
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic catalysis: cis-cyclopropanation of benzoxazoles†
Marta
Meazza
a,
Mark E.
Light
a,
Andrea
Mazzanti
b and
Ramon
Rios
 *a
*a
aFaculty of Natural & Environmental Sciences, University of Southampton, Highfield Campus, Southampton, SO17 1BJ, UK. E-mail: R.Rios-Torres@Southampton.ac.UK; Web: http://www.riosresearchgroup.com
bDepartment of Industrial Chemistry “Toso Montanari”, School of Science, University of Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
First published on 30th October 2015
Abstract
In the present paper we report our latest efforts in pushing the boundaries of synergistic catalysis. We propose the use of 3 different catalytic cycles working in concert for the formation of cis cyclopropane derivatives of benzoxazoles with excellent stereoselectivities. This is the proof of concept that synergistic catalysis could be successfully used in cascade reactions.
Introduction
In the last years huge efforts have been made to develop new activation modes in order to synthesize complex scaffolds in an enantiopure form. The most common approach has been the development of new catalysts to activate an “unreactive” compound A that will react with a highly reactive compound B to render C. However, recently a new approach pushes the boundaries of organic chemistry: synergistic catalysis, where the concurrent activation of both reactants allows the use of both unreactive substrates to form a new C–C bond. Earlier examples of synergistic catalysis merging transition metals and organocatalysts can be found in the pioneering works of Cordova who reported the α-allylic alkylation of unactivated aldehydes with allyl acetates by using both enamine and Pd chemistry.1 Later, Zhang reported an efficient asymmetric variant by using Cordova's synergistic strategy and Pd catalysts with C2-symmetrical chiral metallocene ligands.2 MacMillan and Sibi reported examples of asymmetric synergistic catalysis based on enamine catalysis and metal-bound electrophiles.3On the other hand, synergistic catalysis with α,β-unsaturated carbonyl compounds has attracted much recent interest. The works of Cordova in β-silylation, β-arylation or β-borylation by using iminium and transition-metal activation demonstrated the importance of this approach.4 In this area, List also reported excellent results for allylation and Overman rearrangement reactions by using phosphoric acid derivatives and transition-metal catalysts.5 Very recently, our research group became interested in synergistic catalysis.6
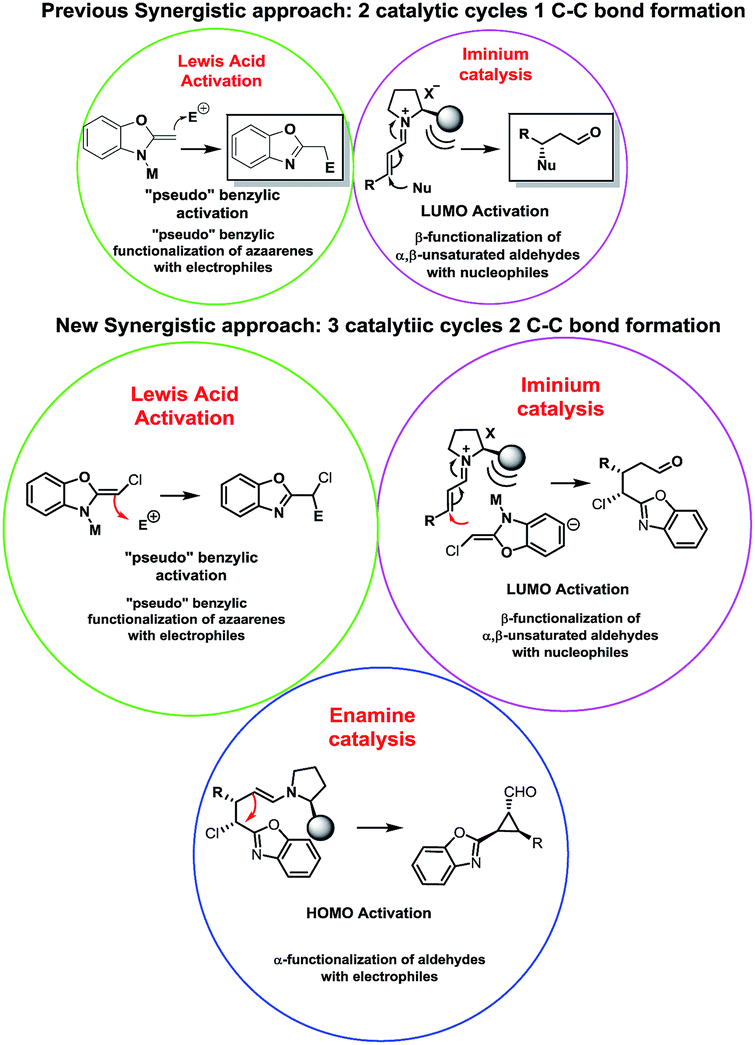
Our research focuses on the development of methodologies for the stereoselective synthesis of alkyl benzoxazoles based on the concurrent activation of alkyl benzoxazoles by metal-based Lewis acid catalysis (inspired by the pioneering work of Lam)7 and organocatalytic activation of several electrophiles.8 It is well known that one of the most important concerns of this approach is the possible autoquenching of both catalysts. However, by careful choice of the catalysts and the reaction conditions, we successfully developed the addition of alkylbenzoxazoles to Morita–Baylis–Hillman (MBH) carbonates and enals in good yields and stereoselectivities (Fig. 1).
 | ||
| Fig. 1 Proposed catalytic cycles. | ||
Despite significant progress and the advantages of using two unreactive substrates, synergistic catalysis presents a clear limitation in terms of range and scope: all these reactions generate only 1 C–C bond, making this approach more expensive and contrary to atom economy (2 catalyst for only 1 bond formation). This drawback attracted our interest and we propose to overcome this limitation by a carefully designed cascade reaction based on a synergistic approach. In our earlier examples we successfully combined metal Lewis activation of azaarenes with iminium catalysis, with good results but with some limitations such as moderate enantioselectivities and low diastereoselectivities due to an epimerization process.6b
We thought that a good option to develop a cascade reaction based on a synergistic approach and, at the same time, to limit the drawbacks of our previous work, should be the design of an organocascade reaction that could trap the enamine “in situ”, rendering a more complex scaffold with high stereoselectivity using the same catalytic system in order to push the boundaries of synergistic catalysis by forming 2 C–C bonds using 3 catalytic cycles.
Due to our experience in organocascade reactions, we envisioned that a cyclopropanation using chloro-alkylbenzoxazoles and enals will fulfil the requirements as a proof of concept of this approach and at the same time will allow us to synthesize an important scaffold in organic chemistry.
Cyclopropanes9 are structural motifs widely present in naturally occurring compounds, including terpenes, fatty acid metabolites, pheromones, etc.,10 and in pharmaceutical compounds with a large spectrum of biological properties.11 Cyclopropanes are also useful as synthetic intermediates that can undergo ring-opening or ring-expansion reactions to form a range of functionalized products.12
Asymmetric cyclopropanation reactions have fascinated organic chemists for decades and exciting advances have recently been reported; however, the development of new methodologies for the stereoselective synthesis of cyclopropanes is still a major challenge. In the literature, several approaches for their enantioselective synthesis are reported, for example, the venerable Simmons–Smith cyclopropanation,13 organometallic methodologies that use Cu, Rh and other transition-metal complexes14 or, with the advent of organocatalysis, several Michael-initiated ring-closing domino reactions, etc.15
Results and discussion
To show the viability of the present reaction initially we studied the addition of chloro-alkylbenzoxazole 1a with cinnamaldehyde. The proposed synergistic mechanism is shown in Fig. 2.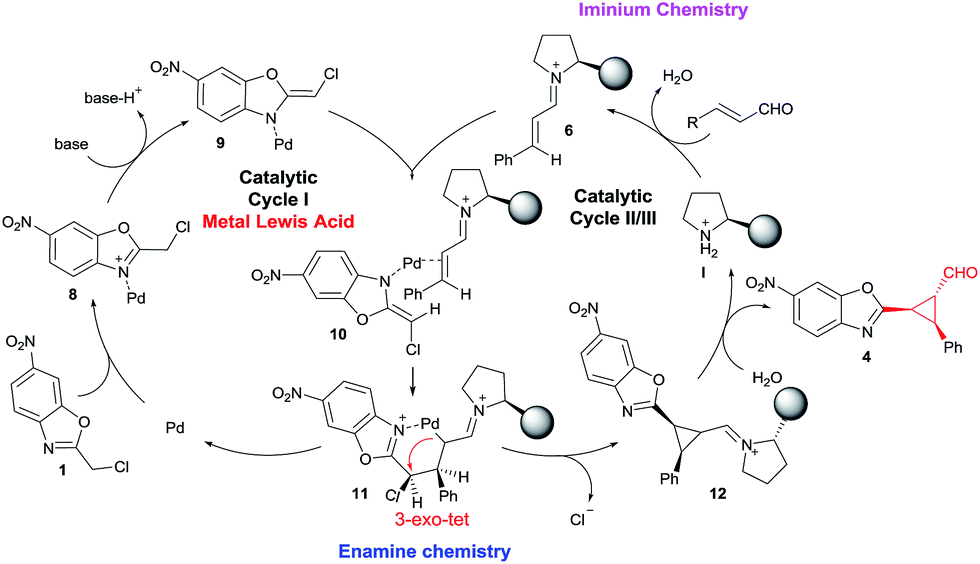 | ||
| Fig. 2 Proposed mechanism. | ||
As shown in Fig. 2, we envisioned that the metallic Lewis acid would interact with the alkylbenzoxazole 1 by coordinating to the nitrogen atom of 1 and increasing the acidity of the proton in the methylene position. The co-catalyzed mechanism involves the stereoselective addition of palladium enolate (9) to chiral iminium intermediate 6, followed by an intramolecular 3-exo-tet cyclization. As suggested by Lam,7 at first a coordination of the palladium to the benzoxazole occurs, followed by deprotonation by the external base leading to species 9. In parallel, iminium intermediate 6 is generated in situ by reactions of enals 2 and chiral amine catalyst I. Next a coordination between the Pd enolate and the double bond of the imine (as Cordova proved in similar synergistic Pd/secondary amine catalysed reactions) takes place to form the intermediate 10. Some stereochemical issues must be discussed here: first the coordination of the Pd to the double bond must be by the Si face of the iminium due the efficient shielding of the Re face of 6 by the bulky chiral group of I; in the case of the Pd enolate 9 the azaallyl ligand possesses E-stereochemistry to minimize steric interaction (as Lam suggested using other substituents) between the Cl substituent and the iminium. In this step the stereochemistry of the process is established: Si-facial nucleophilic attack at the β-carbon takes place, forming the 6 membered cycle C-bound-Pd intermediate 11. Next, a fast intramolecular alkylation between the new Pd enolate formed in α position of the iminium and the methylene chloride with inversion of configuration leads to the product 12 with the cis configuration between the aryl ring and the benzoxazole. Hydrolysis of 12 affords 4 and releases the catalysts, thus completing the catalytic cycle.
After a short screening we found that the best conditions were MeCN as a solvent, Pd(OAc)2 as a Lewis acid, Jorgensen–Hayashi catalyst (I) as secondary amine and 2,6-lutidine as stoichiometric base. To our delight the reaction rendered the final cyclopropane rings in excellent yields and stereoselectivities. The study of NMR spectra confirmed that the major diastereomer obtained in the cyclopropanation reaction has a cis disposition between the aryl ring and the benzoxazole substituent (see ESI†). This result adds, without any doubt, an important value to the present methodology. The synthesis of cis di(hetero)aromatic cyclopropanes via an intermolecular reaction is very challenging. This unusual cis configuration is difficult to obtain by any other enantioselective methodology that forms the C–C bond bearing both aryl groups. Only Mezzetti16 and Katzuki17 using ruthenium and iridium catalyst got the cis compound as a major diastereomer with excellent results in the reaction of styrenes with diazoacetates. Formally, the methodology reported in this paper is stereocomplementary with the MacMillan cyclopropanation that renders trisubstituted cyclopropanes in a trans configuration.18
With the optimized reaction conditions in hand, we investigated the substrate scope of the reaction with respect to enal derivatives. As shown in Scheme 1, the reactions afford the corresponding formyl cyclopropanes in good to excellent yields with moderate to good diastereoselectivities and excellent enantioselectivities in the major diastereomer. Remarkably, in all the examples the major diastereomer could be easily isolated by column chromatography while the other two diastereomers observed, usually are non-separable mixtures. All the diastereomeric ratios are from the final isolated products.
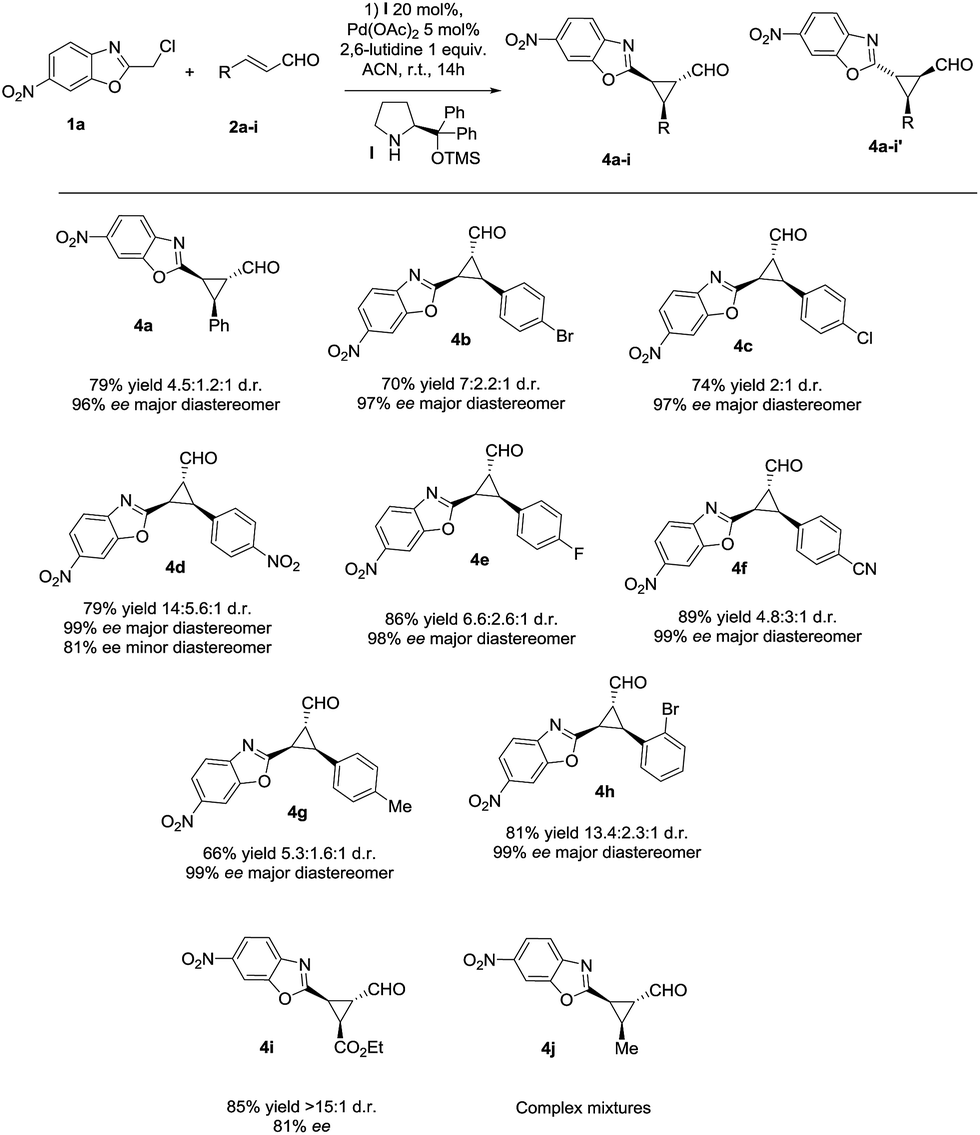 | ||
| Scheme 1 Scope of the reaction with several enals (d.r. determined by 1H NMR of the crude mixture, ee determined by chiral HPLC analysis of the isolated product). | ||
The reaction is compatible with several substituents in the aromatic ring. Electron-withdrawing groups like 4-NO2 (4d) or 4-CN (4f) render the final cyclopropane in good yields (79% and 63%), good diastereoselectivities and excellent enantioselectivities. Several halides were also tested in the reaction giving similar results, for example 4-Br (4b), 4-Cl (4c) and 4-F (4e) render the predicted formyl cyclopropane in good yields and diastereoselectivities (86–70% yield and up to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.) and exceptional enantioselectivities (up to 99% ee). When the most sterically demanding 2-Br cinnamaldehyde derivative was used, the formyl cyclopropane (4h) was obtained with good yields and excellent enantioselectivity in the major diastereomer but with lower diastereoselectivity. To our delight, when we tested the enal derived from glyoxylate, the reaction rendered the cyclopropane (4i) with good yield and excellent diastereoselectivity (only one diastereomer) but lower enantioselectivity. This could be due to an epimerization at C3 position of the product during the reaction.
:1 d.r.) and exceptional enantioselectivities (up to 99% ee). When the most sterically demanding 2-Br cinnamaldehyde derivative was used, the formyl cyclopropane (4h) was obtained with good yields and excellent enantioselectivity in the major diastereomer but with lower diastereoselectivity. To our delight, when we tested the enal derived from glyoxylate, the reaction rendered the cyclopropane (4i) with good yield and excellent diastereoselectivity (only one diastereomer) but lower enantioselectivity. This could be due to an epimerization at C3 position of the product during the reaction.
As shown in Fig. 3, when an ester moiety is placed in C3, this proton becomes quite acidic and could undergo epimerization with the base to form the most stable trans diastereomer, which would be stabilized by a coordination of the metal with the nitrogen from benzoxazole and the carbonyl compound from the ester. If this epimerization occurs, the outcome of the reaction will be one diastereomer with moderate enantioselectivity as observed by us. Finally, the only limitation of the present methodology is with the aliphatic enals that gave complex crude mixtures when subjected to the reaction conditions.
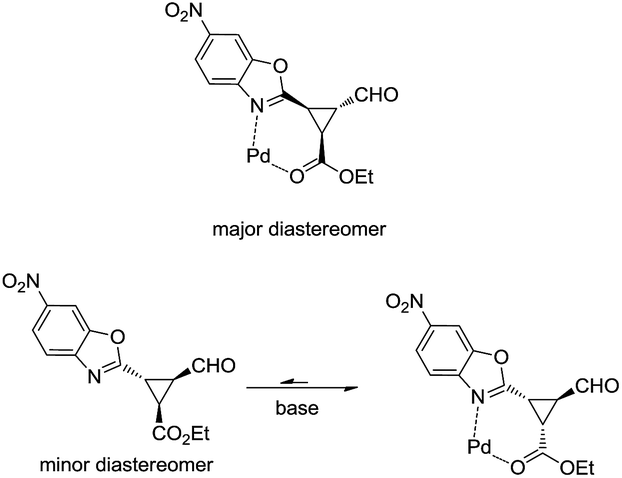 | ||
| Fig. 3 Proposed mechanism for epimerization of 4i. | ||
The relative configuration of the minor diastereomer was confirmed by X-ray crystallography (Fig. 4), while the relative configuration of the major diastereomer was inferred by NMR spectroscopy (see ESI†).
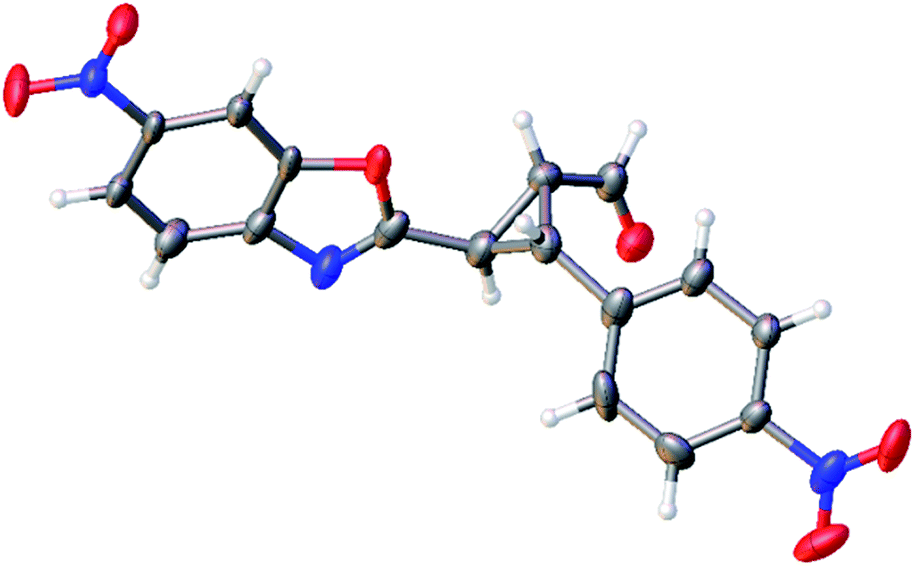 | ||
| Fig. 4 X-ray structure of compound 4d′ (minor diastereomer). The displacement ellipsoids are drawn at the 50% probability level.20 | ||
The absolute configuration of the two diastereomers was determined to be 1R,2R,3S for the major diastereomer 4d (using (S)-I as catalyst) and 1R,2R,3R for the minor diastereomer 4d′ (using (R)-I as catalyst) by TD-DFT simulation of the Electronic Circular Dichroism (ECD) spectra (Fig. 5 and ESI†).19 This absolute configuration and diastereoselectivity of the major diastereomer is in agreement with the mechanism proposed and with the previous works done with this type of catalysts, where the stereochemistry at β-position of the enal is perfectly controlled by catalyst (I).15 The relative and absolute configuration of the minor diastereomer could be rationalized by the reaction with the (Z)-palladium enolate.
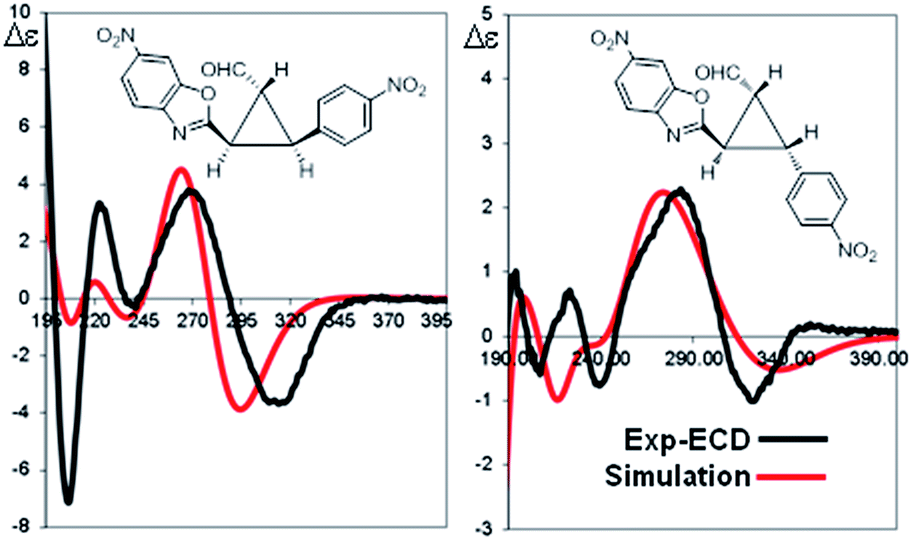 | ||
| Fig. 5 TDDFT simulation of the ECD spectra for the two diastereomers of 4d [(S)-catalyst] and 4d′ [(R)-catalyst]. Shown simulations were obtained with TD-DFT at the M06-2X/6-311++G(2d,p) level of theory. | ||
Next, we investigated the substrate scope of the reaction with respect to the benzoxazole derivative 1. Several benzoxazole derivatives were tested (Scheme 2) and the presence of electron-withdrawing groups in the azaarene ring was crucial for the reactivity. Cl-substituted benzoxazole rendered the final product 5a, 5e and 5f with good yields, good stereoselectivities and excellent enantioselectivities. When the benzoxazole had a –NO2 group at a different position (5b and 5d), slightly lower yields were observed but good diastereoselectivities and excellent enantioselectivities were obtained. Finally, changing the electron-withdrawing –NO2 group to an ester group rendered the final product 5c in excellent yield, good diastereoselectivity and excellent enantioselectivity. The only limitation, as previously noted in our previous reports on synergistic catalysis,6a,b is that an electron-withdrawing group must be present on the benzoxazole ring. Probably, the electron-withdrawing group decreases the pKa value of 1 and allows the deprotonation and the subsequent reaction.
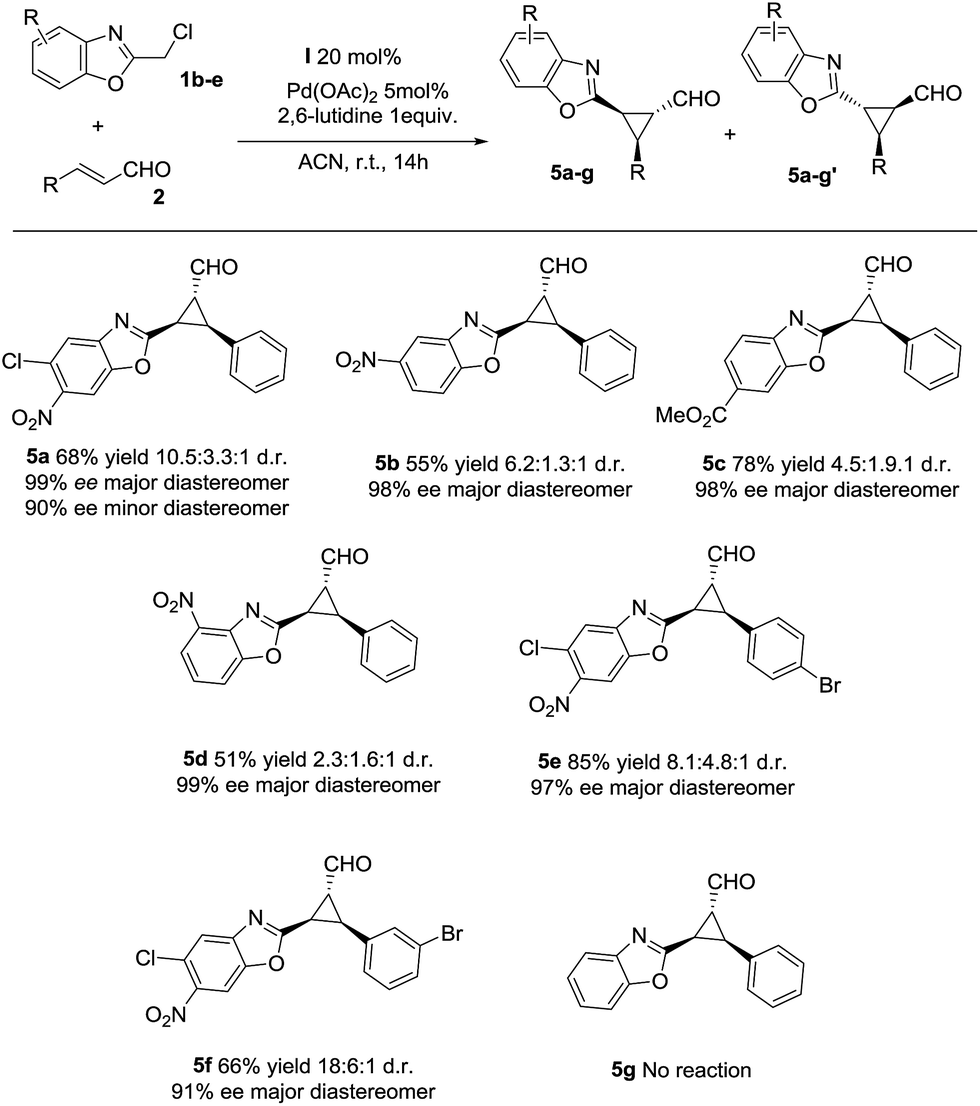 | ||
| Scheme 2 Scope of the reaction with several azaarenes (d.r. determined by 1H NMR of the crude mixture, ee determined by chiral HPLC analysis of the isolated product). | ||
Conclusions
In summary, we have demonstrated that synergistic catalysis can be applied to the development of a cascade reaction by developing a highly enantioselective cyclopropanation of enals with chloro-alkylbenzoxazoles. For the first time, the metal activation of azaarenes was combined with an organocatalytic cascade reaction, pushing the boundaries of this synergistic approach. Two different catalytic cycles, (i) the metallic Lewis acid activation of azaarenes and (ii) the secondary amine activation of enals, worked in accord to afford the final products in good yields, with excellent enantioselectivity and good diastereoselectivity. Moreover this reaction gives access to cis cyclopropanes that are difficult to obtain by any other method. The mechanistic studies, synthetic applications and development of new organocascade reactions based on this concept are currently ongoing in our laboratory.Acknowledgements
M.M. and R.R. acknowledge the European Regional Development Fund (ERDF) for co-financing the AI-CHEM-Chem project (No. 4061) through the INTERREG IV A France (Channel) – England cross-border cooperation Programme. M.M. M.E.L. and R.R. are grateful for EPSRC Core Capability Funding (EP/K039466/1).Notes and references
- (a) I. Ibrahem and A. Cordova, Angew. Chem., Int. Ed., 2006, 45, 1952 CrossRef CAS PubMed; for a pioneering examples in multicatalysis using Rh and Pd see: (b) M. Sawamura, M. Sudoh and Y. Ito, J. Am. Chem. Soc., 1996, 118, 3309 CrossRef CAS, using Eu and Al see: (c) G. M. Sammis, H. Danjo and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 9928 CrossRef CAS PubMed.
- X. Zhao, D. Liu, F. Xie, Y. Liu and W. Zhang, Org. Biomol. Chem., 2011, 9, 1871 CAS.
- See for example: (a) Q. Ding and J. Wu, Org. Lett., 2007, 9, 4959 CrossRef CAS PubMed; (b) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986 CrossRef CAS PubMed; (c) M. Ikeda, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2010, 49, 7289 CrossRef CAS PubMed; (d) A. Yoshida, M. Ikeda, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 592 CrossRef CAS PubMed; (e) M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124 CrossRef CAS PubMed; (f) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260 CrossRef CAS PubMed; (g) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed.
- (a) I. Ibrahem, P. Breistein and A. Cordova, Angew. Chem., Int. Ed., 2011, 50, 12036–12041 CrossRef CAS PubMed; (b) I. Ibrahem, G. Ma, S. Afewerki and A. Cordova, Angew. Chem., Int. Ed., 2013, 52, 878–882 CrossRef CAS PubMed; (c) I. Ibrahem, S. Santoro, F. Himo and A. Cordova, Adv. Synth. Catal., 2011, 353, 245–252 CrossRef CAS.
- (a) G. Jiang and B. List, Angew. Chem., Int. Ed., 2011, 50, 9471 CrossRef CAS PubMed; (b) G. Jiang, R. Halder, Y. Fang and B. List, Angew. Chem., Int. Ed., 2011, 50, 9752 CrossRef CAS PubMed; (c) S. Mukherjee and B. List, J. Am. Chem. Soc., 2007, 129, 11336 CrossRef CAS PubMed; (d) S. Liao and B. List, Angew. Chem., Int. Ed., 2010, 49, 628 CrossRef CAS PubMed; (e) G. Jiang and B. List, Adv. Synth. Catal., 2011, 353, 1667 CrossRef CAS; (f) G. Jiang and B. List, Chem. Commun., 2011, 47, 10022 RSC.
- (a) V. Ceban, P. Putaj, M. Meazza, M. B. Pitak, S. J. Coles, J. Vesely and R. Rios, Chem. Commun., 2014, 50, 660 RSC; (b) M. Meazza, V. Ceban, M. B. Pitak, S. J. Coles and R. Rios, Chem.–Eur. J., 2014, 20, 16853 CrossRef CAS PubMed.
- D. Best, S. Kujawa and H. W. Lam, J. Am. Chem. Soc., 2012, 134, 18193 CrossRef CAS PubMed . For an excellent review about the activation of azaarenes by metal see: D. Best and H. W. Lam, J. Org. Chem., 2014, 79, 831 CrossRef PubMed.
- For examples of our previous works in organocatalysis: (a) X. Companyo, A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyano and R. Rios, Chem. Commun., 2010, 46, 6953 RSC; (b) S. Cihalova, G. Valero, J. Schimer, M. Humpl, M. Dracinsky, A. Moyano, R. Rios and J. Vesely, Tetrahedron, 2011, 67, 8942 CrossRef CAS; (c) A. N. R. Alba, G. Valero, T. Calvet, M. Font-Bardia, A. Moyano and R. Rios, Chem.–Eur. J., 2010, 39, 9884 CrossRef PubMed; (d) X. Companyo, A. Moyano, A. Mazzanti, A. Janecka and R. Rios, Chem. Commun., 2013, 49, 1184 RSC.
- For an excellent review about cyclopropanation see: H. Pellissier, Tetrahedron, 2008, 64, 704 Search PubMed.
- (a) J. Salaun, Top. Curr. Chem., 2000, 207, 1 CrossRef CAS; (b) R. Faust, Angew. Chem., Int. Ed., 2001, 40, 2251 CrossRef CAS; (c) F. Gnad and O. Reiser, Chem. Rev., 2003, 103, 1603 CrossRef CAS PubMed; (d) W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef CAS; (e) Y. Nisii, N. Maruyama, K. Wakasugi and Y. Tanabe, Bioorg. Med. Chem., 2001, 9, 33 CrossRef; (f) J. Pietruszka, Chem. Rev., 2003, 103, 1051 CrossRef CAS PubMed.
- (a) H. W. Liu and C. T. Walsh, The Chemistry of the Cyclopropyl Group, ed. Z. Rappoport, John Wiley, New York, NY, 1997, p. 959 Search PubMed; (b) R. Csuk, M. J. Schabel and Y. von Scholz, Tetrahedron: Asymmetry, 1996, 7, 3505 CrossRef CAS; (c) M. Hogberg, P. Engelhardt, L. Vrang and H. Zhang, Bioorg. Med. Chem. Lett., 2000, 10, 265 CrossRef CAS PubMed; (d) D. K. Mohapatra and A. Datta, J. Org. Chem., 1998, 63, 642 CrossRef CAS PubMed.
- (a) H. N. C. Wong, M.-Y. Hon, C.-W. Tse, Y.-C. Yip, J. Tanko and T. Hudlicky, Chem. Rev., 1989, 89, 165 CrossRef CAS; (b) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (c) R. P. Wurz and A. B. Charette, Org. Lett., 2005, 7, 2313 CrossRef CAS PubMed; (d) Z. Zhang, Q. Zhang, S. Sun, T. Xiong and Q. Liu, Angew. Chem., 2007, 119, 1756 ( Angew. Chem., Int. Ed. , 2007 , 46 , 1726 ) CrossRef; (e) D. Agrawal and V. K. Yadav, Chem. Commun., 2008, 6471 RSC; (f) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (g) F. de Simone and J. Waser, Synthesis, 2009, 3353 CAS.
- (a) H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 6406 CrossRef; (b) H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1959, 81, 4256 CrossRef CAS.
- Selected examples of metal-catalyzed cyclopropanations: (a) A. B. Charette, C. Molinaro and C. Brochu, J. Am. Chem. Soc., 2001, 123, 12160 CrossRef CAS PubMed; (b) H. M. L. Davis, P. R. Bruzinski, D. H. Lake, N. Kong and M. Fall, J. Am. Chem. Soc., 1996, 118, 6897 CrossRef; (c) R. Rios, J. Liang, M.-C. Lo and G. C. Fu, Chem. Commun., 2000, 377 RSC; (d) D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726 CrossRef CAS.
- (a) I. Ibrahem, G.-L. Zhao, R. Rios, J. Vesely, H. Sunden, P. Dziedzic and A. Córdova, Chem.–Eur. J., 2008, 14, 7867 CrossRef CAS PubMed; (b) R. Rios, H. Sunden, J. Vesely, G.-L. Zhao, P. Dziedzic and A. Córdova, Adv. Synth. Catal., 2007, 349, 1028 CrossRef CAS; (c) H. Xie, L. Zu, H. Li, J. Wang and W. Wang, J. Am. Chem. Soc., 2007, 129, 10886 CrossRef CAS PubMed; (d) X. Companyo, A.-N. Alba, F. Cardenas, A. Moyano and R. Rios, Eur. J. Org. Chem., 2009, 3075 CrossRef CAS.
- S. Bachmann, M. Furler and A. Mezzetti, Organometallics, 2001, 20, 2102 CrossRef CAS.
- S. Kanchiku, H. Suematsu, K. Matsumoto, T. Uchida and T. Katzuki, Angew. Chem., Int. Ed., 2007, 46, 3889–3991 CrossRef CAS PubMed.
- R. K. Kunz and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3240 CrossRef CAS PubMed.
- For reviews see: (a) G. Bringmann, T. Bruhn, K. Maksimenka and Y. Hemberger, Eur. J. Org. Chem., 2009, 2717 CrossRef CAS; (b) T. D. Crawford, M. C. Tam and M. L. Abrams, J. Phys. Chem. A, 2007, 111, 12057 CrossRef CAS PubMed; (c) G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603 RSC; (d) A. Mazzanti and D. Casarini, WIREs Computational Molecular Science, 2012, 2, 613 CrossRef CAS.
- Single clear colourless fragment-shaped crystals of 4d′ were recrystallised from a mixture of TCM and hexane by slow evaporation. A suitable crystal (0.09 × 0.08 × 0.05 mm3) was selected and mounted on a MITIGEN holder in perfluoroether oil on a Rigaku AFC12 FRE-HF diffractometer. The crystal was kept at T = 100(2) K during data collection. Using Olex2 (Dolomanov et al., 2009), the structure was solved with the ShelXT (Sheldrick, 2008) structure solution program, using the Direct Methods solution method. The model was refined with version of ShelXL (Sheldrick, 2008) using Least Squares minimisation. Crystal data for C17H11N3O6 (4d′), Mr = 353.29, monoclinic, P21/c (no. 14), a = 9.9877(5) Å, b = 15.1541(7) Å, c = 10.2131(6) Å, β = 103.161(5)°, α = γ = 90°, V = 1505.20(14) Å3, T = 100(2) K, Z = 4, Z′ = 1, μ (MoKα) = 0.121, 14536 reflections measured, 3884 unique (Rint = 0.0709) which were used in all calculations. The final wR2 was 0.3428 (all data) and R1 was 0.1503 (I > 2(I)). Crystallographic data (excluding structure factors) for the structure 4d′ have been deposited with the Cambridge Crystallographic Data Centre with CCDC number 1060573.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1060573. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03597j |
| This journal is © The Royal Society of Chemistry 2016 |