
Saccharification of thermochemically pretreated cellulosic biomass using native and engineered cellulosomal enzyme systems†
Shishir P. S.
Chundawat
*abc,
Chad D.
Paavola‡
d,
Babu
Raman§
e,
Matthieu
Nouailler
f,
Suzanne L.
Chan
g,
Jonathan R.
Mielenz
e,
Veronique
Receveur-Brechot
h,
Jonathan D.
Trent
di and
Bruce E.
Dale
bc
aDepartment of Chemical & Biochemical Engineering, The State University of New Jersey, 98 Brett Road, Engineering Wing C-150A, Piscataway, NJ 08854, USA. E-mail: shishir.chundawat@rutgers.edu; Tel: +848 445 3678
bDOE Great Lakes Bioenergy Research Center (GLBRC), Michigan State University, East Lansing, MI 48824, USA
cChemical Engineering and Materials Science, Michigan State University, 3815 Technology Blvd, Suite 1045, Lansing, MI 48910, USA. E-mail: bdale@egr.msu.edu
dBioengineering Branch, NASA Ames, Moffett Field, CA, USA. E-mail: cdpaavola@gmail.com
eBiosciences Division and BioEnergy Science Center, Oak Ridge National Laboratory, Oak Ridge, TN, USA. E-mail: BRaman@dow.com; biofuels4me@gmail.com
fLISM-UMR 7255 Institut De Microbiologie De La Mediterranee, CNRS and Aix-Marseille University, 31, Chemin Joseph Aiguier, 13402 Marseille Cedex 20, France. E-mail: Matthieu.Nouailler@imm.cnrs.fr
gSETI Institute, Mountain View, CA, USA. E-mail: slchansf@yahoo.com
hAix Marseille Univ, CNRS, INSERM, Institut Paoli-Calmettes, CRCM, Marseille, France. E-mail: veronique.brechot@inserm.fr
iBiomolecular Engineering Department, University of California, Santa Cruz, CA, USA. E-mail: jonathan.d.trent@nasa.gov
First published on 21st October 2016
Abstract
Consolidated bioprocessing (CBP) of pretreated lignocellulosic biomass using microbes like Clostridium thermocellum allows simultaneous polysaccharide saccharification and sugar fermentation to produce fuels or chemicals using a one-pot process. C. thermocellum is a thermophilic bacterium that deconstructs biomass using large multi-enzyme complexes called cellulosomes. Characterization of cellulosomal enzymes tethered to native or engineered scaffoldin proteins has revealed that enzyme complexation is critical to the bacterium's cellulolytic ability. However, we have a limited understanding of the impact of enzyme complexation on the saccharification efficiency of various forms of industrially relevant pretreated biomass substrates. Here, we compared the hydrolytic activity of the most abundant cellulosomal enzymes from C. thermocellum and investigate the importance of enzyme complexation using a model engineered protein scaffold (called ‘rosettasome’). The hydrolytic performance of non-complexed enzymes, enzyme-rosettasome (or rosettazyme) complexes, and cellulosomes was tested on distinct cellulose allomorphs formed during biomass pretreatment. The scaffold-immobilized enzymes always gave higher activity than free enzymes. However, cellulosomes exhibited higher activity than rosettazyme complexes. This was likely due to the greater flexibility of the native versus engineered scaffold, as deciphered using small angle X-ray scattering. Surprisingly, scaffold-tethered enzymes also gave comparable activity on all the cellulose allomorphs tested, which is unlike the preferential activity of non-complexed cellulases seen for certain allomorph forms. Tethered enzyme complexes also gave lower saccharification yields on industrially relevant lignin-rich switchgrass than cellulose alone. In summary, we find that the type of pretreatment can significantly impact the saccharification efficiency of cellulosomal enzymes for various CBP scenarios.
Introduction
Industrial utilization of the sugars derived from cellulosic plant biomass, such as agricultural or forestry wastes and energy crops, is hindered by the tightly hydrogen bonded crystalline cellulose fibrils and the surrounding amorphous matrix of recalcitrant biopolymers.1 Most research on deconstructing lignocellulose derived sugar polymers has focussed on utilizing a suite of carbohydrate active enzymes (CAZymes; currently categorized thoroughly at http://www.cazy.org) secreted by a filamentous fungus called Hypocrea jecorina (or its anamorph Trichoderma reesei). In recent years research has further expanded to include various bacterial enzymes.2–4 There has been significant interest from both an academic and industrial perspective to develop consolidate bioprocessing (CBP) strategies utilizing cellulolytic bacteria to deconstruct lignocellulosic biomass into fermentable sugars that can be directly upgraded into biofuels like ethanol.5–7 One of the challenges identified in these studies was relatively lower cellulose conversions observed at industrially relevant high solids loading based hydrolysis and fermentation of pretreated biomass.6 Recalcitrance of cellulose to enzymatic hydrolysis is thought to primarily arise due to strong hydrogen bonding and stacking forces that stabilizes its highly crystalline structure, among other factors.1,8 Thermochemical pretreatments have been used to reduce the recalcitrant nature of cellulose by altering its native crystalline structure to produce highly amorphous cellulose or non-native cellulose allomorphs that are more readily deconstructed into sugars by commercially available fungal cellulases.9,10 A non-native allomorph called cellulose III has been shown to be hydrolyzed by fungal cellulases (like T. reesei) at rates up to 5-fold higher than native cellulose I allomorphs.9,11 However, a similar detailed understanding of the mechanism of CBP based cellulolytic enzyme complexes on distinct crystalline cellulose allomorphs is far from complete.The anaerobic, thermophilic bacterium Clostridium thermocellum has the highest known growth rate with cellulose as its sole carbon source3,7,12 and is an ideal cellulosomal system for conducting comparative analyses on distinct cellulosic substrates. This organism breaks down insoluble lignocellulose into soluble oligosaccharides using elaborate enzyme complexes known as cellulosomes bound to its surface.13,14 The cellulosomes serve a variety of purposes, including maximizing recruitment of enzymes to substrate, reducing non-productive binding between enzymes and substrate or other surfaces and localization of products close to the cell surface for efficient uptake of soluble reaction products. It has been demonstrated that the individual enzymes have lower activity in solution than they have tethered to the natural cellulosome scaffold or to a number of engineered complexes.15–24 The natural cellulosome in C. thermocellum is assembled on the scaffoldin protein CipA containing nine repeats of a cohesin module that facilitates binding with type I dockerin modules of cellulosomal enzymes.7 CipA also contains a substrate-specific carbohydrate binding module or CBM (e.g., family 3a CBM), and a cell-surface specific type II dockerin module. There are over seventy type I dockerin-containing coding sequences for enzymes and other proteins in the C. thermocellum genome25 and dozens of these have been identified by proteomic analyses of isolated cellulosomes.26 The relative abundance of individual enzymatic components of cellulosomes is dynamically regulated and is closely dependent on the carbon-source used to grow the bacteria.26,27 Raman and co-workers have quantitatively identified the changes in the composition of cellulosomal proteins for a range of biomass substrates.26 Interestingly, they found that the cellulosomal composition for C. thermocellum grown on pure cellulose was distinct from pretreated switchgrass for more than fifty cellulolytic and hemicellulolytic protein sequences identified by quantitative proteomics. However, the impact of these cellulosomal compositional changes on the activity of the cellulosomal complex on well-defined cellulosic substrates has not been assessed. Furthermore, assessments of cellulolytic activity are frequently carried out at very low glucan conversion, making it difficult to assess the practical relevance of such systems to enabling the CBP paradigm for cellulosic biofuels production.
One of the major challenges in understanding the relationship between cellulosomal enzyme composition and their activity on native or pretreated substrates has been the sheer complexity of producing these large multi-module protein complexes. Several engineered protein scaffolds have been created to study the importance of assembly of cellulosomal enzymes into complexes (sometimes called mini- or designer cellulosomes). Designer cellulosomes were first engineered to tether two, three, or four enzymes to a protein scaffold.16,18 Subsequent efforts have created more elaborate structures based on self-assembly of multiple linear scaffolds or multi-subunit ring structures.19,23 However, previous work has predominantly focused on the breakdown of purified native crystalline and amorphous cellulose, rather than more complex lignocellulosic substrates that are industrially relevant.15,23 We recently constructed double ring complexes (called rosettazymes) using a scaffold derived from a chaperonin protein in Sulfolobus shibatae called a rosettasome.19,28,29 These rosettazyme or mini-cellulosome complexes containing four C. thermocellum cellulases, and like previous studies on designer cellulosomes, demonstrated a significant enhancement (up to 2.4-fold) in the activity of complexed clostridial enzymes compared to non-complexed free enzymes on microcrystalline cellulose (or Avicel). These studies have demonstrated that though mini-cellulosomal complexes do not achieve the same level of specific activity as native cellulosomal enzyme complexes, they can provide limited mechanistic insights. Nevertheless, we are still far from fully understanding the role of enzyme complexation on lignocellulosic biomass deconstruction by native cellulosomal complexes.
In an effort to understand the role of complexation on pretreated biomass hydrolysis we have characterized the activity of a mixture of the most abundant cellulosomal enzymes with and without tethering to an engineered rosettasomes scaffold. These included nine of the ten most abundant enzymes found in natural cellulosomes isolated from bacteria grown on complex lignocellulosic substrates, as identified by our team members using quantitative proteomics analysis (Table 1).26 The rosettazyme enzyme composition for hydrolytic activity testing approximately mimicked the compositions of natural cellulosomes grown on either Avicel or switchgrass and examined activities on well-defined crystalline cellulose allomorphs (cellulose I and cellulose III) as well as amorphous cellulose. We have further characterized the activity of enzymes on commercially relevant lignocellulosic substrates such as untreated and dilute acid or ammonia fiber expansion (AFEX¶) pretreated switchgrass. We used small angle X-ray scattering (SAXS) to better understand the structural organization of a rosettazyme complex with all cohesins occupied by a representative single enzyme, Cel9F, to simplify interpretation of the data. We furthermore compared the activity of cellulosomes isolated from C. thermocellum grown on distinct cellulosic substrates (crystalline cellulose and pretreated switchgrass) to industrially relevant Trichoderma derived enzyme cocktails.
| Name | Gene ID | Modules | Relative abundance, Avicel | Relative abundance, switchgrass |
|---|---|---|---|---|
| Man5A | Cthe_0821 | GH5 CBM32 dock1 | 8.2 | 16.9 |
| CbhA | Cthe_0413 | CBM4 GH9 CBM3 dock1 | 5.3 | 4.1 |
| Cel8A | Cthe_0269 | GH8 dock1 | 16.1 | 6.2 |
| Cel5B | Cthe_0536 | GH5 dock1 | 5.8 | 3.9 |
| Cel9F | Cthe_0543 | GH9 CBM3 dock1 | 4.9 | 8.4 |
| Cel9K | Cthe_0412 | CBM4 GH9 dock1 | 6.4 | 9.1 |
| Cel9R | Cthe_0578 | GH9 CBM3 dock1 | 3.8 | 2.8 |
| Cel48S | Cthe_2089 | GH48 dock1 | 36.1 | 34.2 |
| Xyn10Z | Cthe_1963 | CE1 CBM6 dock1 GH10 | 1.2 | 0.4 |
| CtXynGH30 | Cthe_3012 | GH30 CBM6 dock1 | 0.2 | 0 |
| Xyn10C | Cthe_1838 | CBM22 GH10 dock1 | 3.1 | 4.9 |
| Xyn11A | Cthe_2972 | GH11 CBM6 dock1 CE4 | 10 | 9.2 |
Experimental
Cellulosic substrates
Avicel microcrystalline cellulose (PH-101) was purchased from Sigma-Aldrich (St. Louis, MO) and used to make cellulose III and regenerated amorphous cellulose as described previously.9 Switchgrass from the National Renewable Energy Laboratory (NREL, Golden, CO) was pretreated by ammonia fiber expansion (AFEX) and dilute-acid treatment. Conventional AFEX treatment was done at 130 °C, 60% moisture loading, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 NH3-to-dry biomass loading (w/w) for 45 min.30 Dilute acid pretreatment was performed at NREL using the pilot scale (Sunds) reactor as described previously.26,31 Untreated and pretreated biomass was milled to pass through a 200 μm mesh screen size. The compositions of untreated and treated switchgrass are provided in ESI† section 1.
:1 NH3-to-dry biomass loading (w/w) for 45 min.30 Dilute acid pretreatment was performed at NREL using the pilot scale (Sunds) reactor as described previously.26,31 Untreated and pretreated biomass was milled to pass through a 200 μm mesh screen size. The compositions of untreated and treated switchgrass are provided in ESI† section 1.
Production of the rosettazyme complexes
The rosettazyme scaffold was based on one of the rosettasome subunit proteins from Sulfolobus shibatae, genetically engineered to include a cohesin domain, and expressed in E. coli as previously described.19 The individual Clostridium thermocellum cellulases (Cel9F, Cel9K, Cel8A, Cbh9A, Cel5B, Xyn11A, Xyn10C, Xyn10Z, Cthe_0821, Cthe_3012, Cel48S, and Cel9R) were cloned in their full, mature form, including native CBMs and dockerins, into E. coli and purified following expression. All these C. thermocellum glycosyl hydrolase coding genes were amplified by PCR from genomic DNA (ATCC 27405D-5) and cloned into either pET19b (NcoI and XhoI restriction sites) or into pAED4 (NdeI and Acc65I restriction sites). The plasmids were expressed in E. coli BL21 CodonPlus (DE3) RIL cells (Stratagene) using the Studier auto-induction method32 and purified from either the soluble (Cel9F, Cel9K, Cel8A, Cbh9A, Cel5B, Xyn11A, Xyn10C, Xyn10Z, Cthe_0821 and Cthe_3012) or insoluble fraction (Cel48S, Cel9R) of lysed cell pellets. See ESI† section 2 for additional protein purification details.To assemble the rosettazymes, pre-assembled rosettasomes with cohesin-fused double-rings19 were combined with enzymes in molar ratios (Table 1) representative of native cellulosomal compositions as determined previously.26 We have previously shown that the molar ratio of the enzymes associated with rosettazyme complex is similar to the stoichiometry used to form the complexes.19 However, it is currently unknown how the enzymes are distributed on each individual rosettazyme complex and will be the focus of future studies. A total enzyme concentration of 60 μM was combined with the rosettasome scaffold at 8.83 μM in a 1:1 ratio with the cohesins. Non-complexed enzymes were prepared in the same way without inclusion of the scaffold protein. Complexed and non-complexed enzymes were diluted and added to the final reaction mixture in a volume of 60 μL in a total reaction volume of 250 μL.
Production of native bacterial cellulosomes
Cellulosomes from C. thermocellum grown on cellulose (Avicel) or dilute-acid treated switchgrass were isolated as described elsewhere.26 Briefly, the cellulosomes were isolated from the C. thermocellum fermentation extracellular broth using the amorphous cellulose affinity digestion method.33 The purified cellulosomes from replicate experiments (∼1–2 mg mL−1 total protein concentration) were stored in buffer (50 mM Tris, 50 mM CaCl2, 50 mM DTT, pH 7) at −80 °C.Protein concentration determination
Protein concentrations of purified cellulosomes stored in solutions containing dithiothreitol and commercial fungal enzymes in buffers containing reducing sugars, sugar alcohols, and other stabilizers could not be accurately determined by conventional Bradford assays. Conventional Bradford assays can overestimate cellulosomes concentrations by up to 2-fold and underestimate commercial fungal enzyme concentrations by 3–5 fold,34 which can bias enzyme specific activity (IU mg−1 enzyme) by a factor of 6 to 10-fold for fungal cellulases versus bacterial cellulosomes. Proteins concentrations were therefore determined using the 2D Quant Kit (GE Healthcare, Pittsburgh, PA) after precipitation and washing to minimize interference from reducing agents and other small molecules. The washed protein pellet was re-solubilized in a copper containing solution that was colorimetrically quantified as described by the manufacturer. This method agreed well with concentrations of rosettazymes determined by the sum of components each calculated by absorbance at 280 nm using known extinction coefficients for cellulosomal enzymes. This method also agreed well with concentrations of the commercial fungal cellulases determined previously based on quantitative determination of total nitrogen content to estimate protein content.34Enzymatic hydrolysis assays
Enzymatic hydrolysis assays on cellulose and lignocellulosic biomass were carried out based on a protocol described elsewhere in detail.35 Briefly, experiments were done in 0.25 mL reaction volume at 0.1% glucan loading (0.25 mg glucan per well) in 96-well microplates. All assays using fungal cellulases (Danisco Inc., Genencor Division, Rochester, NY) were carried out with a cocktail of 80% Accellerase 1500 (71 mg mL−1 stock protein concentration), 10% Multifect Xylanase (47 mg mL−1), and 10% Multifect Pectinase (91 mg mL−1) on total protein weight basis (mg). For fungal enzymes, the hydrolysis assays were done in pH 4.8 citrate buffer (50 mM) at 50 °C. For bacterial enzymes, the hydrolysis assays were done in pH 6.5 MES buffer (50 mM) at 60 °C. The buffer for natural cellulosome assays included 10 mM CaCl2 and 10 mM DTT and the rosettazyme assays included 25 mM MgCl2 and 1 mM ATP (in addition to the CaCl2 and DTT). Assays were carried out at equivalent total enzyme loading (80, 24, 12 mg enzyme per g glucan loading) to allow for meaningful comparisons between fungal and bacterial enzyme assay results. It was assumed that the total enzyme loading was 80% of the total protein concentration estimated for natural cellulosomes (i.e., scaffold protein cipA contributes approximately 20% of the total cellulosome fraction on a weight basis),26 while the total enzyme loading for the non-complexed bacterial enzymes and fungal cellulases was equivalent to the total protein concentration estimated. To minimize end-product inhibition and hydrolyze glucose and xylose oligomers into monosaccharides, bacterial-derived β-glucosidase and β-xylosidase (Lucigen, Madison, WI) were added at 5% of the total mg of enzyme loaded in each assay. Assays were done in triplicates on two separate days, totalling six replicates for each assay condition. All plates were incubated in a shaker at 200 rpm, with a bead in each well to facilitate mixing, for the necessary duration (24 hours; unless specified otherwise). The total monomeric sugar (glucose and xylose) concentration in the hydrolyzate and hydrolysis yield was determined using a scaled-down glucose and xylose enzymatic detection method, as described elsewhere.36 The specific activity of the cellulosomal enzyme mixtures on cellulose was determined based on slight modifications of our previously reported protocol as described above.36 The non-enzymatic protein scaffold (i.e., cipA or rosettasome) was not taken into consideration for specific activities reported (as μmoles of glucose released per mg total enzyme per minute) in Table 2.| Specific activity (μmol glucose mg−1 enzyme min−1) | Crystalline cellulose I | Crystalline cellulose III | Amorphous cellulose |
|---|---|---|---|
| Non-complexed cellulases mixture | 27.9 ± 2.1 | 13.0 ± 3.1 | 145.5 ± 15.1 |
| Rosettazyme complex | 64.6 ± 7.6 | 58.3 ± 4.4 | 165.5 ± 38.1 |
| Native cellulosomal complex | 165.6 ± 21.6 | 169 ± 33.4 | 177.4 ± 10.8 |
Small angle X-ray scattering (SAXS)
SAXS data was collected for the rosettasome–cohesin scaffold and the rosettazyme at the European Synchrotron Radiation Facility (ID02 beamline, Grenoble, France) as described previously.37 The rosettazyme was prepared by mixing stoichiometric amounts of the rosettasome–cohesin scaffold with purified Cel9F (18 fold molar ratio). Each sample (at 20 °C) in Tris-maleate 50 mM (pH 6), MgCl2 25 mM, ATP 1 mM, CaCl2 1 mM, cellobiose 1.5 mM, and sodium azide 0.02% was injected into a 1.8 mm diameter measurement capillary and continuously circulated in the capillary during X-ray exposure. Ten successive frames of 0.5 seconds were recorded for each protein sample and the corresponding buffer. The frames were examined for bubbles and radiation damage; identical frames were averaged and corrected for the background buffer signal. The sample-to-detector distance was set at 5 meters leading to scattering vector q ranging from 0.003 to 0.11 Å−1 (q = 4π/λsinθ, where 2θ is the scattering angle, and λ the wavelength of the incident X-rays, set at 1 Å). The rosettasome–cohesin samples were analysed at concentrations of 5 and 2 mg mL−1 and rosettazyme samples was analysed at 5, 4, 3 and 2 mg mL−1. The sample-to-detector distance was then set at 1.5 meters, giving access to scattering vector q ranging from 0.007 to 0.35 Å−1, to measure the two samples at 5 mg mL−1. For the rosettasome–cohesin scaffold slight repulsive interparticle interactions were observed at low angle. The data at concentration of 5 mg mL−1 with sample-to-detector distance of 1.5 meters and the data recorded at 2 mg mL−1 with sample-to-detector distance of 5 meters were therefore merged. For the rosettazyme samples no significant effect of protein concentration were observed, and only the data recorded at 1.5 meters at 5 mg mL−1 protein concentration were used for further analysis.
The SAXS data were processed using the program PRIMUS.38 The radius of gyration Rg was inferred at low angles (qRg < 1.3) from the Guinier approximation: I(q) = I(0)e−q2Rg2/3. Indirect Fourier transform of the data and determination of the distance distribution function was computed using GNOM.39Ab initio shape was determined using DAMMIN40 and DAMMIF.41 The atomic structure of the rosettasome (based on homology model for S. shibatae HSP60beta, as described previously42) was docked into the calculated shapes using MASSHA43 and SUPCOMB.44 Similarly, the atomic structures of one subunit of cohesin (pdb code 1OHZ), of cohesin–dockerin, and of the cellulase were positioned and P92 symmetry was applied to completing the 18-subunits rosettazyme. For this purpose we generated atomic models of the dockerin and of Cel9F using PHYRE45 with pdb structures 1OHZ (52% identity) and 1GA2 (61% identity) as templates, respectively. Linkers between the rosettasome and the cohesin, and between the cellulase and the dockerin were built using CHARMM46 and the web interface CHARMM-Gui (http://www.charmm-gui.org/). Normal mode analysis was assessed using the web interface ElNemo (http://www.igs.cnrs-mrs.fr/elnemo/index.html).47 The theoretical scattering profiles of the individually generated structures along the NMA trajectories (5 lowest frequency normal modes) were calculated using CRYSOL.48 GAJOE from the EOM program suite was used to search for an ensemble of these structures that would reproduce the SAXS data.49 Pymol was used to visualize and analyze all protein structures.50
Results
Cellulolytic activity of native bacterial cellulosomes of varying enzyme composition
It is well known that C. thermocellum varies the composition of its cellulosomes depending on growth conditions.26,27 To determine how this variation influences overall cellulase activity, cellulosomes were isolated from C. thermocellum grown on cellulose (Avicel) or dilute-acid-treated switchgrass. These cellulosomes were tested on switchgrass (untreated, AFEX treated, or dilute-acid treated), cellulose I, and cellulose III with total protein loadings ranging between 15 to 100 mg g−1 glucan (Fig. 1; see ESI† section 3 for detailed results). The microcrystalline cellulose hydrolysis yield obtained for native cellulosomes (on a comparable protein loading basis and total assay time) reported here is similar to previously published work.51,52 Interestingly, there was no significant difference in the activity of the two native cellulosome isolates of distinct compositions on the substrates tested and correspondingly no significant differences in the activity of rosettazymes prepared using enzyme ratios mimicking those in the two cellulosomes tested. These results suggest that the previously reported variation in composition and activities of cellulosomes was not detectable under the conditions tested perhaps due to redundancies in the substrate specificity of the scaffolded enzymes. There may also be minor enzymatic components produced by cells that were not isolated by the cellulose affinity cellulosome purification procedure used. C. thermocellum may be regulating cellulosome composition for selective growth advantage not directly related to maximizing cellulose hydrolysis rates. Nevertheless, our results indicate that the cellulosomal compositional differences have little effect on the deconstruction kinetics for pure cellulose or lignocellulosic biomass used in this study.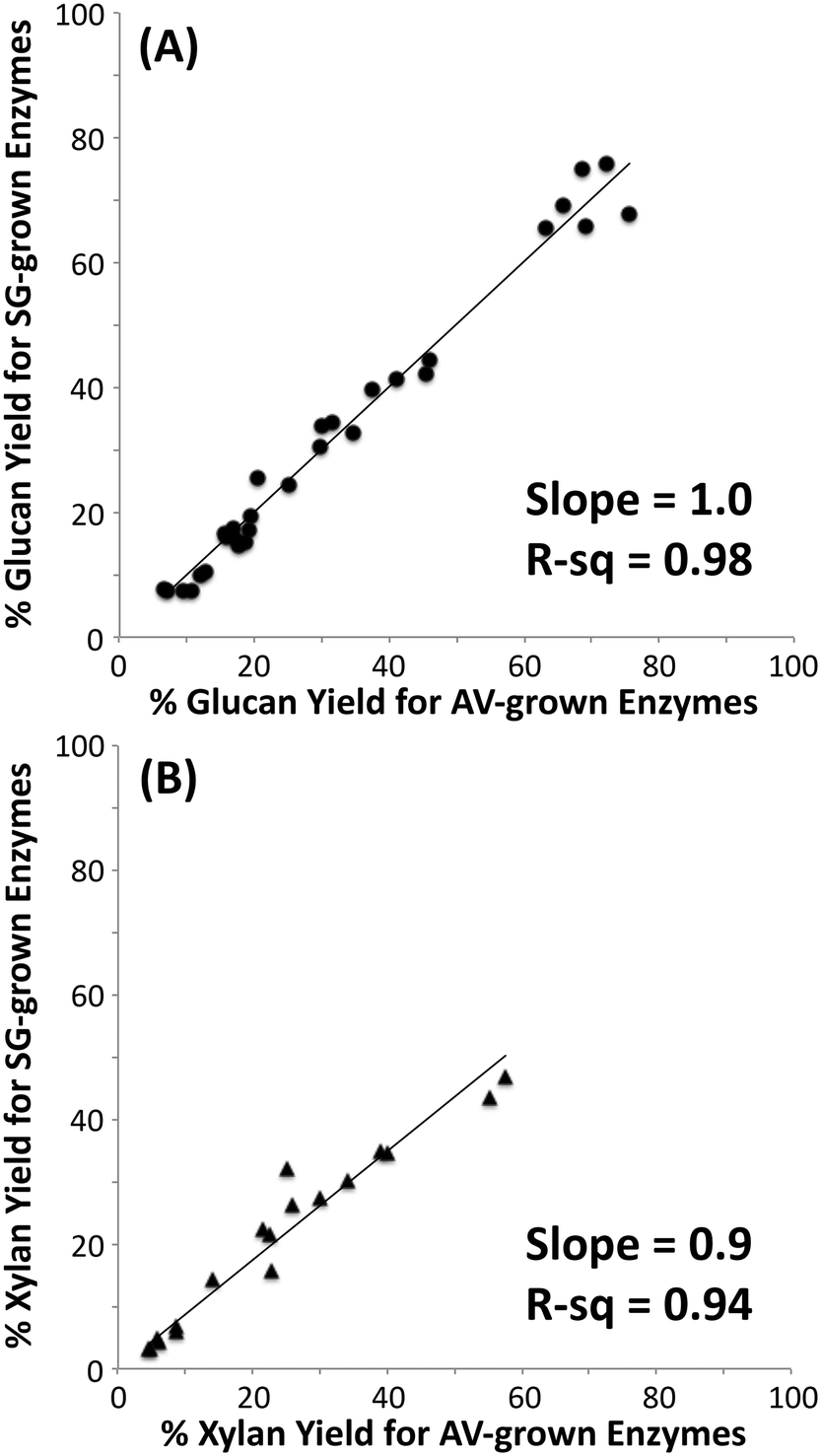 | ||
| Fig. 1 Correlation of hydrolytic activity of cellulosomes isolated from Clostridrium thermocellum grown on Avicel (AV) versus switchgrass (SG) on cellulose I, cellulose III, untreated and pretreated (AFEX and dilute-acid) switchgrass. Glucose yields (A) for cellulose saccharification and xylose yields (B) for hemicellulose saccharification are shown here. Note that xylose yields were determined for only switchgrass based samples. | ||
Hydrolytic activity of native bacterial cellulosomes and fungal cellulases on cellulose allomorphs and lignocellulosic biomass
Since no differences in hydrolysis yields were observed between cellulosomes isolated from C. thermocellum grown on Avicel versus those grown on switchgrass, further work was carried out using the Avicel-grown cellulosomes only. Hydrolysis was also studied at equivalent overall enzyme loadings of commercially available fungal enzymes. Detailed proteomics based composition analyses of the fungal enzyme cocktail34 and individual fungal cellulases activity on various cellulose allomorphs have been published elsewhere.9,11 Cellulosomes surprisingly gave comparable activities on both crystalline and amorphous cellulose although, at the lower protein loadings (12, 24 mg g−1 loadings) tested the activity on amorphous cellulose was marginally lower than both crystalline cellulose allomorphs (Fig. 2). The fungal cellulases, on the other hand, were more effective on amorphous cellulose and cellulose III than cellulose I. At enzyme loadings of 12 and 24 mg g−1 glucan, cellulosomes released significantly less product than fungal enzymes while at 80 mg g−1, the fungal enzyme loading may be beyond the saturation point for a 24 hour reaction.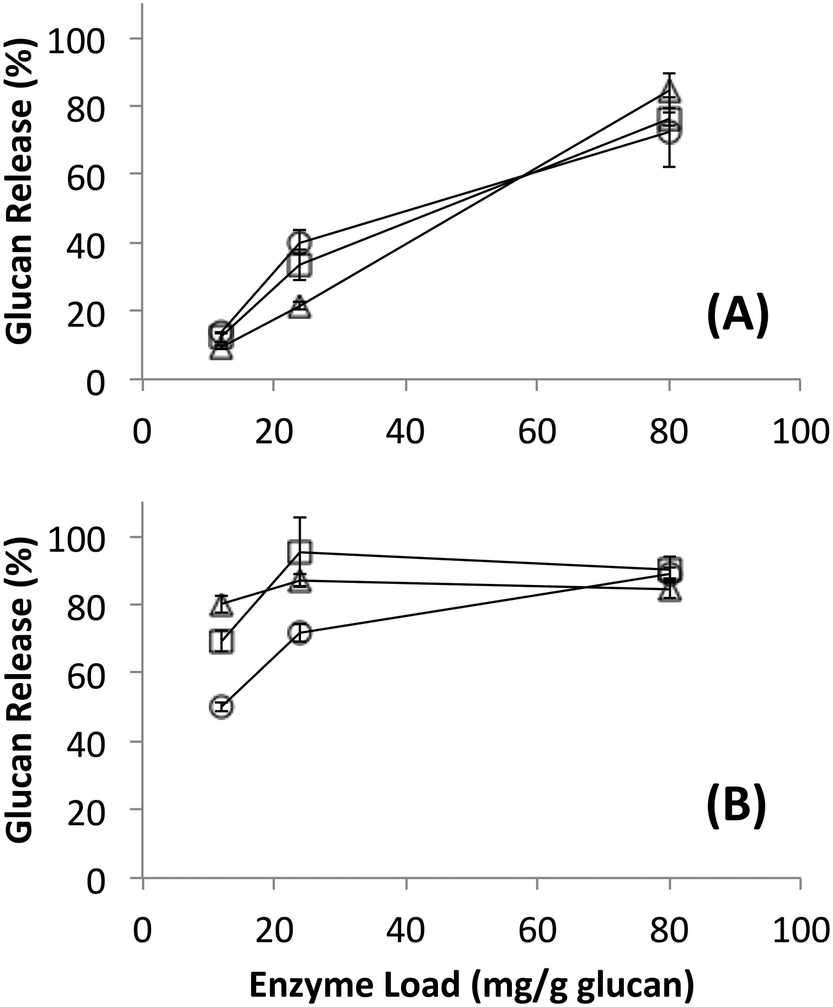 | ||
| Fig. 2 Glucan released during hydrolysis of different forms of cellulose by (A) cellulose affinity digestion purified native bacterial cellulosomes or (B) commercially available fungal enzymes. Substrates were cellulose I (circles), cellulose III (squares) and amorphous cellulose (triangles). Standard deviations for reported mean values (n = 6 replicates) are shown here as well. | ||
Native cellulosomes solubilized somewhat less glucan polysaccharide from AFEX-pretreated versus dilute acid-treated switchgrass (Fig. 3). Unlike dilute acid pretreatment, which removes a significant fraction of hemicellulose, AFEX pretreatment does not extract hemicellulose from the biomass.30,53 In addition the dilute acid-treated switchgrass residues were extensively washed to remove more soluble, low-molecular weight cell wall extractives that may also inhibit cellulosomal enzymes while the AFEX-treated biomass was not washed. In contrast with the relatively low cellulose hydrolysis yield, AFEX-treated switchgrass gave the highest xylan yield (>50%) compared to the other two substrates (5–25%) (Fig. 4). We suspect that inclusion of additional hemicellulases and accessory enzymes (as complexed or non-complexed components) would help increase the overall cellulose and hemicellulose hydrolysis yield for AFEX-treated biomass, as has been reported in our previous work.36,54,55 Finally, the fungal enzyme cocktail (from T. reesei) generally produced a greater degree of conversion at comparable protein loadings compared to cellulosomes on all lignocellulosic substrates tested here. The mixture of free fungal (T. reesei) cellulases released significantly more glucan from pretreated switchgrass materials than natural cellulosomes at 12 and 24 mg g−1 glucan enzyme loads. The fungal enzymes released more xylan from AFEX-treated switchgrass than the cellulosomes but both types of enzymes released similar amounts of xylan from dilute acid-treated switchgrass. Both fungal and cellulosomal enzymes catalyzed very low release of glucan and xylan from untreated switchgrass.
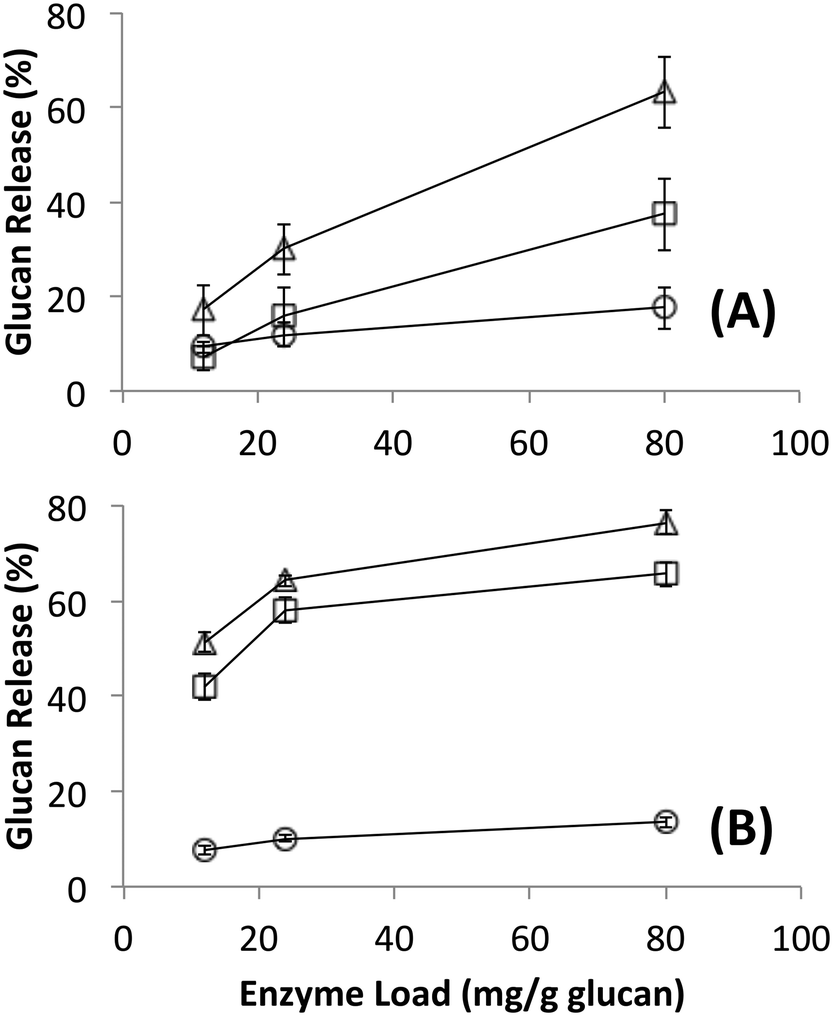 | ||
| Fig. 3 Glucan released during hydrolysis of different forms of switchgrass by (A) cellulose affinity digestion purified native bacterial cellulosomes or (B) commercially available fungal enzymes. Substrates were untreated (circles), AFEX pretreated (squares) and dilute acid pretreated (triangles). Standard deviations for reported mean values (n = 6 replicates) are shown here as well. | ||
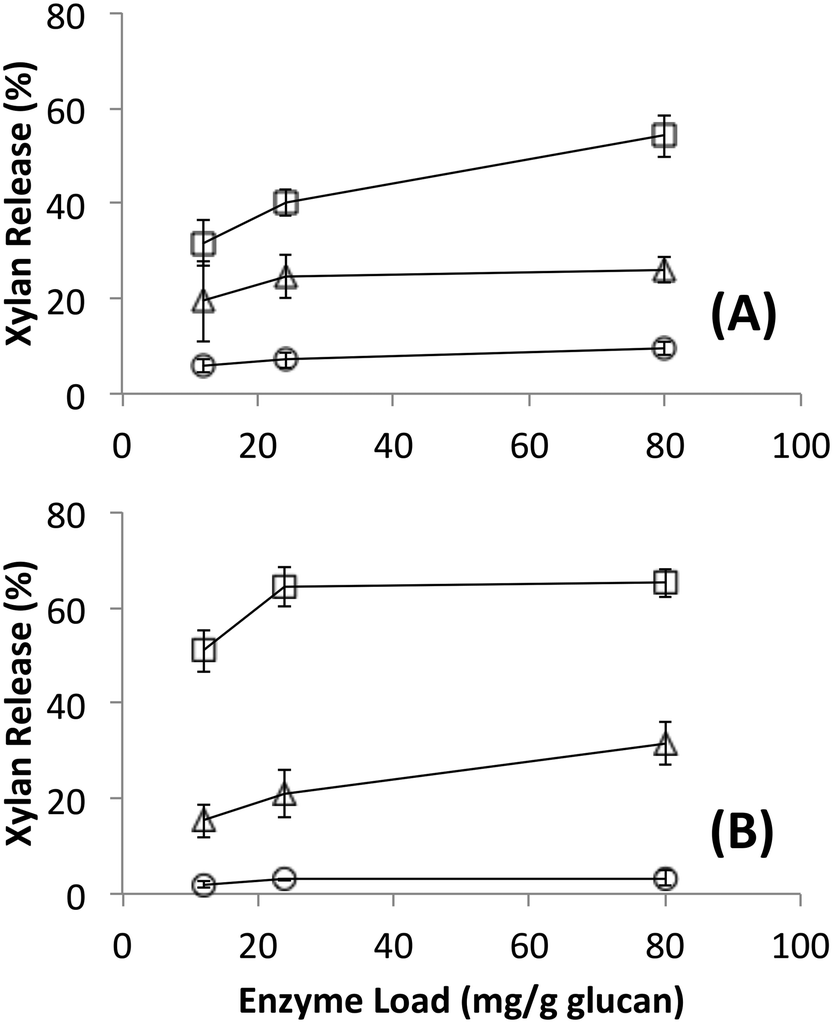 | ||
| Fig. 4 Xylan released during hydrolysis of different forms of switchgrass by (A) affinity purified native bacterial cellulosomes or (B) commercially available fungal enzymes. Substrates were untreated (circles), AFEX pretreated (squares) and dilute acid pretreated (triangles). Standard deviations for reported mean values (n = 6 replicates) are shown here as well. | ||
Hydrolytic activity of free and tethered, engineered cellulosomal enzymes on cellulose allomorphs and lignocellulosic biomass
Since it is challenging to prepare large complexes of well-defined mixtures of enzymes with the native scaffoldin from C. thermocellum, the rosettazyme complex, with its high degree of flexibility between the scaffold and the enzymes may present the opportunity to gain a better understanding of the role of complexation in the activity of cellulosic enzymes. Rosettazymes were prepared with twelve clostridial cellulases at the molar ratios occurring in cellulosomes isolated from C. thermocellum grown on cellulose (Avicel). The twelve-cellulosomal enzymes belonged to glycosyl hydrolase families 5, 8, 9, 10, 11, 30 and 48, which are classified according to the CAZyme database as endo- and exo-based cellulases and hemicellulases (complete description provided in Table 1). Using well-defined cellulosic substrates with varying ultrastructures, rosettazyme activity was compared to free (i.e., non-tethered) dockerin-containing enzymes. The activity of free C. thermocellum cellulases and enzymes complexed in rosettazymes were tested on amorphous cellulose, cellulose I, cellulose III, untreated switchgrass and dilute acid or AFEX pretreated switchgrass (Fig. 5). With crystalline cellulose I, rosettazyme activity was 3 to 4-times higher than free enzymes at a total enzyme loading of 80 mg g−1 glucan. For cellulose III rosettazymes, activity was 6–8-times higher than free enzymes, but on amorphous cellulose there was only a marginal (1.1–1.5-times) improvement in activity over free enzymes. In some previous reports on mini-cellulosome-complexes (with only 2–3 enzymes per scaffold),15,16,18 the complexes had increased activities with cellulose I compared to free enzymes, but for amorphous cellulose the complexes had lower activity than free enzymes.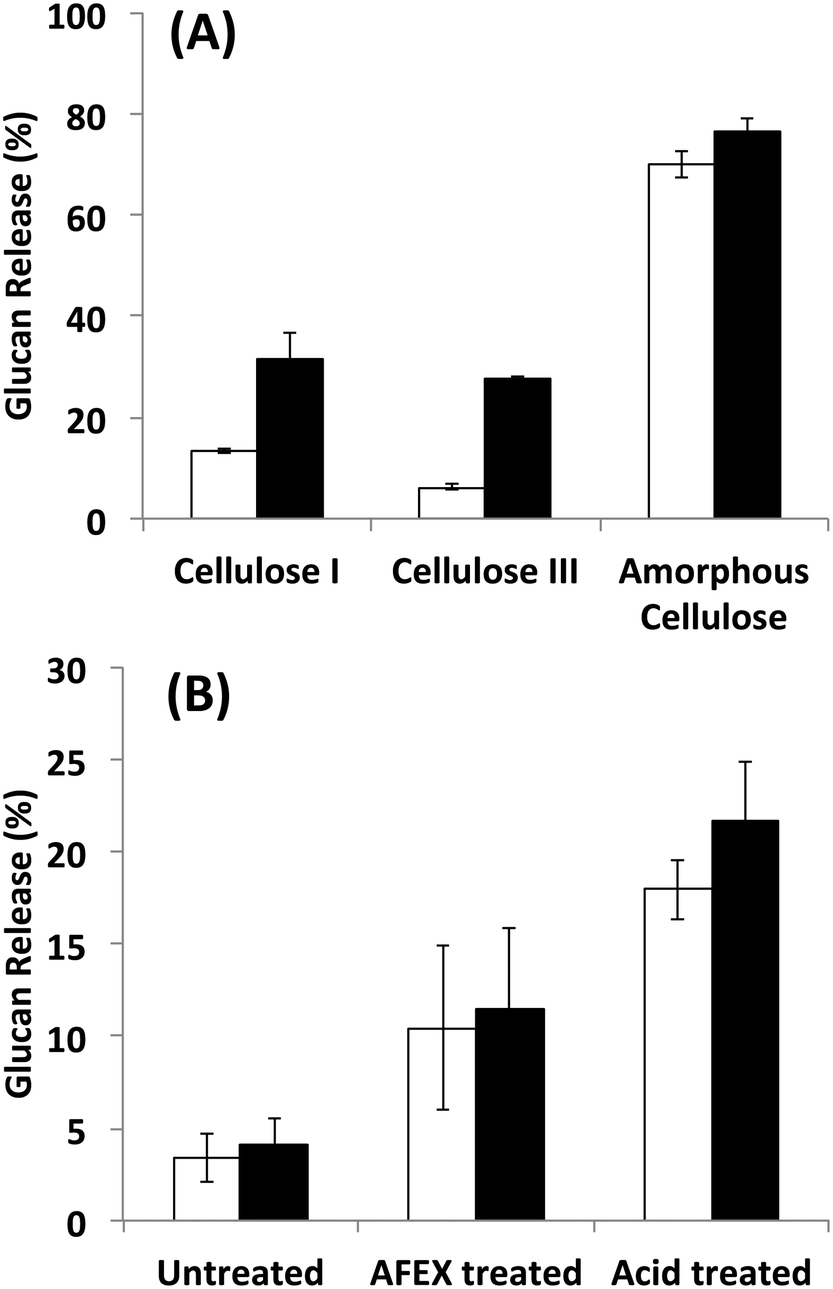 | ||
| Fig. 5 A) Hydrolysis of different forms of cellulose by a mixture of twelve cellulosomal enzymes without tethering (white) or after tethering to the rosettasome scaffold (black) at 80 mg g−1 glucan enzyme loading. B) Hydrolysis of switchgrass with or without pretreatment by a mixture of twelve cellulosomal enzymes with (black) and without (white) tethering. Enzymes were combined in molar ratios similar to Avicel-grown natural cellulosomes as listed in Table 1. Standard deviations for reported mean values (n = 6 replicates) are shown here as well. | ||
The specific activity of the native cellulosomes isolated using the cellulose affinity digestion method on amorphous cellulose is comparable to previously reported values by Krauss and co-workers.56 Interestingly, while the free clostridial enzymes (12-enzyme mixture), rosettazymes (12-enzyme complex) and natural cellulosomes all gave comparable specific activity (μmol glucose released per mg total enzyme per minute) on amorphous cellulose, clear differences were seen for the two crystalline cellulose allomorphs (Table 2). The free clostridial enzymes gave nearly 2-fold higher specific activity on cellulose I versus cellulose III, however, yielded 5-fold lower specific activity on cellulose I compared amorphous cellulose. On the other hand, both native and synthetic cellulosomal complexes yielded similar specific activities on both cellulose allomorphs.
The activity of the rosettazyme complexes was comparable to or slightly higher than the free enzymes on all switchgrass substrates tested. The overall trend in glucan release activity as a function of lignocellulosic substrate was dilute-acid treated switchgrass > AFEX-treated switchgrass > untreated switchgrass.
SAXS-predicted rosettazyme structure
It has not been possible to date to prepare complexes of well-defined mixtures of enzymes with the native scaffoldin from C. thermocellum. Thus, the rosettazyme complex with its high degree of flexibility between the scaffold and the enzymes presents an opportunity to gain a better understanding of the role of cellulase complexation on the activity of cellulolytic bacterial enzymes. The rosettasome, a double-ring structure with 18 subunits, was modified with 18 cohesin-2 modules (from Cthe_3077) to create a self-assembling scaffold capable of binding dockerin containing enzymes.19 The complex was characterized using SAXS with and without a saturating amount of C. thermocellum enzyme Cel9F (Cthe_0543) tethered through the high affinity interaction between the native Cel9F dockerin and the cohesin-2 on the scaffold (Fig. 6).57 The linear Guinier plots indicated the absence of aggregates and the Guinier approximation gave a radius of gyration Rg = 105 ± 5 Å for the cohesin–rosettasome scaffold and Rg = 132 ± 5 Å for the Cel9F-containing rosettazyme complex. The distance distribution functions of the cohesin–rosettasome ring exhibited an asymmetric bell-shape with a peak slightly shifted towards higher radii, and a maximum intramolecular dimensions of Dmax = 350 ± 15 Å. These results are typical of a hollow cylinder and is consistent with the structure of the core rosettasome.28 The distance distribution function of the rosettazyme complex was more symmetric, suggesting that the complex is more cylindrical in shape when tethered to Cel9F. The maximum intramolecular dimensions inferred from this distance distribution functions was Dmax = 400 ± 10 Å.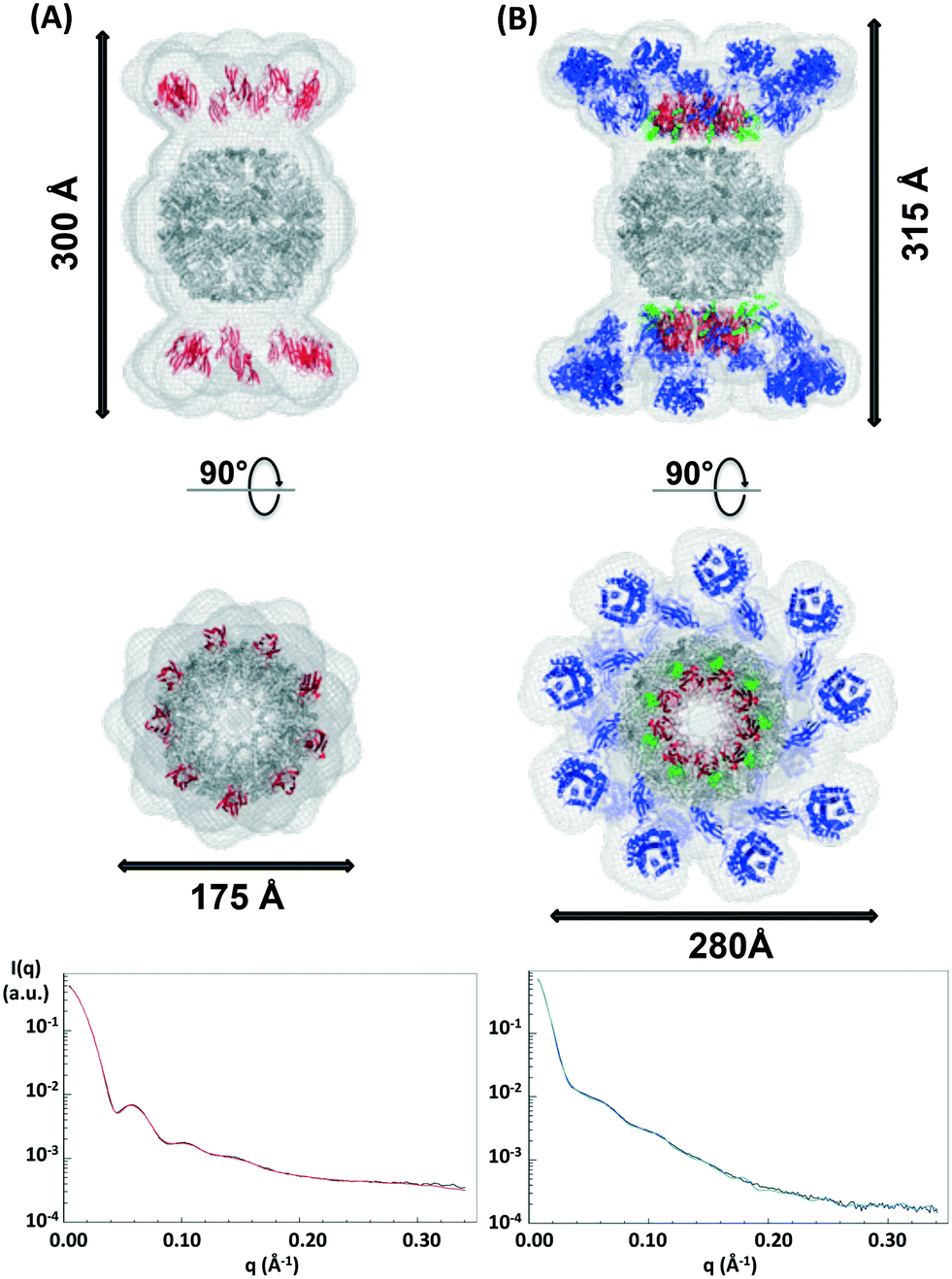 | ||
| Fig. 6 SAXS predicted molecular structure of the rosettasome–cohesin scaffold (A) and rosettazyme (B) superimposed on known atomic structures. The SAXS mesh (light grey) surrounds the ribbon representation of the rosettasome double rings (dark grey), cohesins (red), dockerins (green), and cellulases (blue) prepared using Pymol. The fit of SAXS shape with the experimental data for the rosettasome–cohesins scaffold (red line), rosettazyme (light green line), and corresponding experimental data (black line) are shown here. | ||
SAXS allows approximate determination of the three-dimensional shape of a macromolecule in solution, regardless of the size of the macromolecule or of the complex. Most programs developed for this purpose determine the macromolecule shape by filling the SAXS data predicted volume with a certain number of pseudo-atoms of defined diameter. Considering the size of the rosettasome with and without the 18 enzymes (1.3 MDa and 2.7 MDa, respectively), we used the programs DAMMIN and DAMMIF, to calculate the ab initio shape from the SAXS data profile. Varying parameters such as the estimated particle symmetry, size of pseudo-atoms, expected particle anisometry, and search volume shape did not allow convergence of various model fits to the experimental data for most conditions. This was likely due to the high degrees of freedom possible because of the unusually large size of the complexes. Good fits to the SAXS data were obtained only when the program model parameters were imposed with particle symmetry P92, with a cylindrical search shape, and with an unknown particle anisometry. Under these conditions, the program gave a stable solution on multiple runs and predicted a reliable shape for the cohesin–rosettasome scaffold and the rosettazyme complex. Both models fitted the experimental data well, with χ values of 7.5 and 6.4, respectively.
The SAXS predicted average shape of the cohesin–rosettasome scaffold exhibits a central sphere of ca. 175 Å diameter that perfectly accommodates the double ring of the rosettasome structural model. The two crowns composed of nine large axially-protruding sticks, into which the crystal structure of the cohesin fit, were symmetrically on top of each hemisphere. In this geometry, the cohesins are completely exposed to the solvent. Distance between the rosettasome subunit and the corresponding cohesin is about 30 Å. The linker of 18 amino acids between the domains is therefore extended. These linkers on scaffold-tethered enzymes are certainly flexible, as observed previously in natural cellulosomes.58 We tried to assess this probable flexibility by normal mode analysis (NMA), to follow any putative concerted motions of such systems. Given the high symmetry of the complex, and the small length of the tethering linkers, we wanted to check whether there might be concerted motions governing the flexibility of the whole complex. We first compared the theoretical scattering profiles of each structure generated along the NMA trajectory. However, none of the individual profiles generated corresponded to the experimental scattering curve. We were also unsuccessful in finding the ensemble of these conformations that would reproduce the SAXS data using the program GAJOE. This suggests that the conformational space explored by the linkers is very large, and that the motions of the cohesins through the linker are not concerted. Most probably, the cohesins adopt random motions with steric hindrances as the sole restraints.
The SAXS predicted average shape of the rosettasome–cohesin–cellulase complex corresponded to a rosettasome central sphere with nine enlarged branches on its top and bottom. These branches can accommodate the atomic structures of the cohesin–dockerin–cellulase ensemble. Interestingly, the distance between the rosettasome subunits and the corresponding cohesins is only a few angstroms. As observed for natural cellulosomes, we found here a pleating of the linker tethering the scaffold modules58 when the cellulases are docked onto the cohesins via their dockerin domains. Conversely, the distance spanned by the linker (27 residues) from end to end, between the dockerin and the cellulase, is about 35 Å, suggesting that these linkers can adopt very extended conformations,58 as also observed for natural cellulosomes. The pleating of the cohesin linker can explain the similar heights observed for both the rosettazyme (315 Å) and rosettasome complexes (300 Å). Lastly, the cellulases are arranged in the plane of the cohesins, likely to minimize steric clashes between tethered Cel9F enzymes.
Discussion & conclusion
Designer or mini-cellulosomes make it possible to address several open questions regarding the mechanism of natural cellulosomes such as: how the proximity of different cellulolytic enzymes enhances synergistic activity on cellulose, how the proximity of the endocellulases (lacking CBMs) affects activity and binding avidity, what is the role of cellulosome scaffold flexibility for complex activity etc. Enzyme complexation on a scaffold has been shown previously to improve hydrolytic activity on cellulose and hemicellulose.15,19,59 Most previous studies have been done at low enzyme loadings or for shorter incubation periods giving low overall glucan conversion (<5–10% glucan hydrolysis yield) suggesting that crystalline cellulose hydrolysis was possibly minimal in most cases. Nevertheless, the observed improvement in activity of the complexed enzymes was likely due to multiple reasons such as enzyme proximity and targeting effects as discussed by other authors.18,60 Plasticity of the quaternary structure of cellulosomes (or mini-cellulosomes) has been implicated as one of the primary reason for the effective hydrolytic activity seen for complexed enzymes on cellulose.15,18,37,61 This suggests that the flexibility of the natural cellulosomes quaternary structure to refine its interaction/binding with cellulose fibril surface chains, irrespective of the underlying cellulose ultrastructure, is possibly responsible for the equivalent activity seen on amorphous cellulose and other cellulose allomorphs tested in this study. This could also explain why the rosettazymes, albeit with more limited flexibility, still can achieve similar rates of hydrolysis for various crystalline cellulose allomorphs. These results are unexpected and in direct contrast to the fungal cellulase paradigm for crystalline cellulose deconstruction.Based on our current understanding of how cellulolytic enzymes deconstruct cellulosic biomass it is difficult to understand how complexed enzymes lacking type-A CBMs (as in the case of rosettazymes) might still effectively hydrolyze crystalline cellulose. Interestingly, we find that despite the lack of a cipA CBM3a, the engineered twelve-enzyme rosettazyme complex was remarkably able to give up to 30–40% glucan conversion on both crystalline cellulose I and cellulose III while non-complexed enzymes gave several fold lower yields on either form of crystalline cellulose. Among the enzymes purified from heterologous expression for this study, Cbh9A, Cel9F and Cel9R contain family 3 CBMs; Cbh9A and Cel9K contains family 4 CBMs; Xyn11A, Xyn10Z and Cthe_3012 contain family 6 CBMs; Xyn10C contains a family 22 CBM, and Cthe_0821 contains a family 32 CBM (http://www.cazy.org). However, all these CBMs belong to the type-B CBM category which are thought to target only single glycan chains and not crystalline cellulose surfaces.62 This suggests that enzyme complexation is likely critical to improved substrate targeting analogous to the role of type-A CBMs probably through poorly understood avidity effects between complexed enzyme catalytic units. In recent years, whole genome sequencing of various cellulolytic bacteria has revealed that most cellulases in these organisms lack both CBMs and cohesin–dockerin domains thought to be critical for cellulose deconstruction (based on the well-studied Trichoderma and Clostridium cellulose deconstruction paradigms).4 It is likely that bacterial enzymes or cells are able to bind and decrystallize individual chains from the vicinity of cellulose fibril surface prior to hydrolysing it into soluble cellodextrins.4 However, a detailed investigation is needed to better understand the role of avidity in the binding of large multi-enzyme complexes to cellulosic substrates.
In recent years, work on unnatural cellulosic allomorphs (like cellulose III during certain types of thermochemical pretreatments10) has shown that these substrates are more readily hydrolysed by non-complexed fungal enzymes despite a significant reduction in enzyme binding.9,11 Using such unnatural substrates has allowed us to better understand the role of cellulose ultrastructure on its deconstruction mechanism. Non-complexed enzymes from Trichoderma reesei deconstruct crystalline cellulose based on the classical paradigm of endocellulase–cellobiohydrolase synergy. This model has been expanded to include lytic polysaccharide monooxygenases in recent years.63 However, these models are still limited to aerobic fungal/bacterial non-complexed cellulase systems.7 Most cellulolytic fungal enzyme secretomes34 are abundant in exocellulases (e.g., Cel7A, Cel6A) and endocellulases (e.g., Cel7B, Cel5A, Cel12A, Cel45) that have varying extents of cross-synergistic activity on crystalline cellulose depending on the type of endo–exo combination, enzyme-to-substrate loading and substrate source.1,64,65 Fungal cellulases, like Cel7A or Cel7B from T. reesei, typically contain a type-A family 1 CBM that localizes the catalytic domain in the vicinity of the cellulose surface and facilitates its catalytic action either on readily accessible intact glycosidic bonds (i.e., amorphous regions) or hydrolyzed reducing/non-reducing chain ends.66 Recent work has also implicated the role of cellulose allomorph type on determining the decrystallization work fungal cellulases must perform to extract (or decrystallize) cellobiosyl units from the cellulose surface prior to formation of a catalytically active complex and hydrolysis of the glycosidic linkage1,8,9,11T. reesei cellulase cocktails have been shown to have preferential activity on various cellulose allomorphs in the following order; amorphous cellulose > cellulose III > cellulose II > cellulose I.9 The increased glucan chain flexibility for crystalline cellulose III surface chains, due to altered hydrogen bonding pattern compared to native cellulose I, is thought to be responsible for the enhanced synergistic activity of fungal cellulases seen on cellulose III (see ESI† section 4 for additional assay results). These studies suggest that the decrystallization and hydrolysis of crystalline cellulose for non-complexed fungal cellulases is facilitated by the reduced decrystallization free energy to remove individual glucan chains from the surface of the cellulose fibril.
We have a much more limited understanding, however, of how complexed bacterial cellulases (like cellulosomes from anaerobic cellulolytic bacteria) deconstruct cellulose. Our results show, rather surprisingly, that complexed enzymes (both natural cellulosomes and rosettazymes) had nearly equivalent activity on both cellulose I and III. We have also found that C. thermocellum gave comparable cell growth and substrate consumption rates for both crystalline cellulose I and cellulose III (data not shown). These unpublished in vivo results provide additional support to the in vitro enzyme assay results reported in this study. Overall, these results suggest that the single chain decrystallization and hydrolysis model for cellulose deconstruction must be revisited for multi-enzyme complexes like cellulosomes.1 A similar conclusion can be reached by analysing the work published by Boisset and co-workers who found no significant change in the cellulose Iα/Iβ allomorphs composition of crystalline cellulose hydrolysed by natural cellulosomes.51 Unlike that result, there is a clear preference for fungal non-complexed cellulases hydrolyzing the cellulose Iα over Iβ allomorph.67,68 Beckham and co-workers have also shown that individual cellulose chains can be more readily decrystallized from the surface of cellulose Iα than Iβ further supporting the finding.8 It is clear that the mechanism of cellulose deconstruction for complexed enzymes is distinct from non-complexed enzymes, but there is still a long way to go before we can explain the reasons behind these differences. Specific selective pressures under which each microbial system evolved may be important in ways that are difficult to probe in vitro. For example, in nature cellulosomal enzymes are typically deployed at extremely high local concentrations on bacterial cell surfaces and in the immediate vicinity of cells. Their underlying mechanism also seems to exploit this highly localized enzyme density to decrystallize and hydrolyze cellulose. On the contrary, several non-complexed fungal cellulases are secreted in to the extracellular milieu at fairly low concentrations typically acting at lower localized enzyme densities. Understanding the underlying cellulolytic enzyme mechanism of action at varying substrate concentrations would also have important implications on the commercial scale processing of lignocellulosic biomass. Viikari and co-workers, who recently showed that cellulases lacking CBMs were acting efficiently at high substrate loadings and remained unbound after saccharification for continuous recycle, highlight the relevance of engineering cellulolytic enzymes for practical applications.69
The natural C. thermocellum cellulosome is composed of a scaffoldin that can bind up to nine enzymes and hence might be partially mimicked by the artificial, designer cellulosomes (or rosettazyme) in our case. However, currently the engineered rosettazyme is not able to achieve product release comparable to natural cellulosomes. This is likely due to some combination of the following several reasons; (i) the engineered complex could lack minor enzyme components that are present in the natural cellulosome and that play a critical role in cellulose degradation. Bras and co-workers have identified a cell-surface bound endo-cellulase that synergizes with Cel48S and likely targets the interfacial region of crystalline and disordered cellulose. The model of exo–endo synergy, though mostly applicable to fungal non-complexed enzymes, is not entirely true for cellulosomal enzymes. Recent work has also shown that knocking out a major exo-cellulase (Cel48S) gene from expression doesn't completely retard cellulose degradation by cellulosomes.70 However, it is likely that there are other redundant cellulolytic enzyme activities expressed in vivo but are missing from our in vitro assembled rosettazyme. (ii) It has been shown that the flexibility of the cellulosome is key to its efficacy.18,71,72 Our SAXS study reveals that the rosettazyme exhibits similar flexibility to that which is predicted for cellulosomal interdomain linkers,37,58,73,74 although these similarities are obviously not sufficient to mimic the performance of cellulosomes. It is likely that the circular geometry and rigidity of the rosettasome scaffold does not allow the same meandering and crawling motions on the cellulose surface as is probable for native linear cellulosomes.37,73,74 This rigidity likely provides lesser degree of freedom for precise positioning of tethered enzymes across the cellulosic surface, thus leading to a lower enzymatic efficacy. It is also noted that the two-sided nature of the rosettazyme complex would make it difficult for both sides of the complex to productively engage substrate at the same time. (iii) Another limitation of utilizing rosettazymes has been the lack of control over the exact location of the dockerin containing cellulases on the rosettasome scaffold. Furthermore, though our previous study had shown that the enzymes bind to the rosettasome stoichiometrically,19 it is unclear what is the statistical distribution of the enzymes on the scaffold. Recent studies have suggested that cellulosomal enzymes have preferred localization on the natural cipA scaffold which could play an important role in influencing the activity of the complex.61,75 Though the composition of the rosettazyme complex closely mimicked natural cellulosomes, future work could address the role of varying enzyme ratios (coupled to specificity in localization on the complex) on specific activity. (iv) It is currently unclear if the lack of a CBM3a domain might explain some of the differences seen between the rosettazyme and natural cellulosomes. However, as indicated by Mingardon et al. and our previous study,18,19 additional CBMs on engineered mini-cellulosomes or rosettazymes have reportedly caused reduction in activity on cellulose likely due to superfluous binding to the substrate. Though the rosettazyme complex is able to hydrolyze crystalline cellulose (to fairly high yields) in the absence of CBM3a, it is possible that this module is necessary to achieve higher specific activity for the enzyme complex.
Non-complexed T. reesei-based fungal enzymes have been previously reported to have lower activity than C. thermocellum cellulosomes on crystalline cellulose12 as well as poorer thermostability and ethanol tolerance.76 However, we found that cellulosomes isolated using the traditional cellulose affinity digestion method had comparable activity to fungal enzymes only at higher protein loadings. The glucan conversion per mg cellulosome in 24 h obtained for the natural cellulosomes in our case were comparable to that reported by other researchers.51,52,56 Further purification of the affinity-digestion isolated cellulosome may result in higher specific activity cellulosomal fractions, as recently shown by Resch and co-workers.77,78 It is also possible that cellulosomal enzymes, because they are strongly product inhibited, will not perform well when uncoupled from a cell that absorbs and utilizes hydrolysis products. Lastly, we found that the performance of the complexed, multi-domain cellulases was much poorer on lignin and hemicellulose rich lignocellulosic substrates. Similar poor performance for a large multi-domain cellulase CelA from Caldicellulosiruptor bescii, which is also a highly cellulolytic CBP type organism like C. thermocellum, on lignin-rich cellulosic biomass has been shown.3 Pretreatments that remove both lignin and hemicellulose can help improve the specific activity of cellulosomal enzymes. Future improvements to microbes for CBP and inexpensive low-severity pretreatments might address the outstanding challenges of utilizing complexed cellulosomal type enzymatic systems to deconstruct cellulosic biomass for biofuel production.79
Author contributions
SPSC, CDP & BR conceptualized and designed the study; SPSC, CDP, BR, MN, SLC, VRB performed research; SPSC, CDP, BR, MN, JRM, VRB, JDT, BED analyzed the data; and SPSC, CDP & VRB wrote the paper. All authors reviewed and edited the paper.Competing interests
The authors declare that they have no competing interests.Abbreviations
| CAZymes | Carbohydrate-active enzymes |
| CBM | Carbohydrate binding module |
| AFEX | Ammonia fiber expansion |
| SG | Switchgrass |
| GLBRC | Great Lakes Bioenergy Research Center |
| BESC | BioEnergy Science Center |
| SAXS | Small angle X-ray scattering |
| AV | Avicel |
| Ct | Clostridium thermocellum |
| Tr | Trichoderma reesei |
| Ct-cell | Clostridial cellulosomes |
| Ct-Rose | Clostridial rosettazymes |
| NMA | Normal mode analysis |
| CBP | Consolidated bioprocessing |
| DTT | Dithiothreitol |
Acknowledgements
This study was funded by NASA Ames Research Center and Michigan State University Research Foundation. We also acknowledge partial support by the National Science Foundation Grant #1236120 (CBET – Energy for Sustainability) and the DOE Great Lakes Bioenergy Research Center (supported by the Department of Energy, Office of Science, Office of Biological and Environmental Research, through Cooperative Agreement DE-FC02-07ER64494 between The Board of Regents of the University of Wisconsin System and the Department of Energy; http://www.glbrc.org). BR and JRM were supported through the BioEnergy Science Center (BESC). BESC is a U.S. Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. Oak Ridge National Laboratory is managed by UTBattelle LLC for the US DOE under contract number DE-AC05-00OR22725. SPSC gratefully acknowledges Leonardo Sousa, Nirmal Uppugundla, Mingjie Jin, Venkatesh Balan and other members of the BCRL who provided useful insights, criticisms and technical support during the course of this research. CDP gratefully acknowledges Sigrid Reinsch and Oana Marcu for their work in purification and characterization of selected cellulosomal enzymes. MN and VRB are grateful to the ESRF staff for their valuable assistance during SAXS data collection. We thank colleagues at NREL (Golden, CO) for providing the untreated and dilute-acid treated switchgrass substrates used in this study. We thank Prof. Brian Fox (UW Madison, Department of Biochemistry) for his critical review of the manuscript.References
- S. P. S. Chundawat, G. T. Beckham, M. Himmel and B. E. Dale, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 121–145 CrossRef CAS PubMed.
- T. E. Takasuka, A. J. Book, G. R. Lewin, C. R. Currie and B. G. Fox, Sci. Rep., 2013, 3, 1030 CrossRef PubMed.
- R. Brunecky, M. Alahuhta, Q. Xu, B. S. Donohoe, M. F. Crowley, I. A. Kataeva, S.-J. Yang, M. G. Resch, M. W. W. Adams, V. V. Lunin, M. E. Himmel and Y. J. Bomble, Science, 2013, 342, 1513–1516 CrossRef CAS PubMed.
- P. J. Brumm, Biofuels, 2013, 4, 669–681 CrossRef CAS.
- L. R. Lynd, M. S. Laser, D. Brandsby, B. E. Dale, B. H. Davison, R. Hamilton, M. Himmel, M. Keller, J. D. McMillan, J. Sheehan and C. E. Wyman, Nat. Biotechnol., 2008, 26, 169–172 CrossRef CAS PubMed.
- M. Jin, C. Gunawan, V. Balan and B. E. Dale, Biotechnol. Bioeng., 2012, 109, 1929 CrossRef CAS PubMed.
- L. R. Lynd, P. J. Weimer, W. H. van Zyl and I. S. Pretorius, Microbiol. Mol. Biol. Rev., 2002, 66, 506–577 CrossRef CAS PubMed.
- G. T. Beckham, J. F. Matthews, B. Peters, Y. J. Bomble, M. E. Himmel and M. F. Crowley, J. Phys. Chem. B, 2011, 115, 4118–4127 CrossRef CAS PubMed.
- S. P. S. Chundawat, G. Bellesia, N. Uppugundla, L. Sousa, D. Gao, A. Cheh, U. Agarwal, C. Bianchetti, G. Phillips, P. Langan, V. Balan, S. Gnanakaran and B. E. Dale, J. Am. Chem. Soc., 2011, 133, 11163–11174 CrossRef CAS PubMed.
- L. da Costa Sousa, M. Jin, S. P. S. Chundawat, V. Bokade, X. Tang, A. Azarpira, F. Lu, U. Avci, J. Humpula, N. Uppugundla, C. Gunawan, S. Pattathil, A. M. Cheh, N. Kothari, R. Kumar, J. Ralph, M. G. Hahn, C. E. Wyman, S. Singh, B. A. Simmons, B. E. Dale and V. Balan, Energy Environ. Sci., 2016, 9, 1215–1223 CAS.
- D. Gao, S. P. S. Chundawat, A. Sethi, V. Balan, S. Gnanakaran and B. E. Dale, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10922–10927 CrossRef CAS PubMed.
- E. A. Johnson, M. Sakajoh, G. Halliwell, A. Madia and A. L. Demain, Appl. Environ. Microbiol., 1982, 43, 1125–1132 CAS.
- J. J. Adams, G. Pal, Z. Jia and S. P. Smith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 305–310 CrossRef CAS PubMed.
- Y.-H. P. Zhang and L. R. Lynd, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7321–7325 CrossRef CAS PubMed.
- H.-P. Fierobe, F. Mingardon, A. Mechaly, A. Belaich, M. T. Rincon, S. Pages, R. Lamed, C. Tardif, J.-P. Belaich and E. A. Bayer, J. Biol. Chem., 2005, 280, 16325–16334 CrossRef CAS PubMed.
- H.-P. Fierobe, E. A. Bayer, C. Tardif, M. Czjzek, A. Mechaly, A. Belaich, R. Lamed, Y. Shoham and J.-P. Belaich, J. Biol. Chem., 2002, 277, 49621–49630 CrossRef CAS PubMed.
- F. Mingardon, A. Chanal, A. M. Lopez-Contreras, C. Dray, E. A. Bayer and H.-P. Fierobe, Appl. Environ. Microbiol., 2007, 73, 3822–3832 CrossRef CAS PubMed.
- F. Mingardon, A. Chanal, C. Tardif, E. A. Bayer and H.-P. Fierobe, Appl. Environ. Microbiol., 2007, 73, 7138–7149 CrossRef CAS PubMed.
- S. Mitsuzawa, H. Kagawa, Y. Li, S. L. Chan, C. D. Paavola and J. D. Trent, J. Biotechnol., 2009, 143, 139–144 CrossRef CAS PubMed.
- Y. Vazana, S. Morais, Y. Barak, R. Lamed, E. A. Bayer and J. G. Harry, in Methods in Enzymology, Academic Press, 2012, vol. 510, pp. 429–452 Search PubMed.
- S. Morais, Y. Barak, R. Lamed, D. Wilson, Q. Xu, M. Himmel and E. Bayer, Biotechnol. Biofuels, 2012, 5, 78 CrossRef CAS PubMed.
- S. Morais, Y. Barak, J. Caspi, Y. Hadar, R. Lamed, Y. Shoham, D. B. Wilson and E. A. Bayer, mBio, 2010, 1(5), e00285 CrossRef CAS PubMed.
- S. Moraïs, A. Heyman, Y. Barak, J. Caspi, D. B. Wilson, R. Lamed, O. Shoseyov and E. A. Bayer, J. Biotechnol., 2010, 147, 205–211 CrossRef PubMed.
- S. Morais, Y. Barak, J. Caspi, Y. Hadar, R. Lamed, Y. Shoham, D. B. Wilson and E. A. Bayer, Appl. Environ. Microbiol., 2010, 76, 3787–3796 CrossRef CAS PubMed.
- L. Feinberg, J. Foden, T. Barrett, K. W. Davenport, D. Bruce, C. Detter, R. Tapia, C. Han, A. Lapidus, S. Lucas, J.-F. Cheng, S. Pitluck, T. Woyke, N. Ivanova, N. Mikhailova, M. Land, L. Hauser, D. A. Argyros, L. Goodwin, D. Hogsett and N. Caiazza, J. Bacteriol., 2011, 193, 2906–2907 CrossRef CAS PubMed.
- B. Raman, C. Pan, G. B. Hurst, M. Rodriguez Jr., C. K. McKeown, P. K. Lankford, N. F. Samatova and J. R. Mielenz, PLoS One, 2009, 4, e5271 Search PubMed.
- A. Riederer, T. E. Takasuka, S. Makino, D. M. Stevenson, Y. V. Bukhman, N. L. Elsen and B. G. Fox, Appl. Environ. Microbiol., 2011, 77, 1243–1253 CrossRef CAS PubMed.
- J. D. Trent, E. Nimmesgern, J. S. Wall, F. U. Hartl and A. L. Horwich, Nature, 1991, 354, 490–493 CrossRef CAS PubMed.
- H. K. Kagawa, J. Osipiuk, N. Maltsev, R. Overbeek, E. Quaite-Randall, A. Joachimiak and J. D. Trent, J. Mol. Biol., 1995, 253, 712–725 CrossRef CAS PubMed.
- S. P. S. Chundawat, R. Vismeh, L. Sharma, J. Humpula, L. Sousa, C. K. Chambliss, A. D. Jones, V. Balan and B. E. Dale, Bioresour. Technol., 2010, 101, 8429–8438 CrossRef CAS PubMed.
- D. J. Schell, J. Farmer, M. Newman and J. D. McMillan, Appl. Biochem. Biotechnol., 2003, 105, 69–85 CrossRef PubMed.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed.
- E. Morag Morgenstern, E. A. Bayer and R. Lamed, Enzyme Microb. Technol., 1992, 14, 289–292 CrossRef.
- S. P. S. Chundawat, M. S. Lipton, S. O. Purvine, N. Uppugundla, D. Gao, V. Balan and B. E. Dale, J. Proteome Res., 2011, 10, 4365–4372 CrossRef CAS PubMed.
- S. P. S. Chundawat, V. Balan and B. E. Dale, Biotechnol. Bioeng., 2008, 99, 1281–1294 CrossRef CAS PubMed.
- D. Gao, S. P. S. Chundawat, C. Krishnan, V. Balan and B. E. Dale, Bioresour. Technol., 2010, 101, 2770–2781 CrossRef CAS PubMed.
- M. Hammel, H.-P. Fierobe, M. Czjzek, V. Kurkal, J. C. Smith, E. A. Bayer, S. Finet and V. Receveur-Brechot, J. Biol. Chem., 2005, 280, 38562–38568 CrossRef CAS PubMed.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- D. I. Svergun, J. Appl. Crystallogr., 1992, 25, 495–503 CrossRef.
- D. I. Svergun, Biophys. J., 1999, 76, 2879–2886 CrossRef CAS PubMed.
- D. Franke and D. I. Svergun, J. Appl. Crystallogr., 2009, 42, 342–346 CrossRef CAS PubMed.
- R. A. McMillan, C. D. Paavola, J. Howard, S. L. Chan, N. J. Zaluzec and J. D. Trent, Nat. Mater., 2002, 1, 247–252 CrossRef CAS PubMed.
- P. V. Konarev, M. V. Petoukhov and D. I. Svergun, J. Appl. Crystallogr., 2001, 34, 527–532 CrossRef CAS.
- M. B. Kozin and D. I. Svergun, J. Appl. Crystallogr., 2001, 34, 33–41 CrossRef CAS.
- L. A. Kelley and M. J. E. Sternberg, Nat. Protoc., 2009, 4, 363–371 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- K. Suhre and Y.-H. Sanejouand, Nucleic Acids Res., 2004, 32, W610–W614 CrossRef CAS PubMed.
- D. Svergun, C. Barberato and M. H. J. Koch, J. Appl. Crystallogr., 1995, 28, 768–773 CrossRef CAS.
- P. Bernadó, E. Mylonas, M. V. Petoukhov, M. Blackledge and D. I. Svergun, J. Am. Chem. Soc., 2007, 129, 5656–5664 CrossRef PubMed.
- W. L. DeLano, PyMOL Mol. Graph. Syst., 2002 Search PubMed.
- C. Boisset, H. Chanzy, B. Henrissat, R. Lamed, Y. Shoham and E. A. Bayer, Biochem. J., 1999, 340, 829–835 CrossRef CAS PubMed.
- Y. Lu, Y.-H. P. Zhang and L. R. Lynd, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16165–16169 CrossRef CAS PubMed.
- S. P. S. Chundawat, B. S. Donohoe, L. Sousa, T. Elder, U. P. Agarwal, F. Lu, J. Ralph, M. E. Himmel, V. Balan and B. E. Dale, Energy Environ. Sci., 2011, 4, 973–984 CAS.
- D. Gao, N. Uppugundla, S. Chundawat, X. Yu, S. Hermanson, K. Gowda, P. Brumm, D. Mead, V. Balan and B. Dale, Biotechnol. Biofuels, 2011, 4, 5 CrossRef CAS PubMed.
- D. Gao, S. P. S. Chundawat, T. Liu, S. Hermanson, K. Gowda, P. Brumm, B. E. Dale and V. Balan, BioEnergy Res., 2010, 3, 67–81 CrossRef.
- J. Krauss, V. V. Zverlov and W. H. Schwarz, Appl. Environ. Microbiol., 2012, 78, 4301–4307 CrossRef CAS PubMed.
- A. L. Carvalho, F. M. V. Dias, J. A. M. Prates, T. Nagy, H. J. Gilbert, G. J. Davies, L. M. A. Ferreira, M. J. Romão and C. M. G. A. Fontes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13809–13814 CrossRef CAS PubMed.
- M. Hammel, H.-P. Fierobe, M. Czjzek, S. Finet and V. Receveur-Bréchot, J. Biol. Chem., 2004, 279, 55985–55994 CrossRef CAS PubMed.
- C. C. Lee, R. E. Kibblewhite, C. D. Paavola, W. J. Orts and K. Wagschal, Mol. Biotechnol., 2016, 58, 489–496 CrossRef CAS PubMed.
- A. Ciruela, H. J. Gilbert, B. R. S. Ali and G. P. Hazlewood, FEBS Lett., 1998, 422, 221–224 CrossRef CAS PubMed.
- Y. J. Bomble, G. T. Beckham, J. F. Matthews, M. Nimlos, M. Himmel and M. F. Crowley, J. Biol. Chem., 2011, 286, 5614–5623 CrossRef CAS PubMed.
- A. B. Boraston, D. N. Bolam, H. J. Gilbert and G. J. Davies, Biochem. J., 2004, 382, 769–781 CrossRef CAS PubMed.
- S. J. Horn, G. Vaaje-Kolstad, B. Westereng and V. G. Eijsink, Biotechnol. Biofuels, 2012, 5, 45 CrossRef CAS PubMed.
- B. Henrissat, Cellulose, 1994, 1, 169–196 CrossRef CAS.
- B. Henrissat, H. Driguez, C. Viet and M. Schulein, Bio/Technology, 1985, 3, 722–726 CrossRef CAS.
- S. K. Brady, S. Sreelatha, Y. Feng, S. P. S. Chundawat and M. J. Lang, Nat. Commun., 2015, 6, 10149 CrossRef CAS PubMed.
- N. Hayashi, J. Sugiyama, T. Okano and M. Ishihara, Carbohydr. Res., 1997, 305, 109–116 CrossRef CAS.
- N. Hayashi, J. Sugiyama, T. Okano and M. Ishihara, Carbohydr. Res., 1997, 305, 261–269 CrossRef CAS.
- A. Pakarinen, M. O. Haven, D. T. Djajadi, A. Várnai, T. Puranen and L. Viikari, Biotechnol. Biofuels, 2014, 7, 27 CrossRef PubMed.
- D. G. Olson, S. A. Tripathi, R. J. Giannone, J. Lo, N. C. Caiazza, D. A. Hogsett, R. L. Hettich, A. M. Guss, G. Dubrovsky and L. R. Lynd, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17727–17732 CrossRef CAS PubMed.
- J. J. Adams, M. A. Currie, S. Ali, E. A. Bayer, Z. Jia and S. P. Smith, J. Mol. Biol., 2010, 396, 833–839 CrossRef CAS PubMed.
- B. García-Alvarez, R. Melero, F. M. V. Dias, J. A. M. Prates, C. M. G. A. Fontes, S. P. Smith, M. J. Romão, A. L. Carvalho and O. Llorca, J. Mol. Biol., 2011, 407, 571–580 CrossRef PubMed.
- M. Czjzek, H.-P. Fierobe and V. Receveur-Bréchot, Methods Enzymol., 2012, 510, 183–210 CAS.
- A.-L. Molinier, M. Nouailler, O. Valette, C. Tardif, V. Receveur-Bréchot and H.-P. Fierobe, J. Mol. Biol., 2011, 405, 143–157 CrossRef CAS PubMed.
- R. Borne, E. A. Bayer, S. Pagès, S. Perret and H.-P. Fierobe, FEBS J., 2013, 280, 5764–5779 CrossRef CAS PubMed.
- C. Xu, Y. Qin, Y. Li, Y. Ji, J. Huang, H. Song and J. Xu, Bioresour. Technol., 2010, 101, 9560–9569 CrossRef CAS PubMed.
- M. G. Resch, B. S. Donohoe, J. O. Baker, S. R. Decker, E. A. Bayer, G. T. Beckham and M. E. Himmel, Energy Environ. Sci., 2013, 6, 1858 CAS.
- M. G. Resch, B. S. Donohoe, P. N. Ciesielski, J. E. Nill, L. Magnusson, M. E. Himmel, A. Mittal, R. Katahira, M. J. Biddy and G. T. Beckham, ACS Sustainable Chem. Eng., 2014, 2, 1377–1387 CrossRef CAS.
- L. R. Lynd, W. H. van Zyl, J. E. McBride and M. Laser, Curr. Opin. Biotechnol., 2005, 16, 577–583 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details on relevant methods (biomass composition; gene cloning, protein expression and purification) and experimental results (additional enzyme loading assays) are available as a supplementary pdf document (named “Electronic Supplementary Material_Chundawat.pdf”). See DOI: 10.1039/c6re00172f |
| ‡ Current address for CDP: Eli Lilly and Company, Drop Code 0403, Lilly Corporate Center, Indianapolis, IN 46285, USA. |
| § Current address for BR: Dow AgroSciences, 9330 Zionsville Road, Indianapolis, IN 46268, USA. |
| ¶ AFEX is a trademark of MBI, Lansing (http://www.mbi.org/). |
| This journal is © The Royal Society of Chemistry 2016 |