
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoselective palladium-catalyzed deprotonative arylation/[1,2]-Wittig rearrangement of pyridylmethyl ethers†
Feng
Gao‡
ab,
Byeong-Seon
Kim‡
b and
Patrick J.
Walsh
*b
aDepartment of Medicinal Plants, Agronomy College, Sichuan Agricultural University, 211, Huimin Rd, Wenjiang Region, Chengdu 611130, PR China
bRoy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 S, 34th St., Philadelphia, PA 19104-6323, USA. E-mail: pwalsh@sas.upenn.edu; Web: https://sites.google.com/site/titaniumupenn/ Fax: +1-215-573-6743
First published on 27th October 2015
Abstract
Control of chemoselectivity is one of the most challenging problems facing chemists and is particularly important in the synthesis of bioactive compounds and medications. Herein, the first highly chemoselective tandem C(sp3)–H arylation/[1,2]-Wittig rearrangement of pyridylmethyl ethers is presented. The efficient and operationally simple protocols enable generation of either arylation products or tandem arylation/[1,2]-Wittig rearrangement products with remarkable selectivity and good to excellent yields (60–99%). Choice of base, solvent, and reaction temperature play a pivotal role in tuning the reactivity of intermediates and controlling the relative rates of competing processes. The novel arylation step is catalyzed by a Pd(OAc)2/NIXANTPHOS-based system via a deprotonative cross-coupling process. The method provides rapid access to skeletally diverse aryl(pyridyl)methanol core structures, which are central components of several medications.
1. Introduction
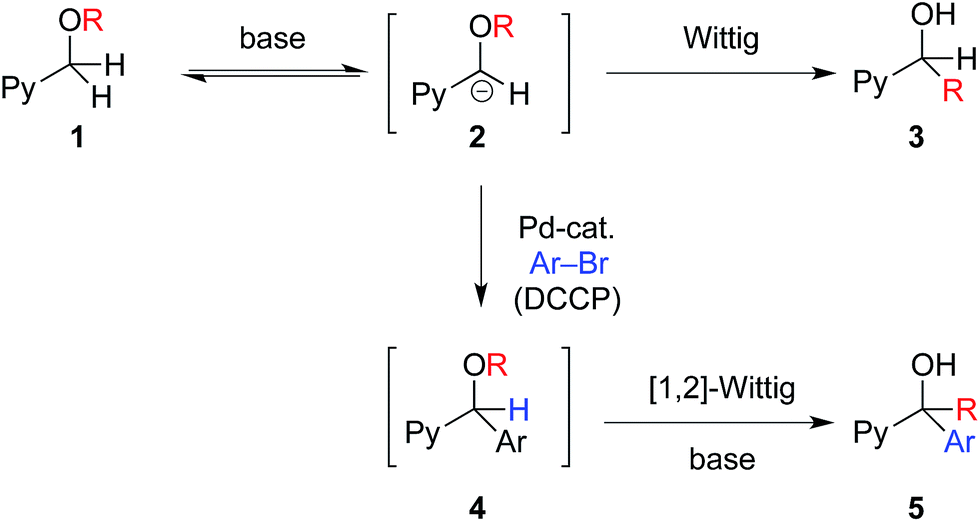
The synthesis of diverse chemical structures is an important endeavor in the discovery of new medications. Forging structural diversity from a common set of organic substrates is extremely desirable in this venture, but is quite difficult to achieve with high selectivity.1 This is particularly challenging when tandem reactions are employed, wherein several chemical transformations are performed without isolation of intermediates, addition of new reagents, or modification of reaction parameters.In considering the synthesis of a family of drug-like molecules based on the aryl(pyridin-2-yl)methanol core (I, Fig. 1), we envisioned a unified approach to access these important structural motifs based on a non-traditional umpolung approach.2 Our strategy (Fig. 2) entailed a palladium-catalyzed deprotonative cross-coupling process (DCCP) with pyridylmethyl ethers 1 and aryl bromides to generate arylated secondary ethers 4 while the tertiary alcohols 5 would be accessed via a tandem DCCP/[1,2]-Wittig rearrangement.
 | ||
| Fig. 1 Structures of drugs derived from the aryl(pyridin-2-yl) core. | ||
 | ||
| Fig. 2 Chemoselective [1,2]-Wittig rearrangement to give 3, arylation to give 4, and tandem arylation/[1,2]-Wittig rearrangement to afford 5. | ||
Upon examination of the reactions in Fig. 2, several challenges are evident. Reversible deprotonation of ether 1 generates the intermediate carbanion 2.3 α-Alkoxy carbanions are known to undergo [1,2]-Wittig rearrangements to generate alcohols 3.4 If anion 2 could be intercepted by a palladium catalyst via rapid transmetallation,5 arylation may outcompete the [1,2]-Wittig rearrangement of 2 to generate the desired aryl(pyridin-2-yl)methyl ether core, 4. Compound 4, possessing a more acidic benzylic hydrogen than 2, may undergo deprotonation under the basic reaction conditions followed by [1,2]-Wittig rearrangement to form tertiary alcohols 5. Because structures 4 and 5 are both valuable intermediates to furnish bioactive molecules and commercialized drugs (Fig. 1), conditions that inhibit and facilitate the tandem [1,2]-Wittig rearrangement of intermediate 4 are crucial for obtaining high selectivity and for the realization of this strategy. If the selectivity could be controlled in the DCCP/[1,2]-Wittig rearrangement in Fig. 2, the core structures of the antihistamines doxylamine,6 carbinoxamine,7 bepotastine,8 triprolidine9 (Fig. 1) and related pyridines10 could be accessed followed by further organic transformations.
Herein we disclose general and highly selective methods for the arylation of pyridylmethyl ethers 1 to give 4 as well as the tandem arylation/[1,2]-Wittig rearrangement to furnish tertiary alcohols 5.
2. Results and discussion
2.1. Optimization of reactions
Given the importance of pyridines in medicinal chemistry and materials science,11 it is not surprising that palladium catalyzed C–H functionalization reactions involving benzylic arylation of pyridines and derivatives have attracted significant attention.12 Difficulties in the direct benzylic arylation of 2-substituted pyridines, however, have been encountered and are attributed to the ability of pyridyl nitrogens to act as ligands.12j,13 Successful strategies to circumvent this potential problem include addition of Lewis acids to bind the pyridyl nitrogen and increase the reactivity of the benzylic C–H's.12j Notably, acceleration of palladium catalyzed reactions of pyridine derivatives by addition of Lewis acids has been described by Nolan,14 Hartwig,15 and Trost.16 Other workarounds include use of substrates with directing groups, such as 2-(2-pyridyl)acetic acids,12h 2-(2-pyridyl)ethanols,12b pyridine N-oxides,12d,12f and N-iminopyridines.12e We previously used 2-benzyl pyridine (pKa = 28.3–28.7 in THF), where the benzylic hydrogens are estimated to be about 6 orders of magnitude more acidic than those in 2-picoline (pKa = 34 in THF).17 2-Benzyl pyridines are also expected to be at least four orders of magnitude more acidic than the pyridylmethyl ether substrates that are the focus of the current investigation.18 Over the last decade, direct C–H functionalization of benzylic ethers has been reported via cross dehydrogenative-coupling (CDC) reactions and photoredox-mediated reactions.19In the course of our studies20 in the DCCP of a variety of weakly acidic substrates, we discovered that palladium complexes of van Leeuwen's NIXANTPHOS ligand21 (Table 1) enable a wide variety of transformations whereas other ligands are much less effective. Thus, we employed a Pd(OAc)2 (5 mol%)/NIXANTPHOS (7.5 mol%) based catalyst to initiate the direct arylation of 2-pyridylmethyl ethyl ether 1a with bromobenzene in combination with 6 bases [LiO–tBu, NaO–tBu, KO–tBu, LiN(SiMe3)2, NaN(SiMe3)2, and KN(SiMe3)2]. Both LiO–tBu and NaO–tBu resulted in no product while KO–tBu afforded a low yield of the arylation/[1,2]-Wittig rearrangement product. Interestingly, LiN(SiMe3)2 generated the arylation/[1,2]-Wittig rearrangement product 5aa exclusively in 48% assay yield (AY, entry 1). Although NaN(SiMe3)2 gave a mixture of arylation 4aa and arylation/[1,2]-Wittig rearrangement 5aa in 25 and 38% AY, respectively (entry 2), KN(SiMe3)2 afforded traces of 4aa and the arylation/[1,2]-Wittig rearrangement product 5aa in 33% AY (entry 3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | M | Solventb | Time (h) | Temp. (°C) | 4aa (%) | 5aa (%) |
| a Reaction conditions: 1a (0.1 mmol), Ph–Br (0.12 mmol), base (0.3 mmol), Pd(OAc)2/NIXANTPHOS (5 mol%/7.5 mol%), solvent (1.0 mL). b Tetrahydrofuran (THF), 1,4-dioxane (dioxane), cyclopentyl methyl ether (CPME), 1,2-dimethoxyethane (DME). c Yield determined by 1H NMR spectroscopy of the crude reaction mixture. d Isolated yield. e Pd(OAc)2/NIXANTPHOS (2.5 mol%/3.75 mol%). | ||||||
| 1 | Li | THF | 12 | 45 | 0 | 48 |
| 2 | Na | THF | 12 | 45 | 25 | 38 |
| 3 | K | THF | 12 | 45 | Trace | 33 |
| 4 | Na | Dioxane | 12 | 45 | 31 | 17 |
| 5 | Na | CPME | 12 | 45 | 33 | 65 |
| 6 | Na | DME | 12 | 45 | 64 | 30 |
| 7 | Na | DME | 12 | 23 | 95 | 0 |
| 8 | Li | Dioxane | 12 | 45 | 16 | 15 |
| 9 | Li | DME | 12 | 45 | 68 | 25 |
| 10 | Li | CPME | 12 | 45 | 38 | 56 |
| 11 | Li | CPME | 24 | 45 | 0 | 85d |
| 12 | Li | CPME | 24 | 45 | 0 | 83 |
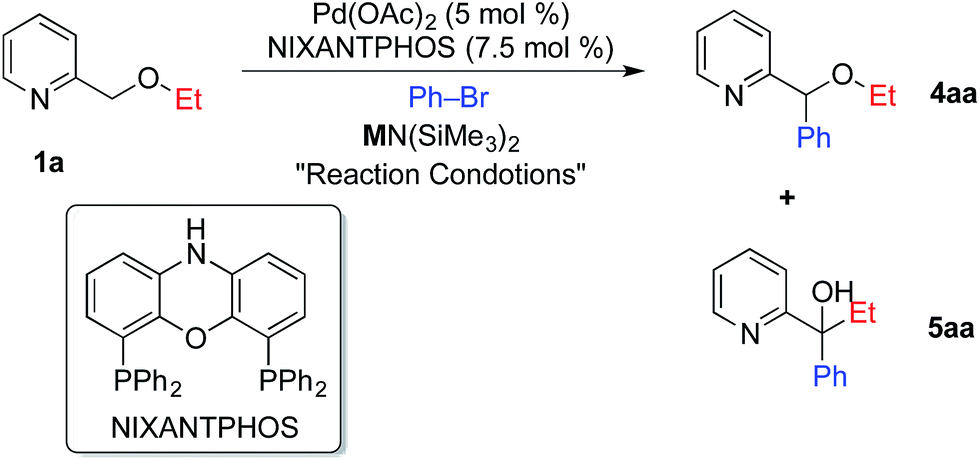
Given the higher yield afforded by NaN(SiMe3)2, we set out to examine different solvents with this base. Employing NaN(SiMe3)2 at 45 °C in dioxane, CPME and DME (entries 4–6) resulted in assay yields of up to 64% arylation (4aa) and 30% arylation/[1,2]-Wittig rearrangement (5aa) in DME. To our surprise, lowering the temperature to 23 °C in DME to minimize the [1,2]-Wittig rearrangement actually prevented it, providing the arylation product in 95% isolated yield (entry 7). With a highly selective protocol for the arylation in place, we desired to perform the tandem arylation/[1,2]-Wittig rearrangement.
The [1,2]-Wittig rearrangement of α-alkoxy carbanions has attracted much attention.22 Despite significant interest, relatively harsh reaction conditions are still the norm.23 There are only a few examples of the [1,2]-Wittig rearrangement in tandem reactions. Recently, Wolfe and co-workers introduced highly stereoselective tandem [1,2]-Wittig rearrangement/aldol or Mannich reactions.24 Dudley and co-workers reported pyridine-directed organolithium additions to enol ethers. The resulting intermediates then undergo anionic rearrangements to afford tertiary pyridyl carbinols.25 These examples indicate that the use of α-alkoxy carbanions in tandem reactions can be an effective strategy to generate multiple C–C bonds and forge complex molecular structures.
To develop the tandem arylation/[1,2]-Wittig rearrangement process, we focused on LiN(SiMe3)2 base in dioxane, DME and CPME (entries 8–10). Of these solvents, CPME gave the best results (4aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5aa, 38:56), but exhibited incomplete [1,2]-Wittig rearrangement after 12 h (entry 10). With a 24 h reaction time, the tandem arylation/[1,2]-Wittig rearrangement product 5aa was isolated in 85% yield (entry 11). As shown in entry 12, the palladium/NIXANTPHOS loading could be lowered to 2.5 and 3.75 mol%, respectively, without a significant drop in yield (83%).
:5aa, 38:56), but exhibited incomplete [1,2]-Wittig rearrangement after 12 h (entry 10). With a 24 h reaction time, the tandem arylation/[1,2]-Wittig rearrangement product 5aa was isolated in 85% yield (entry 11). As shown in entry 12, the palladium/NIXANTPHOS loading could be lowered to 2.5 and 3.75 mol%, respectively, without a significant drop in yield (83%).
2.2. Arylation of 2-pyridylmethyl ethers
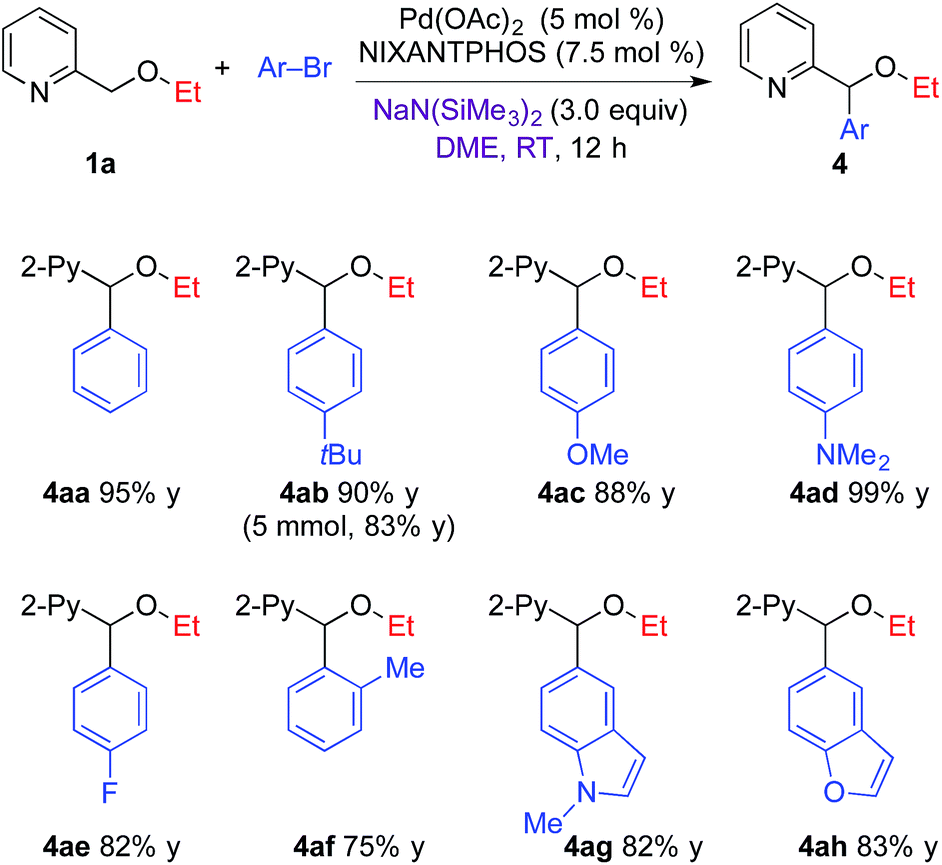
Based on the optimized arylation conditions (Table 1, entry 7), we examined a variety of aryl bromides in the DCCP (Table 2). In general, the direct arylation of 2-pyridylmethyl ethyl ether (1a) exhibited excellent selectivity and good yields with a range of aryl bromides. Aryl bromides bearing electron-donating groups, such as 4-tBu, 4-OMe, or 4-NMe2, resulted in 88–99% yields. 1-Bromo-4-fluorobenzene provided the product in 82% yield. Sterically hindered 2-bromotoluene coupled in 75% yield. Heterocyclic N-methyl 5-bromo indole and 5-bromo benzofuran underwent coupling to provide the products in 82–83% yield. It is noteworthy that when the reaction of 1a and 4-tert-butyl bromobenzene was conducted on a 5 mmol scale, the arylation product 4ab was isolated in 83% yield (1.12 g).| a Reaction conditions: 1a (0.2 mmol), Ar–Br (0.24 mmol), NaN(SiMe3)2 (0.6 mmol), Pd(OAc)2/NIXANTPHOS (5 mol%/7.5 mol%), DME (2.0 mL); isolated yield. |
|---|
|
The substrate scope of 2-pyridylmethyl ethers was next explored (Table 3) with electron-donating 4-bromo-N,N-dimethylaniline (entries 1–4) and with 1-bromo-4-fluorobenzene (entries 5–9). In general, all the 2-pyridylmethyl alkyl or aryl ethers exhibited 80–92% yield and excellent selectivity, with no [1,2]-Wittig product observed by 1H NMR of the crude reaction mixtures. 2-Pyridylmethyl alkyl ethers, including methyl ether (1b), cyclohexyl ether (1c) and tert-butyl ether (1d) showed good reactivity in the arylation reaction with 4-bromo-N,N-dimethylaniline and 1-bromo-4-fluorobenzene to furnish arylated ether products 4bd–4dd (83–92%) and 4be–4de (80–88%). In addition, 2-pyridylmethyl aryl ethers, including phenyl (1e), 4-fluorophenyl (1f) and 4-tert-butylphenyl ether (1g) coupled via DCCP to provide the corresponding products 4ed (82%), 4fe (82%) and 4ge (80%).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | R1 | Py | R2 | Product | Yieldb (%) |
| a Reaction conditions: 1 (0.2 mmol), Ar–Br (0.24 mmol), NaN(SiMe3)2 (0.6 mmol), Pd(OAc)2/NIXANTPHOS (5 mol%/7.5 mol%), DME (2.0 mL). b Isolated yield. | ||||||
| 1 | 1b | Me | 2-Py | NMe2 | 4bd | 83 |
| 2 | 1c | Cy | 2-Py | NMe2 | 4cd | 92 |
| 3 | 1d | tBu | 2-Py | NMe2 | 4dd | 88 |
| 4 | 1e | Ph | 2-Py | NMe2 | 4ed | 82 |
| 5 | 1b | Me | 2-Py | F | 4be | 85 |
| 6 | 1c | Cy | 2-Py | F | 4ce | 88 |
| 7 | 1d | tBu | 2-Py | F | 4de | 80 |
| 8 | 1f | 4-F-C6H4 | 2-Py | F | 4fe | 82 |
| 9 | 1g | 4-tBu-C6H4 | 2-Py | F | 4ge | 80 |

2.3. Tandem arylation/[1,2]-Wittig rearrangement
Having demonstrated the broad scope of the arylation, we then focused on the tandem arylation/[1,2]-Wittig rearrangement. Employing the optimized conditions in Table 1 (entry 12) with LiN(SiMe3)2 in CPME at 45 °C, reaction of 2-pyridylmethyl ethyl ether with aryl bromides was investigated (Table 4). Bromobenzene and 1-bromo-4-tert-butylbenzene furnished the desired products 5aa and 5ab in 85 and 78% yields, respectively. Aryl bromides bearing electron-donating 4-OMe and 4-NMe2 groups led to the coupled/rearranged products 5ac and 5ad in 75 and 72% yield, respectively. Aryl bromides with electron-withdrawing substituents, such as 4-F, 4-Cl and 3-CF3, were well tolerated, providing products 5ae, 5ai and 5aj in 78–88% yields. Heterocyclic 5-bromo-1-methyl-1H-indole and 5-bromobenzofuran, also gave the compounds 5ag and 5ah in 72 and 70% yield. When the reaction of 1a and 1-bromo-4-fluorobenzene was performed on 5 mmol scale with LiN(SiMe3)2 in CPME, the arylation/[1,2]-Wittig rearrangement product 5ae was isolated in 74% yield (0.86 g). 3-Bromophenol was also examined, however, analysis of 1H NMR of the crude reaction mixtures showed no sign of arylation product or tandem product. Less than 10% of the pyridylmethyl ether 1a was recovered, indicating decomposition of starting materials and/or products.| a Reaction conditions: 1a (0.2 mmol), Ar–Br (0.24 mmol), LiN(SiMe3)2 (0.6 mmol), Pd(OAc)2/NIXANTPHOS (2.5 mol%/3.75 mol%), CPME (2.0 mL); isolated yields. |
|---|

|
The scope in the migrating O–R group in the tandem arylation/[1,2]-Wittig rearrangement was investigated with various aryl groups. The 2-pyridylmethyl ethers with primary migrating groups (Table 4), methyl, secondary, and tertiary alkyl ethers (Table 5, entries 1–6) gave arylation/[1,2]-Wittig rearranged products in 60–85% yield. To examine selectivity in the oxidative addition step (aryl bromide vs. chloride) 2-pyridylmethyl tert-butyl ether was coupled with 1-bromo-4-chlorobenzene (entry 6). The arylation/[1,2]-Wittig rearrangement product 5di was obtained in 60% isolated yield, despite the known ability of the catalyst to activate aryl chlorides at rt.20p Amino ethyl-containing R groups also migrated, giving the desired amines in 64–74% yield (entries 7–10).
|
|
||||
|---|---|---|---|---|
| Entry | R | Ar | Product | Yield (%) |
| a Reaction conditions: 1 (0.2 mmol), Ar–Br (0.24 mmol), LiN(SiMe3)2 (0.6 mmol), Pd(OAc)2/NIXANTPHOS (2.5 mol%/3.75 mol%), CPME (2.0 mL); isolated yields; no arylated ethers observed by 1H NMR of crude reaction mixture. b 45 °C. | ||||
| 1b | Me | 4-C6H4-NMe2 | 5bd | 72 |
| 2b | Me | 4-C6H4-F | 5be | 75 |
| 3 | Cy | 4-C6H4-NMe2 | 5cd | 82 |
| 4 | Cy | 4-C6H4-F | 5ce | 85 |
| 5 | tBu | 4-C6H4-NMe2 | 5dd | 65 |
| 6 | tBu | 4-C6H4-Cl | 5di | 60 |
| 7 | CH2CH2NMe2 | C6H5 | 5ha | 74 |
| 8 | CH2CH2NMe2 | 4-C6H4-Cl | 5hi | 74 |
| 9 | CH2CH2N(CH2)4 | C6H5 | 5ia | 64 |
| 10 | CH2CH2N(CH2)4 | 4-C6H4-Me | 5ik | 71 |

2.4. Arylation of 4-pyridylmethyl ethers
Our focus on the arylation and tandem arylation/[1,2]-Wittig rearrangement of 2-pyridylmethyl ethers stems from their utility in the preparation of biologically active compounds (Fig. 1). At this juncture, we were curious if the 2-pyridyl group was required for the deprotonation and arylation outlined above. To probe this question, as well as broaden the scope of our coupling chemistry, we examined reactions of 4-pyridylmethyl alkyl ethers (1j and 1k, Table 6) with various aryl bromides.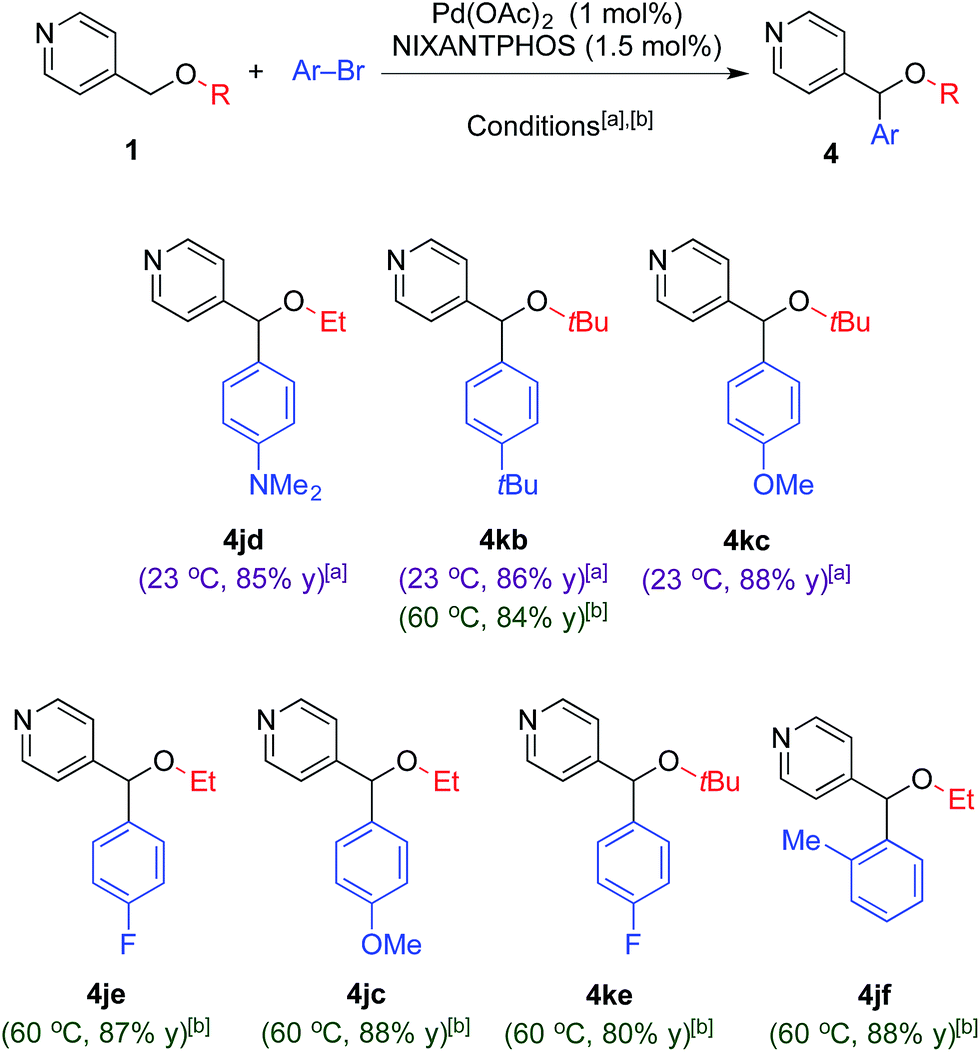
Under our standard arylation conditions [NaN(SiMe3)2 in DME at 23 °C], arylation of 4-pyridylmethyl alkyl ethers (1j and 1k) with aryl bromides provided the arylated products 4jd, 4kb and 4kc in 85–88% yield (Table 6) demonstrating that chelation is not a prerequisite to deprotonation or arylation. The increased reactivity of the 4-pyridyl derivatives over the 2-pyridyl analogues enabled us to reduce the palladium loading to 1 mol% in these cases.
To our surprise, conducting the reaction under the tandem arylation/[1,2]-Wittig rearrangement conditions [LiN(SiMe3)2 in CPME at 60 °C] did not provide the tandem arylation/rearranged products 5, but instead furnished the arylated ether products in 80–88% yield. While the tandem arylation/[1,2]-Wittig reaction of 1j with LiN(SiMe3)2 in CPME at 60 °C resulted in arylation product 4je in Table 6, the same condition at 80 °C for 24 h showed 10% of tandem arylation/[1,2]-Wittig rearrangement product and 72% of arylation product 4je. Higher reaction temperatures resulted in decomposition. These results suggest that [1,2]-Wittig rearrangement of 4-pyridylmethyl ethers is possible, but require higher activation energy. This difference in reactivity between 2- and 4-pyridylmethyl ethers most likely stems from the increased stability of the deprotonated 4-pyridylmethyl ethers. We are aware of a few examples of [1,2]-Wittig rearrangements of 4-pyridyl methyl ethers in the literature.26 The mechanism of [1,2]-Wittig rearrangement proceeds via radical intermediates. The radical intermediates derived from 4-pyridylmethyl ethers are expected to be higher in energy than those generated from 2-pyridylmethyl ethers.27
2.5. Comparative studies
While the scope of the substrates in Tables 2–6 is very good, some substrates were not compatible with the optimized conditions. This occurred when the O–R substituent was allyl or benzyl (Scheme 1A). These substrates underwent [1,2]-Wittig rearrangement faster than arylation, highlighting the fine line in chemoselectivity between the [1,2]-Wittig rearrangement of the carbanion 2 relative to transmetallation to palladium (Fig. 2). Additionally, 2-pyridylmethyl aryl ethers did not undergo the [1,2]-Wittig rearrangement. These observations are in accord with the mechanism of [1,2]-Wittig rearrangement, which proceeds via free radical intermediates.22a The general order of reactivity of migrating groups in the Wittig rearrangement is dictated by the stability of the corresponding radicals: allyl ∼ benzyl > alkyl > aryl.28 | ||
| Scheme 1 Probing the order of the tandem arylation/[1,2]-Wittig rearrangement. (A) [1,2]-Wittig rearrangement of allyl and benzyl ethers is faster than Pd-catalyzed arylation reactions. (B) [1,2]-Wittig rearrangement product does not undergo arylation. (C) [1,2]-Wittig rearrangement takes place in the absence of the Pd-catalyst. | ||
To probe the order of the reactions leading to the tertiary alcohols 5, (pyridin-2-yl)propan-1-ol 3a was prepared by [1,2]-Wittig rearrangement of 1a with n-BuLi in THF at −40 °C.4d When alcohol 3a was subjected to our arylation conditions, no arylation product was observed (Scheme 1B) and starting material was recovered. In contrast, ether 4ad underwent [1,2]-Wittig rearrangement upon exposure to LiN(SiMe3)2 in the absence of the Pd(NIXANTPHOS)-based catalyst in 80% yield (Scheme 1C). Thus, the [1,2]-Wittig rearrangement does not require palladium to occur. Taken together, these results suggest that arylation occurs before [1,2]-Wittig rearrangement.
2.6. Impact of Li and Na on [1,2]-Wittig rearrangements
[1,2]-Wittig rearrangements are known to be influenced by combinations of alkali metals and solvents.23d,29 Although a full mechanistic study of the [1,2]-Wittig rearrangement is beyond the scope of this manuscript, we hoped to gain some insight into the reactivity differences in the [1,2]-Wittig rearrangement with lithium and sodium silylamide bases. Thus, we decided to explore the rearrangement of isolated arylation product 4ab (Table 7). In this study, 2 bases [LiN(SiMe3)2 and NaN(SiMe3)2], 2 solvents [DME and CPME], and 2 additives [12-crown-4 and 15-crown-5] were examined. Reactions were conducted at 45 °C to give higher amounts of [1,2]-Wittig rearrangement product than reactions conducted at room temperature (as in Tables 1–3). Reactions were intentionally quenched before completion to assess reaction progress. The quantities of starting materials and products were determined by 1H NMR integration of reaction mixtures against an internal standard (dibromomethane, see ESI† for details).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | M | Additive | 4ab (%) | 5ab (%) |
| a Reaction conditions: 4ab (0.05 mmol), MN(SiMe3)2 (0.075 mmol), additive (0.075 mmol) in solvent (0.25 mL) at 45 °C. b Yield determined by 1H NMR spectroscopy of the crude reaction mixture. | |||||
| 1 | CPME | Li | — | 37 | 64 |
| 2 | CPME | Na | — | 24 | 75 |
| 3 | DME | Li | — | 72 | 27 |
| 4 | DME | Na | — | 85 | 9 |
| 5 | CPME | Li | 12-Crown-4 | 53 | 47 |
| 6 | CPME | Na | 12-Crown-4 | 90 | 7 |
| 7 | CPME | Na | 15-Crown-5 | 86 | 13 |

The results of this study are illustrated in Table 7. Reactions in CPME promoted by LiN(SiMe3)2 and NaN(SiMe3)2 undergo relatively fast [1,2]-Wittig rearrangements at 45 °C (entries 1 and 2). In DME both bases are less capable of promoting the rearrangement, but NaN(SiMe3)2 is significantly slower than LiN(SiMe3)2 (9 vs. 27% [1,2]-Wittig rearrangement product, entries 3 and 4, respectively). This result suggested that coordinating DME retards the rearrangement with the sodium counterion more so than with the lithium counterion. To probe this hypothesis, reactions were conducted in the presence of 12-crown-4 and 15-crown-5 in the less coordinating CPME. The impact of 12-crown-4/CPME on the LiN(SiMe3)2 reaction is a slight decrease in the conversion (47 vs. 64%, entry 5 vs. 1). In contrast, crown ethers 12-crown-4 and 15-crown-5 strongly inhibited the [1,2]-Wittig rearrangement with NaN(SiMe3)2, resulting in only 7 and 13% product formation (entries 6–7, respectively) compared to 75% in CPME in the absence of crown ether (entry 2). Thus, we conclude that coordinating solvent (DME) and coordinating additives (12-crown-4 and 15-crown-5) both inhibit the reaction with the sodium base to a much greater extent than with the lithium base. These observations are consistent with the results in Table 1.
3. Conclusions
In summary, we have developed a unified approach for the synthesis of structurally diverse pyridines, which are among the most important heterocycles and are found in many natural products and pharmaceutically relevant molecules (Fig. 1). The key to achieving skeletal diversity from a single starting material is to affect either the arylation or tandem arylation/[1,2]-Wittig rearrangement with high selectivity. Despite the remarkable similarity of the reaction parameters for these processes, both being promoted by silylamide bases and a (NIXANTPHOS) Pd-based catalyst, we have successfully identified conditions to strongly favor either reaction. The exceptional selectivity between secondary pyridyl ethers and their rearranged tertiary alcohol isomers is govern by the choice of main group metal (Na vs. Li), solvent, and temperature. We anticipate that these practical methods will be applicable for the synthesis of a broad array of pyridyl-containing ethers and alcohols.Acknowledgements
Financial support for this work was provided by NIH/NIGMS (GM 104349) and the National Science Foundation (CHE-1464744). F. G. thanks the China Scholarship Counsel for fellowships.Notes and references
- B. M. Trost, Science, 1983, 219, 245–250 CAS.
- (a) D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239–258 CrossRef; (b) J. S. Johnson, Angew. Chem., Int. Ed., 2004, 43, 1326–1328 CrossRef CAS PubMed; (c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- (a) I. Suzuki, Pharm. Bull., 1956, 4, 211–216 CrossRef CAS PubMed; (b) I. Suzuki, Pharm. Bull., 1956, 4, 479–482 CrossRef CAS PubMed.
- (a) P. Schorigin, Chem. Ber., 1924, 57, 1634–1637 CrossRef; (b) G. Wittig and L. Löhmann, Liebigs Ann. Chem., 1942, 550, 260–268 CrossRef CAS; (c) Y. Makisumi and S. Notzumoto, Tetrahedron Lett., 1966, 7, 6393–6397 CrossRef; (d) J. F. Garst and C. D. Smith, J. Am. Chem. Soc., 1976, 98, 1526–1537 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed.
- N. Sperber and D. Papa, J. Am. Chem. Soc., 1949, 71, 887–890 CrossRef CAS PubMed.
- V. Barouh, H. Dall, D. Patel and G. Hite, J. Med. Chem., 1971, 14, 834–836 CrossRef CAS PubMed.
- K. A. Lyseng-Williamson, Drugs, 2010, 70, 1579–1591 CrossRef CAS PubMed.
- S. A. Benezra and C.-H. Yang, Anal. Profiles Drug Subst., 1979, 8, 509–528 CAS.
- (a) V. Bonnet, F. Mongin, F. Trécourt, G. Breton, F. Marsais, P. Knochel and G. Quéguiner, Synlett, 2002, 1008–1010 CrossRef CAS; (b) Y. Ducharme, R. W. Friesen, M. Blouin, B. Côté, D. Dubé, D. Ethier, R. Frenette, F. Laliberté, J. A. Mancini, P. Masson, A. Styhler, R. N. Young and Y. Girard, Bioorg. Med. Chem. Lett., 2003, 13, 1923–1926 CrossRef CAS PubMed; (c) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed; (d) D. Ameen and T. J. Snape, MedChemComm, 2013, 4, 893 RSC.
- (a) J. A. Joule and K. Mills, Heterocyclic Chemistry, John Wiley & Sons, Chichester, U.K., 2010 Search PubMed; (b) A. S. Alexander, F. Pozharskii, A. R. Katritzky, Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry and Applications, John Wiley & Sons, Chichester, U.K., 2011 Search PubMed.
- (a) T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 2373–2375 CrossRef CAS PubMed; (b) T. Niwa, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2007, 46, 2643–2645 CrossRef CAS PubMed; (c) M. L. Hlavinka and J. R. Hagadorn, Organometallics, 2007, 26, 4105–4108 CrossRef CAS; (d) L.-C. Campeau, D. J. Schipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266–3267 CrossRef CAS PubMed; (e) J. J. Mousseau, A. Larivée and A. B. Charette, Org. Lett., 2008, 10, 1641–1643 CrossRef CAS PubMed; (f) D. J. Schipper, L.-C. Campeau and K. Fagnou, Tetrahedron, 2009, 65, 3155–3164 CrossRef CAS; (g) P. M. Burton and J. A. Morris, Org. Lett., 2010, 12, 5359–5361 CrossRef CAS PubMed; (h) R. Shang, Z.-W. Yang, Y. Wang, S.-L. Zhang and L. Liu, J. Am. Chem. Soc., 2010, 132, 14391–14393 CrossRef CAS PubMed; (i) G. Song, Y. Su, X. Gong, K. Han and X. Li, Org. Lett., 2011, 13, 1968–1971 CrossRef CAS PubMed; (j) S. Duez, A. K. Steib, S. M. Manolikakes and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 7686–7690 CrossRef CAS PubMed; (k) D. Zhao, M.-X. Zhu, Y. Wang, Q. Shen and J.-X. Li, Org. Biomol. Chem., 2013, 11, 6246–6249 RSC.
- (a) M. Onishi, K. Hiraki, K. Maeda and T. Itoh, J. Organomet. Chem., 1980, 188, 245–253 CrossRef CAS; (b) K. Isobe, Y. Nakamura and S. Kawaguchi, Bull. Chem. Soc. Jpn., 1989, 62, 1802–1808 CrossRef CAS; (c) B. Qian, S. Guo, J. Shao, Q. Zhu, L. Yang, C. Xia and H. Huang, J. Am. Chem. Soc., 2010, 132, 3650–3651 CrossRef CAS PubMed.
- J. K. Huang, C. M. Haar, S. P. Nolan, J. E. Marcone and K. G. Moloy, Organometallics, 1999, 18, 297–299 CrossRef CAS.
- Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7734–7735 CrossRef CAS PubMed.
- (a) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092–14093 CrossRef CAS PubMed; (b) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2009, 131, 12056–12057 CrossRef CAS PubMed.
- (a) R. R. Fraser, T. S. Mansour and S. Savard, J. Org. Chem., 1985, 50, 3232–3234 CrossRef CAS; (b) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- (a) F. G. Bordwell, M. van der Puy and N. R. Vanier, J. Org. Chem., 1976, 41, 1885–1886 CrossRef CAS; (b) F. G. Bordwell and T. Y. Lynch, J. Am. Chem. Soc., 1989, 111, 7558–7562 CrossRef CAS; (c) Substitution of OMe for H in 1-methylfluorene results in a change in the pKa of 0.5 units in ref. 19a.
- (a) M. Ghobrial, K. Harhammer, M. D. Mihovilovic and M. Schnürch, Chem. Commun., 2010, 46, 8836–8838 RSC; (b) S. J. Park, J. R. Price and M. H. Todd, J. Org. Chem., 2012, 77, 949–955 CrossRef CAS PubMed; (c) W. Muramatsu, K. Nakano and C.-J. Li, Org. Lett., 2013, 15, 3650–3653 CrossRef CAS PubMed; (d) W. Muramatsu and K. Nakano, Org. Lett., 2014, 16, 2042–2045 CrossRef CAS PubMed; (e) K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626–629 CrossRef CAS PubMed; (f) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS PubMed; (g) W. Muramatsu and K. Nakano, Org. Lett., 2015, 17, 1549–1552 CrossRef CAS PubMed.
- (a) G. I. McGrew, J. Temaismithi, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2010, 49, 5541–5544 CrossRef CAS PubMed; (b) G. I. McGrew, C. Stanciu, J. Zhang, P. J. Carroll, S. D. Dreher and P. J. Walsh, Angew. Chem., Int. Ed., 2012, 51, 11510–11513 CrossRef CAS PubMed; (c) J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2012, 134, 13765–13772 CrossRef CAS PubMed; (d) T. Jia, A. Bellomo, K. El Baina, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 3740–3743 CrossRef CAS PubMed; (e) S. Montel, T. Jia and P. J. Walsh, Org. Lett., 2013, 16, 130–133 CrossRef PubMed; (f) B. Zheng, T. Jia and P. J. Walsh, Org. Lett., 2013, 15, 1690–1693 CrossRef CAS PubMed; (g) B. Zheng, T. Z. Jia and P. J. Walsh, Org. Lett., 2013, 15, 4190–4193 CrossRef CAS PubMed; (h) G. Frensch, N. Hussain, F. A. Marques and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 2517–2524 CrossRef CAS PubMed; (i) F. Gao, B.-S. Kim and P. J. Walsh, Chem. Commun., 2014, 50, 10661–10664 RSC; (j) N. Hussain, G. Frensch, J. Zhang and P. J. Walsh, Angew. Chem., Int. Ed., 2014, 53, 3693–3697 CrossRef CAS PubMed; (k) M. Li, S. Berritt and P. J. Walsh, Org. Lett., 2014, 16, 4312–4315 CrossRef CAS PubMed; (l) M. Li, B. Yücel, J. Adrio, A. Bellomo and P. J. Walsh, Chem. Sci., 2014, 5, 2383–2391 RSC; (m) S. Montel, L. Raffier, Y. He and P. J. Walsh, Org. Lett., 2014, 16, 1446–1449 CrossRef CAS PubMed; (n) B. Yücel and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 3659–3667 CrossRef PubMed; (o) B. Zheng, T. Jia and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 165–178 CrossRef CAS PubMed; (p) J. Zhang, A. Bellomo, N. Trongsiriwat, T. Jia, P. J. Carroll, S. D. Dreher, M. T. Tudge, H. Yin, J. R. Robinson, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 6276–6287 CrossRef CAS PubMed; (q) J. Mao, K. Eberle, J. Zhang, C. Rodríguez-Escrich, Z. Xi, M. A. Pericàs and P. J. Walsh, Tetrahedron Lett., 2015, 56, 3604–3607 CrossRef CAS PubMed; (r) S.-C. Sha, J. Zhang and P. J. Walsh, Org. Lett., 2015, 17, 410–413 CrossRef CAS PubMed.
- (a) L. A. van der Veen, P. H. Keeven, G. C. Schoemaker, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 872–883 CrossRef CAS; (b) P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed.
- (a) T. Nakai and K. Mikami, Chem. Rev., 1986, 86, 885–902 CrossRef CAS; (b) J. A. Marshall, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 3, pp. 975–1014 Search PubMed; (c) K. Tomooka, H. Yamamoto and T. Nakai, Liebigs Ann., 1997, 1275–1281 CrossRef CAS; (d) J. P. Wolfe, in Comprehensive Organic Synthesis II, ed. P. Knochel, Elsevier, Amsterdam, 2014, pp. 1038–1072 Search PubMed; (e) D. Y. Curtin and S. Leskowitz, J. Am. Chem. Soc., 1951, 73, 2633–2636 CrossRef CAS; (f) D. Y. Curtin and W. R. Proops, J. Am. Chem. Soc., 1954, 76, 494–499 CAS; (g) J. Cast, T. S. Stevens and J. Holmes, J. Am. Chem. Soc., 1960, 3521–3527 RSC; (h) C. Vanderst, W. J. Heus and A. Haasjes, Recl. Trav. Chim. Pays-Bas, 1973, 92, 493–512 Search PubMed; (i) A. Miyashita, Y. Matsuoka, Y. Suzuki, K. Iwamoto and T. Higashino, Chem. Pharm. Bull., 1997, 45, 1235–1242 CrossRef CAS; (j) L. A. Paquette and Q. Zeng, Tetrahedron Lett., 1999, 40, 3823–3826 CrossRef CAS; (k) A. Garbi, L. Allain, F. Chorki, M. Ourévitch, B. Crousse, D. Bonnet-Delpon, T. Nakai and J.-P. Bégué, Org. Lett., 2001, 3, 2529–2531 CrossRef CAS PubMed; (l) O. Kitagawa, S.-I. Momose, Y. Yamada, M. Shiro and T. Taguchi, Tetrahedron Lett., 2001, 42, 4865–4868 CrossRef CAS; (m) I. Vilotijevic, J. Yang, D. Hilmey and L. A. Paquette, Synthesis, 2003, 1872–1874 CAS; (n) T. Hameury, J. Guillemont, L. van Hijfte, V. Bellosta and J. Cossy, Synlett, 2008, 2345–2347 CAS; (o) J. Yang and G. B. Dudley, J. Org. Chem., 2009, 74, 7998–8000 CrossRef CAS PubMed; (p) J. Yang, A. Wangweerawong and G. B. Dudley, Heterocycles, 2012, 85, 1603–1606 CrossRef CAS; (q) T. Nakano, T. Soeta, K. Endo, K. Inomata and Y. Ukaji, J. Org. Chem., 2013, 78, 12654–12661 CrossRef CAS PubMed.
- (a) S. L. Schreiber, M. T. Goulet and G. Schulte, J. Am. Chem. Soc., 1987, 109, 4718–4720 CrossRef CAS; (b) S. L. Schreiber and M. T. Goulet, Tetrahedron Lett., 1987, 28, 1043–1046 CrossRef CAS; (c) K. Tomooka, M. Kikuchi, K. Igawa, M. Suzuki, P. H. Keong and T. Nakai, Angew. Chem., Int. Ed., 2000, 39, 4502–4505 CrossRef CAS; (d) K. Tomooka, H. Yamamoto and T. Nakai, J. Am. Chem. Soc., 1996, 118, 3317–3318 CrossRef CAS; (e) R. E. Maleczka and F. Geng, Org. Lett., 1999, 1, 1115–1118 CrossRef CAS PubMed.
- (a) M. B. Bertrand and J. P. Wolfe, Org. Lett., 2006, 8, 4661–4663 CrossRef CAS PubMed; (b) N. C. Giampietro, J. W. Kampf and J. P. Wolfe, J. Am. Chem. Soc., 2009, 131, 12556–12557 CrossRef CAS PubMed; (c) N. C. Giampietro and J. P. Wolfe, Angew. Chem., Int. Ed., 2010, 49, 2922–2924 CrossRef CAS PubMed.
- J. Yang and G. B. Dudley, Adv. Synth. Catal., 2010, 352, 3438–3442 CrossRef CAS.
- (a) E. Fernández-Mateos, B. Maciá and M. Yus, Adv. Synth. Catal., 2013, 355, 1249–1254 CrossRef; (b) E. Fernández-Mateos, B. Maciá and M. Yus, Eur. J. Org. Chem., 2014, 2014, 6519–6526 CrossRef.
- (a) S. Bank and M. Gernon, J. Org. Chem., 1987, 52, 5105–5111 CrossRef CAS; (b) F. G. Bordwell, J. A. Harrelson Jr and A. V. Satish, J. Org. Chem., 1989, 54, 3101–3105 CrossRef CAS.
- H. Schäfer, U. Schöllkopf and D. Walter, Tetrahedron Lett., 1968, 9, 2809–2814 CrossRef.
- (a) R. E. Maleczka and F. Geng, J. Am. Chem. Soc., 1998, 120, 8551–8552 CrossRef CAS; (b) J. Barluenga, F. J. Fañanás, R. Sanz, C. Marcos and M. Trabada, Org. Lett., 2002, 4, 1587–1590 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, full characterization of new compounds, and calculated structures and energies. See DOI: 10.1039/c5sc02739j |
| ‡ F. G. and B.-S. K. contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |