010f |
| This journal is © The Royal Society of Chemistry 2016 |