
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamic synthesis of solution processable ladder polymers†
Jongbok
Lee
a,
Bharath Bangalore
Rajeeva
b,
Tianyu
Yuan
ac,
Zi-Hao
Guo
a,
Yen-Hao
Lin
a,
Mohammed
Al-Hashimi
d,
Yuebing
Zheng
b and
Lei
Fang
*ac
aDepartment of Chemistry, Texas A&M University, 3255 TAMU, College Station, TX 77843, USA. E-mail: fang@chem.tamu.edu
bDepartment of Mechanical Engineering, Materials Science and Engineering Program, Texas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA
cMaterials Science & Engineering Department, Texas A&M University, 3003 TAMU, College Station, TX 77843, USA
dDepartment of Chemistry, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar
First published on 6th November 2015
Abstract
The synthesis of a carbazole-derived, well-defined ladder polymer was achieved under thermodynamic control by employing reversible ring-closing olefin metathesis. This unique approach featured mild conditions and excellent efficiency, affording the ladder polymer backbone with minimum levels of unreacted defects. Rigorous NMR analysis on a 13C isotope-enriched product revealed that the main-chain contained less than 1% of unreacted precursory vinyl groups. The rigid conformation of the ladder-type backbone was confirmed by photophysical analysis, while the extended rod-like structure was visualized under scanning tunneling microscope. Excellent solubility of this polymer in common organic solvents allowed for feasible processing of thin films using solution-casting techniques. Atomic force microscopy and grazing incident X-ray scattering revealed a uniform and amorphous morphology of these films, in sharp contrast to the polycrystalline thin films of its small molecular counterpart.
Introduction
Fully conjugated ladder polymers with coplanar, sp2 atom-rich backbones represent a captivating class of macromolecules, on account of their well-defined rigid structures, intriguing syntheses, and promising potential in materials applications.1–5 By definition,6 ladder polymers have a distinctive architecture, whereby the chain consists of an uninterrupted sequence of rings, fused together in such a way that adjacent rings share two or more atoms in common, resulting in a constrained chain conformation.7–9 Possessing a backbone of fused aromatic rings, a fully conjugated ladder polymer is intrinsically free of possible torsional disorders that result from σ-bond rotations in between the monomeric units. Without the interruption from these conformational disorders, as a result, the coherent π-conjugation length of a coplanar ladder backbone is much more extended.10 Such a well-defined conformation would afford a faster intra-chain charge/phonon transport,11,12 and a longer exciton diffusion length compared to conjugated polymers with free rotating torsional motions.13 This argument is further corroborated by the unparalleled electronic and thermal conductivity of graphene nanoribbons,14,15 which can be viewed as insoluble ladder polymers composed of only sp2 carbon atoms. Combining the advantages of conventional polymeric materials such as solution processability and structural versatility, ladder polymers emerge as promising candidates for next-generation synthetic organic materials with breakthrough performances.The synthesis of a well-defined ladder polymer, however, is a challenging task because of (i) the potential structural defects originated from moderately efficient ring-closing reactions, and (ii) the often poor solubility of structurally rigid intermediates or products. In an effort to overcome these obstacles, a highly efficient synthetic method and a rationally designed structural characteristic that enables solubility need to be achieved and integrated simultaneously. On one hand, kinetically controlled annulation reactions such as Scholl oxidation and electrophilic cyclization have been widely used16–24 in the preparation of conjugated ladder polymers from pre-organized polymer precursors, affording a number of materials with fascinating optical and electronic properties. On the other hand, thermodynamically controlled annulation reactions, in principle, can offer unique advantages in the synthesis of well-defined ladder polymers: this enables the opportunities of “error-checking” and “proof-reading”25 while pushing the reversible equilibrium to the most stable position. As a result, during the post-polymerization ring-closing step, one could prevent the potential formation of unreacted defects and inter-chain cross-linking, which could be detrimental to the desired materials properties. Despite the early examples and a few subsequent advances involving imine condensation,18,26,27 this promising strategy has not been extensively explored up to date. Herein, we report the synthesis of a fully conjugated ladder polymer with an extremely low level of unreacted defects, by taking advantage of thermodynamically controlled ring-closing olefin metathesis (RCM) reaction. The carefully designed structure gave good solubility of the ladder polymer in common organic solvents, which allowed for excellent solution processability, and extensive solution- and solid-state characterization.
Results and discussion
We envisioned that the RCM reaction held28–33 the promise for the synthesis of conjugated ladder polymers, because (i) it can produce C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds by releasing about 28 kcal mol−1 of enthalpy in forming a stable aromatic benzene ring,31,33 and (ii) its mild condition enables a wide substrate scope and excellent functional group tolerance.34 Given a properly designed precursor, RCM reactions should lead to an uncross-linked, stable, and conjugated ladder polymer with minimum unreacted defects.
C double bonds by releasing about 28 kcal mol−1 of enthalpy in forming a stable aromatic benzene ring,31,33 and (ii) its mild condition enables a wide substrate scope and excellent functional group tolerance.34 Given a properly designed precursor, RCM reactions should lead to an uncross-linked, stable, and conjugated ladder polymer with minimum unreacted defects.
Guided by these principles, a synthetic route (Fig. 1a) was designed and executed. This synthesis involved two essential steps: first, step-growth polymerization of two divinyl-functionalized monomers affords a conjugated polymer with pre-organized pendant vinyl groups. In the second step, the RCM reaction forms the bridging aromatic rings and hence leads to a coplanar ladder polymer. A carbazole unit was employed as the primary building block in this design (Fig. 1b), motivated by the following facts: firstly, it is inexpensive and feasible to prepare functionalized carbazole derivatives with excellent regioselectivity.35,36 For example, in the designed carbazole precursor 1, two boronic ester functions were installed on the 2,7-positions for the step-growth polymerization step while the 3,6-positions were functionalized with vinyl groups for RCM annulation. Secondly, the presence of an easy-to-alkylate nitrogen position on the carbazole unit allows for the installation of an α-branched alkyl group (1-octylnonyl in this case) to enhance the solubility of the rigid ladder polymer product. Such a side-chain would extend in a perpendicular direction with respect to the π-system, so that solubility could be drastically enhanced by breaking the potentially too strong intermolecular π–π stacking interactions.37,38 Finally, carbazole-containing organic materials have demonstrated high performance in applications associated with photovoltaics39,40 and light emitting diodes.41,42 Thus, successful synthesis of ladder polymers derived from carbazole can be readily translated into applications in these fields.
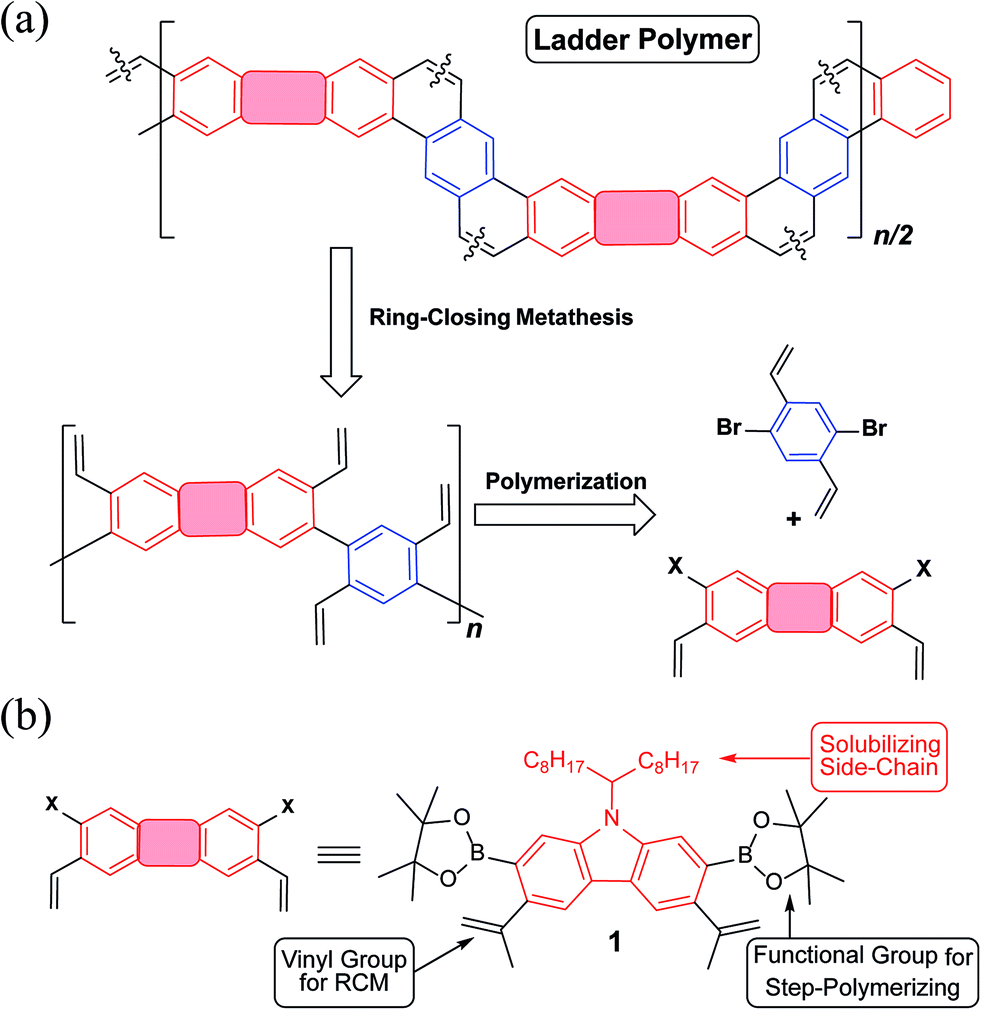 | ||
| Fig. 1 (a) A general retrosynthetic analysis for the designed ladder polymer; (b) structural formula of the carbazole-derived monomer 1. | ||
Precursor 1 was synthesized from 2,7-dibromocarbazole in 60% overall yield on a ∼3 grams scale (see ESI†). Before exploring the synthesis of the target polymer, a small molecular model compound 3 was synthesized to validate the strategy and to assist future characterization (Scheme 1a). 2-Bromostyrene and compound 1 were cross-coupled by Suzuki reaction to give a tetra-vinyl derivative 2. Butylated hydroxytoluene (BHT) was used in this reaction as a radical scavenger to inhibit free-radical polymerization of the styrene derivatives. During the subsequent RCM step, it was critical to achieve quantitative conversion of the vinyl groups. This was particularly important for the later synthesis of polymer P2 (Scheme 1b). A subset of conditions for the RCM reaction was tested on 2 (Table S1, ESI†). In the most efficient method identified, the catalyst was added with a syringe pump over 4 h at reflux temperature to give product 3 in 96% isolated yield. According to a liquid chromatogram (see ESI†), the conversion of the RCM reaction was close to quantitative. In this transformation, the reversible nature of RCM prevented undesired side reactions such as intermolecular cross olefin metathesis of the vinyl groups, and afforded the thermodynamically most stable product in nearly quantitative conversion. The structure of 3 was characterized unambiguously by 1H–1H NOESY NMR spectroscopy (Fig. S6, ESI†) and single crystal X-ray diffraction (Fig. 2a). The crystal structure clearly demonstrated that the annulated aromatic rings extended in a coplanar geometry. The 1-octylnonyl side-chain was perpendicular to the π-backbone in the solid state. As a result, the potential strong intermolecular π–π stacking interactions were suppressed, leading to good solubility in common organic solvents, such as chloroform, tetrahydrofuran and toluene. In the 1H NMR spectra of 2 and 3 (Fig. 3a), as expected, the vinyl peaks at 5.0–7.0 ppm in 2 disappeared in 3 after the RCM reaction, while other peaks shifted downfield. Moreover, after RCM, the IR spectrum of 3 revealed the disappearance of the alkenyl C–H and CC stretching peaks originally present in the spectrum of 2 (Fig. S4, ESI†).
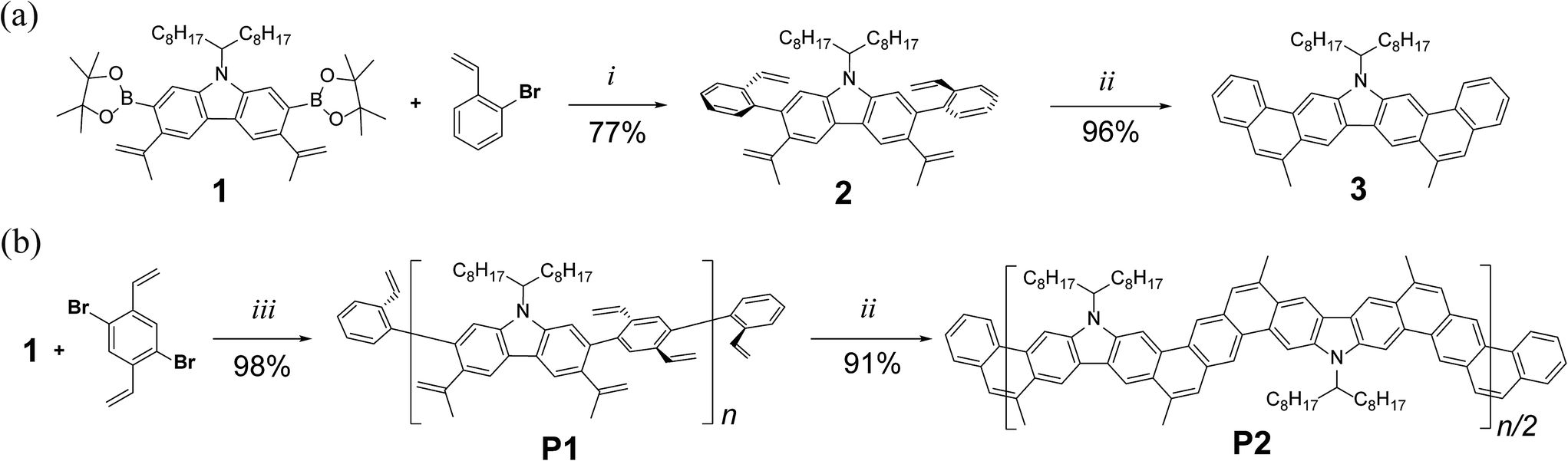 | ||
| Scheme 1 (a) Synthesis of small molecular model compound 3; (b) synthesis of ladder polymer P2. Reaction conditions: (i) Pd(PPh3)4, K2CO3, aliquat 336, BHT, PhMe, H2O, 100 °C, 24 h. (ii) Grubbs' 2nd generation catalyst, PhMe, reflux, 6 h. (iii) Pd(PPh3)4, K2CO3, aliquat 336, BHT, PhMe, H2O, 100 °C, 24 h; then 2-bromostyrene and 2-vinylphenylboronic acid. | ||
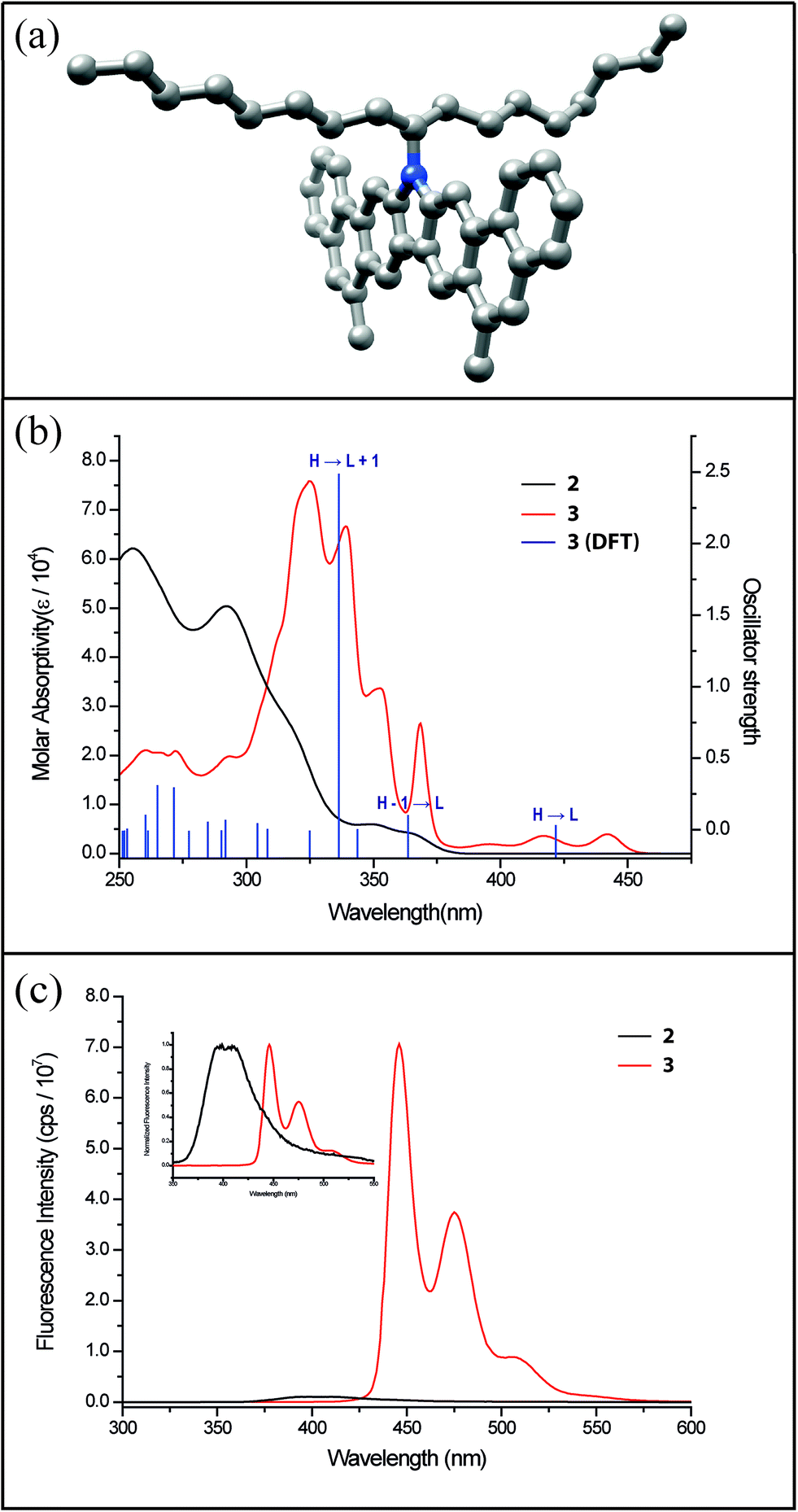 | ||
| Fig. 2 (a) Solid-state structure of compound 3 obtained from single-crystal X-ray diffraction; (b) UV-vis absorption spectra of compound 2 (black) and 3 (red) in CHCl3, with the TDDFT calculated electronic transitions of 3 shown as blue bar; (c) fluorescent emission spectra of 2 (black) and 3 (red) with absolute intensity scale. Inset is the same spectra with normalized intensity. | ||
 | ||
Fig. 3 (a) Partial 1H NMR spectra of 2 and 3. Dotted lines represent the change of chemical shift of each resonance peak after RCM reaction. Proton resonance peaks for c, c′, f and g in the box all disappeared after converting 2 into 3; (b) partial 1H NMR spectra of P1 and P2 showing the similar change of chemical shift. Proton resonance peaks for the terminal vinyl groups c, c′, f, and g in the box all disappeared after the RCM reaction; (c) 13C NMR spectra of P1* (S/N = 94 after 413 scans) and P2* (S/N = 367 after 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 816 scans). The carbons labeled with “*” are 99% 13C isotope enriched. 13C resonance peaks for the terminal vinyl carbon β in the box disappeared after the RCM reaction. 816 scans). The carbons labeled with “*” are 99% 13C isotope enriched. 13C resonance peaks for the terminal vinyl carbon β in the box disappeared after the RCM reaction. | ||
The comparison of optical properties between 2 and 3 clearly illustrated (Fig. 2b) the impact of aromatic ring-closure on the electronic structures. UV-vis absorption spectrum of the annulated compound 3 was red-shifted compared to that of 2 because of the more extended conjugation and a covalently enforced coplanar π-electron system. Moreover, both the absorption and emission spectra of 3 showed distinctive vibrational progressions that were indicative of a rigid conjugated system with insignificant conformational fluctuation.43 Interestingly, the absorption spectrum of 3 showed a weak, symmetrically forbidden HOMO–LUMO transition similar to that of C60.44 Time-dependant density functional theory (TDDFT) [B3LYP/6-311G(d,p)] was performed to obtain the calculated electronic transitions of 3, which were in excellent agreement with the experimental data (Fig. 2b). Furthermore, the fluorescent emission spectrum of 3 (Fig. 2c) was also red-shifted and enhanced in terms of intensity relative to that of 2. This phenomenon corroborated again the extended π-conjugation of 3. Overall, the successful synthesis and unambiguous characterization of the small molecular model compound provided a firm basis for the preparation and investigation of ladder polymer P2 using a similar strategy.
The synthetic route to the target ladder-type polymer P2 is outlined in Scheme 1b. The step-growth Suzuki polymerization between 1 and 1,4-dibromo-2,5-divinylbenzene afforded a conjugated polymer with pendant vinyl groups, which was then end-capped by subsequent addition of 2-bromostyrene and 2-vinylphenylboronic acid to give P1. The end-capping groups not only quenched the active bromide and boronic ester functions, but also provided complementary vinyl groups for the polymer chain ends to undergo RCM reaction in the next step. BHT was again added as a radical scavenger in this step to prevent vinyl cross-linking between the polymer chains. The reaction was carried out on a 1 g scale to afford P1 in 98% yield. The crude polymer was collected by precipitation from methanol, followed by washing with acetone. Further purification was accomplished using preparative size exclusion chromatography (SEC) with CHCl3 as the eluent to remove the low molecular weight oligomers, affording a narrowly distributed batch of P1 in 75% isolated yield. SEC analysis of the purified P1 revealed a molecular weight (Mn = 20 kg mol−1, PDI = 1.88) that was comparable to that of polymers used as high performance organic solar cell donor materials45–49 (Fig. S2, ESI†).
In the subsequent RCM step, the optimized conditions screened from the synthesis of 3 were employed to convert P1 into the ladder-type product P2 in 91% yield. The resulting product P2 was purified by precipitation and Soxhlet extraction. The molecular weight of P2 (Mn = 15 kg mol−1, PDI = 2.00) was slightly smaller than its precursor P1 because the higher molecular weight fraction was partially removed during these purification steps due to its lower solubility at high concentration (Fig. S3, ESI†). Despite the lower solubility of P2 compared to P1, it was still feasible to prepare a solution of purified P2 in CHCl3 at a concentration of 5 mg mL−1. This good solubility allowed for extensive NMR investigation, SEC analysis, and solution processing into thin films.
The comparison of 1H NMR spectra of P1 and P2 resembled that of model compounds 2 and 3: the resonance peaks associated with the terminal protons on the vinyl groups disappeared after RCM (Fig. 3b). In addition, IR spectra of P2 also showed the disappearance of the alkenyl C–H and CC stretching after RCM, similar to 3 (Fig. S5, ESI†). Despite these promising results, however, the possibility of unreacted vinyl groups in P2 could not be ruled out by these characterization methods. 1H NMR resonance signals were too broad to be useful for a rigorous quantification of defects, especially if the amount of unreacted vinyl groups was less than 5%. Such a structural defect, however, could be critical to the electronic, optical, and mechanical properties of conjugated ladder polymers.50–52 In this context, a 13C isotope labeling method53 was employed to track the terminal vinyl carbons using much sharper 13C NMR resonance peaks. Therefore, a much sensitive analysis of the unreacted defects of P2 could be performed. The same synthetic procedures afforded 13C labeled P1*, in which a methyl carbon α and the terminal alkenyl carbon β are 99% 13C isotope enriched (Fig. 3c). The 13C NMR spectrum of P1* showed the two expected intense sharp peaks for these 13C isotope enriched carbons at 116.0 ppm and 24.1 ppm with good signal/noise ratio (S/N = 94 after 413 scans). P1* was then subjected to the optimized RCM conditions to afford P2*. The resonance signal associated with the terminal vinyl carbon at 116.0 ppm disappeared completely while the peak at 21.0 ppm retained an excellent signal/noise ratio (S/N = 367 after 17816 scans). This result corroborated that the unreacted vinyl defect in P2* was less than 1%. Considering that the degree of polymerization of P2 and P2* was around 23–27, the average possible defect site in a single polymer chain was in fact much less than one. Based on these numbers, we can conclude that most of the polymer chains should be free of defect but there might still be a small fraction of the polymer chains possessing one or more unreacted defects.
UV-vis absorption and fluorescent emission spectra of P1 and P2 were recorded in CHCl3 solution (Fig. 4a and b). Similar to that of 3, both absorption and emission spectra of P2 were red-shifted compared to that of P1, as a result of the much more delocalized and larger conjugated π-system. In addition, HOMO–LUMO transition of P2 was weak and optically forbidden, similar to that of 3. Meanwhile, the almost zero wavelength difference (Fig. S30, ESI†) between HOMO–LUMO transition of absorption and LUMO–HOMO transition of emission, and distinctive vibrational progression were in accordance with the highly rigid nature of the P2 backbone, which prevented any significant conformational change between the excited and the ground states.43 Unlike the unstructured and low emission of P1 (quantum yield < 1%), the emission spectrum of P2 was composed of well-structured vibrational progressions with much higher quantum yield (15%). Furthermore, it was observed that solid-state UV-vis absorption of P1 was red-shifted compared to that in the solution phase. Such a red-shift can be attributed to solid-state packing-induced coplanarization of the P1 backbone, similar to that of the well studied regioregular poly(3-alkylthiophene).54,55 In contrast, the solution-phase and solid-state absorption spectra of P2 were almost identical because P2 in solution was already coplanar and rigid, hence solid-state packing could not change the spectra by alternating its conformation (Fig. 4c and d). These photophysical observations suggest that ladder polymer P2 possesses a highly rigid backbone and well-extended π-conjugation.
 | ||
| Fig. 4 (a) UV-vis absorption spectra of P1 (black) and P2 (red) in CHCl3 and as thin films; (b) fluorescence emission spectra of P1 (black) and P2 (red) in CHCl3 with absolute intensity scale. Inset is the same spectra with normalized intensity. Graphical illustration of the conformational change of (c) P1 and (d) P2 between in solution and in the solid state. | ||
It is expected that sp2 atom rich, aromatic ladder-type polymers have a backbone that is stable at high temperatures, resembling fused ring carbon materials like carbon nanotubes and graphene nanoribbons.56,57 Thermogravimetric analysis (TGA, Fig. S32 and S33, ESI†) of P1 showed a clear weight loss in the range of 380–480 °C, corresponding to the thermal cleavage of the sp3 1-octylnonyl side-chains. Upon further increasing the temperature the non-ladder type backbone of P1 saw a continuous weight loss, affording only a 34% carbonization yield at 900 °C. In comparison, polymer P2 also experienced a weight loss due to the alkyl chain cleavage at around 348–480 °C. The remaining backbone, however, was stable up to 900 °C, giving a carbonization yield of 52%. The result is in good agreement with the weight percentage of the aromatic backbone (57%). The high thermal stability of the P2 backbone was a result of its ladder-type nature and promised potential in applications as pre-functionalized precursor for sp2 carbon materials.58,59 Differential scanning calorimetric (DSC, Fig. S34 and S35, ESI†) analysis of P1 showed an irreversible exothermic transition at 129 °C. This transition was attributed to a thermally triggered cross-linking reaction between the vinyl groups, which indeed converted P1 into an insoluble material after just one heating cycle. In comparison, P2 possesses a thermally stable backbone, showing no thermal transition before its side-chain cleavage temperature in the DSC measurement.
Solution processing of P2 into thin films would be a key step for future exploration of its applications. Compared to its small molecular counterpart 3, polymeric P2 was expected to possess better processability for uniform thin films. Thin film morphologies of 3 and P2 on SiO2 substrates were investigated after spin-casting or solution-shearing casting from solutions in toluene (Fig. 5). For small molecule 3, both methods afforded polycrystalline thin films, which were composed of randomly distributed microcrystals with sizes in the range of 1–3 μm. Atomic force microscopy (AFM) revealed a very rough surface for this polycrystalline film with root mean square (RMS) roughness of 5.39 nm. In contrast, P2 can be processed into uniform thin films by either method. No observable feature can be identified under optical microscope. AFM images demonstrated an amorphous morphology with much lower roughness (RMS = 0.45 nm). Grazing incidence wide-angle X-ray scattering (GIWAXS) revealed highly crystalline scattering peaks for the thin film of 3 and no observable diffraction features for P2 (Fig. 5e and f). These results suggest that the polymer chains were packed in an amorphous manner on SiO2 substrates despite its rigid backbone. The excellent film formation ability of P2 enables future investigation of its material properties.
 | ||
| Fig. 5 Film morphology comparison of 3 and P2. The thin films were deposited on UV-ozone cleaned silicon wafers by spin-casting solutions (2 mg mL−1 in toluene) at a rate of 2000 rpm. (a) Optical microscope image of 3 with observed microcrystals (1–3 μm) and (b) that of P2 with no optical feature; (c) AFM images of microcrystals of 3 (RMS = 5.39 nm) and (d) amorphous morphology of P2 (RMS = 0.45 nm); (e) GIWAXS patterns of 3 and (f) P2. Both samples were measured at an incident angle of 0.2° and 30 second exposure time, and both images have the same color scale. | ||
In order to further characterize the solid-state dimension, conformation, and self-assembly of P2, scanning tunneling microscopic (STM) images were recorded on highly ordered pyrolytic graphite (HOPG) (Fig. 6). A solution of P2 (0.3 mg mL−1) in chloroform was drop-casted onto heated HOPG substrate and analyzed by STM. The images exhibited uniform and fully extended rod-like morphology, indicating self-assembly of the rigid polymer chain of P2 on the HOPG substrate.20,60 The highly ordered self-assembly was likely a result of the strong π–π interaction between the HOPG substrate and the aromatic ladder-type backbone.61,62 These rods under STM showed alternating sections of high and low signal with a periodic length at around 1.27 nm. According to the single crystal structure of 3 and DFT calculations on model oligomers resembling P2, the feature length of each repeating unit on P2 was close to 1.26 nm, matching well with the experimental results. The periodic distance between the rods under STM was around 0.7 nm, while the width of calculated polymer backbone without alkyl chain is close to 0.68 nm. Taking consideration of the additional contribution to the width from the side-chains, it is likely that the neighbouring P2 rods were partially stacked, in a manner similar to the literature reported graphene nanoribbons.60 In sharp contrast, STM analysis of P1 showed no ordered features, probably a result of low surface interaction between the non-ladder backbone and the HOPG substrate (Fig. S38 and S39, ESI†).
 | ||
| Fig. 6 (a) STM image of P2 on highly-ordered pyrolytic graphite; (b) section profile along the arrow line drawn in (a). The STM image was obtained with a tunneling current set point of 0.9 nA and sample bias of 50 mV. The distance between consecutive convex spots along the X-axis is 1.27 nm, and average distance between peaks along the Y-axis is 0.70 nm. (c) Theoretical dimensions of a oligomeric model P2 backbone from DFT calculation (B3LYP/6-311G). | ||
Conclusion
In conclusion, a highly efficient synthetic approach to a conjugated ladder-type polymer has been established on the basis of thermodynamically controlled ring-closing olefin metathesis reaction. The resulting polymer possesses a highly rigid backbone with less than one defect per chain on average, meanwhile maintaining good solubility and solution processability. The feasible and versatile nature of this method enables an efficient and impactful way to the synthesis of novel polymer materials with desirable features that will potentially be useful for thin film devices.Experimental section
Synthesis of P1
To a 100 mL Schlenk flask was added 1 (1.03 g, 1.40 mmol), 1,4-dibromo-2,5-divinylbenzene (0.40 g, 1.40 mmol), Pd(PPh3)4 (0.16 g, 10 mol%), K2CO3 (1.16 g, 8.40 mmol), aliquat 336 (0.65 mL, 0.35 mmol), and several crystals of BHT under N2. Degassed toluene (40 mL) and water (8 mL) were added and further degassed 3 times by freeze–pump–thaw. The reaction mixture was stirred at 100 °C for 24 h in dark, before it was cooled down to room temperature. 2-Bromostyrene (0.77 mL, 5.60 mmol) was added into the flask, and the mixture was stirred at 100 °C for 24 h. After 24 h, 2-vinylphenylboronic acid (1.73 g, 11.2 mmol) was added into the flask, and the mixture was stirred at 100 °C for another 24 h. The resulting product was precipitated from methanol, filtered, and washed with acetone. The solid was dried under vacuum to afford P1 (0.84 g, 98%, Mn = 10 kg mol−1, PDI = 2.78 by SEC). P1 was further purified by preparative recycling SEC to remove oligomers to afford a purified batch with higher Mn and lower PDI (0.64 g, 75%, Mn = 20 kg mol−1, PDI = 1.88 by SEC).Synthesis of P2
To a 100 mL Schlenk flask was added P1 (120 mg, 0.20 mmol) and Grubbs' 2nd generation catalyst (8 mg, 5 mol%) under N2. Subsequently, degassed toluene (12 mL) was added, and the reaction mixture was stirred at reflux temperature. Immediately, another portion of Grubbs' 2nd generation catalyst (26 mg, 15 mol%) in degassed toluene (8 mL) was added for 4 h using syringe pump. After that, the reaction mixture was stirred for an additional 2 h at reflux temperature before cooling down to room temperature. The resulting product was then precipitated from methanol and filtered. The product was further washed via Soxhlet extraction with acetone and hexane, before extracted with chloroform. The chloroform solution was filtered and condensed under reduced pressure. The desired product was precipitated from methanol. The precipitate was filtered and dried under vacuum to afford P2 (100 mg, 91%, Mn = 15 kg mol−1, PDI = 2.00 by SEC).Acknowledgements
This work was supported by the National Priorities Research Program award (NPRP7-285-1-045) from the Qatar National Research Fund. The authors thank Dr Nattamai Bhuvanesh for X-ray crystallography support, Dr Vladimir Bakhmoutov for NMR spectroscopy support, Dr Xu Zhou, Fang-Dong Zhuang, and Dr Jian Pei (Peking University) for fluorescence quantum yield measurement, and Dr Joseph Strzalka (Argonne National Laboratory) for GIWAXS film characterization support. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.References
- F. E. Arnold and R. L. van Deusen, Macromolecules, 1969, 2, 497–502 CrossRef CAS.
- A.-D. Schluter, Adv. Mater., 1991, 3, 282–291 CrossRef.
- U. Scherf and K. Müllen, Makromol. Chem., Rapid Commun., 1991, 12, 489–497 CrossRef CAS.
- A. L. Briseno, S. C. B. Mannsfeld, P. J. Shamberger, F. S. Ohuchi, Z. Bao, S. A. Jenekhe and Y. Xia, Chem. Mater., 2008, 20, 4712–4719 CrossRef CAS.
- J. D. Plumhof, T. Stöferle, L. Mai, U. Scherf and R. F. Mahrt, Nat. Mater., 2014, 13, 247–252 CrossRef CAS PubMed.
- R. G. Jones, R. Stepto, E. S. Wilks, M. Hess, T. Kitayama and W. Val Metanomski, Compedium of polymer terminology and nomenclature IUPAC recommendations 2008, The Royal Society of Chemistry, Cambridge, UK, 2008 Search PubMed.
- S. Liu, Z. Jin, Y. C. Teo and Y. Xia, J. Am. Chem. Soc., 2014, 136, 17434–17437 CrossRef CAS PubMed.
- M. B. Goldfinger, K. B. Crawford and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 4578–4593 CrossRef CAS.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- F. C. Grozema, P. T. van Duijnen, Y. A. Berlin, M. A. Ratner and L. D. A. Siebbeles, J. Phys. Chem. B, 2002, 106, 7791–7795 CrossRef CAS.
- P. Prins, F. C. Grozema, J. M. Schins, S. Patil, U. Scherf and L. D. A. Siebbeles, Phys. Rev. Lett., 2006, 96, 146601 CrossRef CAS PubMed.
- E. J. Dell, B. Capozzi, K. H. DuBay, T. C. Berkelbach, J. R. Moreno, D. R. Reichman, L. Venkataraman and L. M. Campos, J. Am. Chem. Soc., 2013, 135, 11724–11727 CrossRef CAS PubMed.
- M. Samiullah, D. Moghe, U. Scherf and S. Guha, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205211 CrossRef.
- Y.-W. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347–349 CrossRef CAS PubMed.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS PubMed.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed.
- Z. H. Chen, J. P. Amara, S. W. Thomas and T. M. Swager, Macromolecules, 2006, 39, 3202–3209 CrossRef CAS.
- R. L. van Deusen, J. Polym. Sci., Part B: Polym. Phys., 1966, 4, 211–214 CrossRef CAS.
- A. D. Schluter, M. Loffler and V. Enkelmann, Nature, 1994, 368, 831–834 CrossRef.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS PubMed.
- U. Scherf and K. Müllen, Polymer, 1992, 33, 2443–2446 CrossRef CAS.
- U. Scherf, J. Mater. Chem., 1999, 9, 1853–1864 RSC.
- S. A. Patil, U. Scherf and A. Kadashchuk, Adv. Funct. Mater., 2003, 13, 609–614 CrossRef CAS.
- S. Qiu, P. Lu, X. Liu, F. Shen, L. Liu, Y. Ma and J. Shen, Macromolecules, 2003, 36, 9823–9829 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- M. M. Durban, P. D. Kazarinoff, Y. Segawa and C. K. Luscombe, Macromolecules, 2011, 44, 4721–4728 CrossRef CAS.
- Y. X. Yao and J. M. Tour, Macromolecules, 1999, 32, 2455–2461 CrossRef CAS.
- R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446–452 CrossRef CAS.
- T. J. Donohoe, A. J. Orr and M. Bingham, Angew. Chem., Int. Ed., 2006, 45, 2664–2670 CrossRef CAS PubMed.
- A. Iuliano, P. Piccioli and D. Fabbri, Org. Lett., 2004, 6, 3711–3714 CrossRef CAS PubMed.
- M. C. Bonifacio, C. R. Robertson, J. Y. Jung and B. T. King, J. Org. Chem., 2005, 70, 8522–8526 CrossRef CAS PubMed.
- S. K. Collins, A. Grandbois, M. P. Vachon and J. Côté, Angew. Chem., Int. Ed., 2006, 45, 2923–2926 CrossRef CAS PubMed.
- N. A. Vermeulen, O. Karagiaridi, A. A. Sarjeant, C. L. Stearn, J. T. Hupp, O. K. Fahra and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 14916–14919 CrossRef CAS PubMed.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed.
- J. F. Morin and M. Leclerc, Macromolecules, 2001, 34, 4680–4682 CrossRef CAS.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef CAS PubMed.
- Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed.
- H. Langhals, Helv. Chim. Acta, 2005, 88, 1309–1343 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- J. Li and A. C. Grimsdale, Chem. Soc. Rev., 2010, 39, 2399–2410 RSC.
- Y. Zou, T. Ye, D. Ma, J. Qin and C. Yang, J. Mater. Chem., 2012, 22, 23485–23491 RSC.
- Z. M. Hudson, Z. Wang, M. G. Helander, Z. H. Lu and S. Wang, Adv. Mater., 2012, 24, 2922–2928 CrossRef CAS PubMed.
- J. Gierschner, J. Cornil and H. J. Egelhaaf, Adv. Mater., 2007, 19, 173–191 CrossRef CAS.
- Z. Wei-ya, X. Si-shen, Q. Sheng-fa, W. Gang and Q. Lu-xi, J. Phys.: Condens. Matter, 1996, 8, 5793 CrossRef.
- J. W. Kingsley, P. P. Marchisio, H. Yi, A. Iraqi, C. J. Kinane, S. Langridge, R. L. Thompson, A. J. Cadby, A. J. Pearson, D. G. Lidzey, R. A. L. Jones and A. J. Parnell, Sci. Rep., 2014, 4, 5286 CAS.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456–4462 CrossRef CAS PubMed.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- S. Wakim, S. Beaupre, N. Blouin, B.-R. Aich, S. Rodman, R. Gaudiana, Y. Tao and M. Leclerc, J. Mater. Chem., 2009, 19, 5351–5358 RSC.
- A. C. Grimsdale and K. Müllen, Macromol. Rapid Commun., 2007, 28, 1676–1702 CrossRef CAS.
- S. Y. Cho, A. C. Grimsdale, D. J. Jones, S. E. Watkins and A. B. Holmes, J. Am. Chem. Soc., 2007, 129, 11910–11911 CrossRef CAS PubMed.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- H. Roex, P. Adriaensens, D. Vanderzande and J. Gelan, Macromolecules, 2003, 36, 5613–5622 CrossRef CAS.
- T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- A. R. Aiyar, J.-I. Hong, R. Nambiar, D. M. Collard and E. Reichmanis, Adv. Funct. Mater., 2011, 21, 2652–2659 CrossRef CAS.
- K. Kim, W. Regan, B. Geng, B. Alemán, B. M. Kessler, F. Wang, M. F. Crommie and A. Zettl, Phys. Status Solidi RRL, 2010, 4, 302–304 CrossRef CAS.
- T. Yamamoto, K. Watanabe and E. Hernández, in Carbon Nanotubes, ed. A. Jorio, G. Dresselhaus and M. Dresselhaus, Springer, Heidelberg, 2008, vol. 111, ch. 5, pp. 165–195 Search PubMed.
- J. P. McGann, M. Zhong, E. K. Kim, S. Natesakhawat, M. Jaroniec, J. F. Whitacre, K. Matyjaszewski and T. Kowalewski, Macromol. Chem. Phys., 2012, 213, 1078–1090 CrossRef CAS.
- P. Bajaj and A. K. Roopanwal, J. Macromol. Sci., Polym. Rev., 1997, 37, 97–147 CrossRef.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. de Feyter and K. Müllen, Nat. Chem., 2014, 6, 126–132 CrossRef CAS PubMed.
- Y. B. Zheng, B. K. Pathem, J. N. Hohman, J. C. Thomas, M. Kim and P. S. Weiss, Adv. Mater., 2013, 25, 302–312 CrossRef CAS PubMed.
- D. den Boer, M. Li, T. Habets, P. Iavicoli, A. E. Rowan, R. J. M. Nolte, S. Speller, D. B. Amabilino, S. de Feyter and J. A. A. W. Elemans, Nat. Chem., 2013, 5, 621–627 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, NMR spectra, and crystallographic data (CIF). CCDC 1040797. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02385h |
| This journal is © The Royal Society of Chemistry 2016 |