
Progress in reactor engineering of controlled radical polymerization: a comprehensive review
Xiaohui
Li
ab,
Erlita
Mastan
b,
Wen-Jun
Wang
a,
Bo-Geng
Li
a and
Shiping
Zhu
*b
aCollege of Chemical and Biological Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, PR China
bDepartment of Chemical Engineering, McMaster University, Hamilton, Ontario L8S 4L7, Canada. E-mail: shipingzhu@mcmaster.ca
First published on 24th November 2015
Abstract
Controlled radical polymerization (CRP) represents an important advancement in polymer chemistry. It allows synthesis of polymers with well-controlled chain microstructures. Reactor engineering is essential in bringing lab-scale chemistry to industrial realization. This paper reviews the research progress in reactor engineering of CRP, namely, atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer radical polymerization (RAFT), and nitroxide-mediated or stable free radical polymerization (NMP or SFRP). Research activities in semi-batch reactors, tubular reactors, and continuous stirred-tank reactors (CSTR) of both homogeneous (bulk and solution) and heterogeneous (emulsion, mini-emulsion, heterogeneous catalyst, etc.) CRP systems are summarized. Typical examples are selected and discussed in detail. Perspectives on the current status and future development are also provided.
1. Introduction
Free radical polymerization is one of the most commonly employed processes for large-scale production of polymers, owing to its versatility in polymerizing a wide range of monomer types under facile conditions. However, in a conventional free radical polymerization (FRP) system, a polymer chain grows to completion in a matter of seconds. The almost-instantaneous growth makes it difficult to impose control over the polymer architectures. In addition, each polymer chain experiences different growth environments, depending on the time it is initiated during the polymerization period. This different experience coupled with the presence of various side reactions, such as termination and transfer, result in polymers possessing a broad molecular weight distribution (MWD).1,2 Meanwhile, there is a growing market demand for polymers with well-controlled architectures and narrow MWD to obtain highly tailored properties required in specific applications.Living polymerization was discovered by Szwarc in 1956.3 The advent of living polymerization, including anionic4–7 and cationic,8–10 has made it possible to synthesize polymer materials with well-controlled and complex architectures. In an ideal living polymerization system, all polymer chains are initiated at the beginning of polymerization and continue to propagate throughout the whole polymerization period, providing the same experience for all chains. The side reactions, such as chain transfer and termination, are absent in this system when conducted under appropriate conditions. As a result, the polymers synthesized by living polymerization possess narrow MWD. However, living polymerization has two main disadvantages: (1) only a limited variety of monomers can be polymerized through this mechanism (due to incompatibility between the active center of propagating chains and functionalized monomers); (2) the required experimental conditions are stringent to avoid undesirable side reactions (water, oxygen, and other impurities must not be present). These two disadvantages severely limit the use of living polymerization in industry.
Researchers have been trying to combine the advantages of FRP and living polymerization.11–14 As a result, controlled radical polymerization (CRP) was discovered in the 1980's. CRP, also referred to as reversible deactivation radical polymerization (RDRP),15 provides a middle ground between these two extremes of chain-growth polymerization mechanisms. In CRP, polymerization proceeds in a controlled manner, with active chains growing throughout the polymerization, along with suppression of side reactions. The improved livingness and control in CRP, when compared to FRP, produces narrowly distributed polymer chains, which are expected to have MWD values between those produced by living polymerization and FRP.16,17 This method also allows polymer chains to be extended to form block copolymers more efficiently. Furthermore, because the lifetime of propagating radical chains is extended from a matter of seconds to hours, this allows design and control of detailed chain microstructural properties from end to end along the backbone, by various engineering means. It also provides sufficient time for polymer chains to come into contact with each other, which is especially useful for branching and cross-linking polymerization. Therefore, CRP has opened a path to control polymer architectures. Polymer materials with controlled MW and narrow MWD, along with well-defined architectures, have been extensively synthesized by CRP in homogeneous and heterogeneous systems.2,18–21
What's surprising when conducting a literature review in CRP is the big gap between the limited industrial applications and the extensive research works in the lab.22 The majority of research works on CRP are conducted in small-scale batch reactors, usually in round-bottom flasks. Reactor engineering of CRP can help overcome this gap and bridge lab-scale research to industrial applications.23 The use of various reactor configurations must be evaluated for the different CRPs and targeted products to ensure that appropriate choices are made. There have been some major works done in utilizing reactor engineering concepts to further the development of CRP. The pioneering work on investigating the use of semi-batch reactors for CRP was reported by Matyjaszewski's group in 1997 under constant comonomer feeding rate operation.24–26 Their work demonstrated that a semi-batch reactor allows better control over copolymer composition distribution (CCD) than a batch reactor. However, constant feeding rate operation still lacks versatility to control CCD and to tailor the synthesis of copolymers with more sophisticated CCD. Zhu's group first introduced programmed feeding rate operation for CRP in a semi-batch reactor, whereby the comonomer feeding rate was controlled according to a mathematical model.27–33 By using the model-based design to vary the feeding rate, the CCD could be designed at will and tailored as required to a higher degree of precision. The development of CRP into continuous processes was pioneered by Zhu's group.34–36 This first work for continuous CRP utilized packed column reactors in order to overcome the drawback of batch reactors and to eliminate the need for costly catalyst separation.
In this section, the basic reaction mechanisms of various types of CRP will be briefly discussed, followed by introduction of the reactor types commonly employed in the polymer industry. The last part of this section provides an outline of this review.
1.1. Controlled radical polymerization (CRP)
CRP can be divided into three main types based on the reaction mechanism:22,37,38 (1) reversible deactivation by atom transfer;37,39–41 (2) reversible deactivation by degenerative transfer;42–45 and (3) reversible deactivation by coupling.46–48 The three most studied types of CRP following each of these mechanisms are atom transfer radical polymerization (ATRP),37,39–41 reversible addition–fragmentation chain transfer (RAFT) polymerization,42,49–51 and nitroxide-mediated polymerization (NMP).46,52,53 For brevity's sake, this review only covers these three types of CRP.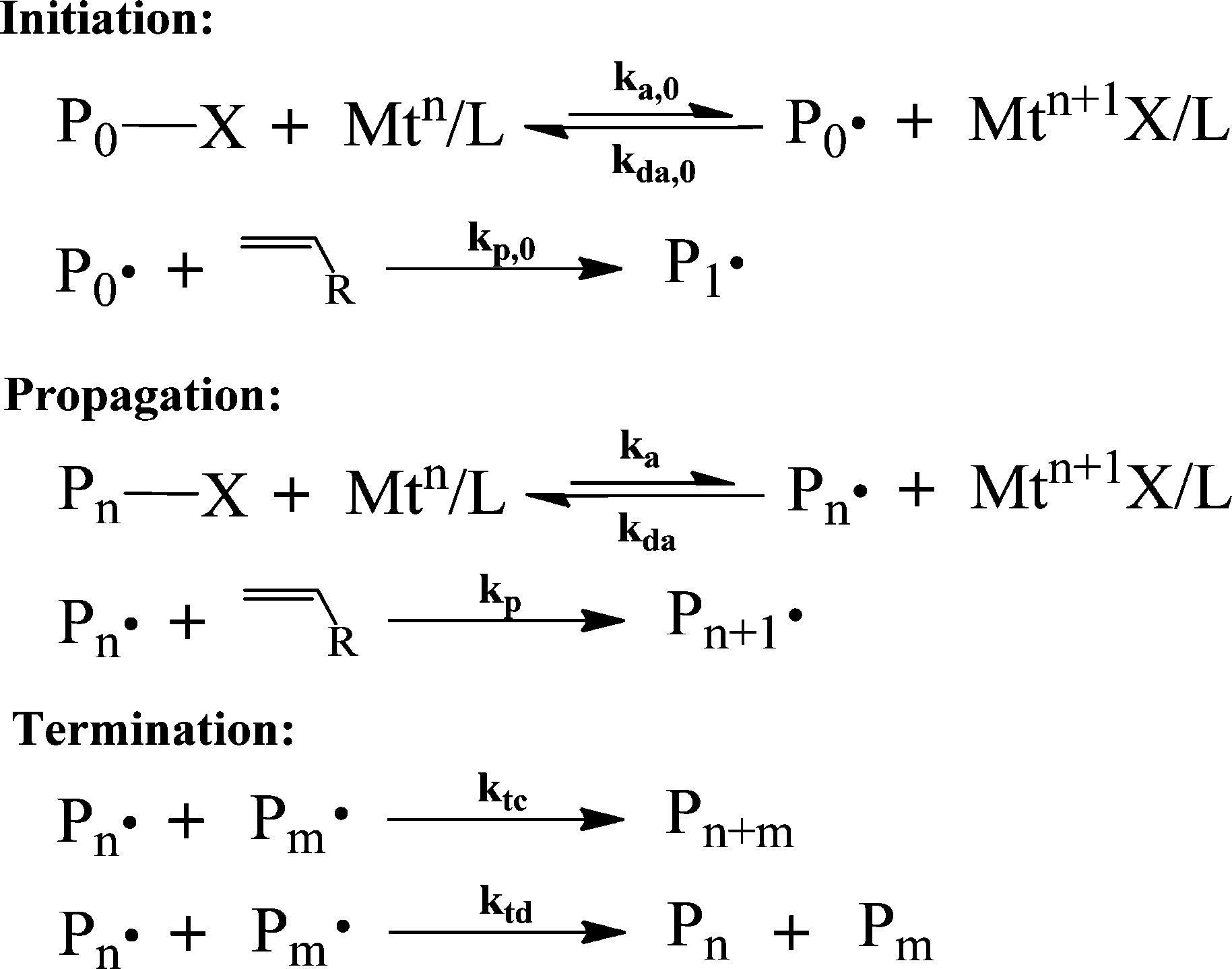 | ||
| Scheme 1 The general mechanism of ATRP.37,54,55 | ||
During the activation–deactivation of dormant-radical pairs, the metal complex, Mtn/L, forms a higher oxidation state complex (Mtn+1X/L). The metal complexes, Mtn/L and Mtn+1X/L, consist of a transition metal (Mt) and a ligand (L), which is commonly nitrogen-based. The transition metal that is commonly used in ATRP is copper (Cu), however other metals (e.g., Fe, Ru, Pd) have also been used.55,56 These metal complexes act as a catalyst and a deactivator in the ATRP system.
As a result of the activation–deactivation reactions, a dynamic equilibrium between P·n and PnX is established. In this equilibrium, the deactivation reaction is much more favored than the activation reaction (kda ≫ ka), resulting in a much higher concentration of dormant chains than that of propagating radical chains. In addition to these reactions, the propagating radical chains can also be terminated by either combination (ktc) or disproportionation (ktd) to form dead chains.
ATRP has played a very important role in the development of polymer chemistry to produce highly tailored polymer architectures. However, the high catalyst loading and residual transition metal present in the final polymer products limit the commercial exploitation of ATRP, due to the high post-polymerization separation cost needed. Therefore, one of the main questions in ATRP commercialization is how to reduce the amount of copper, while maintaining a moderate polymerization rate and an acceptable level of control. In recent years, researchers have developed a series of modified ATRPs requiring a smaller amount of catalyst to overcome this problem. The new ATRP are: (1) simultaneous reverse and normal initiation (SR&NI);57 (2) activator generated by electron transfer (AGET);58,59 (3) initiators for continuous activator regeneration (ICAR);60–62 (4) activators regenerated by electron transfer (ARGET);63–66 (5) single-electron transfer-living radical polymerization (SET-LRP);67–71 (6) electrochemically mediated ATRP (eATRP)72 and photochemically mediated ATRP (photoATRP).73 Albeit being referred to as SET-LRP in some works, Matyjaszewski's group used both experimental and simulation approaches to show that the polymerization occurs based on the mechanism of supplemental activator and reducing agent (SARA) ATRP, not the SET-LRP mechanism.74 However, the term SET-LRP will be used in this review to maintain consistency with the original articles. Among these new variants of ATRP, normal ATRP, ARGET ATRP, and SET-LRP are the most commonly used in semi-batch and continuous reactors. Several universities and companies have participated in the commercialization effort of ATRP.22,75
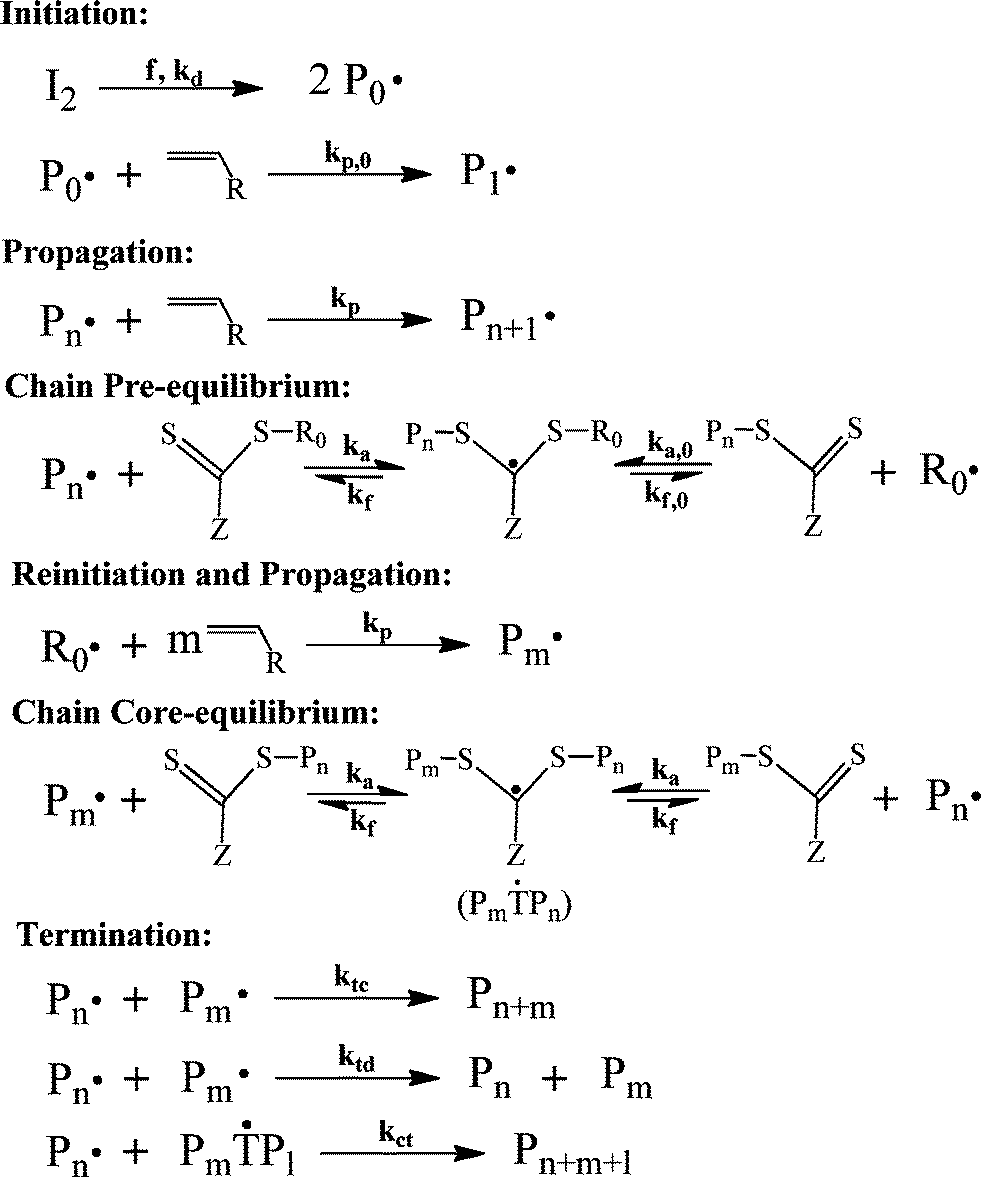 | ||
| Scheme 2 The general mechanism of RAFT.42,79 | ||
There are two theories in choosing the value of reaction rate constants in RAFT polymerization, namely slow fragmentation and intermediate termination theories.80–83 As the name suggests, the slow fragmentation theory proposes that the fragmentation reaction of the intermediate radical occurs at a slow rate. Moreover, this theory assumes that the intermediate radical does not undergo any other reaction but fragmentation. This results in a long lifetime of the intermediate radicals (i.e., kct = 0 and low kf).80 On the other hand, the intermediate termination theory proposes that the intermediate radical chains cannot propagate with monomers, but undergo fast fragmentation and can also cross terminate with propagating radical chains (i.e., kct ≠ 0 and high kf).81,83 Therefore, dead chains are generated by self-termination of propagating radical chains (kt) and cross-termination between propagating radical chains and intermediate radical chains (kct).79
Compared to the ATRP system, conducting polymerization by RAFT is simpler. A controlled polymerization can usually be obtained by a simple addition of RAFT agents into the conventional free radical polymerization system. Furthermore, some RAFT agents have already been made commercially available.22 Similar to ATRP, industrial development of RAFT technologies has also been reported.84–86
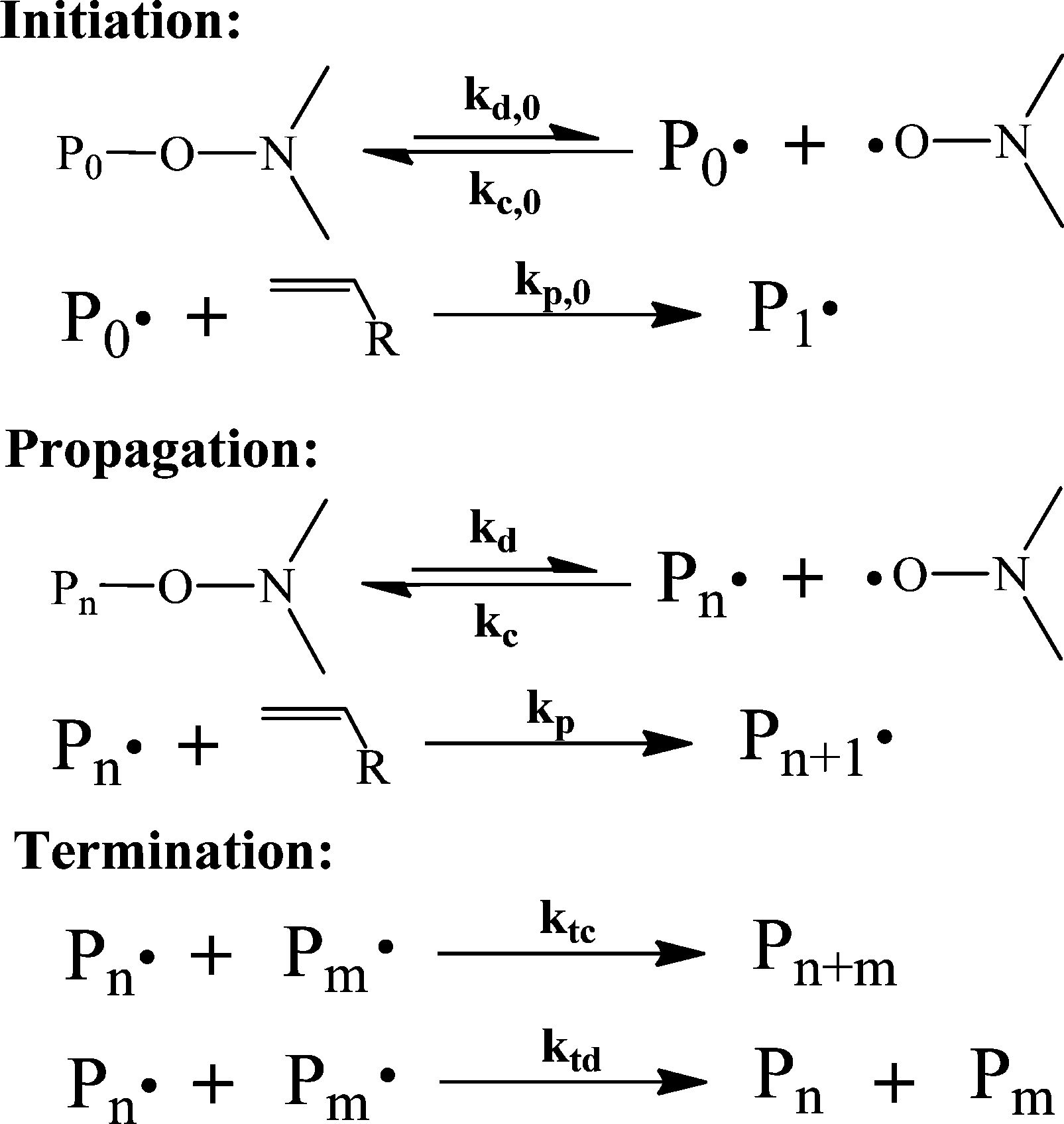 | ||
| Scheme 3 The general mechanism of NMP.88 | ||
By comparing the mechanisms of ATRP, RAFT and NMP, it is clear that the core of CRP lies in the equilibrium or reversible transfer between dormant and propagating radical chains. Owing to this reversible transfer, propagating chains have a longer lifetime, i.e., continuous growth throughout the course of polymerization. Moreover, termination is suppressed and all the polymer chains grow at the same time, and are thus exposed to the same growth environment. This results in a linear growth of molecular weight (MW) with respect to conversion and a narrow molecular weight distribution (MWD).
1.2. Reactor type
The type of reactors used (continuous, semi-batch, and batch reactors) has a significant impact on the polymerization behavior and the resulting polymer properties, such as MWD and copolymer composition distribution (CCD). This is because different reactors have different residence time distributions and concentration profiles, as shown in Scheme 4. Therefore, one must carefully consider the reactor type (or the use of multiple reactors in various configurations) in order to efficiently and precisely synthesize various polymers. For example, the semi-batch reactor is widely used in the production of copolymers to avoid composition drifting. The semi-batch reactor is not included in Scheme 4 because of the endless possible curves that can be obtained by varying the flow rate.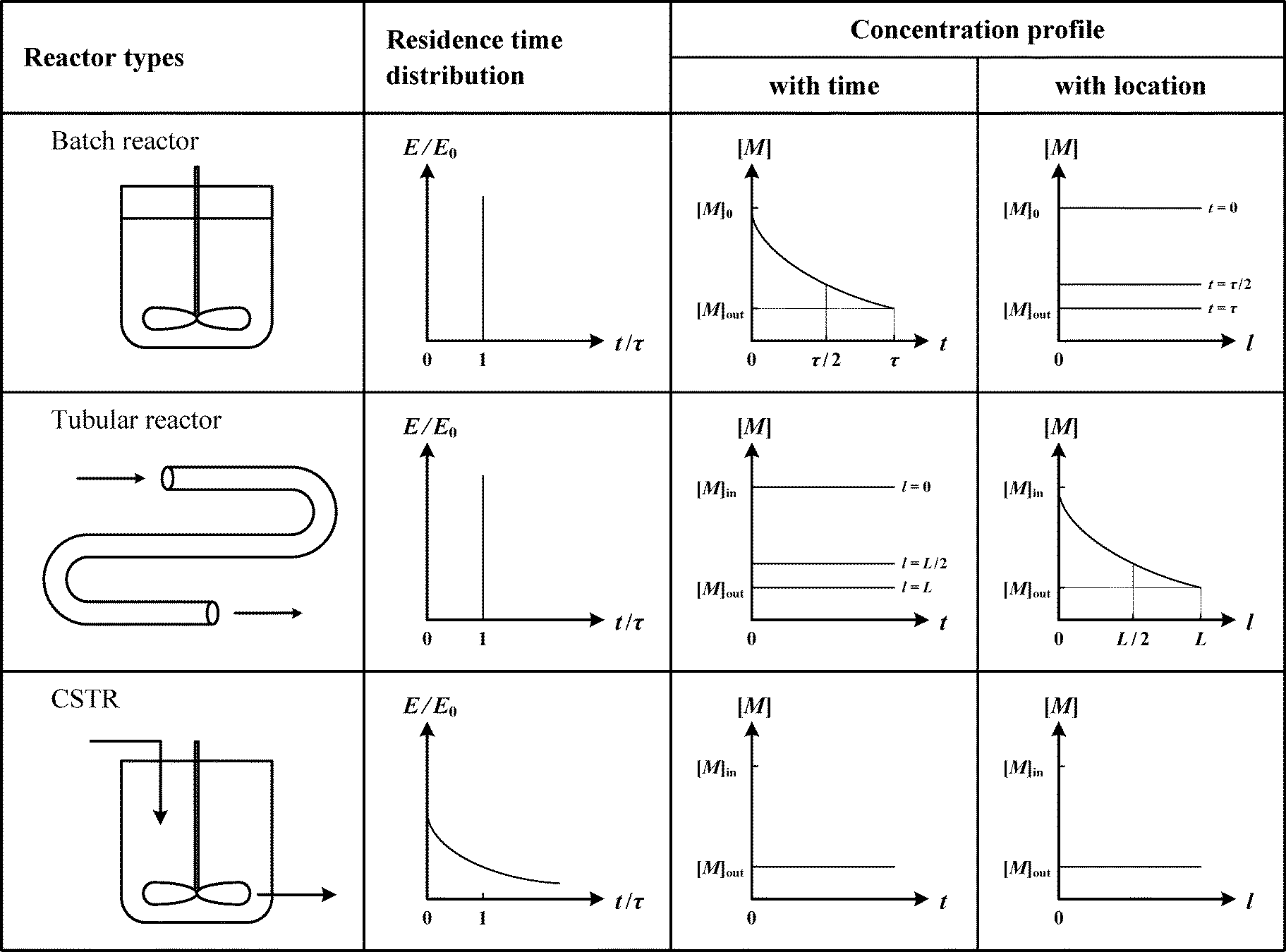 | ||
| Scheme 4 Residence time distribution (RTD) and concentration profiles of ideal reactors. The symbols t, τ, l, and L represent the reaction time, residence time, location inside the reactor, and total length of the tubular reactor, respectively. E/E0 and [M] represent the fraction of the reaction mixture having residence time between t and (t + dt) and the concentration of a reactant, respectively. | ||
Ideal continuous reactors that are usually considered in the polymerization industry are the tubular reactor (also referred to as the plug-flow tubular reactor, PFTR) and the continuous stirred tank reactor (CSTR). The residence time distribution (RTD) is an important parameter for continuous reactors, because it greatly influences the resulting MWD. The effect of RTD depends on the residence time (RT) of the polymer chain relative to the time needed to form a complete chain, i.e., it depends on the polymerization mechanism. For FRP, an individual chain completes in seconds, which means that the RT of the polymer chain is much longer than the time needed for that chain to fully grow. Therefore, the MW of an individual chain is not affected by its RT. The MWD of the instantaneous chain population generated at a small time interval is not affected much by the RTD. However, for CRP, the RT of the polymer chain is smaller than the formation time of the chain, since polymer chains continue to grow as long as they are in the reactor. Therefore, the RTD has a strong impact on the resulting MWD for CRP.
The type of reactors not only affects the MWD of polymers, but also affects the resulting CCD. It is well known that CCD is mainly dependent on the mole ratio of two monomers and their reactivities, and therefore varies with reactor types due to different concentration profiles (see Scheme 4). In a batch reactor or in a tubular reactor, for a given feeding ratio of two monomers, “drifting” in CCD is usually observed due to different monomer reactivities. However, in CSTR, CCD is uniform along the polymer chain due to the constant mole ratio of two monomers at any given instance.
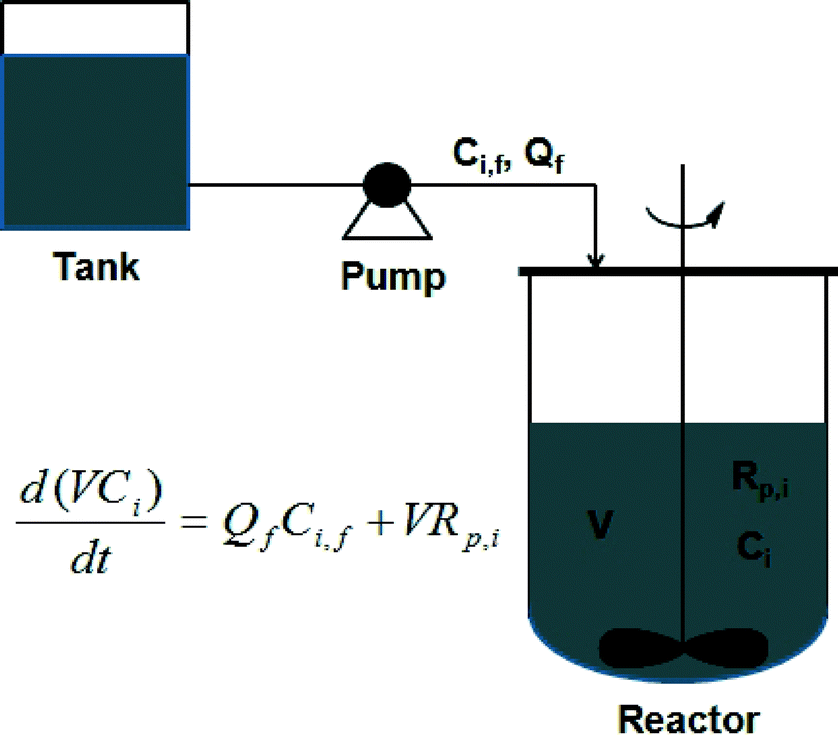 | ||
| Scheme 5 Schematic of a semi-batch reactor. | ||
One of the main advantages of using a semi-batch reactor is the ability to control the copolymer composition distribution (CCD). In a batch reactor, CCD drifts due to different reactivities of monomers. On the other hand, in a semi-batch reactor, the problem of CCD drifting can be readily solved by feeding comonomers into the reactor to maintain a fixed concentration ratio of monomers in the reactor. Furthermore, similar to linear copolymerization, monomer feeding is also effective to control topologies in nonlinear copolymerization.
Another advantage of using a semi-batch reactor is the control over branching or cross-linking density distribution. This is achieved by feeding monomers to maintain a certain concentration ratio of monovinyl monomer and divinyl monomer. Moreover, gelation can be avoided by feeding divinyl monomers to maintain a low concentration of divinyl monomers in the reactor. Control of CCD and topologies in semi-batch reactors has been well investigated.91–94 Polymer products with low molecular weight can also be produced by feeding monomer and initiator with a defined ratio.
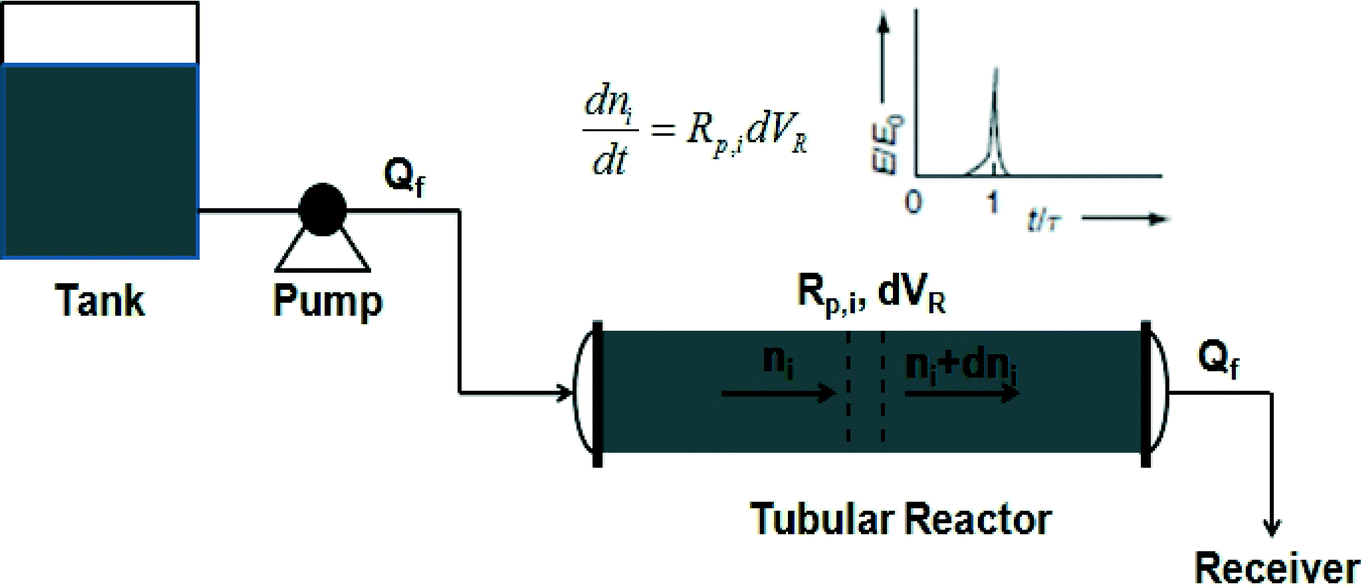 | ||
| Scheme 6 Schematic of a tubular reactor. | ||
One of the biggest selling points of using a tubular reactor is the excellent heat removal due to its large surface-to-volume ratio. Therefore, in terms of safety, a tubular reactor is more advantageous than a batch reactor and a continuous stirred-tank reactor (CSTR). However, a tubular reactor usually faces mixing problems, which may broaden the RTD away from the Dirac delta function, as shown in Scheme 6. This affects the properties of polymers produced in a tubular reactor, which deviate from those produced in a batch reactor. Solutions available for the mixing problem include the design of modified tubular reactors, including loop reactors, wicker-tube reactors and pulsed-flow reactors.
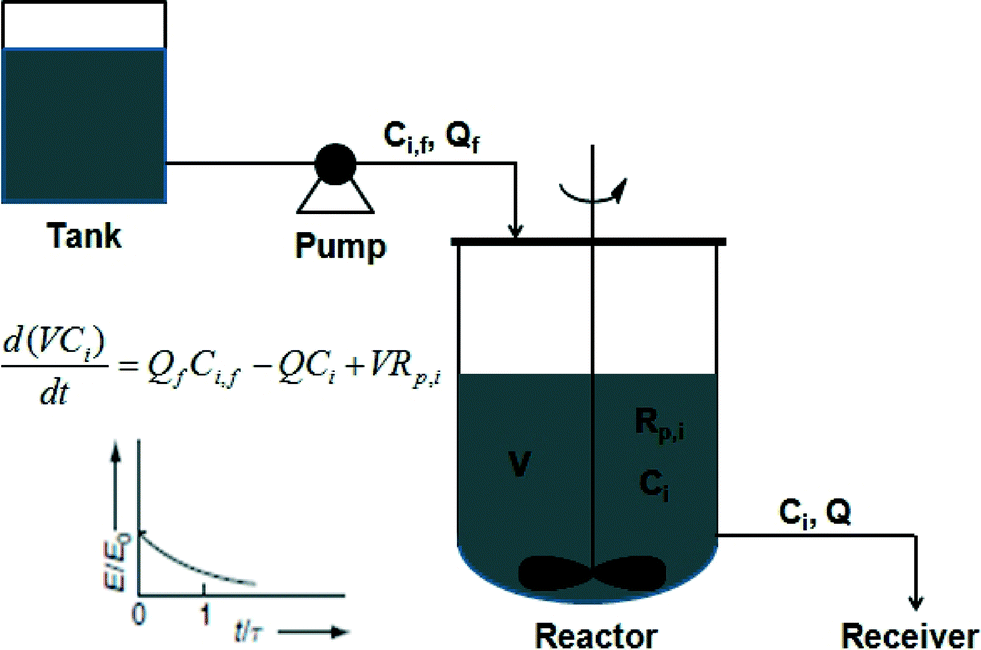 | ||
| Scheme 7 Schematic of a continuous stirred tank reactor (CSTR). | ||
The RTD of a CSTR is an exponential decay function. Due to the broader RTD in CSTR than those in batch and tubular reactors, the polymers synthesized in CSTR experienced different reaction time from one another. Therefore, these polymer chains are expected to possess a broader MWD. The RTD of a large number of CSTRs in series approaches that of a tubular reactor. Also, the RTD of a tubular reactor with large volume reflux or recycling approaches that of a CSTR.
1.3. Review scope
To date, there are only a few reviews discussing polymerization in various types of reactors. Steinbacher and McQuade reviewed flow microreactor technologies that produce various polymer materials including beads, microcapsules and fibers.95 Schork and Guo summarized mini-emulsion polymerization works in semi-batch and continuous reactors.96 Microreactors for polymer synthesis by ionic and radical polymerization methods were respectively reviewed by Frey et al.97,98 and Nagaki et al.99 Recently, copper-mediated CRPs in continuous flow reactors have been reviewed by Cunningham et al.100This work provides a comprehensive review of the progress in reactor engineering of CRP systems including ATRP, RAFT, and NMP. Homogeneous and heterogeneous CRP processes in semi-batch reactors, tubular reactors, and CSTRs are summarized and discussed in detail. The differences of semi-batch reactors and continuous reactors compared to batch reactors are highlighted based on the published experimental data. Perspectives on the future of reactor engineering of CRP are also offered.
2. Semi-batch reactor
The feasibility of controlling copolymer composition distributions (CCDs) by semi-batch reactors has been well demonstrated. Coupling the advantage of a semi-batch reactor with the slow chain growth of CRP has provided the opportunity to control the copolymer composition to have various CCDs. Most copolymerization experiments in semi-batch reactors used constant feeding of monomers (CF) to control the CCD. As the name implies, this process involves feeding monomers to the reactor at a constant feeding rate. However, the degree of control of CCDs synthesized by using CF is not very precise. In recent years, a model-based monomer feeding policy (MMFP) has been developed to produce polymers with pre-designed CCDs, as shown in Scheme 8.27–33 In MMFP, a kinetic model for the controlled radical copolymerization (CRcoP) process is first developed and then correlated with the batch experimental data for parameter estimation. The model is then combined with a semi-batch reactor model for targeting the pre-designed CCD using the comonomer feeding rate as an operating variable. The obtained comonomer feeding rate is actually controlled by a computer-programmed pump; thus this process is also referred to as programmed feeding (PF) in this review. Using programmed feeding, polymer products with the pre-designed CCD can be produced with a high degree of precision in semi-batch CRcoP. This precise control over the polymer structures for targeted properties represents an emerging trend in polymer reaction engineering.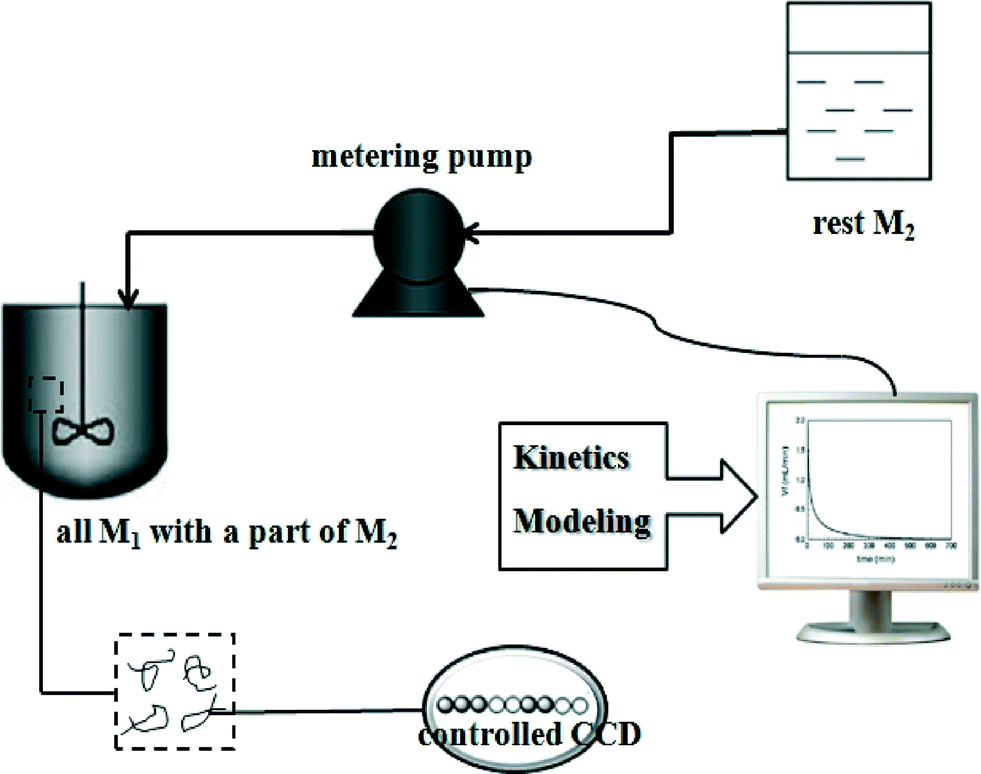 | ||
| Scheme 8 Model-based monomer feeding policy.27 | ||
To date, many different variations of CCDs have been obtained by utilizing semi-batch CRcoP. These CCDs can be divided into eight groups as shown in Scheme 9: (1) uniform (U); (2) linear gradient (LG); (3) S-shape gradient (SG) including hyperbolic, parabolic and sigmoidal; (4) di-block (DB-1); (5) di-block with a gradient block (DB-2); (6) tri-block (TB-1); (7) tri-block with one middle gradient block (TB-2); and (8) tri-block with two terminal gradient blocks (TB-3).
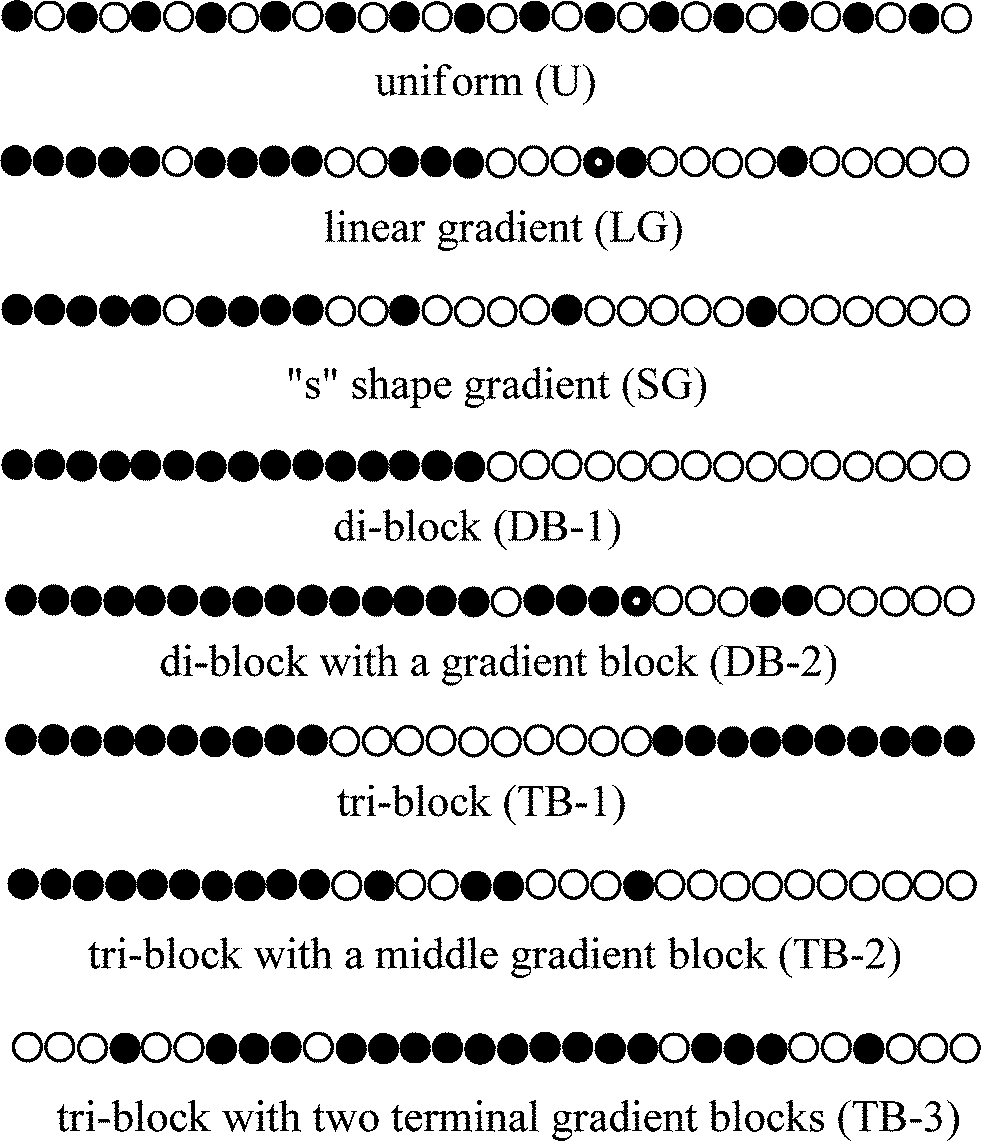 | ||
| Scheme 9 Different CCDs by semi-batch CRcoP. | ||
2.1. ATRP
Similar syntheses were reported by the same group for 2-(dimethylamino)ethyl methacrylate/n-butyl methacrylate (DMAEMA/BMA) and BA/isobornyl acrylate (BA/IBA) copolymers. The copolymers with an LG profile were synthesized by constant feeding in semi-batch reactors, while the random and block copolymer counterparts were synthesized in batch reactors (reactions S-ATRP-1 and S-ATRP-2 in Table 1).101,102 Luo et al. has also used constant feeding in semi-batch solution ATRP to synthesize LG tert-butyl acrylate/2,2,3,3,4,4,4-heptafluorobutyl methacrylate (tBA/HFBMA) copolymers by using HFBMA as the feeding monomer in the solution ATRP of tBA (reaction S-ATRP-3).103
| Reaction no. | Monomers M1/M2 | Homogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | T | t | X | M n | PDI | Feeding policy | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Reaction conditions = total mole ratios of M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RX:C:L; M = monomer; RX = ATRP initiator; C = catalyst; L = ligand; F1t = targeted mole fraction of M1 (M2 is feeding monomer); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; DMSO = dimethyl sulfoxide. :RX:C:L; M = monomer; RX = ATRP initiator; C = catalyst; L = ligand; F1t = targeted mole fraction of M1 (M2 is feeding monomer); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; DMSO = dimethyl sulfoxide. |
|||||||||||||
| S-ATRP-1 | DMAEMA/BMA | Solution | 200:1:1:2 |
0.74 [0.80] | LG [random] | Water/2-propanol | 25 | N/A [210] | N/A [87.2] | 20.96 [30.87] | 1.30 [1.27] | CF batch | 101 |
| S-ATRP-2 | BA/IBA | Solution | 533:1:4.38:3.6 |
0.59 | LG | Anisole/diphenyl ether | 50 | 378 | 82.3 | 74.80 | 1.13 | CF | 102 |
| S-ATRP-3 | tBA/HFBMA | Solution | 100:1:1:2 |
0.50 | LG | Toluene | 80 | 480 | 72.0 | 10.66 | 1.45 | CF | 103 |
| S-ATRP-4 | tBMA/MMA | Solution | 200:1:1:2 |
0.50 | LG [random] | p-Xylene | 100 | 590 570] | 94.0 [96.0] | 26.00 [25.00] | 1.12 [1.14] | PF batch | 31 |
| S-ATRP-5 | tBMA/MMA | Solution | 200:1:1:2 |
0.50 | SG | p-Xylene | 100 | 670 | 94.0 | 24.00 | 1.14 | PF | 31 |
| S-ATRP-6 | MMA/tBMA | Solution | 200:1:1:2 |
0.50 | U | p-Xylene | 100 | N/A | 93.9 | 21.70 | 1.17 | PF | 32 |
| S-ATRP-7 | tBMA/MMA | Solution | 200:1:1:2 |
0.50 | DB-1 | p-Xylene | 100 | N/A | 93.0 | 21.00 | 1.24 | PF | 32 |
| S-ATRP-8 | tBMA/MMA | Solution | 200:1:1:2 |
0.50 | TB-2 | p-Xylene | 100 | N/A | 99.0 | 25.00 | 1.18 | PF | 32 |
| S-ATRP-9 | HEMA/DMAEMA | Solution | 300:1:1.4:1.4 |
0.40–0.67 | LG | DMSO | 50 | 600 | N/A | 38.0–48.0 | 1.04–1.07 | PF | 104 |
| S-ATRP-10 | HEMA/DMAEMA | Solution | 300:1:1.4:1.4 |
0.31–0.68 [0.64] | SG [random] | DMSO | 50 | 600 | N/A | 36.0–101.0 [34.00] | 1.06–1.08 [1.05] | PF batch | 104 |
| S-ATRP-11 | MMA/HEMA-TMS | Solution | 525:1:1.65:3.31 |
0.48 | LG | Xylene/anisole | 90 | 420 | 47.7 | 56.70 | 1.22 | CF | 106 |
| S-ATRP-12 | MMA/HEMA-TMS | Solution | 450:1:1.37:2.62 |
0.56 | LG | Toluene | 90 | 420 | 47.1 | 43.00 | 1.12 | CF | 108 |
| S-ATRP-13 | HEMA-TMS/MMA | Solution | 450:1:1.37:2.62 |
0.44 | LG | Toluene | 90 | 420 | 53.6 | 50.00 | 1.05 | CF | 108 |
| S-ATRP-14 | St | Solution | 50:1:1–0.1:1 [50:1:1:1] |
1.00 | Homo | Toluene | 110 | 720–900 [360] | 80.0–96.0 [93.0] | 6.00–11.5 [6.70] | 1.05–1.16 [1.07] | CF batch | 109 |
| S-ATRP-15 | BA | Solution | 40:1:1:1 |
1.00 | Homo | Toluene | 90 [90] | 420 [240] | 97.0 [99.0] | 7.60 [4.80] | 1.62 [1.18] | CF batch | 110 |
| Reaction no. | Monomer M1/M2 | Heterogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | T | t | X | M n | PDI | Feeding policy | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RX:C:L; M = monomer; RX = ATRP initiator; C = catalyst; L = ligand; F1t = targeted mole fraction of M1 (M2 is feeding monomer); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; Homo = homopolymer; DMF = N,N-dimethylformamide. |
|||||||||||||
| S-ATRP-16 | BA/tBA | Mini-emulsion | 200:1:0.2:0.2 |
0.50 | SG [random] | — | 80 | N/A | 50.0 [55.0] | 12.0 [12.5] | 1.22 [1.18] | CF batch | 111 |
| S-ATRP-17 | BMA/MMA | Mini-emulsion | 300:1:0.2:0.2 |
0.67 | SG | — | 75 | N/A | 85.0 | N/A | 1.20–1.26 | CF | 111 |
| S-ATRP-18 | BA/St | Mini-emulsion | 200:1:0.2:0.2 |
0.50 | SG | — | 80 | N/A | 100 | 22.00 | 1.22 | CF | 111 |
| S-ATRP-19 | tBA/MMA | Heterogeneous catalyst | 200:1:0.1:0.1, 200:1:0.25:0.25, 200:1:0.5:0.5 |
0.50 | LG | DMF | 25 | 330 | 56.8–68.9 | 13.77–16.02 | 1.38–1.45 | CF | 112 |
Albeit useful to produce LG copolymers, constant feeding cannot control the CCD at will and to a high precision. Programmed feeding (PF) was developed by Zhu's group to produce copolymers with more sophisticated CCDs and better precision.27–33 Zhao et al. used PF to produce a series of tert-butyl methacrylate/MMA (tBMA/MMA) copolymers with uniform (U), LG, S-shape gradient (SG), tri-block with a middle gradient block (TB-2) and di-block (DB-1) composition profiles in semi-batch solution ATRP (reactions S-ATRP-4 to S-ATRP-8).32,33 Good agreements between the experimental CCDs and the theoretically targeted CCDs were obtained, clearly showing the power of PF for precise copolymer production. Similar successes were reported by Gallow et al. They synthesized a series of 2-hydroxyethyl methacrylate/DMAEMA (HEMA/DMAEMA) copolymers with LG and SG composition profiles by PF via semi-batch solution ATRP (reactions S-ATRP-9 and S-ATRP-10).104,105
Copolymers with different composition profiles can be further used as backbones for synthesis of molecular brushes. The properties of these brushes depend on the profile of the copolymer backbones. Novel molecular brushes with an LG backbone composition profile were synthesized by Matyjaszewski's group106 and Luo's group.107 The methyl methacrylate/HEMA-TMS (MMA/HEMA-TMS) copolymer backbone with an LG profile was synthesized by constant feeding of HEMA-TMS during the solution ATRP of MMA (reactions S-ATRP-11 and S-ATRP-12).106,108 Inverse CCD was obtained when MMA was chosen as the feeding monomer during ATRP of HEMA-TMS (S-ATRP-13).108 MMA/HEMA-TMS copolymer backbones with random and block profiles were also synthesized in batch and sequential batch reactors by Luo et al. The same group also compared the solution properties of molecular brushes with LG, random, and block backbone profiles.107
Other than controlling CCDs, semi-batch reactors also have been employed to synthesize polymers with low MW, which are used as coating resins. Fu et al. first used a semi-batch reactor to produce low-MW polystyrenes (PSt) with Mn = 6.0–11.5 kg mol−1.109 Both the polymerization rate and initiator efficiency in the semi-batch reactor were found to be lower than those in a counterpart batch reactor. For example, in the semi-batch reactor, a conversion of about 90% was obtained after 600 min of reaction with an initiator efficiency of 0.30. By comparison, the same conversion was achieved after only 360 min with an initiator efficiency of 0.75 (reaction S-ATRP-14 in Table 1) in the batch reactor.109 They attributed the decrease to the higher initiator concentration at the beginning of polymerization in the semi-batch reactor. The initiator efficiency can be improved by initially charging a small amount St to the reactor or by decreasing the final monomer content, at the cost of slowing down the polymerization.109 The same group also synthesized low-MW poly(butyl acrylate) (PBA) in a similar system (reaction S-ATRP-15).110 For controlling CCDs in semi-batch reactors, Fu et al. also synthesized a series of low-MW St/BA copolymers with U, LG, and DB-2 composition profiles.110
Numerous investigations have been conducted to establish the relationships between the properties of copolymers with their composition profiles. A comparison of the thermal properties of LG copolymers with those of random and block copolymers was conducted by Matyjaszewski et al. for DMAEMA/BMA and BA/IBA copolymers systems.101,102 Luo et al. also studied the effect of CCD on the glass transition temperature (Tg) of MMA/HEMA-TMS copolymers.108 Both groups found that the LG copolymers exhibit broad Tg, while the random and block copolymers possess narrow Tg and two distinct Tg's, respectively. The effect of copolymer composition profiles on the pH responsivity and micelle formation of MAA/MMA copolymers (formed by hydrolyzing tBMA/MMA under acidic conditions) was reported and found to be significant.32 Moreover, the cloud points of HEMA/DMAEMA copolymers in solution were shown to greatly depend on their composition profiles.104,105
The works summarized above clearly demonstrate the high potential in exploiting novel properties of polymer products by designing and controlling CCDs via constant feeding and programmed feeding in semi-batch reactors. Moreover, these works allow investigation of the structure–property relationships. With the relationships, it is possible to further design the CCD to produce polymer products with tailor-made properties.
The recently developed SET-LRP, having the advantages of low catalyst loading, fast polymerization rate, and low polymerization temperature, has attracted some attention.67–70 Zhou and Luo synthesized a series of LG MMA/tBA copolymer by constant feeding via semi-batch SET-LRP with Cu(0) and conventional ATRP ligands as the catalyst system at 25 °C (S-ATRP-19).112 Programmed feeding for precise production in heterogeneous ATRP systems has not been reported to date.
2.2. RAFT
| Reaction no. | Monomer M1/M2 | Homogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | T | t | X | M n | PDI | Feeding policy | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RAFT:I; M = monomer; RAFT = RAFT agent; I = conventional initiator; F1t = targeted mole fraction of M1 (M2 is feeding monomer); T = polymerization temperature; t = polymerization time; X = total monomer conversion; Mn = number-average molecular weight; PDI = polydispersity index; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions.a Sodium acetate/acid acetate buffer solution with pH = 5. |
|||||||||||||
| S-RAFT-1 | St/AcGalEA | Solution | 232:1:0.12 [217:1:0.13] |
0.81 [0.80] | DB-2 [random] | DMAc | 90 | 450 [1440] | 49.8 [55.0] | 10.27 [13.3] | 1.18 [1.26] | CF batch | 113 |
| S-RAFT-2 | AA/TFEMA | Solution | 200:1:0.25 |
0.50 | N/A | 1,4-Dioxane | 60–80 | 480 | 54.2–78.7 | N/A | N/A | CF | 114 |
| S-RAFT-3 | BA/St | Solution | 200:1:0.40 |
0.25 | U | Toluene | 70 | 2160 | 80.0 | 15.00 | 1.35 | PF | 29 |
| S-RAFT-4 | BA/St | Solution | 200:1:0.25 |
0.25 | LG | Toluene | 70 | 2160 | 70.0 | 15.75 | 1.24 | PF | 29 |
| S-RAFT-5 | BA/St | Solution | 333:1:0.22 |
0.50 | SG | Toluene | 88 | N/A | N/A | 29.00 | 1.30 | PF | 30 |
| S-RAFT-6 | BA/St | Solution | 333:1:0.22 |
0.50 | TB-2 | Toluene | 88 | N/A | N/A | 31.25 | 1.30 | PF | 30 |
| S-RAFT-7 | AM/BisAM | Solution | 630:1:0.50 |
0.95 | Branched | 60 | 120–300 [110] | 95.5–99.5 [68.0] | 180.0–245.0 [145.0] | 6.85–8.15 [4.50] | CF batch | 116 | |
| S-RAFT-8 | AM/BisAM | Solution | 610–630:1:0.50 |
0.95–0.98 | Branched | 60 | 210 | 95.0–98.0 | 80.00–180.0 | 2.00–8.00 | CF | 116 | |
Programmed feeding for precise production in homogeneous RAFT systems was first developed by Sun et al.29,30 A series of St/BA copolymers with various pre-designed CCDs were successfully produced using PF in semi-batch RAFT (reactions S-RAFT-3 to S-RAFT-6).29,30 The effects of composition profiles on the thermal properties of these products were also carefully investigated by DSC analysis.30 Their results clearly indicated that programmed feeding was feasible for design and precise control over CCDs to produce polymer products with tailor-made properties.
Semi-batch reactor technologies are most employed in linear CRP systems. In CRP, individual chains grow slowly, providing ample time for chains to come into contact with each other, which could also facilitate inter-chain reactions for nonlinear polymers. Wang et al. first employed semi-batch technologies in vinyl/divinyl CRP to control gelation and to synthesize hyperbranched polymer products.115–117 A large amount of RAFT agents (mole ratio of RAFT to divinyl monomers greater than 0.5) is usually required in batch RAFT, to synthesize branched polymers without gelation.118–121 In Wang's work, a series of hyperbranched polyacrylamides (PAMs) were synthesized, free of gels, by constantly feeding divinyl monomer N,N′-methylenebis(acrylamide) (BisAM) in a semi-batch solution RAFT copolymerization (reactions S-RAFT-7 and S-RAFT-8).115–117 Using this strategy, hyperbranched polymers were successfully produced with a low mole ratio of RAFT agents to divinyl monomers (no more than 0.1).115–117 Furthermore, a more uniform branching density distribution was obtained in the semi-batch reactor than in the counterpart batch reactor.116
| Reaction no. | Monomer M1/M2 | Heterogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | T | t | X | M n | PDI | Feeding policy | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RAFT:I; M = monomer; RAFT = RAFT agent; I = conventional initiator; F1t = targeted mole fraction of M1 (M2 is feeding monomer); T = polymerization temperature; t = polymerization time; X = total monomer conversion; Mn = number-average molecular weight; PDI = polydispersity index; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; Homo = homopolymer.a BA and St are both feeding monomers. |
|||||||||||||
| S-RAFT-9 | St | Emulsion | N/A | 1.00 | Homo | — | 85 | N/A | N/A | 16.60–90.60 | 2.10–3.30 | CF | 43 |
| S-RAFT-10 | BA | Emulsion | N/A | 1.00 | Homo | — | 85 | N/A | 70.0–100 | 20.6–81.00 | 1.40–2.30 | CF | 43 |
| S-RAFT-11 | MMA/St | Mini-emulsion | 260–300:1:0.30 |
0.25–0.90 | DB-2 | — | 60 | N/A | 95.0–100 | 22.67–24.32 | 1.27–1.44 | CF | 122 |
| S-RAFT-12 | St/Bu | Mini-emulsion | N/A | 0.25 | DB-2 | — | 70 | 660 | 92.0 | 40.70 | 1.41 | CF | 123 |
| S—RAFT-13 | BMA/DFMA | Mini-emulsion | 81:1:0.3 |
0.78 | DB-1 | — | 75 | 720 | 90.9 | 10.25 | 1.22 | CF | 124 |
| S-RAFT-14 | BA/Sta | Emulsion | N/A | N/A | TB-1 | — | 70 | 190–240 | 90.0–97.0 | 76.80–338.1 | 1.41–3.19 | CF | 125 |
| S-RAFT-15 | AA/TFEMA | Emulsion | 233:1:0.5 |
0.57 | SG | 5% acetone aqueous | 70 | 300 | 93.5 | 200.0 | 1.71 | CF | 126 |
| S-RAFT-16 | BA/St | Mini-emulsion | 333:1:0.33 |
0.50, 0.75 [0.50] | U [random] | — | 70 | 420, 540 [300] | 82.0, 88.0 [80.0] | 36.00, 40.00 [37.00] | 1.04, 1.13 [1.15] | PF batch | 33 |
| S-RAFT-17 | St/BA | Mini-emulsion | 333:1:0.33 |
0.50, 0.75 | LG | — | 70 | 420, 540 | 73.3, 85.8 | 30.88, 40.00 | 1.07, 1.08 | PF | 33 |
| S-RAFT-18 | TEGDMA/St | Mini-emulsion | 100:1:0.1 |
0.005 | — | — | 70 | 600 | 75.0 | 13.24 | 2.87 | PF | 127 |
Several research works have investigated the control of CCDs in RAFT (mini)emulsion systems. Luo and Liu synthesized a series of DB-2 MMA/St copolymers by constant feeding of St at 8 ml h−1 after complete copolymerization of MMA and St (reaction S-RAFT-11).122 Similar to Luo and Liu's work, Wang et al. synthesized DB-2 St/butadiene (St/Bu) copolymers by constant feeding of Bu at 80 ml h−1 after 1 hour of RAFT mini-emulsion polymerization of St (reaction S-RAFT-12).123 Zhang et al. synthesized DB-1 butyl methacrylate/dodecafluoroheptyl methacrylate (BMA/DFMA) copolymers by constant feeding of DFMA at 2 ml h−1 after complete RAFT polymerization of BMA (reaction S-RAFT-13).124 In the work of Zhu et al., a series of TB-1 St/BA copolymers with various MW were synthesized by first feeding BA at a constant rate of 20 g h−1 until completion of St, followed by constant feeding of St at the same rate until completion of BA (reaction S-RAFT-14).125 Their data also showed that thermoplastic elastomer products can be produced by semi-batch RAFT emulsion block polymerization.125 Recently, Chen et al. synthesized SG acrylic acid/2,2,2-trifluoroethyl methacrylate (AA/TFEMA) copolymers by constant feeding of TFEMA in an emulsifier-free RAFT emulsion polymerization (reaction S-RAFT-15).126
The use of PF for precise production has well been demonstrated in homogeneous CRP systems.29–32 However, high product separation costs and poor heat transfer limit their commercial exploitation. These problems could be countered by employing heterogeneous systems (such as emulsion and mini-emulsion). Li et al. was the first to develop programmed feeding in heterogeneous RAFT systems. A series of U and LG St/BA copolymer products were successfully produced by programmed feeding in semi-batch RAFT mini-emulsion polymerization (reactions S-RAFT-16 and S-RAFT-17).33 Also, Li et al. extended this PF strategy for the control of topology in heterogeneous systems. A series of hyperbranched polystyrenes having uniform branching density distributions were produced via semi-batch RAFT mini-emulsion polymerization (reaction S-RAFT-18).127
2.3. NMP
| Reaction no. | Monomer M1/M2 | Homogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | T | t | X | M n | PDI | Feeding policy | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:PT; M = monomer; PT = NMP initiator; F1t = targeted mole fraction of M1 (M2 is feeding monomer); T = polymerization temperature; t = polymerization time; X = total monomer conversion; Mn = number-average molecular weight; PDI = polydispersity index; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; Homo = homopolymer. |
|||||||||||||
| S-NMP-1 | St | Solution | N/A | 1.00 | Homo | Xylene | 138 | 400–1300 | 40.0–98.0 | 2.000–10.50 | 1.20–1.50 | CF | 129 |
| S-NMP-2 | BA/DMA | Solution | 500:1 |
0.51 | LG, SG | Toluene | 112 | 240–800 | 55.1–83.7 | 24.00–35.00 | 1.24–1.48 | CF | 130 |
| S-NMP-3 | BA/St | Bulk | 480:1 |
0.42 | LG, SG | — | 120 | 430, 240 | 77.8, 73.7 | 51.80 40.70 | 1.21 1.16 | CF | 132 |
| S-NMP-4 | ODA/MA | Solution | 500:1 |
0.27 | SG | Toluene | 112 | 215 | 44.5 | 21.35 | 1.21 | CF | 133 |
| S-NMP-5 | BA/MMA | Bulk | 500:1 |
0.50 | SG [random] | — | 115 | 720 [390] | 91.0 [70.0] | 61.90 [48.60] | 1.32 [1.24] | CF batch | 134 |
| S-NMP-6 | AA/St | Solution | 230:1 |
N/A | DB-2 | 1,4-Dioxane | 120 | N/A | N/A | 14.00–15.30 | N/A | CF | 135 |
| S-NMP-7 | AA/St | Solution | 230:1 400:1 |
N/A | TB-3 | 1,4-Dioxane | 120 | N/A | 63.0 70.0 | 12.55 17.00 | 1.25 1.33 | CF | 136 |
| S-NMP-8 | St/MS | Solution | N/A | 0.42 | LG | Cyclohexane | 90 | 840 | 100 | 84.60 | 1.33 | CF | 137 |
| S-NMP-9 | St/MMA | Bulk | N/A | 0.51 | LG | — | 93 | 480 | 100 | 55.20 | 1.44 | CF | 138 |
| S-NMP-10 | St/tBA | Bulk | N/A | 0.55–0.72 | LG | — | 115 | 480 | 95.0–100 | 38.60–91.80 | 1.32–1.48 | CF | 141 |
| S-NMP-11 | St/AS | Bulk | N/A | 0.56 | LG | — | 115 | 300 | 100 | 93.80 | 1.40 | CF | 143 |
| S-NMP-12 | St/BA | Bulk | N/A | 0.60 | LG | — | 100 | 480 | N/A | 72.00 | N/A | CF | 148 |
| S-NMP-13 | St/BMA | Bulk | N/A | 0.49–0.71 | LG | 115 | 130–180 | N/A | 57.80–83.00 | 1.37–1.39 | CF | 140 | |
| S-NMP-14 | St/AS | Bulk | N/A | 0.25–0.76 | DB-2 | — | 90 | N/A | N/A | 48.00–67.00 | N/A | CF | 142 |
| S-NMP-15 | St/MMA | Bulk | N/A | 0.55 | SG | — | 93 | 540 | N/A | 102.0 | 1.58 | ICF | 151, 152 |
| S-NMP-16 | St/BA | Bulk | N/A | 0.60 | SG | — | 100 | 540 | 100 | 95.00 | 1.37 | ICF | 153 |
| S-NMP-17 | St/AS | Bulk | N/A | 0.58 | DB-2 | — | 90 | 360 | 100 | 53.80 | 1.11 | ICF | 154 |
Torkelson et al. contributed significantly to the control of CCDs in homogeneous NMP systems. A series of LG St/4-methylstyrene (St/MS) (reaction S-NMP-8),137 St/MMA (reaction S-NMP-9),138 St/tBA (reaction S-NMP-10),139–141 St/4-acetoxystyrene (St/AS) (reaction S-NMP-11),140,142–147 St/BA (reaction S-NMP-12),140,148 St/BMA (reaction S-NMP-13),140 and St/4-vinylpyridine (St/4VP)140,147 copolymers were synthesized by constant feeding methods.137–148 A series of DB-2 St/AS copolymers were also synthesized by constant feeding of AS at the beginning of bulk NMP of St using PSt as the macroinitiator or by constant feeding of AS after 2 hours of bulk NMP of St (reaction S-NMP-14).142,149,150
In semi-batch reactors, constant feeding is often used to produce LG copolymer products. Torkelson's group first developed a new feeding policy, referred to as increasing constant feeding (ICF) in this review, to produce SG (such as sigmoidal gradient) copolymer products. For example, SG St/MMA copolymers can be produced by constant feeding of MMA at 10 ml h−1 for the first 3 hours, 15 ml h−1 for the next 3 hours, and 20 ml h−1 for the final 3 hours during NMP of St (reaction S-NMP-15).151,152 By using a similar feeding policy, a series of St/BA (reaction S-NMP-16)140,144,145,147,148,153 copolymers were also synthesized by Torkelson et al. The synthesis of St/AS copolymers with a DB-2 composition profile by ICF of AS at 0.03 ml min−1 for the first 2 hours and 0.08 ml min−1 for the final 4 hours with PSt macroinitiators was also reported (reaction S-NMP-17).154 The properties of these copolymer products with different composition profiles were also thoroughly investigated. The use of programmed feeding for precise control over CCD has not been reported for homogeneous NMP systems.
| Reaction no. | Monomer M1/M2 | Heterogeneous system | Reaction conditions | F1t | Copolymer profile | Solvent | T | t | X | Mn | PDI | Feeding policy | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:PT; M = monomer; PT = NMP initiator; F1t = targeted mole fraction of M1 (M2 is feeding monomer); T = polymerization temperature; t = polymerization time; X = total monomer conversion; Mn = number-average molecular weight; PDI = polydispersity index; N/A = not available in the literature; Homo = homopolymer.a Both BMA and St are feeding monomers.b Both BMA and St are feeding monomers, and MA is added to improve initiator efficiency.163 |
|||||||||||||
| S-NMP-18 | BA | Emulsion | 270:1 |
1.00 | Homo | — | 112 | 390–600 | 70.0–97.0 | 27.00–42.50 | 1.20–1.55 | CF | 158 |
| S-NMP-19 | BA | Emulsion | N/A | 1.00 | Homo | — | 120 | 450 | 94.0–100 | 43.28–81.85 | 2.06–4.46 | CF | 159 |
| S-NMP-20 | BA | Microemulsion | 155–618:1 |
1.00 | Homo | — | 120 | 360 | 78.0–100 | 16.60–53.30 | 1.38–3.26 | CF | 160 |
| S-NMP-21 | BMA/Sta | Emulsion | 147:1 |
0.86 | N/A | — | 90 | 1320 | 30.0 | 11.00 | 1.55 | CF | 163 |
| S-NMP-22 | BMA/St/MAb | Emulsion | 266–279:1 |
0.87–0.89 | N/A | — | 90 | 1320–1386 | 36.4–67.5 | 16.18–38.40 | 1.48–1.92 | CF | 163 |
From an industrial point of view, surfactant-free NMP emulsion systems are attractive. However, the problems of multi-step polymerization, broad particle size distribution (PSD) and/or bimodal PSD have limited their commercial potential.161,162 Recently, Thomson et al. reported a one-step surfactant-free NMP emulsion copolymerization of BMA with a small amount of St. By constant feeding of BMA and St mixture, stable surfactant-free emulsion systems with monomodal PSDs were successfully prepared (reactions S-NMP-21 and S-NMP-22).163
2.4. Summary
Semi-batch reactors have been most frequently employed to control CCDs by feeding comonomers. Table 7 summarizes different kinds of polymer products with different CCDs, which were made by semi-batch CRP. It is well known that copolymers produced by one-step polymerization in batch reactors only possess random profiles. The synthesis of di-block copolymers usually requires two-step polymerization in batch reactors with macroinitiators synthesized in the first step. However, the purification of the macroinitiator from the first step is both time consuming and costly. Synthesis of di-block copolymers in semi-batch reactors does not require any purification of the macroinitiators. In semi-batch reactors, di-block copolymers (poly[M1-b-M2], DB-1) can be produced by feeding M2 upon completion of M1. Tri-block copolymers (poly[M1-b-M2-b-M3], TB-1) can also be produced by the same feeding policy in semi-batch reactors. Therefore, semi-batch reactors are preferred to batch reactors in synthesizing copolymers with pre-specified composition profiles.| System | Homo | U | DB-1 | DB-2 | LG | SG | TB-1 | TB-2 | TB-3 |
|---|---|---|---|---|---|---|---|---|---|
| Homo = homopolymer; U = uniform; DB-1 = di-block; DB-2 = di-block with a gradient block; LG = linear gradient; SG = ‘s’ shape gradient; TB-1 = tri-block; TB-2 = tri-block with a middle gradient block; TB-3 = tri-block with two terminal gradient blocks. | |||||||||
| Homogeneous ATRP | S-ATRP-14 S-ATRP-15 | S-ATRP-6 | S-ATRP-7 | S-ATRP-1 S-ATRP-2 S-ATRP-3 S-ATRP-4 S-ATRP-9 S-ATRP-11 S-ATRP-12 S-ATRP-13 | S-ATRP-5 S-ATRP-10 | S-ATRP-8 | |||
| Heterogeneous ATRP | S-ATRP-19 | S-ATRP-16 S-ATRP-17 S-ATRP-18 | |||||||
| Homogeneous RAFT | S-RAFT-3 | S-RAFT-1 | S-RAFT-4 | S-RAFT-5 | S-RAFT-6 | ||||
| Heterogeneous RAFT | S-RAFT-9 S-RAFT-10 | S-RAFT-16 | S-RAFT-13 | S-RAFT-11 S-RAFT-12 | S-RAFT-17 | S-RAFT-15 | S-RAFT-14 | ||
| Homogeneous NMP | S-NMP-1 | S-NMP-6 S-NMP-14 S-NMP-17 | S-NMP-2 S-NMP-3 S-NMP-8 S-NMP-9 S-NMP-10 S-NMP-11 S-NMP-12 S-NMP-13 | S-NMP-2 S-NMP-3 S-NMP-4 S-NMP-5 S-NMP-15 S-NMP-16 | S-NMP-7 | ||||
| Heterogeneous NMP | S-NMP-18 S-NMP-19 S-NMP-20 | ||||||||
The methods for producing copolymers with the other types of CCDs in semi-batch reactors are summarized according the literature as follows: uniform copolymers (poly[M1-u-M2], U) are produced by feeding both M1 and M2, or by feeding the fast comonomers. Di-block copolymers with a gradient block (poly[M1-b-(M1-grad-M2)], DB-2) are produced by feeding M2 after M1 is polymerized for some time. Linear gradient copolymers (poly[M1-lg-M2], LG) and ‘s’ shape gradient copolymers (poly[M1-sg-M2], SG) are both produced by feeding M2 from the beginning of M1 polymerization. Tri-block copolymers with a gradient middle block (poly[M1-b-(M1-grad-M2)-b-M2], TB-2) are produced by feeding M2 after polymerizing M1 for some time, while tri-block copolymers with two gradient terminal blocks (poly[(M1-grad-M2)-b-M1-(M1-grad-M2)], TB-3) are produced by feeding M2 after M1 is homopolymerized for some time using difunctional initiators. As shown in Table 7, semi-batch reactors are most employed to produce LG and SG copolymers, while reports on tri-block copolymers are very rare.
Feeding policies for control of CCDs can be divided into three kinds: constant feeding (CF), increasing constant feeding (ICF), and programmed feeding (PF). CF is the most commonly used feeding policy to control CCDs. ICF was developed to produce SG (such as sigmoidal gradient) copolymers. However, CF and ICF cannot control CCDs at will to a high degree of precision. Zhu's group first developed PF to produce polymer products with pre-designed CCDs, allowing precise control of CCDs. The influence of CCD on copolymer properties is significant and it has been demonstrated with various monomer combinations. It is clear that control over CCD is invaluable in producing polymers having tailored properties. Therefore, further research should focus on targeting product properties, i.e., to produce polymer products with tailor-made properties, based on structure–property relationships.
3. Tubular reactor
Owing to the narrow residence time distribution (RTD), the polymerization kinetics in a tubular reactor is similar to that in a batch reactor. Tubular reactors are popular in industry due to their excellent capacity for heat removal. In this chapter, the progress in ATRP, RAFT, and NMP in tubular reactors is reviewed. Moreover, comparisons of polymers produced from tubular reactors with those from batch reactors are also discussed wherever applicable.3.1. ATRP
| Reaction no. | Monomer M1/M2 | Homogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | T | t | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RX:C:L; M = monomer; RX = ATRP initiator; C = catalyst; L = ligand; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; DMSO = dimethyl sulfoxide; DMF = dimethylformamide. |
|||||||||||||
| T-ATRP-1 | MMA | Solution | 100:1:1:2.2 |
1.00 | Homo | Toluene | N/A batch | 90 | 27–300 [240] | 13.7–89.9 [80.5] | 5.85–13.80 [9.820] | 1.05–1.17 [1.24] | 164, 166 |
| T-ATRP-2 | MMA | Solution | 50–200:1:1:2.2 |
1.00 | Homo | Toluene | N/A | 90 | 27–300 | 13.7–89.9 | 4.26–13.80 | 1.05–1.36 | 164 |
| T-ATRP-3 | MMA | Solution | 100:1:1:2.2 |
1.00 | Homo | Toluene | N/A | 60–100 | 120–165 | 16.1–83.7 | 3.27–12.60 | 1.04–1.27 | 164 |
| T-ATRP-4 | BA | Solution | 39.6:1:0.15:0.49 |
1.00 | Homo | Acetonitrile | 170.7 batch | 80 | 320 [360] | 71.0 [82.0] | 4.160 [4.797] | 1.09 [1.09] | 165 |
| T-ATRP-5 | St | Solution | 50.2:1:0.50:1.0 |
1.00 | Homo | Toluene | 170.7 | 110 | 240 [380] | 50.0 [75.0] | 2.807 [4.426] | 1.14 [1.13] | 165 |
| T-ATRP-6 | BMA | Solution | 100:1:0.005:0.005 |
1.00 | Homo | Anisole | 300 batch | 90 | 540–600 [360] | 19.0–96.0 [97.0] | 2.80–13.80 [13.90] | 1.28–1.34 [1.31] | 167 |
| T-ATRP-7 | HPMA | Solution | N/A | 1.00 | Homo | Water/methanol | N/A | N/A | 12–120 | 17.0–92.0 | 2.77–6.24 | 1.19–1.32 | 169 |
| T-ATRP-8 | MMA | Solution | 300:1:0.5:1 |
1.00 | Homo | Anisole | 280 batch | 50 | N/A [300] | 44.0 [65.0] | 14.0 [21.80] | 1.28 [1.38] | 170 |
| T-ATRP-9 | MA | Solution | 47:1:0.02:0.12 |
1.00 | Homo | DMSO | 20 | 15 | N/A | 80.0 | 2.500 | 1.16 | 174 |
| T-ATRP-10 | EO/HPMA | Solution | 100:1:1:2 |
N/A | DB-1 | Water/methanol | N/A | N/A | 16–188 | 27.0–69.0 | 5.60–10.20 | 1.17–1.24 | 171 |
| Batch | [210] | [63.0] | [8.80] | [1.25] | 171 | ||||||||
| T-ATRP-11 | MMA/LMA | Solution | 255:1:0.5:1 |
N/A | DB-1 | Anisole | 90 | 50 | N/A | 33.0 | 42.90 | 1.46 | 170 |
| T-ATRP-12 | MMA/BMA | Solution | N/A | N/A | DB-2 | Toluene | N/A | 90 | 305–365 | N/A | 15.50–16.30 | 1.09–1.14 | 164 |
| T-ATRP-13 | MMA/BzMA | Solution | N/A | N/A | DB-2 | Toluene | N/A | 90 | 305–365 | N/A | 15.30–15.70 | 1.22–1.32 | 164 |
| T-ATRP-14 | MMA/BIEM | Solution | 300:1:3.0:1.1 |
0.95 | Branched | DMF | 60 batch | 60 | 120 | 64.0 [58.0] | 5.30 [3.10] | 2.15 [2.01] | 175 |
| T-ATRP-15 | DMAEMA/BIEM | Solution | N/A | 0.90 | Branched | DMF | 120 | 75 | 120 | 77.0 | 2.218 | 2.50 | 179 |
| 74.5 | 3.618 | 2.20 | |||||||||||
| 0.95 | |||||||||||||
The living characteristics of ATRP of St and BA in tubular reactors were also investigated by Cunningham et al. (reactions T-ATRP-4 and T-ATRP-5).165 The initial polymerization rates in tubular reactors were higher than those in batch reactors due to the operation mode, while Mn and PDI were similar. Moreover, they showed that the tubular reactor gave a narrow RTD, indicating a nearly ideal plug flow condition. PDI after 71% conversion was 1.09 for the BA system, while PDI at 50% conversion was 1.14 for the St system.165 The same group employed a modified ATRP technology (ARGET ATRP) in a tubular reactor with tin(II) 2-ethylhexanoate (Sn(EH)2) as the reducing agent.167,168 A low amount Cu catalysts (ppm level), a stoichiometric ratio of ligand to Cu, and unpurified monomers and solvents were used in their experiments.167 A much slower polymerization rate was observed in the tubular reactor than in the batch reactor when the reducing agent used was 10% of the initiator (83% vs. 19% conversion in batch and tubular reactors, respectively). However, increasing the ratio to 40% resulted in comparable polymerization rates, with 97% vs. 96% conversion in batch and tubular reactors, respectively (reaction T-ATRP-6).167 This work demonstrated the industrial feasibility of ARGET ATRP.
Wu et al. reported controlled ATRP of 2-hydroxypropylmethacrylate (HPMA) in a continuous microfluidic reactor (reaction T-ATRP-7).169 Chastek et al. reported solution ATRP of MMA in a continuous microfluidic reactor and in a batch reactor for comparison (reaction T-ATRP-8).170 Other studies by the same group extended this concept to synthesize di-block171 and brush172,173 polymers. Additionally, Wenn et al. performed UV-induced SET-LRP of methyl acrylate (MA) in a tubular UV-reactor; the polymerization gave good control over polymer molecular weight (reaction T-ATRP-9).174
In tubular reactors, DB-1 copolymers are usually produced by employing macroinitiators or by using two tubular reactors. Wu et al. synthesized a series of ethylene oxide/2-hydroxypropyl methacrylate (EO/HPMA) DB-1 copolymers with PEO as macroinitiators in a microchannel reactor (reaction T-ATRP-10).171 Compared to a batch reactor, the polymerization rate was somewhat lower at the beginning in the microchannel reactor, but it became slightly higher as the polymerization progressed. The PDI's observed from both reactors were very similar.171 The same method was used by Chastek et al. to synthesize DB-1 MMA/lauryl methacrylate (MMA/LMA) copolymers by using PMMA macroinitiators in a microfluidic reactor (reaction T-ATRP-11).170 On the other hand, Haddleton et al. reported the use of two tubular reactors in series to synthesize DB-2 MMA/BMA and MMA/benzyl methacrylate (MMA/BzMA) (reactions T-ATRP-12 and T-ATRP-13).164
Many studies have been conducted on non-linear polymerization in tubular reactors. Bally et al. first reported branching polymerization by CRP in a tubular reactor. A series of branched polymers was synthesized by self-condensing vinyl copolymerization (SCVCP) through solution ATRP of MMA and BIEM in a tubular microreactor.175 The polymerization rate and branching efficiency were both higher in the tubular microreactor than those in a batch reactor under the same experimental conditions.175 For example, after 120 minutes of polymerization, the conversion and branching efficiency of MMA was 64% and 44% in the tubular microreactor but only 58% and 28% in the batch reactor, respectively (reaction T-ATRP-14).175 Their study demonstrated that tubular microreactors can be used for better control of the branching process in CRP systems than batch reactors. Moreover, tubular microreactors offer excellent heat transfer and fast mixing, resulting in improved polymer products.176–178 Furthermore, Parida et al. also used a coil flow inverter microreactor to synthesize branched poly(2-(dimethylamino)ethyl methacrylate)) (PDMAEMA) by ATRP, and it was found that the branching efficiency was in the order of coil flow inverter microreactor > normal coiled tube microreactor > batch reactor (reaction T-ATRP-15).179
Well-controlled MMA polymerization by catalyst-supported ATRP in a column reactor was demonstrated by Zhu's group (reaction T-ATRP-16 in Table 9).34 A linear relationship between Mn and monomer conversion at different RT was observed, indicating a well-controlled polymerization. The PDI was about 1.8 at 90% conversion, which was slightly higher than the value observed in a batch reactor.188 The higher PDI in a tubular reactor may be due to back-mixing in this column reactor or due to some trapped polymers inside the silica-gel pores, thus broadening the RTD.34 A high stability of the column packed with silica gel and a high retention of catalyst reactivity were also reported in this work. Steady-state operation was maintained at a monomer conversion of 80% for more than 120 h with a feeding rate of 1.2 ml h−1. In addition, the effects of both polymerization temperature and targeted chain length were investigated in the column reactor (reactions T-ATRP-17 and T-ATRP-18).36 The polymerization was faster when conducted at a higher polymerization temperature or when conducted with a lower targeted chain length. The same conclusions were also reported by Haddleton et al.164
| Reaction no. | Monomer M1/M2 | Heterogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | T | t | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RX:C:L; M = monomer; RX = ATRP initiator; C = catalyst; L = ligand; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions.a [M]:[RX] = 100:1 and [C]:[L] = 1:1; DMSO = dimethyl sulfoxide; NM2P = N-methyl-2-pyrrolidone, AA = ascorbic acid. |
|||||||||||||
| T-ATRP-16 | MMA | Heterogeneous catalyst | 1.00 | Homo | Toluene | 38–300 | 90 | N/A | 20.0–90.0 | 3.500–11.00 | 1.50–1.80 | 34 | |
| T-ATRP-17 | MMA | Heterogeneous catalyst | [M]:[RX] = 100:1 |
1.00 | Homo | Toluene | 60–560 | 70,80 | N/A | 8.00–80.0 | 3.050–12.00 | 1.30–1.75 | 36 |
| T-ATRP-18 | MMA | Heterogeneous catalyst | [M]:[RX] = 100:1, 200:1 |
1.00 | Homo | Toluene | 120–460 | 80 | N/A | 8.00–80.0 | 3.800–19.40 | 1.38–1.63 | 36 |
| T-ATRP-19 | MMA/BMA | Heterogeneous catalyst | N/A | N/A | DB-2 | Toluene | N/A | 80 | 27000–34500 | N/A | 11.00–18.00 | 1.70–1.84 | 36 |
| T-ATRP-20 | MA | Heterogeneous catalyst | [M]:[RX]:[L] = 100:1:0.05 |
1.00 | Homo | DMSO | 4–16 | 23–25 | 50–176 | 43.0–67.0 | 4.930–6.660 | 1.22–1.44 | 195 |
| T-ATRP-21 | MA | Heterogeneous catalyst | [M]:[RX]:[L] = 100:1:0.05, 100:1:0.01 |
1.00 | Homo | DMSO | 16 | 23–25 | 136, 144 | 67.0, 47.0 | 6.660, 4.390 | 1.44, 1.30 | 195 |
| T-ATRP-22 | MA | Heterogeneous catalyst | [M]:[RX]:[L]:[AA] = 200:1:0.01:0.02 |
1.00 | Homo | DMSO | 8–62 batch | 23–25 [30] | 210–350 [60] | 27.0–78.0 [96.0] | 4.670–13.84 [16.59] | 1.27–1.47 [1.21] | 196 |
| T-ATRP-23 | MA | Heterogeneous catalyst | [M]:[RX]:[L]:[AA] = 200:1:0.01:0.01–0.04 |
1.00 | Homo | DMSO | 35 | 23–25 | 227 | 65.0–67.0 | 11.80–12.23 | 1.42–1.47 | 196 |
| T-ATRP-24 | AN | Heterogeneous catalyst | [M]:[RX]:[L] = 200:1:0 |
1.00 | Homo | NM2P | N/A | 25 | 80 | 41.2 | 6.430 | 1.28 | 197 |
| T-ATRP-25 | MA | Heterogeneous catalyst | [M]:[RX] = 50:1 |
1.00 | Homo | DMSO | 13–80 | 25 | N/A | 69.0–90.0 | 3.200–4.200 | 1.14–1.17 | 198 |
The synthesis of DB-2 MMA/BMA copolymers using two column reactors in series was demonstrated by Zhu et al. DB-2 MMA/BMA copolymers with Mn = 11 kg mol−1 and PDI = 1.70 were produced by feeding BMA at 1.2 ml h−1. When the feeding rate of BMA was increased to 1.8 ml h−1, the concentration of BMA in the second column reactor increased, resulting in faster polymerization. Moreover, the DB-2 MMA/BMA copolymers produced in the system with a faster feeding rate possess Mn = 18 kg mol−1 and PDI = 1.84 (reaction T-ATRP-19).36
There are numerous studies in the literature on SET-LRP conducted in batch reactors at ambient temperature. Chan et al. first employed SET-LRP in a continuous reactor.195 SET-LRP of MA was carried out in a copper tubular reactor, with the copper tubing acting as the catalyst source. A high surface area of the copper tubular reactor resulted in a high catalytic efficiency. A monomer conversion of up to 67% was obtained with only 16 min of residence time (RT) (reaction T-ATRP-20).195 When the ligand concentration was decreased five times, the conversion was only reduced from 67 to 47% at the same 16 min RT (reaction T-ATRP-21).195 This is important for cost reduction in commercial applications. The chain extension experiments showed that the polymers prepared via SET-LRP in the copper tubular reactor possessed higher livingness than those in a batch reactor.195 However, using the copper tubular reactor as the catalyst source is associated with possible damage to the reactor, with copper continuously dissolved from the reactor wall into the reaction mixture. To overcome this problem, Chan et al. combined a short copper tubular reactor with a long stainless steel tubular reactor.196 The short copper tubular reactor was used to initiate the polymerization by providing soluble copper species. The majority of the polymerization occurred in the long stainless steel tubular reactor. In systems with the copper tubular reactor alone, the conversion could go as high as 53% with a RT of only 16 min.195 On the other hand, in the combined reactor, a conversion of 55% could only be achieved when the RT was increased to 62 min.196 In order to enhance the polymerization rate, ascorbic acid (AA) was added to the stainless steel tubular reactor (reactions T-ATRP-22 and T-ATRP-23).196 Similar to the work of Cunningham's, Chen et al. used SET-LRP of acrylonitrile (AN) in an iron tubular reactor by using an iron tube as the catalyst source without the use of a ligand (reaction T-ATRP-24).197 Additionally, Burns et al. performed SET-LRP of MA in a PTFE tubular reactor with a copper wire threaded through the tubing (reaction T-ATRP-25).198
3.2. RAFT
| Reaction no. | Monomer M1/M2 | Homogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | T | t | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RAFT:I; M = monomer; RAFT = RAFT agent; I = conventional initiator; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; EtOAc = ethyl acetate; EHA = 2-ethylhexyl acrylate. |
|||||||||||||
| T-RAFT-1 | NIPAM | Solution | 200:1:0.1 |
1.00 | Homo | 1,4-Dioxane | 60 batch | 90 | 60 | 88.0 [40.0] | 21.50 [N/A] | 1.15 [N/A] | 199 |
| T-RAFT-2 | NIPAM | Solution | 200:1:0.6 |
1.00 | Homo | EtOAc | 60 batch | 70–100 | 120 | 79.0–94.0 [83.0–99.0] | 20.50–23.60 [19.50–22.20] | 1.17–1.32 [1.13–1.24] | 200 |
| T-RAFT-3 | DMA | Solution | 200:1:0.6 |
1.00 | Homo | Dioxane | 60 batch | 80 | 120 | 98.0 [99.0] | 15.90 [16.60] | 1.16 [1.12] | 200 |
| T-RAFT-4 | BA | Solution | 200:1:0.6 |
1.00 | Homo | EtOAc | 60 batch | 80 | 120 | 85.0 [87.0] | 24.80 [24.90] | 1.27 [1.25] | 200 |
| T-RAFT-5 | VAc | Solution | 62.5:1:0.25 |
1.00 | Homo | EtOAc | 60 batch | 100 | 120 | 81.0 [87.0] | 4.570[4.62 0] | 1.28 [1.26] | 200 |
| T-RAFT-6 | St | Solution | [RAFT]:[I] = 4:3 |
1.00 | Homo | Toluene | 10–40 | 120 | N/A | N/A | 12.00–15.00 | 1.20–1.24 | 204 |
| T-RAFT-7 | BA | Solution | 10:1:0.05 |
1.00 | Homo | Butyl acetate | N/A batch | 100 | 5 [10] | 54.0 [N/A] | 1.100 [1.400] | 1.20 [1.14] | 206 |
| T-RAFT-8 | BA/tBA | Solution | 80:1:0.05 |
N/A | DB-1 | Butyl acetate | N/A batch | 100 | 5 [100] | N/A | 8.300 [8.100] | 1.14 [1.36] | 206 |
| T-RAFT-9 | BA/tBA/EHA | Solution | 80:1:0.05 |
N/A | Multiblock (=3) | Butyl acetate | N/A | 100 | 5 | N/A | 10.70 | 1.28 | 206 |
| Batch | [10] | [9.300] | [1.93] | 206 | |||||||||
| T-RAFT-10 | BA/tBA/EHA/BA | Solution | 120:1:0.05 |
N/A | Multiblock (=4) | Butyl acetate | N/A | 100 | 10 | N/A | 16.50 | 1.32 | 206 |
| T-RAFT-11 | BA/tBA/EHA/BA/tBA | Solution | 130:1:0.05 |
N/A | Multiblock (=5) | Butyl acetate | N/A | 100 | 10 | N/A | 31.20 | 1.46 | 206 |
RAFT agents are often added to conventional free radical polymerization to provide a better control at the cost of the polymerization rate. However, increasing the reaction temperature and pressure represents a possible solution to counter this problem, i.e., increasing the polymerization rate without losing control over polymer molecular weight.202,203 Koch and Busch first developed RAFT polymerization in tubular reactors at elevated temperature and pressure.204 A series of RAFT polymerization runs of St at 120 °C and 50 bar with different residence times were carried out (reaction T-RAFT-6).204 PDIs of PSt were found to be between 1.20 and 1.24 at elevated temperature and pressure, showing well-controlled polymerization.
RAFT block copolymerization of homogeneous systems was also reported using tubular reactors.204–206 Koch and Busch synthesized DB-1 St/MA and St/MMA copolymers by RAFT polymerization using PSt as macro-RAFT agents at elevated temperature and pressure in tubular reactors.204 Hornung et al. synthesized various DB-1 copolymers by combining two tubular reactors in series.205 Most work reporting block copolymerization using CRP limited the synthesis of block copolymers with no more than three blocks, regardless if it was conducted in batch, semi-batch, or continuous reactors. This is because of an accumulation of dead chains in each block addition, which broadens the MWD. The synthesis of block copolymers with more than three blocks yet possessing a narrow MWD is still challenging. Vandenbergh et al. first reported the synthesis of multiblock copolymers by RAFT polymerization in a continuous tubular microreactor.206 The polymerization for each block was all kept at 100 °C within 5 to 20 min. The first step was RAFT polymerization of BA to produce PBA as a macroRAFT agent (reaction T-RAFT-7).206 This macroRAFT agent was used in sequential block polymerization (reactions T-RAFT-8 to T-RAFT-11),206 with the final polymer products having five blocks (poly[BA-b-tBA-b-EHA-b-BA-tBA]) with cumulative Mn = 31.20 kg mol−1 and PDI = 1.46 (reaction T-RAFT-11).206 Similar polymerization steps were conducted in a batch reactor under the same reaction conditions as in the continuous microreactor. However, only tri-block copolymers (poly[BA-b-tBA-b-EHA]) with Mn = 9.30 kg mol−1 and PDI = 1.93 could be produced. Their work clearly demonstrated how the continuous tubular microreactor could benefit consecutive polymerization while maintaining good control and high livingness.
In ATRP systems, polymers are produced with deep color due to a large residual amount of copper catalyst. Similar to ATRP systems, RAFT-derived polymers also display deep color because of thiocarbonylthio end groups. Removal and modification of the end groups are often required.207 Hornung et al. reported a radical-induced RAFT end group removal at 100 °C in a tubular reactor.208 RAFT polymerizations of AM, MA and St were first conducted in a batch reactor to produce RAFT-derived polymers at 70–100 °C. RAFT end group removal was then conducted in a continuous tubular reactor in either organic solvents or water at 100 °C. Recently, Hornung et al. also reported both RAFT polymerization and end-group removal in a flow tubular reactor by a sequential two-step process.209 Vandenbergh and Junkers reported end group modification of RAFT-derived polymers in a continuous microreactor.210 RAFT-derived PBA polymers were modified by aminolysis/thiol–ene reactions and it took only 20 min. Moreover, the reactor could be easily scaled up from the production of hundred grams to kilograms per day. Furthermore, Vandenbergh et al. reported the modification of ATRP-derived and RAFT-derived polymers and produced DB-1 copolymers via click chemistry in tubular microreactors.211
| Reaction no. | Monomer M1/M2 | Heterogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | T | t | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RAFT:I; M = monomer; RAFT = RAFT agent; I = conventional initiator; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions; DMF = dimethylformamide. |
|||||||||||||
| T-RAFT-12 | St | Mini-emulsion | 300:1:0.1 |
1.00 | Homo | — | 79–424 batch | 70 | 420 [480] | 65.0 [62.0] | 23.00 [17.50] | 1.50 [1.40] | 212 |
| T-RAFT-13 | St | Mini-emulsion | 300:1:0.1 |
1.00 | Homo | — | 55–226 batch | 70 | 240 [420] | 52.0 [68.0] | 18.00 [N/A] | 1.60 [1.20] | 213 |
| T-RAFT-14 | St | Mini-emulsion | 500:1:1 |
1.00 | Homo | — | 70–140 batch | 70 | N/A | 22.0–92.0 [24.0–93.0] | 11.85–46.40[10.39–48.44] | 1.26–1.66 [1.21–1.65] | 214 |
| T-RAFT-15 | MMA | Emulsion | 200:1:0.5 |
1.00 | Homo | Water/DMF | 28.9–50.5 | 90 | N/A | 7.90–89.8 | 8.500–23.50 | 1.05–1.07 | 215 |
| T-RAFT-16 | MMA | Emulsion | 200:1:0.5 |
1.00 | Homo | Water/DMF | 43.3 | 90 | N/A | 39.1–75.3 | 23.20–27.50 | 1.05–1.31 | 215 |
| T-RAFT-17 | MMA | Emulsion | 300–600:1:0.5 |
1.00 | Homo | Water/DMF | 48.1 | 90 | N/A | 63.4–79.5 | 32.50–61.00 | 1.14–1.31 | 215 |
A detailed kinetic comparison was reported for a similar reactor system (reaction T-RAFT-13).213 In the work, a chain extension experiment was conducted to demonstrate the livingness of polymers produced from a tubular reactor. Subsequent polymerization in a batch reactor was conducted to produce DB-1 copolymers without additional initiators. Russum et al. extended the work on mini-emulsion RAFT polymerization of St to a single tubular reactor.214 The kinetics and flow characteristics of the RAFT mini-emulsion polymerization were thoroughly investigated in the tubular reactor (reaction T-RAFT-14).214 Their experimental results indicated that the flow regime and RTD play important roles in the resulting PDI. The polymerization conducted in a near-ideal flow regime produced polymers having a similar PDI to the ones obtained from a batch reactor.
However, in mini-emulsion, ultrasonication is usually used to prepare monomer droplets, and a large amount of surfactant is added to stabilize the droplets, which limits the commercial application of the system in a continuous process. Recently, Li et al. reported a surfactant-free RAFT emulsion polymerization of MMA with 4-cyano-4-(thiobenzoylthio)pentanoic acid (CTBCOOH) playing dual roles as a RAFT agent and as an emulsion stabilizer in a tubular reactor.215 The effects of residence time, feeding rate and targeted molecular weight were all investigated, and a chain extension experiment was also carried out, which showed a controlled and living polymerization (reactions T-RAFT-15 to T-RAFT-17).215
3.3. NMP
| Reaction no. | Monomer M1/M2 | Homogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | t | T | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:PT:NR; M = monomer; PT = NMP initiator; NR = nitroxide radical; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a tubular reactor at similar experimental conditions; 2-MPA = 2-methoxypropyl acetate.a BA conversion. |
|||||||||||||
| T-NMP-1 | St | Solution | 288:1:0.05 |
1.00 | Homo | Toluene | 100–380 batch | 100–380 | 140 | 45.0–72.0 [52.0–72.0] | 15.00–23.00[15.00–22.00] | 1.20–1.38 [1.18–1.38] | 216 |
| T-NMP-2 | BA | Solution | 288:1:0.05 |
1.00 | Homo | Toluene | 65–380 batch | 65–380 | 140 | 7.00–28.0 [12.0–98.0] | 2.500–8.800 [5.000–30.00] | 1.20–1.30 [1.25–2.80] | 216 |
| T-NMP-3 | St | Solution | 200:1:0 |
1.00 | Homo | 2-MPA | 30–300 batch | 30–300 [300] | 105 | 7.00–48.0 [39.0] | 2.000–8.800 [7.500] | 1.09–1.37 [1.16] | 217 |
| T-NMP-4 | St | Solution | 100:1:0 |
1.00 | Homo | 2-MPA | 30–300 batch | 30–300 [300] | 140 | 26.0–87.0 [78.0] | 6.200–18.50 [16.70] | 1.12–1.19 [1.26] | 217 |
| T-NMP-5 | BA | Solution | 100:1:0 |
1.00 | Homo | 2-MPA | 60, 120 | N/A | 120 | 77.0, 89.0 | 10.10 11.30 | 1.41, 1.35 | 217 |
| T-NMP-6 | MMA | Solution | 100:1:0 |
1.00 | Homo | 2-MPA | 30–300 | 30–300 | 90 | 38.0–62.0 | 6.100–11.60 | 1.53–1.94 | 217 |
| T-NMP-7 | St | Solution | N/A | 1.00 | Homo | — | 75 batch | 75–345 [75] | 135 | 7.00–9.00 [17.0–17.8] | 1.490–2.285 [3.126–3.186] | 1.17–1.18 [1.14–1.17] | 218 |
| T-NMP-8 | BA/St | Solution | 200:1:0 |
0.50 | DB-2 | 2-MPA | 240 | N/A | 120–140 | 76.0 | 16.30 | 1.26 | 217 |
| T-NMP-9 | BA/St | Solution | N/A | N/A | DB-1 | Toluene | 380 batch | N/A | 125 | 96.0–99.0a [99a] | 26.60–36.60 [33.60] | 1.40–1.73 [1.74] | 219 |
| T-NMP-10 | BA/St | Solution | N/A | N/A | DB-1 | Toluene | 380 | N/A | 125–140 | 92.0–98.0a | 28.40–36.60 | 1.28–1.52 | 220 |
| T-NMP-11 | BA/St | Solution | N/A | N/A | DB-1 | Toluene | 340 | N/A | 125–140 | 97.0–98.0a | 39.70–45.70 | 1.40–1.47 | 220 |
| T-NMP-12 | BA/St | Solution | N/A | N/A | DB-1 | Toluene | 290 | N/A | 125–140 | 98.0–99.0a | 44.50–49.60 | 1.42–1.46 | 220 |
Fukuyama et al. also reported NMP of St (reactions T-NMP-3 and T-NMP-4),217 BA (reaction T-NMP-5),217 and MMA (reaction T-NMP-6)217 in tubular microreactors. They reported better control in the tubular microreactor than in a batch reactor. For example, with a residence time of 300 min, PSt with Mn = 8.8 kg mol−1 and PDI = 1.09 was prepared at 48% conversion in the tubular microreactor. Meanwhile, the batch reactor gave a conversion of only 39% after 300 minutes of reaction, with Mn = 7.5 kg mol−1 and PDI = 1.16 under the same experimental conditions (reaction T-NMP-3).217 Enright et al. also investigated the polymerization kinetics of bulk NMP of St in a tubular reactor and in a batch reactor (reaction T-NMP-7).218
There are several reports on the synthesis of DB-1 copolymers by homogeneous NMP in tubular reactors.217,219,220 Fukuyama et al. synthesized DB-2 BA/St copolymers via solution NMP by combining two tubular reactors in series (reaction T-NMP-8).217 Rosenfeld et al. studied the effects of micromixer type (reaction T-NMP-9)219 and geometry (reactions T-NMP-10 to T-NMP-12)220 on the properties of DB-1 copolymers, synthesized in two tubular reactors in series. Multilamination and bilamination micromixers were the two types compared in the study. A higher polymerization rate of the comonomer (St in Rosenfeld's work) and a lower PDI were found with the multilamination micromixer. Moreover, combining the two micromixer types in the tubular reactors resulted in better control than in a batch reactor.219 The work also showed that the micromixer geometry, varied by the number of microchannels and characteristic lengths, played an important role in the resulting properties of DB-1 copolymers, which could be optimized to obtain a more efficient mixing.220
| Reaction no. | Monomer M1/M2 | Heterogeneous system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | T | t | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:PT:NR; M = monomer; PT = NMP initiator; NR = nitroxide radical; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a tubular reactor at similar experimental conditions.a St conversion. |
|||||||||||||
| T-NMP-13 | St | Mini-emulsion | N/A | 1.00 | Homo | — | N/A batch | 135 | 200 [210] | 87.0 [94.0] | 24.00 [27.00] | 1.30 [1.32] | 221 |
| T-NMP-14 | St | Mini-emulsion | N/A | 1.00 | Homo | — | 180 | 135 | N/A | 82.6–99.1 | 15.50–25.23 | 1.19–1.34 | 218 |
| T-NMP-15 | St/BA | Mini-emulsion | N/A | N/A | DB-1 | — | 180 | 135 | N/A | 85.6–99.9a | 20.50–39.30 | 1.25–2.02 | 218 |
| T-NMP-16 | St/BA | Mini-emulsion | N/A | N/A | TB-1 | — | 120 | 135 | N/A | 91.5a | 58.60 | 2.95 | 218 |
The synthesis of DB-1 and TB-1 St/BA copolymers was also reported in a complete continuous tubular reactor by using macroinitiators.218 DB-1 St/BA copolymers having Mn = 20.5–39.3 kg mol−1 and PDI = 1.25–2.02 were produced with PSt as the macroinitiator (reaction T-NMP-15).218 The volume-average diameter of the final particles ranged from 160 to 200 nm. TB-1 St/BA/St copolymers having Mn = 58.60 kg mol−1 and PDI = 2.95 were produced with DB-1 St/BA copolymer as the macroinitiator (reaction T-NMP-16)218 and the volume-average diameter of the final particles was 213 nm. Both PDI and particle size increased with increasing number of blocks.
3.4. Summary
Numerous studies have reported the investigation of homogeneous and heterogeneous CRP systems using tubular reactors, with pioneering work done by Zhu's group for ATRP systems. The kinetics observed in tubular reactors were in general similar to those in batch reactors. Moreover, the characteristics of living polymerization in batch reactors were present in tubular reactors. These studies also showed that the mean residence time (RT) and residence time distribution (RTD) play an important role in the polymerization kinetics.Tubular reactors are efficient for conducting continuous CRP. For example, the column reactor packed with silica-gel supported catalysts and the copper tubular reactor used as a catalyst source both improved the catalyst efficiency significantly in ATRP systems. The removal or modification of the RAFT end group takes only a few minutes to complete in tubular reactors. The large surface-to-volume ratio of tubular reactors offers excellent heat transfer ability, allowing better control over highly exothermic reactions than in a batch reactor.
Table 14 summarizes the polymers having various composition profiles produced from CRP using tubular reactors, with a majority of the studies conducted for homopolymers, followed by di-block copolymers, and then multi-block copolymers having three or more blocks.
| System | Homopolymer | Di-block | Multi-block (≥3) |
|---|---|---|---|
| ATRP (homogeneous) | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-4 T-ATRP-5 T-ATRP-6 T-ATRP-7 T-ATRP-8 T-ATRP-9 | T-ATRP-10 T-ATRP-11 T-ATRP-12 T-ATRP-13 | |
| ATRP (heterogeneous) | T-ATRP-16 T-ATRP-17 T-ATRP-18 T-ATRP-20 T-ATRP-21 T-ATRP-22 T-ATRP-23 T-ATRP-24 T-ATRP-25 | T-ATRP-19 | |
| RAFT (homogeneous) | T-RAFT-1 T-RAFT-2 T-RAFT-3 T-RAFT-4 T-RAFT-5 T-RAFT-6 T-RAFT-7 | T-RAFT-8 | T-RAFT-9 T-RAFT-10 T-RAFT-11 |
| RAFT (heterogeneous) | T-RAFT-12 T-RAFT-13 T-RAFT-14 T-RAFT-15 T-RAFT-16 T-RAFT-17 | ||
| NMP (homogeneous) | T-NMP-1 T-NMP-2 T-NMP-3 T-NMP-4 T-NMP-5 T-NMP-6 T-NMP-7 | T-NMP-8 T-NMP-9 T-NMP-10 T-NMP-11 T-NMP-12 | |
| NMP (heterogeneous) | T-NMP-13 T-NMP-14 | T-NMP-15 | T-NMP-16 |
Tubular reactors are advantageous for production of block copolymers. For example, in batch reactors, the synthesis of di-block copolymers usually involves two steps. However, di-block copolymers can be prepared by using two tubular reactors in series. Moreover, the di-block copolymer properties can be improved by optimizing the geometry of the micromixer. Conducting CRP in tubular reactors also allows production of block copolymers having five blocks with a PDI less than 1.5. In comparison, the block copolymers having only three blocks from batch reactors give a PDI of 1.9. It should be noted that Table 14 only summarizes linear polymerization in tubular reactors. There was also one paper from Serra et al. that reported branching polymerization by ATRP. The polymerization rate and branching efficiency were improved in tubular reactors in comparison to those in batch reactors.
It is evident from Table 14 that tubular reactors are employed mostly in ATRP, much less in NMP. This may be due to the high temperature used in NMP. It is also clear that homogeneous systems are mostly studied in tubular reactors, because they are less complicated than heterogeneous systems. Among the heterogeneous systems, mini-emulsion polymerization was mostly studied, with little reported work on emulsion polymerization to date.
4. CSTR
Reports on CRP systems using CSTRs were scattered, with only a few examples with ATRP and RAFT polymerization. Selective CSTR works on ATRP and RAFT are summarized in Tables 15 and 16, respectively.| Reaction no. | Monomer M1/M2 | Polymerization system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | T | t | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RX:C:L; M = monomer; RX = ATRP initiator; C = catalyst; L = ligand; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; DMSO = dimethyl sulfoxide; DMF = dimethylformamide.a Two CSTRs in series. |
|||||||||||||
| C-ATRP-1 | MA | Heterogeneous catalyst | [M]:[RX]:[L] = 100:1:0.05 |
1.00 | Homo | DMSO | 30–90 | 30 | 210–630 | 40.9–65.2 | 4.67–6.73 | 1.78–1.79 | 222 |
| C-ATRP-2 | MA | Heterogeneous catalyst | [M]:[RX]:[L] = 100:1:0.01 |
1.00 | Homo | DMSO | 60 | 30 | 420 | 52.3–61.7 | 6.33–6.67 | 1.72–1.81 | 222 |
| C-ATRP-3 | MA | Heterogeneous catalyst | [M]:[RX]:[L] = 100:1:0.01, 100:1:0.05 |
1.00 | Homo | DMSO | 60 | 30 | 420 | 52.3, 56.2 | 6.33, 5.87 | 1.72, 1.78 | 222 |
| C-ATRP-4a | MA | Heterogeneous catalyst | [M]:[RX]:[L] = 100:1:0.05 |
1.00 | Homo | DMSO | 60 | 30 | 840 | 50.0–55.069.0–73.0 | 6.090 7.343 | 1.70 1.55 | 222 |
| C-ATRP-5 | BA | Solution | 100:1:0.005:0.005 |
1.00 | Homo | DMF | 60–120 batch | 90 | 420–840 [360] | 38.7–56.1 [95.0] | 5.85–8.50 [11.46] | 1.82–1.92 [1.36] | 223 |
| C-ATRP-6 | MMA | Solution | 100:1:0.005:0.005 |
1.00 | Homo | Anisole/DMF | 60–120 batch | 90 | 420–840 [360] | 33.7–53.5 [91.0] | 10.09–12.12 [12.30] | 1.89–1.96 [1.46] | 223 |
| Reaction no. | Monomer M1/M2 | Polymerization system | Reaction conditions | F 1t | Copolymer profile | Solvent | RT | T | t | X | M n | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction conditions = total mole ratios of M:RAFT:I; M = monomer; RAFT = RAFT agent; I = conventional initiator; F1t = targeted mole fraction of M1; RT = residence time (min); T = polymerization temperature (°C); t = polymerization time (min); X = total monomer conversion (%); Mn = number-average molecular weight (kg mol−1); PDI = polydispersity index; Homo = homopolymer; N/A = not available in the literature; [] = results in a batch reactor, compared with results in a semi-batch reactor at similar experimental conditions.a Three CSTRs in series.b Four CSTRs in series. |
|||||||||||||
| C-RAFT-1 | St | Solution | [RAFT]:[I] = 4:3 |
1.00 | Homo | Toluene | 10–40 | 120 | N/A | N/A | 10.00–18.50 | 1.70–1.92 | 204 |
| C-RAFT-2a | St | Mini-emulsion | 300:1:1 |
1.00 | Homo | — | 106 | 70 | 2620 | 25.0 | 10.00 | 2.60 | 224 |
| 106 | 70 | 2620 | 45.0 | 22.00 | 1.88 | ||||||||
| 106 | 70 | 2620 | 60.0 | 30.00 | 1.63 | ||||||||
| C-RAFT-3a | St | Mini-emulsion | 300:1:1 |
1.00 | Homo | — | 120 | 70 | 2340 | 17.0 | 4.500 | 1.77 | 225 |
| 120 | 70 | 2340 | 30.0 | 7.400 | 1.75 | ||||||||
| 120 | 70 | 2340 | 41.0 | 9.700 | 1.65 | ||||||||
| C-RAFT-4b | St/BA | Mini-emulsion | 480:1:1 |
0.635 | Block | — | 133 | 74 | 3600 | 25.0 | 25.00 | 2.60 | 226 |
| 133 | 74 | 3600 | 42.0 | 31.00 | 2.13 | ||||||||
| 133 | 71 | 3600 | 71.0 | 46.00 | 2.70 | ||||||||
| 133 | 71 | 3600 | 86.0 | 53.00 | 2.65 | ||||||||
4.1. ATRP
Chan et al. employed a CSTR system for copper-mediated CRP.222 In the work, SET-LRP of MA with copper wire was conducted in CSTR at 30 °C. The effects of residence time (RT), copper surface area, and ligand concentration on the kinetics of SET-LRP of MA were thoroughly investigated. The increase of RT led to the increase of conversion and Mn, while it had a minor effect on PDI at the steady state. For example, when the RT was increased from 30 to 90 min, the conversion increased from 40.9 to 65.8% and Mn increased from 4.67 to 6.73 kg mol−1, while PDI remained unchanged at about 1.78 to 1.79 (reaction C-ATRP-1).222A higher copper surface area resulted in a higher rate of polymerization. When the copper surface area was increased from 5.88 to 23.52 cm2, the steady-state conversion increased from 52.3 to 61.7% with Mn ranging from 6.19 to 6.67 kg mol−1 and PDI remained around 1.72 to 1.81 (reaction C-ATRP-2).222 Reducing the ligand concentration led to a slight drop in the polymerization rate, but only had a little effect on the control of molecular weight. For example, a reduction of the ligand concentration by five-fold with RT = 60 min and a copper surface area of 5.88 cm2 resulted in a decrease of the steady-state conversion from 56.2 to 52.3%, while Mn and PDI were in the range of 5.87 to 6.33 kg mol−1 and 1.72 to 1.78, respectively (reaction C-ATRP-3).222
Furthermore, SET-LRP of MA was also conducted in a train of two CSTRs in series. From the first CSTR to the second CSTR, the conversion increased from 50.0–55.0% to 69.0–73.0%, Mn increased from 6.090 to 7.434 kg mol−1, and PDI decreased from 1.70 to 1.55 (reaction C-ATRP-4).222 Chain extension experiments in a batch reactor revealed that the polymer chains in the final product were still living.
Recently, Chan et al. conducted ARGET ATRP in CSTR.223 Solution ARGET ATRP of BA and MMA were both conducted in CSTRs with RT = 60, 90, and 120 min at 90 °C. Steady states were reached within four mean residence times. When RT increased from 60 to 120 min, BA conversion increased from 38.7 to 56.1%, while Mn increased from 5.85 to 8.50 kg mol−1 and PDI increased from 1.82 to 1.92. The same increase in RT led to an increase of MMA conversion from 33.7 to 53.5% and Mn from 10.09 to 12.12 kg mol−1, but a decrease of PDI from 1.96 to 1.89 (reactions C-ATRP-5 and C-ATRP-6).223 Compared with the batch reactor counterparts, faster rates of polymerization were observed in CSTRs. This is due to a higher steady-state concentration of the reducing agent in CSTR, in contrast to a gradual depletion of the reducing agent in the batch reactor. Chain extension experiments indicated good livingness despite the broad MWD from CSTR systems.
4.2. RAFT
Koch and Busch reported the homogeneous RAFT of St in CSTR at elevated temperature and pressure.204 The effect of RT on polymerization kinetics was investigated. At 120 °C and 120 bar, when RT was increased from 10 to 40 min, the PSt yield increased by about 13 to 20%, Mn increased from 10 to 18.5 kg mol−1, while PDI decreased from 1.92 to 1.70 (reaction C-RAFT-1).204The only study reported in the literature about heterogeneous RAFT using CSTR was by Schork's group.224–226 Smulders et al. investigated the kinetics of RAFT mini-emulsion polymerization of styrene in a train of three CSTRs at 70 °C (reaction C-RAFT-2).224 Increasing the number of CSTRs increased the conversion and Mn, and decreased the PDI. However, a steady state could not be achieved in this study, as evidenced from the increase of conversion over time. They attributed the unsteady state to the possible continuous formation of oligomeric RAFT agents, which promoted the polymerization rate. Compared to a batch reactor, particle nucleation efficiencies were lower in CSTRs, because there existed a larger difference in the polymer contents among particles, that could promote coalescence of monomer droplets and particles.
In a subsequent study, Qi et al. further investigated the unsteady state observed in continuous RAFT mini-emulsion in CSTRs. They considered two different factors that might cause the unsteady state, namely the reaction mechanism itself and the equipment design. By modifying the equipment design and conducting RAFT under similar experimental conditions to those in the previous work, a steady-state behavior was achieved (reaction C-RAFT-3).225 Therefore, the previously unsteady state was found to be caused by equipment design and operation, not the reaction mechanism itself.
Furthermore, Smulders et al. also reported the synthesis of block copolymers by CRP in CSTR. A series of St/BA block copolymers were successfully produced by RAFT mini-emulsion polymerization in a train of four CSTRs (reaction C-RAFT-4).226 Their results not only demonstrated the feasibility of block copolymer production using a CSTR train, but also showed that the copolymer composition and block chain length could be easily regulated by the monomer feeding rate, the injection point of the comonomer, and/or the polymerization temperature.226
4.3. Summary
There are only a few reported studies on CRP conducted in CSTRs. However, the kinetic characteristics of CRP in CSTR can be drawn from these studies. Similar to tubular reactors, both RTD and RT play important roles in polymerization processes. The steady state can usually be achieved after several RTs. Living characteristics can be retained when CRP is conducted in CSTRs. However, broader MWDs should be expected compared to the polymers produced in batch and tubular reactors, due to the broader RTD in CSTR systems. Both polymerization rate and molecular weight increased with increasing number of CSTRs in a CSTR train, while an opposite trend was observed for PDI. Block copolymers can also be produced by using CSTRs in series. Furthermore, the copolymer composition and block chain length can be controlled by the monomer feeding rate and the injection point of the comonomer.Homogenous and heterogeneous ATRP in CSTRs have only been reported by Cunningham's group. The literature of RAFT mini-emulsion polymerization in CSTRs is mainly contributed by Schork's group. To date, little work on NMP in CSTR has been reported. It is not easy to develop CRP from batch reactors to CSTRs. Despite the limited references, the feasibility of conducting CRP in CSTRs is clearly demonstrated.
5. Progress in reaction engineering of CRP
Since CRP was first developed in semi-batch reactors in 1997 by Matyjaszewski's group, and the development in continuous reactors first reported in 2000 by Zhu's group, there has been significant progress made in the reactor engineering of CRP. Numerous fundamental research works on CRP with various reactor configurations have been reported. Semi-batch reactors are mainly used to control copolymer composition distribution (CCD), as summarized in Table 7. More importantly, programmed feeding policies have been developed to precisely synthesize copolymers with pre-defined CCDs at will. Many researchers have also investigated the effects of CCDs on copolymer properties. The development of the relationships between polymer chain structures and polymer material properties becomes essential and has been making good progress. On the other hand, low molecular weight polymers (such as coatings) have also been produced by CRP using semi-batch reactors. The kinetics of CRP in tubular reactors and CSTRs under different operating conditions have also been thoroughly investigated, which provides fundamental insight and practical guidance to optimize polymerization processes and/or to improve polymer properties in continuous reactors. The feasibility of CRP in continuous reactors has been well demonstrated.Table 17 summarizes the reported residence time (RT) of continuous reactors (tubular reactor and CSTR). Four RT intervals are defined, (1) RT ≤ 40 min, (2) 40 < RT ≤ 100 min, (3) 100 < RT ≤ 200 min and (4) RT > 200 min. The number of studies on tubular reactors conducted in each RT range is comparable. However, for CSTRs, most studies are conducted in the ranges of 40–100 min and 100–200 min. Moreover, CRP with a RT longer than 200 min in CSTR is rare.
| Reactor type | Residence time (minutes) | |||
|---|---|---|---|---|
| ≤40 | 40–100 | 100–200 | >200 | |
| Tubular | T-ATRP-9 T-ATRP-16 T-ATRP-20 T-ATRP-21 T-ATRP-22 T-ATRP-23 T-ATRP-25 | T-ATRP-11 T-ATRP-14 T-ATRP-16 T-ATRP-17 T-ATRP-22 T-ATRP-25 | T-ATRP-4 T-ATRP-5 T-ATRP-15 T-ATRP-16 T-ATRP-17 T-ATRP-18 | T-ATRP-6 T-ATRP-8 T-ATRP-16 T-ATRP-17 T-ATRP-18 |
| T-RAFT-6 T-RAFT-15 | T-RAFT-1 T-RAFT-2 T-RAFT-3 T-RAFT-4 T-RAFT-5 T-RAFT-12 T-RAFT-13 T-RAFT-14 T-RAFT-15 T-RAFT-16 T-RAFT-17 | T-RAFT-12 T-RAFT-13 T-RAFT-14 | T-RAFT-12 T-RAFT-13 | |
| T-NMP-3 T-NMP-4 T-NMP-6 | T-NMP-1 T-NMP-2 T-NMP-3 T-NMP-4 T-NMP-5 T-NMP-6 T-NMP-7 | T-NMP-1 T-NMP-2 T-NMP-3 T-NMP-4 T-NMP-5 T-NMP-6 T-NMP-14 T-NMP-15 T-NMP-16 | T-NMP-1 T-NMP-2 T-NMP-3 T-NMP-4 T-NMP-6 T-NMP-8 T-NMP-9 T-NMP-10 T-NMP-11 T-NMP-12 | |
| CSTR | C-ATRP-1 | C-ATRP-1 C-ATRP-2 C-ATRP-3 C-ATRP-4 C-ATRP-5 C-ATRP-6 | C-ATRP-5 C-ATRP-6 | |
| C-RAFT-1 | C-RAFT-2 C-RAFT-3 C-RAFT-4 | |||
The data of conversion, number-average molecular weight and polydispersity index discussed in Tables 1–6, Tables 8–13, Tables 15 and 16 are summarized in Tables 18–20, respectively. Generally speaking, the conversions obtained in semi-batch reactors are higher than those obtained in continuous reactors, as shown in Table 18. In semi-batch reactors, most CRP can reach high conversions, ranging from 80 to 100%, with only a few reported conversions lower than 40%. In tubular reactors and CSTRs, the conversions are mostly in the range of 40–60% and 60–80%.
| Reactor type | Conversion (%) | |||
|---|---|---|---|---|
| ≤40 | 40–60 | 60–80 | 80–100 | |
| Semi-batch | S-ATRP-11 S-ATRP-12 S-ATRP-13 S-ATRP-16 S-ATRP-19 | S-ATRP-3 S-ATRP-14 S-ATRP-19 | S-ATRP-2 S-ATRP-4 S-ATRP-5 S-ATRP-6 S-ATRP-7 S-ATRP-8 S-ATRP-14 S-ATRP-15 S-ATRP-17 S-ATRP-18 | |
| S-RAFT-1 S-RAFT-2 | S-RAFT-2 S-RAFT-3 S-RAFT-4 S-RAFT-10 S-RAFT-17 S-RAFT-18 | S-RAFT-7 S-RAFT-8 S-RAFT-10 S-RAFT-11 S-RAFT-12 S-RAFT-13 S-RAFT-14 S-RAFT-15 S-RAFT-16 S-RAFT-17 | ||
| S-NMP-1 S-NMP-21 S-NMP-22 | S-NMP-1 S-NMP-2 S-NMP-4 S-NMP-22 | S-NMP-1 S-NMP-2 S-NMP-3 S-NMP-7 S-NMP-18 S-NMP-20 S-NMP-22 | S-NMP-1 S-NMP-2 S-NMP-5 S-NMP-8 S-NMP-9 S-NMP-10 S-NMP-11 S-NMP-16 S-NMP-17 S-NMP-18 S-NMP-19 S-NMP-20 | |
| Tubular | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-6 T-ATRP-7 T-ATRP-8 T-ATRP-9 T-ATRP-10 T-ATRP-11 T-ATRP-16 T-ATRP-17 T-ATRP-18 T-ATRP-22 | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-5 T-ATRP-6 T-ATRP-7 T-ATRP-8 T-ATRP-10 T-ATRP-16 T-ATRP-17 T-ATRP-18 T-ATRP-20 T-ATRP-21 T-ATRP-22 T-ATRP-24 | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-4 T-ATRP-6 T-ATRP-7 T-ATRP-9 T-ATRP-10 T-ATRP-14 T-ATRP-15 T-ATRP-16 T-ATRP-17 T-ATRP-18 T-ATRP-20 T-ATRP-21 T-ATRP-22 T-ATRP-23 T-ATRP-25 | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-6 T-ATRP-7 T-ATRP-16 T-ATRP-25 |
| T-RAFT-14 T-RAFT-15 T-RAFT-16 | T-RAFT-7 T-RAFT-13 T-RAFT-14 T-RAFT-15 T-RAFT-16 | T-RAFT-2 T-RAFT-12 T-RAFT-14 T-RAFT-15 T-RAFT-16 T-RAFT-17 | T-RAFT-1 T-RAFT-2 T-RAFT-3 T-RAFT-4 T-RAFT-5 T-RAFT-14 T-RAFT-15 | |
| T-NMP-2 T-NMP-3 T-NMP-4 T-NMP-6 T-NMP-7 | T-NMP-1 T-NMP-3 T-NMP-4 T-NMP-6 | T-NMP-1 T-NMP-4 T-NMP-5 T-NMP-6 T-NMP-8 | T-NMP-4 T-NMP-5 T-NMP-9 T-NMP-10 T-NMP-11 T-NMP-12 T-NMP-13 T-NMP-14 T-NMP-15 T-NMP-16 | |
| CSTR | C-ATRP-5 C-ATRP-6 | C-ATRP-1 C-ATRP-2 C-ATRP-3 C-ATRP-4 C-ATRP-5 C-ATRP-6 | C-ATRP-1 C-ATRP-2 C-ATRP-4 | |
| C-RAFT-2 C-RAFT-3 C-RAFT-4 | C-RAFT-2 C-RAFT-3 C-RAFT-4 | C-RAFT-4 | C-RAFT-4 | |
| Reactor type | M n (kg mol−1) | |||
|---|---|---|---|---|
| ≤10 | 10–50 | 50–100 | >100 | |
| Semi-batch | S-ATRP-14 S-ATRP-15 | S-ATRP-1 S-ATRP-3 S-ATRP-4 S-ATRP-5 S-ATRP-6 S-ATRP-7 S-ATRP-8 S-ATRP-9 S-ATRP-10 S-ATRP-12 S-ATRP-13 S-ATRP-14 S-ATRP-16 S-ATRP-18 S-ATRP-19 | S-ATRP-2 S-ATRP-10 S-ATRP-11 | S-ATRP-10 |
| S-RAFT-1 S-RAFT-3 S-RAFT-4 S-RAFT-5 S-RAFT-6 S-RAFT-9 S-RAFT-10 S-RAFT-11 S-RAFT-12 S-RAFT-13 S-RAFT-16 S-RAFT-17 S-RAFT-18 | S-RAFT-8 S-RAFT-9 S-RAFT-10 S-RAFT-14 | S-RAFT-7 S-RAFT-8 S-RAFT-14 S-RAFT-15 | ||
| S-NMP-1 | S-NMP-1 S-NMP-2 S-NMP-3 S-NMP-4 S-NMP-6 S-NMP-7 S-NMP-10 S-NMP-14 S-NMP-18 S-NMP-19 S-NMP-20 S-NMP-21 S-NMP-22 | S-NMP-3 S-NMP-5 S-NMP-8 S-NMP-9 S-NMP-10 S-NMP-11 S-NMP-12 S-NMP-13 S-NMP-14 S-NMP-16 S-NMP-17 S-NMP-19 S-NMP-20 | S-NMP-15 S-NMP-16 | |
| Tubular | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-4 T-ATRP-5 T-ATRP-6 T-ATRP-7 T-ATRP-9 T-ATRP-10 T-ATRP-14 T-ATRP-15 T-ATRP-16 T-ATRP-17 T-ATRP-18 T-ATRP-20 T-ATRP-21 T-ATRP-22 T-ATRP-24 T-ATRP-25 | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-6 T-ATRP-8 T-ATRP-10 T-ATRP-11 T-ATRP-12 T-ATRP-13 T-ATRP-16 T-ATRP-17 T-ATRP-18 T-ATRP-19 T-ATRP-22 T-ATRP-23 | ||
| T-RAFT-5 T-RAFT-7 T-RAFT-8 T-RAFT-15 | T-RAFT-1 T-RAFT-2 T-RAFT-3 T-RAFT-4 T-RAFT-6 T-RAFT-9 T-RAFT-10 T-RAFT-11 T-RAFT-12 T-RAFT-13 T-RAFT-14 T-RAFT-15 T-RAFT-16 T-RAFT-17 | T-RAFT-17 | ||
| T-NMP-2 T-NMP-3 T-NMP-4 T-NMP-6 T-NMP-7 | T-NMP-1 T-NMP-4 T-NMP-5 T-NMP-6 T-NMP-8 T-NMP-9 T-NMP-10 T-NMP-11 T-NMP-12 T-NMP-13 T-NMP-14 T-NMP-15 | T-NMP-16 | ||
| CSTR | C-ATRP-1 C-ATRP-2 C-ATRP-3 C-ATRP-4 C-ATRP-5 | C-ATRP-6 | ||
| C-RAFT-3 | C-RAFT-1 C-RAFT-2 C-RAFT-4 | C-RAFT-4 | ||
| Reactor type | PDI | |||
|---|---|---|---|---|
| ≤1.1 | 1.1–1.5 | 1.5–2.0 | >2.0 | |
| Semi-batch | S-ATRP-9 S-ATRP-10 S-ATRP-13 S-ATRP-14 | S-ATRP-1 S-ATRP-2 S-ATRP-3 S-ATRP-4 S-ATRP-5 S-ATRP-6 S-ATRP-7 S-ATRP-8 S-ATRP-11 S-ATRP-12 S-ATRP-14 S-ATRP-16 S-ATRP-17 S-ATRP-18 S-ATRP-19 | S-ATRP-15 | |
| S-RAFT-16 S-RAFT-17 | S-RAFT-1 S-RAFT-3 S-RAFT-4 S-RAFT-5 S-RAFT-6 S-RAFT-10 S-RAFT-11 S-RAFT-12 S-RAFT-13 S-RAFT-14 S-RAFT-16 | S-RAFT-8 S-RAFT-10 S-RAFT-14 S-RAFT-15 | S-RAFT-7 S-RAFT-8 S-RAFT-9 S-RAFT-10 S-RAFT-14 S-RAFT-18 | |
| S-NMP-1 S-NMP-2 S-NMP-3 S-NMP-4 S-NMP-5 S-NMP-7 S-NMP-8 S-NMP-9 S-NMP-10 S-NMP-11 S-NMP-13 S-NMP-16 S-NMP-17 S-NMP-18 S-NMP-20 S-NMP-22 | S-NMP-15 S-NMP-16 S-NMP-18 S-NMP-20 S-NMP-21 S-NMP-22 | S-NMP-19 S-NMP-20 | ||
| Tubular | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-4 T-ATRP-12 | T-ATRP-1 T-ATRP-2 T-ATRP-3 T-ATRP-5 T-ATRP-6 T-ATRP-7 T-ATRP-8 T-ATRP-9 T-ATRP-10 T-ATRP-11 T-ATRP-12 T-ATRP-13 T-ATRP-17 T-ATRP-18 T-ATRP-20 T-ATRP-21 T-ATRP-22 T-ATRP-23 T-ATRP-24 T-ATRP-25 | T-ATRP-16 T-ATRP-17 T-ATRP-18 T-ATRP-19 | T-ATRP-14 T-ATRP-15 |
| T-RAFT-15 T-RAFT-16 | T-RAFT-1 T-RAFT-2 T-RAFT-3 T-RAFT-4 T-RAFT-5 T-RAFT-6 T-RAFT-7 T-RAFT-8 T-RAFT-9 T-RAFT-10 T-RAFT-11 T-RAFT-12 T-RAFT-14 T-RAFT-16 T-RAFT-17 | T-RAFT-13 T-RAFT-14 | ||
| T-NMP-3 | T-NMP-1 T-NMP-2 T-NMP-3 T-NMP-4 T-NMP-5 T-NMP-7 T-NMP-8 T-NMP-9 T-NMP-10 T-NMP-11 T-NMP-12 T-NMP-13 T-NMP-14 T-NMP-15 | T-NMP-6 T-NMP-9 T-NMP-10 T-NMP-15 | T-NMP-15 T-NMP-16 | |
| CSTR | C-ATRP-1 C-ATRP-2 C-ATRP-3 C-ATRP-4 C-ATRP-5 C-ATRP-6 | |||
| C-RAFT-1 C-RAFT-2 C-RAFT-3 | C-RAFT-2 C-RAFT-4 | |||
The number-average molecular weight of polymers produced by CRP using various reactors mostly fall in the range of 10–50 kg mol−1. Semi-batch reactors can produce higher molecular weight (Mn > 100 kg mol−1). Such high molecular weight products have not been reported in tubular reactors and CSTRs. However, the number of studies reporting the synthesis of low molecular weight polymers (Mn< = 10 kg mol−1) using tubular reactors and CSTRs is higher than that using semi-batch reactors. A majority of the CRP systems conducted in various reactor types discussed in this review showed good control in the polymerization with the reported PDI from 1.1 to 1.5. However, polymers produced in CSTRs generally possessed higher PDI (PDI > 1.5) than those from semi-batch and tubular reactors.
6. Outlook and recommendations
It becomes evident that ATRP is one of the most studied CRP types in each reactor category. Approximately half of the reports on CRP are based on ATRP. It is interesting to note that ATRP was also the first CRP system conducted in semi-batch and continuous reactors. The chemicals involved in ATRP are readily available from commercial sources. Moreover, metal complexes used to mediate the ATRP process can now be reduced to the ppm level. The number of reports on RAFT is much lower than that on ATRP but is comparable to NMP. However, little work has been done in NMP using CSTRs to date.There are more studies on CRP using semi-batch reactors than tubular or CSTR reactors. In terms of the ease of operation, the semi-batch reactor is easier to operate than the continuous reactor. Furthermore, it is more feasible to control CCDs using semi-batch reactors by varying the monomer feeding. A narrow MWD of polymer products is a key point for CRP systems, but continuous reactors can give broader MWD than semi-batch reactors due to the broader RTD in continuous reactors. However, polymer products with more consistent chain properties can be produced from continuous reactors than from semi-batch reactors.
Continuous reactors are preferred in industrial settings for large-scale production. Continuous CRP processes using various types of reactors must be further developed, particularly for CSTR. The challenges related to the continuous systems need to be tackled. For example, how do we control CCD in continuous reactors? It is well known that polymer products with uniform CCD (U) and block (DB-1) copolymers can be produced in continuous reactors, but how do we produce polymer products with novel CCDs (such as LG and SG)? It would represent significant progress if copolymers with pre-defined CCDs can be precisely synthesized through continuous processes. Fortunately, kinetic modeling works on different kinds of CRPs in continuous reactors have provided a great foundation for precise control of CCDs in continuous processes.227–231
More than half of the reported CRP works involve homogeneous systems. However, heterogeneous systems, such as emulsion polymerization, are favored in industrial applications, because of low energy consumption, low cost of product separation, and green environment with water usually used as solvent. It should be pointed out that heterogeneous CRP systems using different reactors are mainly conducted as mini-emulsion. Compared to emulsion, mini-emulsion requires extra energy in ultrasonication. Therefore, the emphasis in further research on heterogeneous CRP with different reactors should be on continuous emulsion polymerization.
Precise control over CCD through programmed feeding policies has been well developed. The goal for the programmed feeding policy has been on targeted CCDs. In applications, tailored made material properties of polymer products, not chain structures, are most desirable. Further development in this area should target on the desired material properties directly, through design and control of chain structures based on the relationships between the chain structure and polymer properties. On the other hand, there are only a few reports on the control of chain topologies. Reports on nonlinear CRP (such as branching and cross-linking) using different types of reactors are rare. Nonlinear CRP in continuous reactors represents an important research area, yet to be explored.
Abbreviations
| ATRP | Atom transfer radical polymerization |
| AGET ATRP | Activator generated by electron transfer ATRP |
| ARGET ATRP | Activator regenerated by electron transfer ATRP |
| ICAR ATRP | Initiator for continuous activator regeneration ATRP |
| SARA ATRP | Supplemental activator and reducing agent ATRP |
| SET-LRP | Single-electron transfer living radical polymerization |
| CCD | Copolymer composition distribution |
| CF | Constant feeding |
| ICF | Increasing constant feeding |
| CRP | Controlled Radical Polymerization |
| CRcoP | Controlled radical copolymerization |
| CSTR | Continuous-stirred tank reactor |
| FRP | Conventional free radical polymerization |
| MMFP | Model-based monomer feeding policy |
| NMP | Nitroxide-mediated polymerization |
| PF | Programmed feeding |
| PFTR | Plug flow tubular reactor |
| RAFT | Reversible addition–fragmentation chain transfer |
| RT | Residence time |
| RTD | Residence time distribution |
| MWD | Molecular weight distribution |
| MW | Molecular weight |
| U | Uniform |
| LG | Linear gradient |
| SG | “S” shape gradient |
| DB-1 | Di-block |
| DB-2 | Di-block with a gradient block |
| TB-1 | Tri-block |
| TB-2 | Tri-block with one middle gradient block |
| TB-3 | Tri-block with two terminal gradient blocks |
| PDI | Polydispersity index (also referred to as dispersity, Đ) |
Chemical abbreviations
| 2-MPA | 2-Methoxypropyl acetate |
| AA | Acrylic acid |
| AcGalEA | 2-(2′,3′,4′,6′-Tetra-o-acetyl-D-galactosyloxy)ethyl acrylate |
| AM | Acrylamide |
| AN | Acrylonitrile |
| AS | 4-Acetoxystyrene |
| BA | n-Butyl acrylate |
| BIEM | 2-(2-Bromoisobutyryloxy)-ethyl methacrylate |
| BisAM | N,N′-Methylenebis(acrylamide) |
| BMA | n-Butyl methacrylate |
| Bu | Butadiene |
| BzMA | Benzyl methacrylate |
| DFMA | Dodecafluoroheptyl methacrylate |
| DMA | N,N-Dimethylacrylamide |
| DMAEMA | 2-(Dimethylamino)ethyl methacrylate |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EHA | 2-Ethylhexyl acrylate |
| EO | Ethylene oxide |
| EtOAc | Ethyl acetate |
| HEMA | 2-Hydroxyethyl methacrylate |
| HEMA-TMS | 2-(Trimethylsilyl)ethyl methacrylate |
| HFBMA | 2,2,3,3,4,4,4-Heptafluorobutyl methacrylate |
| HPMA | 2-Hydroxypropylmethacrylate |
| IBA | Isobornyl acrylate |
| LMA | Lauryl methacrylate |
| MA | Methyl acrylate |
| MMA | Methyl methacrylate |
| MS | 4-Methylstyrene |
| NIPAM | N-Isopropyl acrylamide |
| NM2P | N-Methyl-2-pyrrolidone |
| ODA | Octadecyl acrylate |
| St | Styrene |
| tBA | tert-Butyl acrylate |
| tBMA | tert-Butyl methacrylate |
| TFEMA | 2,2,2-Trifluoroethyl methacrylate |
| VAc | Vinyl acetate |
Acknowledgements
The authors acknowledge the National Research Council of China (NSFC) and the Natural Science and Engineering Research Council (NSERC) of Canada for supporting their research. XH Li also thanks Zhejiang University for supporting his visit to McMaster University.References
- G. Moad and D. H. Solomon, The chemistry of free radical polymerization, Pergamon, Oxford, Tarrytown, N.Y, 1st edn, 1995 CAS.
- J. Qiu, B. Charleux and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 2083–2134 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- M. Szwarc, in Living Polymers and Mechanisms of Anionic Polymerization, Springer Berlin Heidelberg, 1983, ch. 1, vol. 49, pp. 1–177 Search PubMed.
- H. L. Hsieh and R. P. Quirk, Anionic Polymerization: Principles and Practical Applications, Marcel Dekker Inc, New York, 1996 Search PubMed.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed.
- Y. Shen, F. Zeng, S. Zhu and R. Pelton, Macromolecules, 2001, 34, 144–150 CrossRef CAS.
- M. Miyamoto, M. Sawamoto and T. Higashimura, Macromolecules, 1984, 17, 265–268 CrossRef CAS.
- M. Sawamoto, in Cationic Polymerizations: Mechanisms, Synthesis & Applications, ed. K. Matyjaszewski, Controlled polymer synthesis by cationic polymerization, CRC Press, 1996, vol. 35 Search PubMed.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
- D. Colombani, Prog. Polym. Sci., 1997, 22, 1649–1720 CrossRef CAS.
- K. Matyjaszewski, in Controlled Radical Polymerization, ed. K. Matyjaszewski, ACS Publications, 1998, ch. 2, vol. 685, pp. 2–30 Search PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- K. Matyjaszewski and A. H. Müller, Prog. Polym. Sci., 2006, 31, 1039–1040 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CAS.
- H. Tobita, Macromol. Theory Simul., 2006, 15, 12–22 CrossRef CAS.
- E. Mastan, D. Zhou and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 639–651 CrossRef CAS.
- T. E. Patten and K. Matyjaszewski, Adv. Mater., 1998, 10, 901–915 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- M. Destarac, Macromol. React. Eng., 2010, 4, 165–179 CrossRef CAS.
- S. Zhu, Macromol. React. Eng., 2010, 4, 163–164 CrossRef CAS.
- S. Arehart, D. Greszta and K. Matyjaszewski, in Gradient copolymers of styrene and n-butyl acrylate through atom transfer radical polymerization, Abstr. Pap. Am. Chem. Soc., American Chemical Society 1155 16th st, NW, Washington DC 20036, 1997, POLY-329 Search PubMed.
- D. Greszta and K. Matyjaszewski, in Gradient copolymers of styrene and acrylonitrile via atom transfer radical polymerization, Abstr. Pap. Am. Chem. Soc., American Chemical Society 1155 16th st, NW, Washington DC 20036, 1997, POLY-331 Search PubMed.
- K. Matyjaszewski, M. J. Ziegler, S. V. Arehart, D. Greszta and T. Pakula, J. Phys. Org. Chem., 2000, 13, 775–786 CrossRef CAS.
- R. Wang, Y. Luo, B. Li, X. Sun and S. Zhu, Macromol. Theory Simul., 2006, 15, 356–368 CrossRef CAS.
- R. Wang, Y. Luo, B.-G. Li and S. Zhu, AIChE J., 2007, 53, 174–186 CrossRef CAS.
- X. Sun, Y. Luo, R. Wang, B.-G. Li, B. Liu and S. Zhu, Macromolecules, 2007, 40, 849–859 CrossRef CAS.
- X. Sun, Y. Luo, R. Wang, B.-G. Li and S. Zhu, AIChE J., 2008, 54, 1073–1087 CrossRef CAS.
- Y. Zhao, Y.-W. Luo, C. Ye, B.-G. Li and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 69–79 CrossRef CAS.
- Y. Zhao, Y.-W. Luo, B.-G. Li and S. Zhu, Langmuir, 2011, 27, 11306–11315 CrossRef CAS.
- X. Li, W.-J. Wang, F. Weng, B. Li and S. Zhu, Ind. Eng. Chem. Res., 2013, 53, 7321–7332 CrossRef.
- Y. Shen, S. Zhu and R. Pelton, Macromol. Rapid Commun., 2000, 21, 956–959 CrossRef CAS.
- S. Zhu, Y. Shen and R. Pelton, Supported Catalysts for Use in Atom Transfer Radical Polymerization and Continuous Processes for Atom Transfer Radical Polymerization, WO 0162803, 2001 Search PubMed.
- Y. Shen and S. Zhu, AIChE J., 2002, 48, 2609–2619 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- A. J. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- D. Charmot, P. Corpart, H. Adam, S. Zard, T. Biadatti and G. Bouhadir, Macromol. Symp., 2000, 150, 23–32 CrossRef CAS.
- S. Yamago, K. Iida and J.-I. Yoshida, J. Am. Chem. Soc., 2002, 124, 2874–2875 CrossRef CAS PubMed.
- P. Lacroix-Desmazes, R. Severac and B. Boutevin, Macromolecules, 2005, 38, 6299–6309 CrossRef CAS.
- M. K. Georges, R. P. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- B. B. Wayland, G. Poszmik, S. L. Mukerjee and M. Fryd, J. Am. Chem. Soc., 1994, 116, 7943–7944 CrossRef CAS.
- Y. Piette, A. Debuigne, V. Bodart, N. Willet, A.-S. Duwez, C. Jérôme and C. Detrembleur, Polym. Chem., 2013, 4, 1685–1693 RSC.
- G. Moad, Y. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- J. Huang, W.-J. Wang, B. Li and S. Zhu, Macromol. Mater. Eng., 2013, 298, 391–399 CrossRef CAS.
- P. M. Kazmaier, K. Daimon, M. K. Georges, G. K. Hamer and R. P. Veregin, Macromolecules, 1997, 30, 2228–2231 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- S. Zhu, Macromol. Theory Simul., 1999, 8, 29–37 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, J. Mol. Catal. A: Chem., 2006, 254, 155–164 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- J. Gromada and K. Matyjaszewski, Macromolecules, 2001, 34, 7664–7671 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Macromolecules, 2005, 38, 4139–4146 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2005, 127, 3825–3830 CrossRef CAS PubMed.
- A. Plichta, W. Li and K. Matyjaszewski, Macromolecules, 2009, 42, 2330–2332 CrossRef CAS.
- D. Konkolewicz, A. J. Magenau, S. E. Averick, A. Simakova, H. He and K. Matyjaszewski, Macromolecules, 2012, 45, 4461–4468 CrossRef CAS.
- A. Mohammad Rabea and S. Zhu, Ind. Eng. Chem. Res., 2014, 53, 3472–3477 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., 2006, 118, 4594–4598 CrossRef.
- W. Jakubowski, B. Kirci-Denizli, R. R. Gil and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 32–39 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Macromol. Chem. Phys., 2008, 209, 1797–1805 CrossRef CAS.
- X. Li, W.-J. Wang, B. Li and S. Zhu, Macromol. React. Eng., 2011, 5, 467–478 CrossRef CAS.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- N. H. Nguyen, H.-J. Sun, M. E. Levere, S. Fleischmann and V. Percec, Polym. Chem., 2013, 4, 1328–1332 RSC.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2015 DOI:10.1021/acs.chemrev.5b00191.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. Atilla Tasdelen and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2878–2888 CrossRef CAS.
- D. Konkolewicz, Y. Wang, P. Krys, M. Zhong, A. Isse, A. Gennaro and K. Matyjaszewski, Polym. Chem., 2014, 5, 4396–4417 RSC.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- T. P. Le, G. Moad, E. Rizzardo and S. H. Thang, Polymerization with living characteristics, US Pat., 09/762,833, 1998 Search PubMed.
- R. T. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977–6980 CrossRef CAS.
- P. Corpart, D. Charmot, S. Zard, X. Franck and G. Bouhadir, Method for block polymer synthesis by controlled radical polymerisation from dithiocarbamate compounds, US Pat., 09/582,390, 1999 Search PubMed.
- Y. Luo, R. Wang, L. Yang, B. Yu, B. Li and S. Zhu, Macromolecules, 2006, 39, 1328–1337 CrossRef CAS.
- C. Barner-Kowollik, M. L. Coote, T. P. Davis, L. Radom and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2828–2832 CrossRef CAS.
- A. R. Wang, S. Zhu, Y. Kwak, A. Goto, T. Fukuda and M. S. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2833–2839 CrossRef CAS.
- K. Suzuki, Y. Nishimura, Y. Kanematsu, Y. Masuda, S. Satoh and H. Tobita, Macromol. React. Eng., 2012, 6, 17–23 CrossRef CAS.
- A. R. Wang and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1553–1566 CrossRef CAS.
- C. Such, E. Rizzardo, A. Serelis, B. Hawkett, R. Gilbert, C. Ferguson and R. Hughes, Aqueous dispersions of polymer particles, US Pat., CA 2470522, 2003 Search PubMed.
- B. Hawkett, C. Such, D. Nguyen, J. Farrugia and O. Mackinnon, Surface polymerisation process and polymer product using RAFT agent, US Pat., PCT/AU2007/000437, 2006 Search PubMed.
- D. Taton, M. Destarac and S. Z. Zard, Macromolecular Design by Interchange of Xanthates: Background, Design, Scope and Applications, in: Handbook of RAFT Polymerization, WILEY-VCH, 2008 Search PubMed.
- D. H. Solomon, E. Rizzardo and P. Cacioli, Polymerization process and polymers produced thereby, US Pat., 06/629,929, 1986 Search PubMed.
- Y. Chong, F. Ercole, G. Moad, E. Rizzardo, S. H. Thang and A. G. Anderson, Macromolecules, 1999, 32, 6895–6903 CrossRef CAS.
- J. Nicolas, L. Mueller, C. Dire, K. Matyjaszewski and B. Charleux, Macromolecules, 2009, 42, 4470–4478 CrossRef CAS.
- P. Gérard, L. Couvreur, S. Magnet, J. Ness and S. Schmidt, Controlled/living radical polymerization: progress in RAFT, DT, NMP & OMRP, 2009, pp. 361–373 Search PubMed.
- G. Arzamendi and J. M. Asua, J. Appl. Polym. Sci., 1989, 38, 2019–2036 CrossRef CAS.
- G. Arzamendi and J. M. Asua, Makromol. Chem., Macromol. Symp., 1990, 35–36, 249–268 CrossRef CAS.
- T. Enright and S. Zhu, Macromol. Theory Simul., 2000, 9, 196–206 CrossRef CAS.
- A. Möck, A. Burgath, R. Hanselmann and H. Frey, Macromolecules, 2001, 34, 7692–7698 CrossRef.
- J. L. Steinbacher and D. T. McQuade, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6505–6533 CrossRef CAS.
- F. J. Schork and J. Guo, Macromol. React. Eng., 2008, 2, 287–303 CrossRef CAS.
- D. Wilms, J. Klos and H. Frey, Macromol. Chem. Phys., 2008, 209, 343–356 CrossRef CAS.
- C. Tonhauser, A. Natalello, H. Löwe and H. Frey, Macromolecules, 2012, 45, 9551–9570 CrossRef CAS.
- A. Nagaki and J. Yoshida, in Controlled Polymerization and Polymeric Structures, ed. A. Abe, K.-S. Lee, L. Leibler and S. Kobayashi, Controlled Polymerization in Flow Microreactor Systems, Springer Berlin Heidelberg, 2012, ch. 179, pp. 1–50 Search PubMed.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3081–3096 CrossRef CAS.
- S. B. Lee, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2003, 4, 1386–1393 CrossRef CAS PubMed.
- W. Jakubowski, A. Juhari, A. Best, K. Koynov, T. Pakula and K. Matyjaszewski, Polymer, 2008, 49, 1567–1578 CrossRef CAS.
- J. Chen, J.-J. Li and Z.-H. Luo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1107–1117 CrossRef CAS.
- K. C. Gallow, Y. K. Jhon, W. Tang, J. Genzer and Y.-L. Loo, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 629–637 CrossRef CAS.
- K. C. Gallow, Y. K. Jhon, J. Genzer and Y.-L. Loo, Polymer, 2012, 53, 1131–1137 CrossRef CAS.
- H. G. Börner, D. Duran, K. Matyjaszewski, M. da Silva and S. S. Sheiko, Macromolecules, 2002, 35, 3387–3394 CrossRef.
- J.-J. Li, Y.-N. Zhou and Z.-H. Luo, Soft Matter, 2012, 8, 11051–11061 RSC.
- Y.-N. Zhou, J.-J. Li and Z.-H. Luo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3052–3066 CrossRef CAS.
- Y. Fu, A. Mirzaei, M. F. Cunningham and R. A. Hutchinson, Macromol. React. Eng., 2007, 1, 425–439 CrossRef CAS.
- Y. Fu, M. F. Cunningham and R. A. Hutchinson, in Semibatch atom transfer radical copolymerization of styrene and butyl acrylate, Macromol. Symp., Wiley Online Library, 2007, pp. 151–163 Search PubMed.
- K. Min, J. Kwon Oh and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1413–1423 CrossRef CAS.
- Y.-N. Zhou and Z.-H. Luo, Polym. Chem., 2013, 4, 76–84 RSC.
- P. Escalé, S. S. Ting, A. Khoukh, L. Rubatat, M. Save, M. H. Stenzel and L. Billon, Macromolecules, 2011, 44, 5911–5919 CrossRef.
- Y. Chen, Y. Zhang, Y. Wang, C. Sun and C. Zhang, J. Appl. Polym. Sci., 2013, 127, 1485–1492 CrossRef CAS.
- W.-J. Wang, D. Wang, B. Li and S. Zhu, Macromolecules, 2010, 43, 4062–4069 CrossRef CAS.
- D. Wang, X. Li, W.-J. Wang, X. Gong, B. Li and S. Zhu, Macromolecules, 2012, 45, 28–38 CrossRef CAS.
- D. Wang, W.-J. Wang, B. Li and S. Zhu, AIChE J., 2013, 59, 1322–1333 CrossRef CAS.
- C.-D. Vo, J. Rosselgong, S. P. Armes and N. C. Billingham, Macromolecules, 2007, 40, 7119–7125 CrossRef CAS.
- L. Tao, J. Liu, B. Tan and T. P. Davis, Macromolecules, 2009, 42, 4960–4962 CrossRef CAS.
- Q. Yu, S. Xu, H. Zhang, Y. Ding and S. Zhu, Polymer, 2009, 50, 3488–3494 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919–5924 CrossRef CAS.
- Y. Luo and X. Liu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6248–6258 CrossRef CAS.
- Z. X. Wang, Q. H. Zhang, Y. T. Yu, X. L. Zhan, F. Q. Chen and J. H. Xiong, Chin. Chem. Lett., 2010, 21, 1497–1500 CrossRef CAS.
- Q. H. Zhang, X. L. Zhan, F. Q. Chen, Y. Shi and Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1585–1594 CrossRef CAS.
- Y. Luo, X. Wang, Y. Zhu, B.-G. Li and S. Zhu, Macromolecules, 2010, 43, 7472–7481 CrossRef CAS.
- Y. Chen, W. Luo, Y. Wang, C. Sun, M. Han and C. Zhang, J. Colloid Interface Sci., 2012, 369, 46–51 CrossRef CAS PubMed.
- X. Li, W.-J. Wang, B.-G. Li and S. Zhu, Macromol. React. Eng., 2015, 9, 90–99 CrossRef CAS.
- Y. Wang, F. Naulet, M. F. Cunningham and R. A. Hutchinson, in Nitroxide-mediated semibatch polymerization for the production of low-molecular weight solvent-borne coatings resins, ACS Symp. Ser., ACS Publications, 2003, pp. 466–480 Search PubMed.
- Y. Wang, R. A. Hutchinson and M. F. Cunningham, Macromol. Mater. Eng., 2005, 290, 230–241 CrossRef CAS.
- K. Karaky, L. Billon, C. Pouchan and J. Desbrières, Macromolecules, 2007, 40, 458–464 CrossRef CAS.
- K. Karaky, C. Derail, G. Reiter and L. Billon, in Tuning the surface/bulk properties by the control of the amphiphilic profile in gradient copolymer, Macromol. Symp., Wiley Online Library, 2008, pp. 31–40 Search PubMed.
- K. Karaky, E. Péré, C. Pouchan, J. Desbrières, C. Dérail and L. Billon, Soft Matter, 2006, 2, 770–778 RSC.
- K. Karaky, G. Clisson, G. Reiter and L. Billon, Macromol. Chem. Phys., 2008, 209, 715–722 CrossRef CAS.
- N. Cherifi, A. Issoulie, A. Khoukh, A. Benaboura, M. Save, C. Derail and L. Billon, Polym. Chem., 2011, 2, 1769–1777 RSC.
- O. Borisova, L. Billon, M. Zaremski, B. Grassl, Z. Bakaeva, A. Lapp, P. Stepanek and O. Borisov, Soft Matter, 2012, 8, 7649–7659 RSC.
- O. Borisova, L. Billon, M. Zaremski, B. Grassl, Z. Bakaeva, A. Lapp, P. Stepanek and O. Borisov, Soft Matter, 2011, 7, 10824–10833 RSC.
- M. K. Gray, H. Zhou, S. T. Nguyen and J. M. Torkelson, Polymer, 2004, 45, 4777–4786 CrossRef CAS.
- R. W. Sandoval, D. E. Williams, J. Kim, C. B. Roth and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2672–2682 CrossRef CAS.
- C. L. Wong, J. Kim and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2842–2849 CrossRef CAS.
- M. M. Mok, J. Kim, C. L. Wong, S. R. Marrou, D. J. Woo, C. M. Dettmer, S. T. Nguyen, C. J. Ellison, K. R. Shull and J. M. Torkelson, Macromolecules, 2009, 42, 7863–7876 CrossRef CAS.
- W. Yuan, M. M. Mok, J. Kim, C. L. H. Wong, C. M. Dettmer, S. T. Nguyen, J. M. Torkelson and K. R. Shull, Langmuir, 2010, 26, 3261–3267 CrossRef CAS PubMed.
- M. K. Gray, H. Zhou, S. T. Nguyen and J. M. Torkelson, Macromolecules, 2004, 37, 5586–5595 CrossRef CAS.
- J. Kim, M. M. Mok, R. W. Sandoval, D. J. Woo and J. M. Torkelson, Macromolecules, 2006, 39, 6152–6160 CrossRef CAS.
- M. M. Mok, J. Kim and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 48–58 CrossRef CAS.
- M. M. Mok, S. Pujari, W. R. Burghardt, C. M. Dettmer, S. T. Nguyen, C. J. Ellison and J. M. Torkelson, Macromolecules, 2008, 41, 5818–5829 CrossRef CAS.
- M. Mok, J. Kim, S. Marrou and J. Torkelson, Eur. Phys. J. E: Soft Matter Biol. Phys., 2010, 31, 239–252 CrossRef CAS PubMed.
- M. M. Mok and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 189–197 CrossRef CAS.
- M. M. Mok, K. A. Masser, J. Runt and J. M. Torkelson, Macromolecules, 2010, 43, 5740–5748 CrossRef CAS.
- J. Kim, H. Zhou, S. T. Nguyen and J. M. Torkelson, Polymer, 2006, 47, 5799–5809 CrossRef CAS.
- J. Kim, R. W. Sandoval, C. M. Dettmer, S. T. Nguyen and J. M. Torkelson, Polymer, 2008, 49, 2686–2697 CrossRef CAS.
- Y. Tao, J. Kim and J. M. Torkelson, Polymer, 2006, 47, 6773–6781 CrossRef CAS.
- C. L. Wong, J. Kim, C. B. Roth and J. M. Torkelson, Macromolecules, 2007, 40, 5631–5633 CrossRef CAS.
- M. M. Mok, C. J. Ellison and J. M. Torkelson, Macromolecules, 2011, 44, 6220–6226 CrossRef CAS.
- D. Woo, J. Kim, M.-H. Suh, H. Zhou, S. T. Nguyen, S.-H. Lee and J. M. Torkelson, Polymer, 2006, 47, 3287–3291 CrossRef CAS.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Angew. Chem., 2004, 116, 6312–6315 CrossRef.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2005, 38, 9963–9973 CrossRef CAS.
- P. A. Lovell, M. S. El-Aasser and P. Lovell, Emulsion polymerization and emulsion polymers, Wiley New York, 1997 Search PubMed.
- J. Nicolas, B. Charleux and S. Magnet, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4142–4153 CrossRef CAS.
- M. E. Thomson, J. S. Ness, S. C. Schmidt, N. Macy, T. F. L. McKenna and M. F. Cunningham, Polym. Chem., 2013, 4, 1803–1814 RSC.
- W. S. J. Li and M. F. Cunningham, Polym. Chem., 2014, 5, 3804–3816 RSC.
- R. W. Simms, M. D. Hoidas and M. F. Cunningham, Macromolecules, 2008, 41, 1076–1079 CrossRef CAS.
- C. Dire, S. Magnet, L. Couvreur and B. Charleux, Macromolecules, 2009, 42, 95–103 CrossRef CAS.
- M. E. Thomson, A.-M. Manley, J. S. Ness, S. C. Schmidt and M. F. Cunningham, Macromolecules, 2010, 43, 7958–7963 CrossRef CAS.
- T. Noda, A. J. Grice, M. E. Levere and D. M. Haddleton, Eur. Polym. J., 2007, 43, 2321–2330 CrossRef CAS.
- M. Müller, M. F. Cunningham and R. A. Hutchinson, Macromol. React. Eng., 2008, 2, 31–36 CrossRef.
- D. M. Haddleton, M. C. Crossman, B. H. Dana, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, Macromolecules, 1999, 32, 2110–2119 CrossRef CAS.
- N. Chan, S. Boutti, M. F. Cunningham and R. A. Hutchinson, Macromol. React. Eng., 2009, 3, 222–231 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Macromol. React. Eng., 2010, 4, 369–380 CrossRef CAS.
- T. Wu, Y. Mei, J. T. Cabral, C. Xu and K. L. Beers, J. Am. Chem. Soc., 2004, 126, 9880–9881 CrossRef CAS PubMed.
- T. Q. Chastek, K. Iida, E. J. Amis, M. J. Fasolka and K. L. Beers, Lab Chip, 2008, 8, 950–957 RSC.
- T. Wu, Y. Mei, C. Xu, H. Byrd and K. L. Beers, Macromol. Rapid Commun., 2005, 26, 1037–1042 CrossRef CAS.
- C. Xu, T. Wu, C. M. Drain, J. D. Batteas and K. L. Beers, Macromolecules, 2005, 38, 6–8 CrossRef CAS.
- C. Xu, S. E. Barnes, T. Wu, D. A. Fischer, D. M. DeLongchamp, J. D. Batteas and K. L. Beers, Adv. Mater., 2006, 18, 1427–1430 CrossRef CAS.
- B. Wenn, M. Conradi, A. D. Carreiras, D. M. Haddleton and T. Junkers, Polym. Chem., 2014, 5, 3053–3060 RSC.
- F. Bally, C. A. Serra, C. Brochon and G. Hadziioannou, Macromol. Rapid Commun., 2011, 32, 1820–1825 CrossRef CAS PubMed.
- C. A. Serra and Z. Chang, Chem. Eng. Technol., 2008, 31, 1099–1115 CrossRef CAS.
- F. Bally, C. A. Serra, V. Hessel and G. Hadziioannou, Chem. Eng. Sci., 2011, 66, 1449–1462 CrossRef CAS.
- F. Bally, C. A. Serra, V. Hessel and G. Hadziioannou, Macromol. React. Eng., 2010, 4, 543–561 CrossRef CAS.
- D. Parida, C. A. Serra, D. K. Garg, Y. Hoarau, F. Bally, R. Muller and M. Bouquey, Macromolecules, 2014, 47, 3282–3287 CrossRef CAS.
- W. Jakubowski, in Progress in Controlled Radical Polymerization: Mechanisms and Techniques, ed. K. Matyjaszewski, S. Sumerlin Brent and V. Tsarevsky Nicolay, American Chemical Society, 2012, ch. 13, vol. 1100, pp. 203–216 Search PubMed.
- Y. Shen, H. Tang and S. Ding, Prog. Polym. Sci., 2004, 29, 1053–1078 CrossRef CAS.
- A. J. Clark, J. V. Geden and S. Thom, J. Org. Chem., 2006, 71, 1471–1479 CrossRef CAS PubMed.
- S. Ding, Y. Xing, M. Radosz and Y. Shen, Macromolecules, 2006, 39, 6399–6405 CrossRef CAS.
- S. Faucher and S. Zhu, Macromol. Rapid Commun., 2004, 25, 991–994 CrossRef CAS.
- D. Fournier, S. Pascual, V. Montembault and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5316–5328 CrossRef CAS.
- S. C. Hong and K. Matyjaszewski, Macromolecules, 2002, 35, 7592–7605 CrossRef CAS.
- G. Kickelbick, H.-J. Paik and K. Matyjaszewski, Macromolecules, 1999, 32, 2941–2947 CrossRef CAS.
- Y. Shen, S. Zhu, F. Zeng and R. H. Pelton, Macromolecules, 2000, 33, 5427–5431 CrossRef CAS.
- Y. Shen, S. Zhu, F. Zeng and R. Pelton, Macromol. Chem. Phys., 2000, 201, 1387–1394 CrossRef CAS.
- Y. Shen, S. Zhu and R. Pelton, Macromolecules, 2001, 34, 5812–5818 CrossRef CAS.
- Y. Shen, S. Zhu, F. Zeng and R. Pelton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1051–1059 CrossRef CAS.
- S. Ding, J. Yang, M. Radosz and Y. Shen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 22–30 CrossRef CAS.
- S. Faucher and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 553–565 CrossRef CAS.
- S. Faucher and S. Zhu, Macromolecules, 2006, 39, 4690–4695 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Macromol. Rapid Commun., 2011, 32, 604–609 CrossRef CAS PubMed.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Polym. Chem., 2012, 3, 1322–1333 RSC.
- H. Chen, M. Zhang, M. Yu and H. Jiang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4721–4724 CrossRef CAS.
- J. A. Burns, C. Houben, A. Anastasaki, C. Waldron, A. A. Lapkin and D. M. Haddleton, Polym. Chem., 2013, 4, 4809–4813 RSC.
- C. Diehl, P. Laurino, N. Azzouz and P. H. Seeberger, Macromolecules, 2010, 43, 10311–10314 CrossRef CAS.
- C. H. Hornung, C. Guerrero-Sanchez, M. Brasholz, S. Saubern, J. Chiefari, G. Moad, E. Rizzardo and S. H. Thang, Org. Process Res. Dev., 2011, 15, 593–601 CrossRef CAS.
- C. H. Hornung, X. Nguyen, G. Dumsday and S. Saubern, Macromol. React. Eng., 2012, 6, 458–466 CrossRef CAS.
- T. Arita, M. Buback and P. Vana, Macromolecules, 2005, 38, 7935–7943 CrossRef CAS.
- T. Arita, M. Buback, O. Janssen and P. Vana, Macromol. Rapid Commun., 2004, 25, 1376–1381 CrossRef CAS.
- S. C. Koch and M. Busch, Chem. Ing. Tech., 2011, 83, 1720–1727 CrossRef CAS.
- C. H. Hornung, X. Nguyen, S. Kyi, J. Chiefari and S. Saubern, Aust. J. Chem., 2013, 66, 192–198 CrossRef CAS.
- J. Vandenbergh, T. de Moraes Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- C. H. Hornung, A. Postma, S. Saubern and J. Chiefari, Macromol. React. Eng., 2012, 6, 246–251 CrossRef CAS.
- C. H. Hornung, A. Postma, S. Saubern and J. Chiefari, Polymer, 2014, 55, 1427–1435 CrossRef CAS.
- J. Vandenbergh and T. Junkers, Polym. Chem., 2012, 3, 2739–2742 RSC.
- J. Vandenbergh, T. Tura, E. Baeten and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1263–1274 CrossRef CAS.
- J. P. Russum, C. W. Jones and F. J. Schork, Macromol. Rapid Commun., 2004, 25, 1064–1068 CrossRef CAS.
- J. P. Russum, C. W. Jones and F. J. Schork, Ind. Eng. Chem. Res., 2005, 44, 2484–2493 CrossRef CAS.
- J. P. Russum, C. W. Jones and F. J. Schork, AIChE J., 2006, 52, 1566–1576 CrossRef CAS.
- Z. Li, W. Chen, Z. Zhang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2015, 6, 1937–1943 RSC.
- C. Rosenfeld, C. Serra, C. Brochon and G. Hadziioannou, Chem. Eng. Sci., 2007, 62, 5245–5250 CrossRef CAS.
- T. Fukuyama, Y. Kajihara, I. Ryu and A. Studer, Synthesis, 2012, 44, 2555–2559 CrossRef CAS.
- T. E. Enright, M. F. Cunningham and B. Keoshkerian, Macromol. React. Eng., 2010, 4, 186–196 CrossRef CAS.
- C. Rosenfeld, C. Serra, C. Brochon, V. Hessel and G. Hadziioannou, Chem. Eng. J., 2008, 135, S242–S246 CrossRef CAS.
- C. Rosenfeld, C. Serra, C. Brochon and G. Hadziioannou, Lab Chip, 2008, 8, 1682–1687 RSC.
- T. E. Enright, M. F. Cunningham and B. Keoshkerian, Macromol. Rapid Commun., 2005, 26, 221–225 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Polym. Chem., 2012, 3, 486–497 RSC.
- N. Chan, J. Meuldijk, M. F. Cunningham and R. A. Hutchinson, Ind. Eng. Chem. Res., 2013, 52, 11931–11942 CrossRef CAS.
- W. W. Smulders, C. W. Jones and F. J. Schork, AIChE J., 2005, 51, 1009–1021 CrossRef CAS.
- G. Qi, C. W. Jones and J. F. Schork, Ind. Eng. Chem. Res., 2006, 45, 7084–7089 CrossRef CAS.
- W. W. Smulders, C. W. Jones and F. J. Schork, Macromolecules, 2004, 37, 9345–9354 CrossRef CAS.
- M. Zhang and W. H. Ray, J. Appl. Polym. Sci., 2002, 86, 1047–1056 CrossRef CAS.
- A. Zargar and F. J. Schork, Ind. Eng. Chem. Res., 2009, 48, 4245–4253 CrossRef.
- W. Wang, Y.-N. Zhou and Z.-H. Luo, Ind. Eng. Chem. Res., 2014, 53, 11873–11883 CrossRef CAS.
- W. Wang, Y.-N. Zhou, L. Shi and Z.-H. Luo, Polym. Eng. Sci., 2015, 55, 1030–1038 CAS.
- W. Wang, Y.-N. Zhou and Z.-H. Luo, Macromol. React. Eng., 2015, 9, 418–430 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |