
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA flexible approach to Pd-catalyzed carbonylations via aroyl dimethylaminopyridinium salts†
Jeffrey S.
Quesnel
 ,
Alexander
Fabrikant
and
Bruce A.
Arndtsen
*
,
Alexander
Fabrikant
and
Bruce A.
Arndtsen
*
Department of Chemistry, McGill University, 801 Sherbrooke St. W., Montreal, QC, Canada H3A 0B8. E-mail: bruce.arndtsen@mcgill.ca; Fax: +1-514-398-3797; Tel: +1-514-398-6999
First published on 30th September 2015
Abstract
4-Dimethylaminopyridine (DMAP) is shown to undergo Pd/PtBu3 catalyzed coupling with aryl halides and carbon monoxide to form electrophilic aroyl–DMAP salts. The reaction is easily scalable to prepare multigram quantities with low catalyst loadings, while the precipitation of these salts as they form leads to products with low impurities. These reagents rapidly react with a variety of nucleophiles, including those that contain potentially incompatible functional groups under standard carbonylative conditions.
Introduction
Activated carboxylic acid derivatives play a fundamental role in synthetic chemistry. Their use ranges from simple esterification and amidation reactions to Friedel–Crafts acylation, peptide coupling, heterocycle synthesis, polymer chemistry, or metal-catalyzed bond-forming reactions.1–5 Classic approaches to generate these reagents involve either the use of stoichiometric activating agents (e.g. DCC, HOBt, etc.), or, for even greater electrophilicity, the conversion of carboxylic acids into acyl halides. While effective, these can require large amounts of high energy, costly, or sometimes toxic activating agents (thionyl chloride, oxalyl chloride, or PCl3), and generate significant waste. These concerns have led to the search for more efficient methods to construct amides, esters, and related compounds.6An alternative platform to prepare carbonyl-containing products is through the palladium-catalyzed carbonylation of organic halides.7,8 Introduced over 40 years ago, these reactions provide a synthetic route to ketones, esters, amides, and related carboxylic acid derivatives using simple CO as the carbonyl unit. In spite of its atom economy, there remain a number of inherent drawbacks to carbonylations. For example, the putative palladium–aroyl intermediates are much less electrophilic than common acylating reagents such as acid chlorides, necessitating pressing reaction conditions even for relatively simple ester or amide syntheses.8 Reactions with weak or sterically encumbered nucleophiles are often unknown. Secondly, competitive and undesired side reactions, or catalyst poisoning, can occur with substrates containing functionalities that can be activated by transition metals. The often high catalyst loadings and coordinating ability of the carbonylated products also leads to practical concerns of cost, waste of precious metal, and product contamination with palladium. The latter can be a significant challenge in preparing APIs (active pharmaceutical ingredients).9
Some efforts to alleviate the scope limitations of carbonylations have been reported through the work of Buchwald, Skrydstrup, Manabe, Grushin, and others via the in situ formation of phenoxyesters,10 thioesters,11N-hydroxysuccinimido esters,12 and related derivatives13 as precursors to carbonyl-containing derivatives (Fig. 1a). Alternatively, we recently described the catalytic synthesis of acid chlorides from aryl halides, CO, and chloride salts (Fig. 1b).14,15 In contrast to the moderate reactivity of esters and thioesters, the high electrophilicity of acid chlorides allows the application of carbonylations to a range of nucleophiles. Nevertheless, a challenge encountered in this latter transformation is the reactivity of the acid chloride itself. Unlike most palladium-catalyzed coupling reactions, the acid chloride product is more reactive than the aryl halide substrate towards the catalyst, and its build-up strongly inhibits the reaction. Thus, heating, high pressures, or high catalyst loading are required to achieve good conversion. Acid chlorides are also sensitive reagents with similar physical properties (e.g. boiling and melting points) to the aryl halide starting materials, making isolation a challenge.
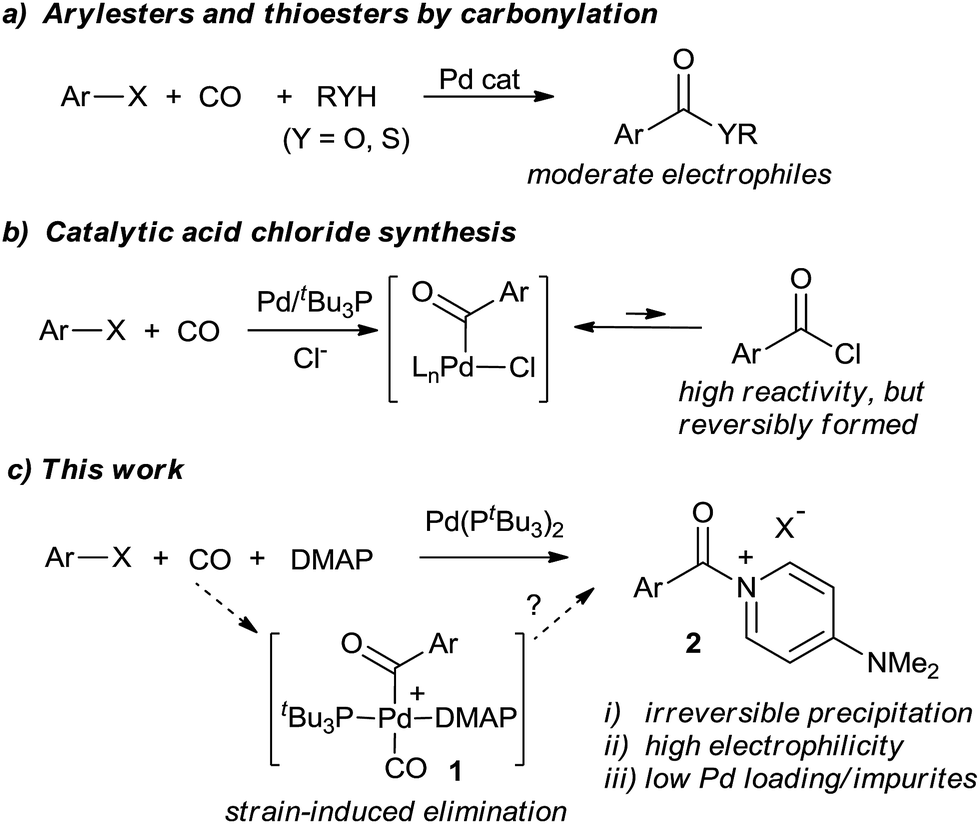 | ||
| Fig. 1 Palladium-catalyzed formation of active carboxylic acid derivatives via carbonylation. | ||
In considering the above, we became interested in designing a more efficient catalytic route to synthesize activated acylating reagents that is: (a) not inhibited by product build-up, and therefore can allow for rapid and efficient catalysis, (b) leads to the formation of easily isolated products, yet (c) creates potent acylating reagents with the reactivity of acid chlorides. We report herein the design of such a method via the use of 4-dimethylaminopyridine (DMAP, Fig. 1c). Acyl–DMAP reagents are potent electrophiles commonly generated in situ to facilitate nucleophilic attack, although these compounds are rarely synthesized as isolated products.16,17 The use of carbonylations to generate cationic products such as aroyl–DMAP salts has to our knowledge not been previously reported. The latter is likely due to the challenge of achieving reductive elimination of a cationic product from 1 relative to those with anionic nucleophiles (e.g. RO−, R2N−). As described below, the use of highly sterically encumbered palladium catalysts can provide a facile method to carbonylate aryl halides into aroylated–DMAP salts. These cationic products are easily separable, even during catalysis, which can therefore allow for a rapid reaction, low catalyst loadings, and generate activated carboxylic acid derivatives for subsequent reactions. This has allowed the application of Pd-catalyzed carbonylations to a range of new classes of nucleophilic substrates.
Results and discussion
Initial examination of this carbonylation involved the reaction of phenyl iodide, DMAP, and CO. Our postulate was the catalytic formation of acid chloride in the presence of DMAP would lead to the formation of the ionic 2a, and could drive the reaction forward under mild conditions. As anticipated from our previous report, the combination of Pd2dba3·CHCl3/PtBu3 with Bu4NCl as a chloride source did indeed allow the formation of 2a in modest yield (Table 1, entry 1).14a In attempting to improve the efficiency of this reaction, we questioned if stoichiometric chloride is required for this reaction, and if DMAP may instead itself bind to palladium for reductive elimination. The omission of chloride leads to minimal product formation with most of the catalysts examined (entries 2–13), likely due to the difficult reductive elimination step of a cationic product. This includes a number of ligands which are active in other palladium-catalyzed coupling (entries 3–6, 12) and carbonylation reactions (entries 8–11, 13).8,18 However, the use of the largest cone angle trialkyl phosphine, PtBu3, which would generate a highly sterically encumbered palladium–aroyl complex 1 for reductive elimination (vide infra), results in a dramatic increase in activity, and the near quantitative formation of DMAP salt 2a (entry 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | L | Yieldb (%) | Entry | L | Yieldb (%) |
| a PhI (102 mg, 0.50 mmol), DMAP (73 mg, 0.60 mmol), [Pd] = Pd2dba3·CHCl3 (13 mg, 25 μmol), L (50 μmol), CO (1 atm), THF (0.75 mL). b NMR yield of benzamide when quenched with BnNH2 (55 mg, 0.51 mmol), EtNiPr2 (100 μL, 0.57 mol). c 1.0 equiv. d Isolated yield of 2a. | |||||
| 1 | PtBu3/Bu4NClc | 33 | 8 | dppf | 3 |
| 2 | — | 3 | 9 | dcpe | 0 |
| 3 | PPh3 | — | 10 | dppp | 1 |
| 4 | P(o-tolyl)3 | 2 | 11 | dppb | 1 |
| 5 |
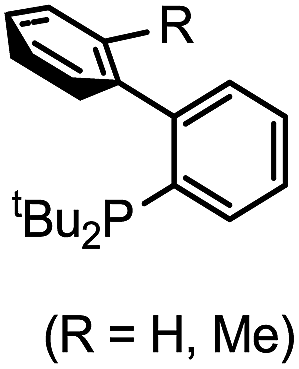
|
3 | 12 |
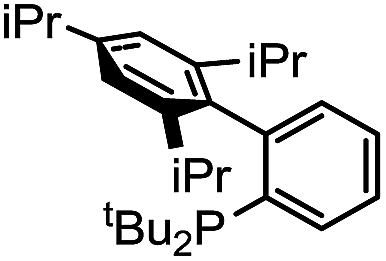
|
1 |
| 6 |
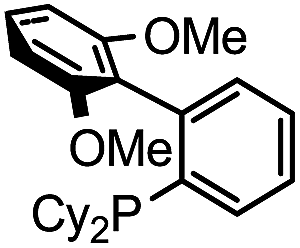
|
8 | 13 |
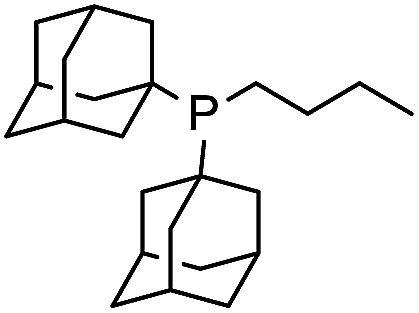
|
6 |
| 7 | PCy3 | 10 | 14 | PtBu3 | 97d |
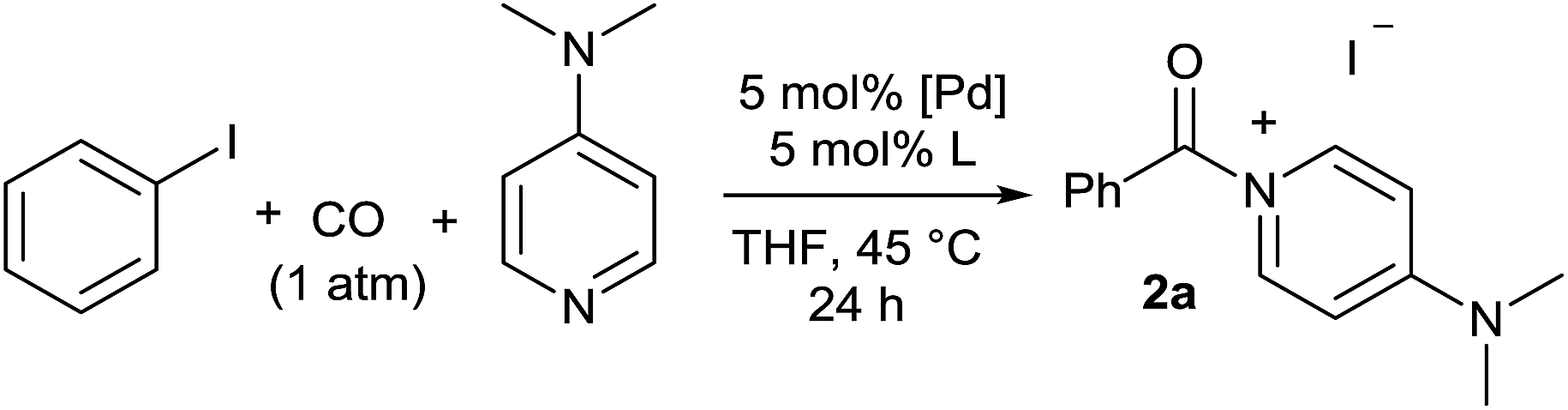
As shown in Table 2, this reaction can be used to prepare a diverse array of aroylated products from the corresponding aryl iodides under mild conditions.19 This includes both electron-poor substrates (2c, 2e, 2f), those with reactive functional groups (2c, 2l) and deactivated electron-rich aryl iodides (2b, 2g). Similarly, steric encumbrance does not appear to hinder the reaction, with 2-methoxy and 2-tolyl reagents forming products in high yield (2g, 2h).
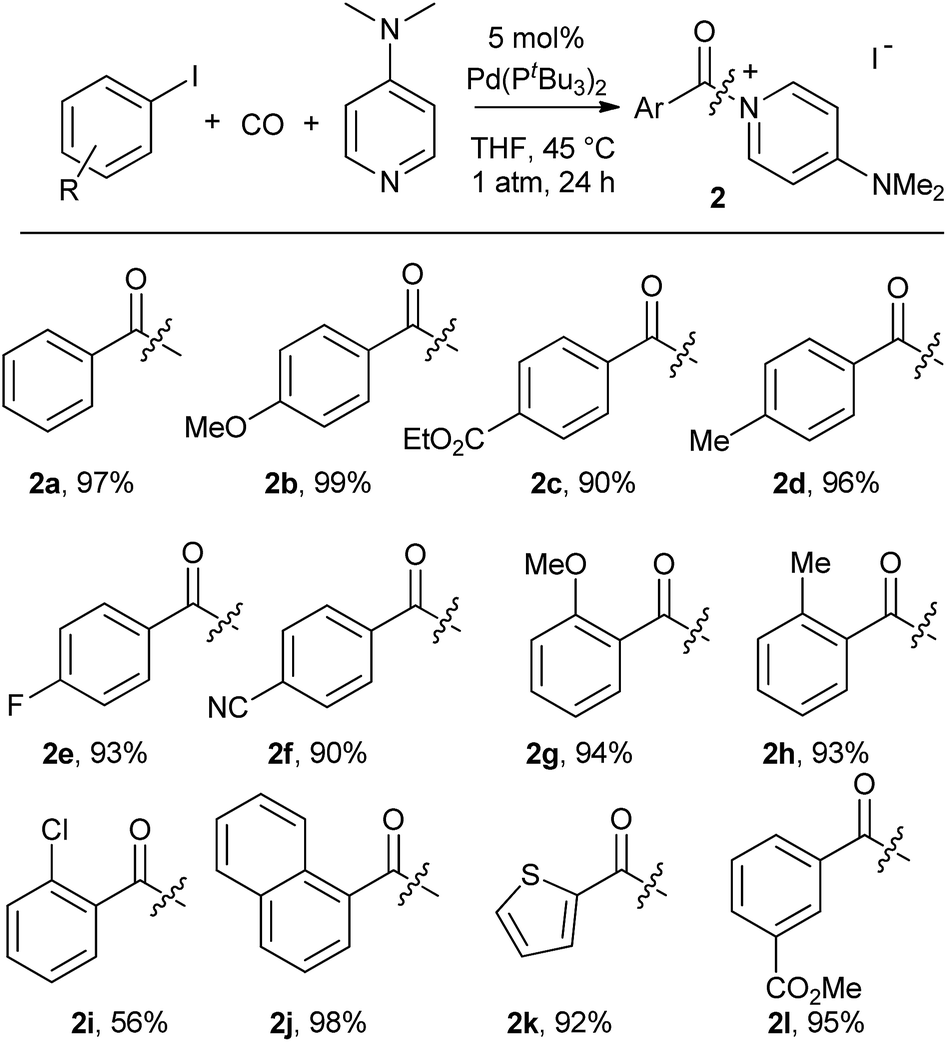
A feature of this transformation is the insolubility of the aroylated DMAP product 2 in the reaction solvent. The product can therefore be isolated at the end of the reaction by simple filtration. These solid reagents hydrolyse relatively slowly in their solid state, making them suitable candidates for medium to long term storage.20 The precipitation of 2 as it forms also limits the potential for product inhibition. As such, the reaction is scalable to generate multigram quantities of product, and with as low as 0.5 mol% palladium catalyst under more pressing conditions (Table 3). In addition, the precipitation of the DMAP salt leaves the palladium catalyst in solution, and provides in some instances a route to generate carbonylated products with low palladium contamination. Analysis of 2a formed with 5 mol% Pd and a slight excess of PhI by ICP-OES shows 98 ppm Pd in the product (or the removal of 99.5% Pd), without any special precautions beyond filtration. Similar results are observed with the other aryl iodides (100–500 ppm: 2b–2f),21 although higher Pd content is noted with more sterically encumbered aryl iodides (ca. 2000 ppm in 2g, 2h, 2j).22 Subsequent conversion of these products to esters or amides (Table 5) leads to isolated products with negligible palladium (<10 ppm).23
|
|
||
|---|---|---|
| Product | Time (h) | Yield (%) |
| a Aryl iodide (10 mmol), DMAP (1.46 g, 12 mmol), Pd(PtBu3)2 (25 mg, 50 μmol), CO (20 atm), THF (10 mL), 80 °C, 24 h. | ||
| 2a | 7 | 99 |
| 2b | 24 | 99 |
| 2c | 7 | 97 |
| 2d | 13 | 99 |
| 2e | 30 | 93 |
| 2f | 64 | 98 |
| 2g | 4 | 99 |
| 2j | 24 | 99 |
| 2l | 7 | 97 |
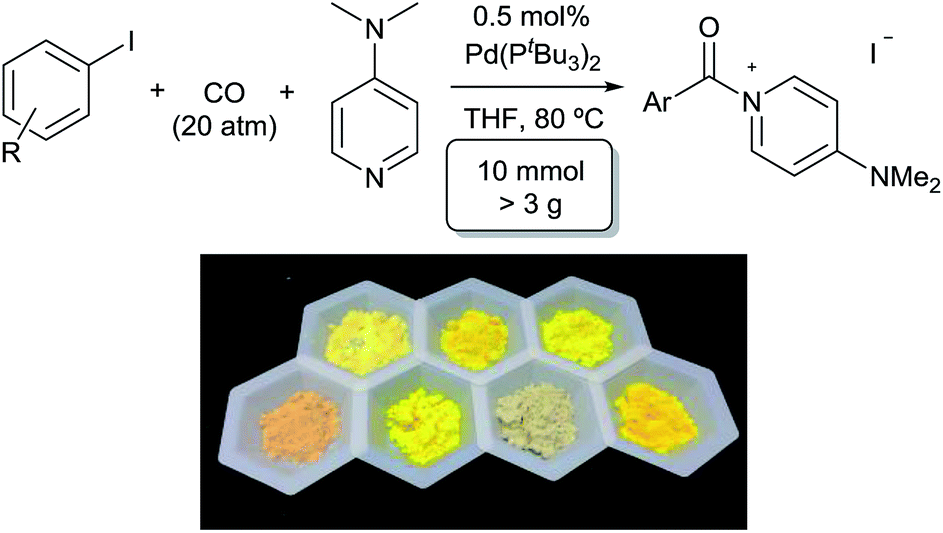
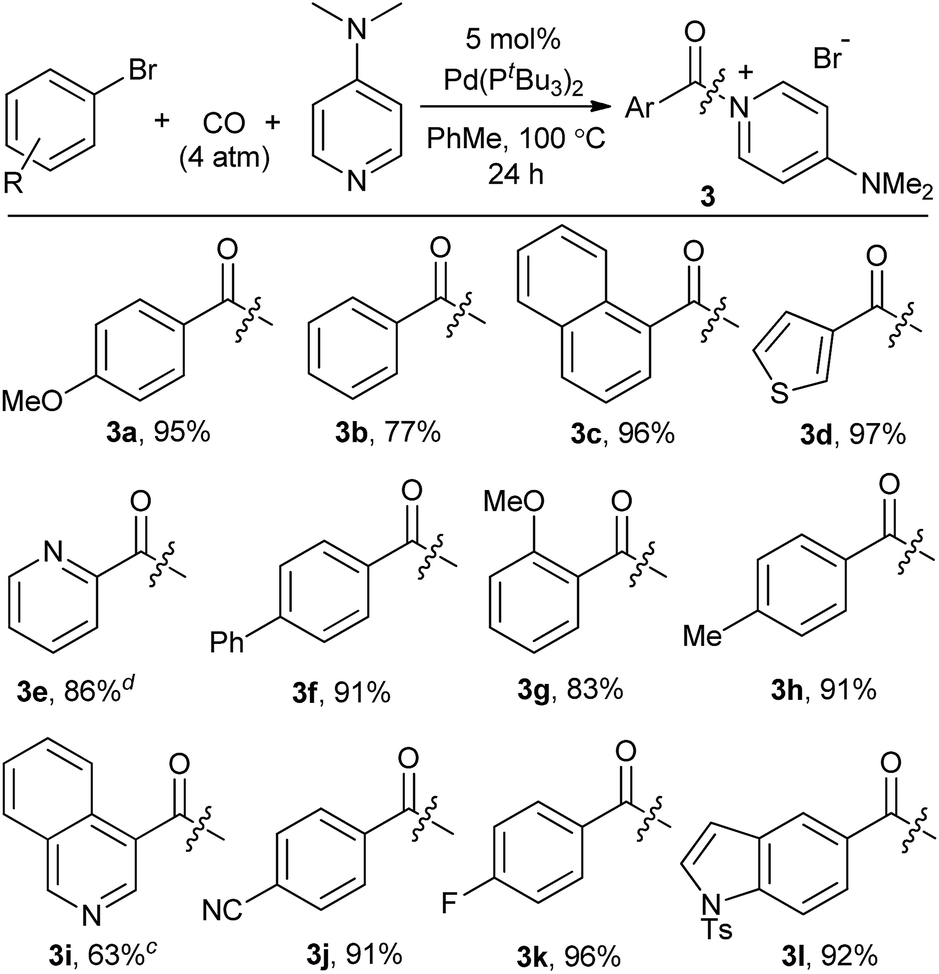
We next turned our attention to more challenging, yet widely available, aryl bromide substrates. We have recently reported the carbonylation of aryl bromides to acid chlorides, although in this case very pressing conditions are required and the high reactivity of the acid chloride product towards palladium relative to the reagent (aryl bromides) often leads to incomplete conversion.14b As shown in Table 4, bromide substrates can be similarly converted to aroyl–DMAP salts with the Pd(PtBu3)2 catalyst in high yield.24 The products are again isolated by filtration and no formal purification steps are required. In addition to substituted arenes, the use of bromide reagents permits the use of various heteroaromatic compounds (e.g. quinoline-, thiophene-, indole- and pyridine-containing substrates, 3d, 3e, 3i, 3l), which are generally more accessible than the corresponding iodides.
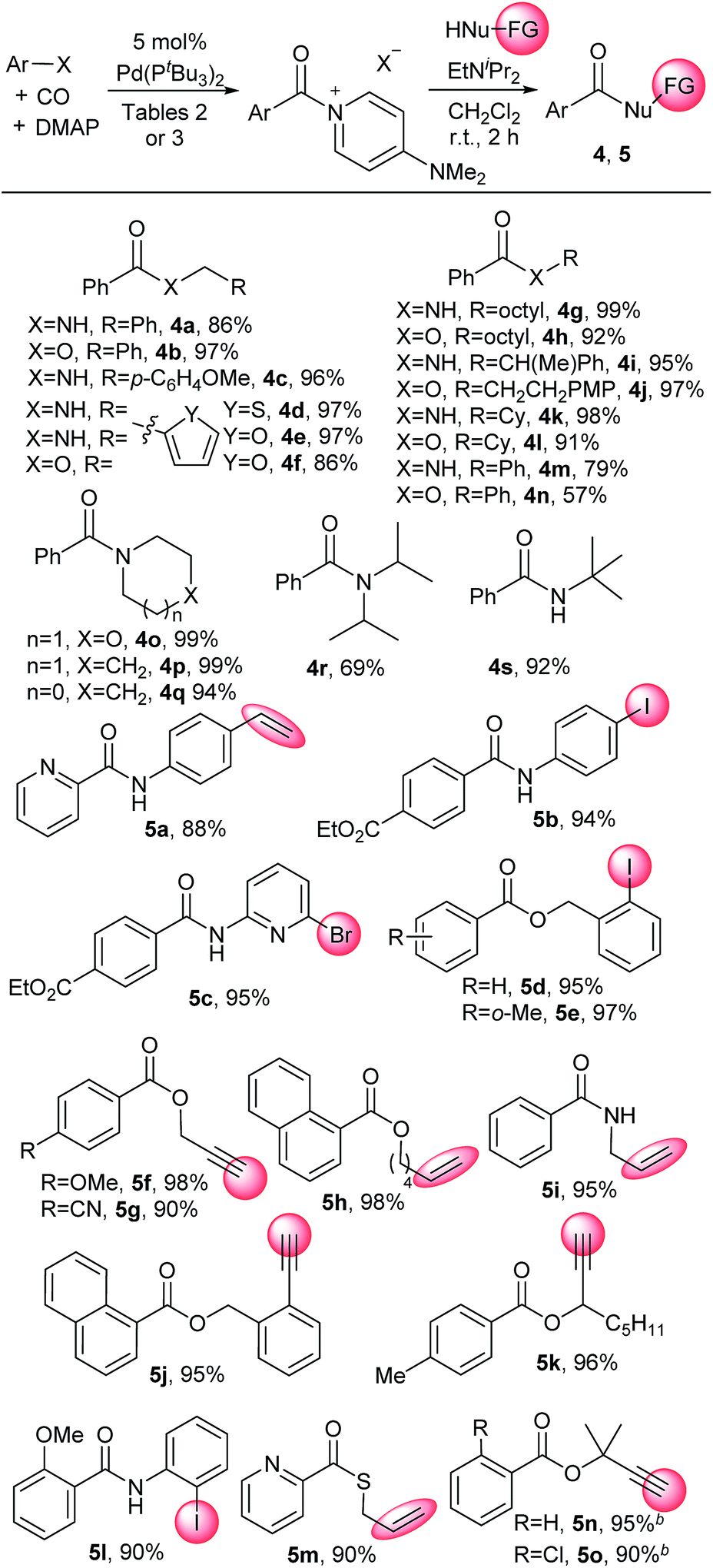
Aroyl–DMAP salts are potent electrophiles commonly employed as intermediates to activate neutral acylating agents towards coupling with nucleophiles. This reactivity makes it convenient to apply carbonylation reactions to a diverse array of nucleophiles that can be challenging or not viable as substrates in palladium catalysis. For example, the addition of amines or alcohols to catalytically generated 2 furnishes the corresponding amides and esters without long reaction times or heating common in carbonylations (Table 5). Substrates include both primary amines and alcohols (e.g.4a, 4b, 4g–l) and more sterically encumbered nucleophiles such as tert-butylamine (4s), diisopropyl amine (4r) or tertiary alcohols (5n, 5o). The latter are often difficult to employ in carbonylation chemistry.25 Weaker nitrogen nucleophiles such as aniline and phenol are viable substrates (4m, 4n). These reactions are rapid, with many nucleophiles (e.g. aliphatic amines and alcohols) proceeding to completion within seconds. As these DMAP salts are only slightly soluble in the reaction solvent (dichloromethane), dissolution of the aroylating reagent becomes a visual indicator of reaction progression. This approach also allows for the use of nucleophiles containing functional groups that can otherwise lead to unwanted side reactions under typical carbonylative conditions. As examples, terminal alkynes (e.g.5f, 5g), alkenes, including styrene (e.g.5a), or sulfur-containing reagents (5m) can be employed on the nucleophile without competitive side reactions, as can aryl iodides or heteroaryl bromides containing substrates (e.g.5b–e, 5l). Overall, this provides a method to perform carbonylations under mild conditions with a broad range of nucleophiles.
We have performed preliminary studies to probe the mechanism of this catalytic reaction. Kinetic analysis shows that catalysis is accelerated by both phosphine and CO concentration, but inhibited by excess DMAP (see ESI†). A plausible reaction mechanism that accounts for these factors is shown in Scheme 1. Oxidative addition of aryl iodide and CO insertion to palladium are established to be rapid, and generate the three-coordinate complex 1b with PtBu3.14a However, control experiments with a pre-synthesized 1b (X = I) demonstrate that DMAP can displace PtBu3 from this complex at high concentrations. We therefore postulate that the beneficial effects of added PtBu3, and inhibition of catalysis by excess DMAP, arise from this equilibrium, as excess DMAP can create a Pd(II) complex unable to undergo reductive elimination. As noted in Table 1, the PtBu3 ligand is required to allow the catalytic formation of 2 under mild conditions. This is analogous to that we have noted in the generation of weak C–Cl bonds in acid chlorides14 and presumably arises from the tertiary steric bulk at phosphorus in this ligand, which can induce reductive elimination with even a weak, neutral nucleophile such as DMAP as a mechanism to relieve strain. The association of CO to 1b, and the precipitation of the product 2, likely further facilitate this step.26
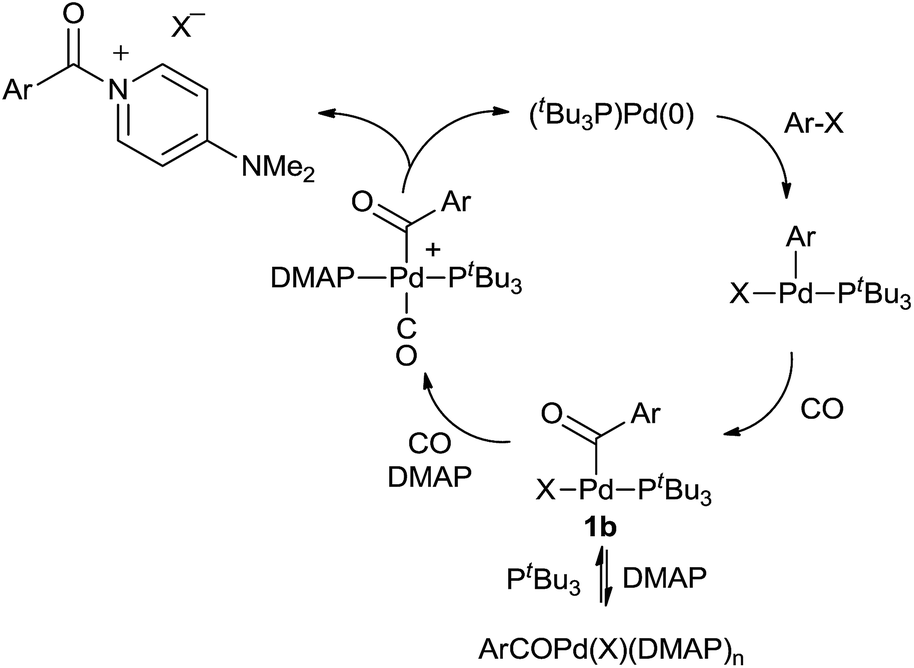 | ||
| Scheme 1 Proposed mechanism of catalytic aroyl–DMAP formation. | ||
Conclusions
In summary, a palladium-catalyzed, carbonylative synthesis of aroyl-DMAP salts has been developed. The precipitation of these products avoids catalyst inhibition, allows the reaction to be performed in high yields with low palladium loadings, and generates potent aroylating reagents with negligible impurities upon simple filtration. These products react with a variety of nucleophiles under mild conditions, including those with functional groups that can be incompatible under standard carbonylative conditions or are poorly nucleophilic. These features suggest the potential general utility of aroyl–DMAP formation in carbonylation chemistry, especially with problematic substrates. Experiments directed towards exploiting this reactivity in new classes of carbonylation reactions are currently in progress.Acknowledgements
We thank NSERC, the Canadian Foundation for Innovation (CFI), and the FQRNT supported Centre for Green Chemistry and Catalysis for funding this research.Notes and references
- (a) G. A. Olah, Friedel–Crafts and Related Reactions, Interscience Publishers, New York, 1963 Search PubMed; (b) G. A. Olah, Friedel–Crafts Chemistry, John Wiley-Interscience, New York, 1973 Search PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- (a) I. J. Turchi and M. J. S. Dewar, Chem. Rev., 1975, 75, 389 CrossRef CAS; (b) J. B. Wright, Chem. Rev., 1951, 48, 397 CrossRef CAS PubMed; (c) N. S. Isaacs, Chem. Soc. Rev., 1976, 5, 181 RSC; (d) G. W. Gribble, in Mesoionic Ring Systems from the Chemistry of Heterocyclic Compounds, ed. A. Padwa and W. H. Pearson, Wiley-Vch, 2002, vol 59, pp. 682–753 Search PubMed; (e) D. M. D'Souza and T. J. J. Müller, Chem. Soc. Rev., 2007, 36, 1095 RSC.
- (a) H. H. Yang, Kevlar Aramid Fiber, J. Wiley, Chichester, 1993 Search PubMed; (b) H. G. Chae and S. J. Kumar, Appl. Polym. Sci., 2006, 100, 791 CrossRef CAS.
- R. K. Dieter, Tetrahedron, 1999, 55, 4177 CrossRef CAS.
- (a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC; (b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC; (c) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (d) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed.
- (a) A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318 CrossRef CAS; (b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327 CrossRef CAS.
- For reviews see: (a) H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed; (b) C. F. J. Barnard, Organometallics, 2008, 27, 5402 CrossRef CAS; (c) R. Grigg and S. P. Mutton, Tetrahedron, 2010, 66, 5515 CrossRef CAS; (d) A. Brennführer, X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC.
- For select examples and reviews see: (a) J. Magano, in Transition Metal-Catalyzed Couplings in Process Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2003, pp. 313–355 Search PubMed; (b) J. Bien, G. Lane and M. Oberholzer, in Organometallics in Process Chemistry, Springer Berlin Heidelberg, 2004, vol. 6, pp. 263–283 Search PubMed; (c) C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS.
- (a) D. A. Watson, X. Fan and S. L. Buchwald, J. Org. Chem., 2008, 73, 7096 CrossRef CAS PubMed; (b) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2012, 14, 5370 CrossRef CAS PubMed.
- M. N. Burhardt, R. H. Taaning and T. Skrydstrup, Org. Lett., 2013, 15, 948 CrossRef CAS PubMed.
- (a) R. Lou, M. vanAlstine, X. Sun and M. P. Wentland, Tetrahedron Lett., 2003, 44, 2477 CrossRef CAS; (b) A. M. de Almeida, T. L. Andersen, A. T. Lindhardt, M. V. de Almeida and T. Skrydstrup, J. Org. Chem., 2015, 80, 1920 CrossRef CAS PubMed.
- Aroyl azides (a) F. M. Miloserdov and V. V. Grushin, Angew. Chem., Int. Ed., 2012, 51, 3668 CrossRef CAS PubMed, Acyl fluorides (b) T. Okano, N. Harada and J. Kiji, Bull. Chem. Soc. Jpn., 1992, 65, 1741 CrossRef CAS; (c) T. Sakakura, M. Chaisupakitsin, T. Hayashi and M. Tanaka, J. Organomet. Chem., 1987, 334, 205 CrossRef CAS; (d) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2013, 15, 5370 CrossRef CAS PubMed, Weinreb amides (e) J. R. Martinelli, D. M. M. Freckmann and S. L. Buchwald, Org. Lett., 2006, 8, 4843 CrossRef CAS PubMed, N-acyl pyrroles (f) S. Ho, G. Bondarenko, D. Rosa, B. Dragisic and A. Orellana, J. Org. Chem., 2012, 77, 2008 CrossRef CAS PubMed.
- (a) J. S. Quesnel and B. A. Arndtsen, J. Am. Chem. Soc., 2013, 135, 16841 CrossRef CAS PubMed; (b) J. S. Quesnel, L. V. Kayser, A. Fabrikant and B. A. Arndtsen, Chem.–Eur. J., 2015, 20, 9550 CrossRef PubMed.
- For acid chloride syntheses from non-aryl halide electrophiles: (a) J. Tsuji, M. Morikawa and J. Kiji, J. Am. Chem. Soc., 1964, 86, 4851 CrossRef CAS; (b) W. T. Dent, R. Long and G. H. J. Whitfield, J. Chem. Soc., 1964, 1588 RSC; (c) M. C. Bonnet, N. Carmona and I. Tkatchenko, J. Mol. Catal. A: Chem., 1999, 143, 181 CrossRef CAS; (d) T. A. Cernak and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 3124 CrossRef CAS PubMed.
- (a) G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed., 1978, 17, 569 CrossRef; (b) E. F. V. Scriven, Chem. Soc. Rev., 1983, 12, 129 RSC; (c) U. Ragnarsson and L. Grehn, Acc. Chem. Res., 1998, 31, 494 CrossRef CAS; (d) D. J. Berry, C. V. Digiovanna, S. S. Metrick and R. Murugan, ARKIVOC, 2001, 2, 944 Search PubMed; (e) A. C. Spivey and S. Arseniyadis, Angew. Chem., Int. Ed., 2004, 43, 5436 CrossRef CAS PubMed; (f) N. de Rycke, F. Couty and O. R. P. David, Chem.–Eur. J., 2011, 17, 12852 CrossRef CAS PubMed.
- (a) E. Guibe-Jampel, G. Le Corre and M. Wakselman, Tetrahedron Lett., 1979, 20, 1157 CrossRef; (b) J. A. King and G. L. Bryant, J. Org. Chem., 1992, 57, 5136 CrossRef CAS; (c) M. S. Wolfe, Synth. Commun., 1997, 27, 2975 CrossRef CAS; (d) R. Weiss, M. Bess, S. M. Huber and F. W. Heinemann, J. Am. Chem. Soc., 2008, 130, 4610 CrossRef CAS PubMed.
- W. Fang, H. Zhu, Q. Deng, S. Liu, X. Liu, Y. Shen and T. Tu, Synthesis, 2014, 46, 1689 CrossRef.
- For ease of reaction set-up and manipulation, (PtBu3)2Pd was employed as a catalyst, although the combination of Pd2dba3/PtBu3 (Table 1) can also be used. In addition, the reaction can be performed on the bench using standard Schlenk techniques with only slightly diminished yields (using Pd2dba3/tBu3PHCl as the catalyst precursor). See ESI† for details.
- This was judged by exposing 2a to the open atmosphere for 10 days where after which 11% hydrolysis was observed (by 1H NMR analysis).
- ICP-OES palladium content analysis show removal of 98–99% Pd in: 2b = 450 ppm (65 °C, 4 atm); 2c = 270; 2d = 199; 2e = 522 ppm. General conditions employed: ArI (0.6 mmol), DMAP (0.5 mmol), Pd(PtBu3) (5 mol%), CO (1 atm), THF (1 mL), 45 °C, 24 h.
- 2g = 2132 ppm; 2h = 2635 ppm; 2j = 1780 ppm. We postulate that the higher Pd content may arise from the diminished ability of these aryl iodides to sequester palladium in solution.
- The products were obtained using the procedure in Table 5 and purified by standard silica gel column chromatography. ICP-OES palladium content analysis: 4a = 7 ppm; 5f < 1 ppm; 5h < 1 ppm.
- The reaction of aryl bromides can also be performed at 1 atm CO at slightly diminished yields (e.g.3a, 84% yield at 1 atm CO).
- M. Majek and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 2270 CrossRef CAS PubMed and references therein.
- It is also possible that the sterically encumbered palladium intermediate undergoes aroyl-halide reductive elimination, followed by reaction of this product with DMAP. At present, we cannot distinguish between these possibilities.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and NMR spectra for compounds. See DOI: 10.1039/c5sc02949j |
| This journal is © The Royal Society of Chemistry 2016 |