
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a peripherally conjugated 5-6-7 nanographene†
Marika
Żyła
a,
Elżbieta
Gońka
a,
Piotr J.
Chmielewski
a,
Joanna
Cybińska
ab and
Marcin
Stępień
*a
aWydział Chemii, Uniwersytet Wrocławski, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: marcin.stepien@chem.uni.wroc.pl
bDepartment of Nanotechnology, Wrocław Research Centre EIT+, ul. Stabłowicka 147, 54-066 Wrocław, Poland
First published on 29th September 2015
Abstract
A heteroaromatic nanographene containing a unique assembly of five-, six- and seven-membered rings is synthesized using oxidative coupling of an indole-containing precursor. Near-infrared absorption and emission properties of the nanographene core are enhanced by peripheral expansion and ring fusion at all oxidation levels. The dicationic state shows distinct aromaticity originating from a peripheral π-conjugated circuit. A partially coupled intermediate, trapped in the synthesis of the 5-6-7 nanographene, is explored as a reference system, showing an unexpected reduction of the optical band gap due to intramolecular charge transfer.
Introduction
Doping large aromatic structures with heteroatoms is one of the key exploration areas in current nanographene research.1,2 While structurally precise doping of graphene sheets is yet to be achieved, bottom-up approaches developed for the synthesis of extended polycyclic aromatic hydrocarbons (PAHs) have been investigated as potential routes to doped nanographenes and nanoribbons. Heteroaromatic five-membered (N,3–12 P,13 S14–23) and six-membered rings (B,18,24–26 N,24–38 O,29,30 S29,30) have recently been incorporated into large PAH-like structures, yielding systems with distinct electronic and optical properties. In nanoribbons, atomically precise introduction of nitrogens has been found to produce band shifts corresponding to n-type doping, and enabled charge transport tuning and the fabrication of heterojunctions and heterostructures.39–41 At present, knowledge of the site-specific effects of heteroatom doping is limited, and such influences are best explored by preparing structurally new nanographene molecules. However, established synthetic routes to PAHs are often incompatible with heterocyclic precursors, and need to be refined or replaced in order to encompass new structural designs.An elegant route to heteroatom-doped coronene derivatives involves the oxidative coupling of star-shaped aromatic precursors containing heterocyclic groups.17,27,34,36 Such reactions have been modeled after the classical synthesis of hexa-peri-hexabenzocoronenes (p-HBCs).42 A remarkable example of this approach is provided by the FeCl3-mediated coupling of hexapyrrolylbenzenes,43 which was found to produce hexapyrrolohexaazabenzocoronenes (HPHACs, 1, Scheme 1).3,44 HPHACs and the related HPHAC–HBC hybrids4 are fluorescent in their neutral oxidation states and can be converted into strongly NIR-absorbing cations by reversible multistep oxidation. The work on hybrid systems showed, however, that the effectiveness of coupling is dependent on the precursor design, notably on the choice of subunits and peripheral substitution.4
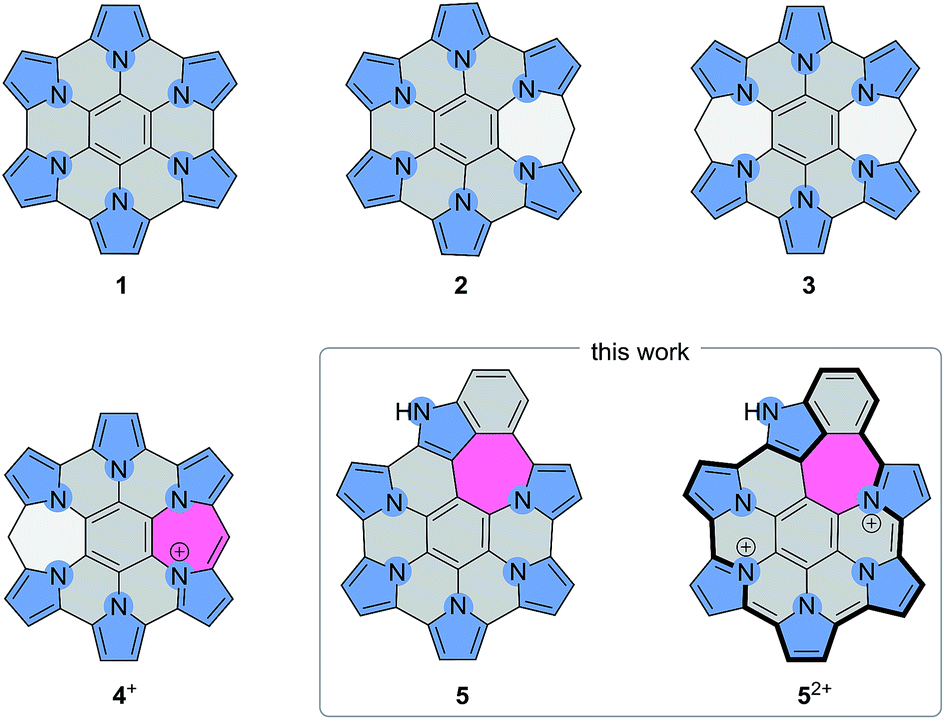 | ||
| Scheme 1 Pyrrole-fused azacoronene systems. | ||
We have recently reported HPHAC analogues containing non-conjugated bridges on the periphery (2–3).11 Up to four electrons could be removed reversibly from these systems, leading to cationic species with extended NIR absorptions. In these oxidations, the benzylidene bridges were found to be remarkably resistant to dehydrogenation, and complete peripheral π conjugation could not be achieved. Stereospecific oxidation of a single bridge could however be induced in 3, to provide the monocation 4+, the first example of a heteroaromatic nanographene structure with a conjugated 7-membered ring. The inclusion of 7-membered rings in PAH and related structures is known to affect the curvature and electronic conjugation of fused frameworks, however, the construction of such systems is still a non-routine task.45–49 In conjunction with heteroatom doping, non-benzenoid fusion is an attractive method of increasing structural diversity in nanographenes. The rich chemistry of peripherally expanded azacoronenes 2–3 inspired us to seek synthetic designs that would produce systems with fully conjugated and non-trivial ring fusion patterns. Here we show that heteroaromatic nanographene structures containing a combination of 5-, 6-, and 7-membered rings (“5-6-7 nanographenes”) can be obtained by the direct oxidative coupling of star-shaped precursors containing pyrrole and indole arms. The electronic structure of the new nanographene is compared with a partially coupled reference system. We demonstrate for the first time how the absorption and emission properties of such nanographenes can be tailored by peripheral expansion and ring fusion.
Results and discussion
The requisite indole-containing precursors 8a–b were prepared in two steps from N-substituted indoles 6a–b (Scheme 2). 6a–b were coupled with pentafluorobenzene, using Pd/Ag-mediated direct double C–H activation,50 to yield C6F5-substituted derivatives 7a–b. The latter intermediates were subjected to quintuple nucleophilic substitution with β-substituted pyrroles,9,51,52 to yield the star-shaped 8a–b in up to 63% yield. In contrast to our previous work, which had used p-butoxyphenyl substituents on the periphery of nanographenes 2–3,11p-chlorophenyl groups were introduced to the structures of 8a–b. The electron-withdrawing character of the latter substituents and their limited solubilizing effect in nonpolar solvents provided better stability and easier isolation, respectively, of the subsequent coupling products.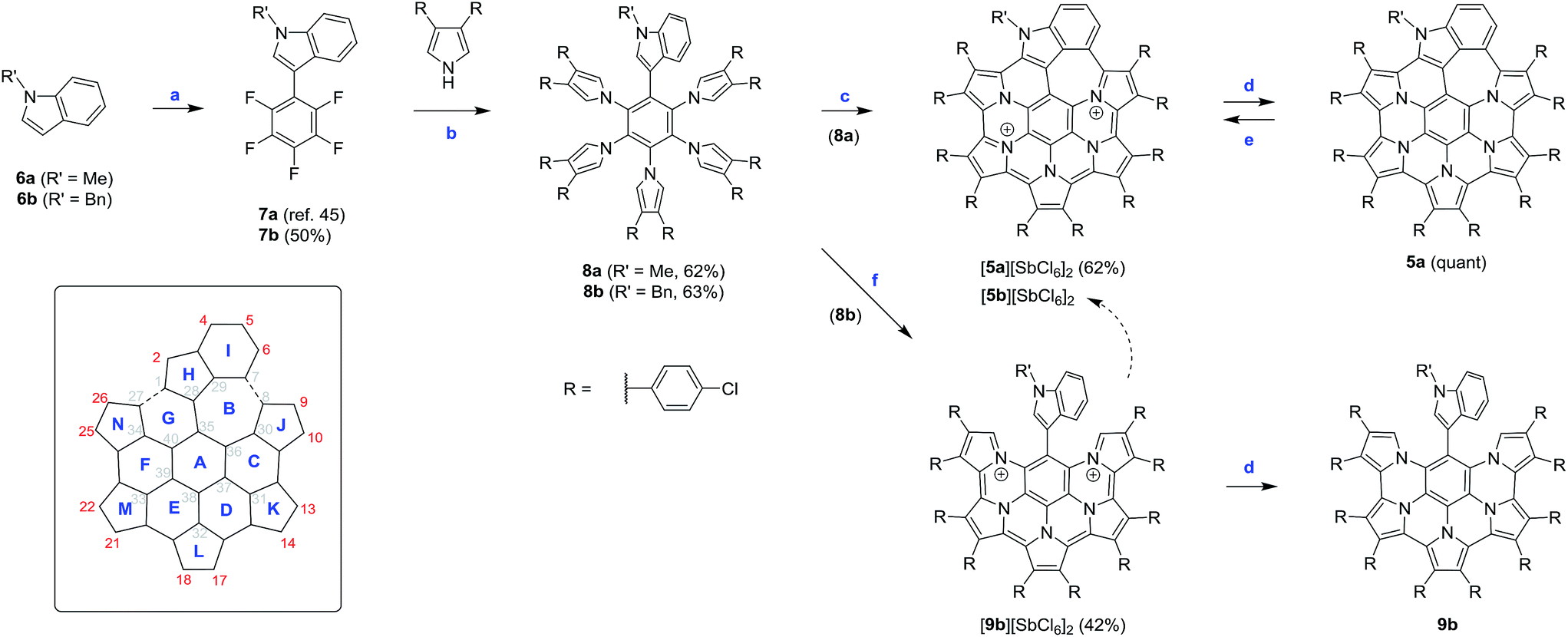 | ||
| Scheme 2 Synthetic work and the labeling scheme for 5 and 9. Reagents and conditions: (a) Ag2CO3 (1.5 equiv.), Pd(OAc)2 (cat.), AcOH (1 equiv.), pentafluorobenzene (3 equiv.), DMF/DMSO, 120 °C. (b) 1. NaH (5 equiv.), diarylpyrrole (5 equiv.), DMF, ice bath, 2. 7a or 7b, 50 °C, overnight. (c) BAHA (12 equiv.), Et2O/THF, RT. (d) Zn amalgam or Zn powder, DCM or CDCl3, quant. (e) BAHA, (NO)[SbF6], or DDQ; DCM or MeCN, quant. (f) BAHA (10 equiv.), Et2O/THF, RT, 42%. | ||
Oxidative coupling reactions are well suited for the rapid development of complex aromatic molecules; however, their use is often limited by reactivity and selectivity problems.53 In dichloromethane solutions, 8a was not reactive towards DDQ, but it was easily oxidized by ionic reagents such as FeCl3,3,4,11 Fe(ClO4)3, Ag[SbF6], or (NO)[SbF6]. Disappointingly, all these oxidants produced mixtures of products that could not be separated without decomposition. It was subsequently found that 8a was cleanly oxidized by tris(4-bromophenyl)ammoniumyl hexachloroantimonate (BAHA),54 a one-electron, radical-cation reagent with preparative applications in oligopyrrole chemistry.55–57 The oxidation, performed with 12 equiv. of BAHA in a diethyl ether/THF mixture, led directly to the dicationic species 5a2+, which was isolated in the form of the dark-brown hexachloroantimonate salt in 62% yield. The isolation was particularly convenient because [5a][SbCl6]2 precipitated from the solution, and after an additional crystallization from dichloromethane/hexane was found to be free of the oxidant byproducts. The reaction with BAHA strongly favors the formation of 5a2+ and small amounts of the salt could be isolated even when 2 equiv. of the oxidant were used. [5a][SbCl6]2 was quantitatively converted to the neutral species 5a by reduction with zinc amalgam.
Interestingly, when the benzyl-substituted derivative 8b was subjected to the same BAHA-induced oxidation, the outcome of the reaction was different. The green precipitate that formed was found to contain mainly the partly coupled product [9b][SbCl6]2. Under optimized conditions, involving oxidation with 10 equiv. of BAHA in THF/diethyl ether, the above salt was isolated in 42% yield. In analogy to [5a][SbCl6]2, compound [9b][SbCl6]2 was cleanly reduced by zinc amalgam, to yield the corresponding neutral species 9b. A benzene-containing molecule, similar to 9b, was previously reported by Takase et al.4 but the accessibility of higher oxidized states was not disclosed for that system. Dication 9b2+ is moderately stable in solution, undergoing gradual conversion to 5b2+ in the presence of air.
The extent of oxidative coupling and the charge of the products was determined using ESI mass spectrometry. In the mass spectra recorded for the oxidation products of 8a–b, peaks corresponding to singly (5a+, 5b+) and doubly charged species (5a2+, 5b2+) could be identified on the basis of the observed m/z ratio and isotope patterns. The partially coupled dication 9b2+ produced only peaks corresponding to 5b2+ and 5b+, indicative of instantaneous dehydrogenation in the ESI source.
The spectroscopic features of 5a2+ are consistent with extensive π-electron conjugation. In dichloromethane solutions, the dication has an intense brown color, with electronic absorptions covering the entire visible range and reaching beyond 1800 nm in the near infrared (Fig. 1). With the lowest-energy absorption at 1525 nm, the NIR part of the spectrum of 5a2+ shows an overall red shift relative to the reported spectrum of an aryl substituted HPHAC dication (12+),3 correlating with the more extended π-conjugation in the indole-containing nanographene. In the spectrum of 9b2+, major NIR absorptions are located in the 1000–1150 nm range. However, a weak maximum at 1530 nm, observed consistently for various samples of 9b2+, indicated that the optical HOMO–LUMO gaps of the two dications are almost identical (0.81 eV). This unexpected result was rationalized using TD-DFT calculations (vide infra).
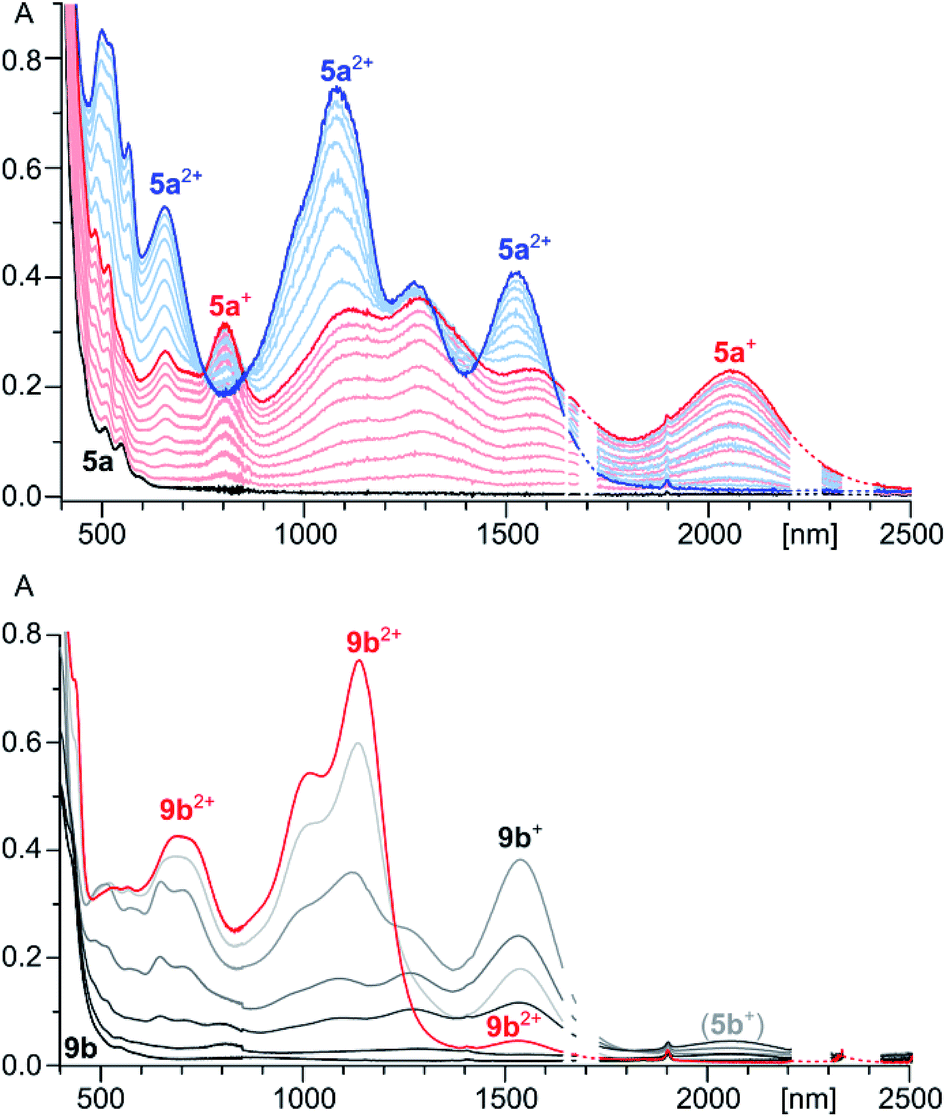 | ||
| Fig. 1 Top: vis-NIR spectra of 5a+ (red) and 5a2+ (blue) obtained by spectrophotometric titration of 5a (black) with BAHA (dichloromethane, 293 K). Bottom: vis-NIR spectrum of 9b2+ (red trace, SbCl6 salt, dichloromethane). Gray traces correspond to stepwise in situ reduction of 9b2+ with Zn powder. Apart from 9b+ and 9b, small amounts of 5b+ are also formed in contact with air. | ||
5a has an orange color in solution and displays noticeable orange fluorescence. With several weak bands in the 500–600 nm range (ε = 900–2700 M−1 cm−1), the absorption spectrum of 5a resembles the optical spectra of other neutral HPHAC systems, although a red shift of the lowest energy transition is clearly observable.3,4,11 The fluorescence emission spectrum of 5a displays a vibronically resolved pattern with major maxima at 620, 675 and 740 nm (ΔE ∼ 1300 cm−1, Fig. 2). The fluorescence excitation spectrum of 5a accurately reproduces the absorption profile above 400 nm, including the three weak bands with λ > 500 nm. The emission quantum yield of 5a (ca. 0.5%) is in the range reported for related nanographene systems (0.1–1.8%) and the small radiative rate constants kf = ΦF/τF ≈ 106 s−1 are also consistent with the semi-allowed character of the S0 → S1 transitions.4 The moderate Stokes shift in 5a (ca. 680 cm−1) indicates a relatively small reorganization of the nanographene chromophore in the excited state. In spite of the small Stokes shift, the fluorescence of 5a is noticeably red-shifted in comparison with previously reported HPHAC-like systems.3,4 In DCM solutions, 9b has a yellow color and shows yellow fluorescence corresponding to a broadened emission (λmax ≈ 565 nm), which becomes well-resolved at low temperatures. The λ0–0 peak (510 nm at 77 K in frozen DCM solution) corresponds to a Stokes shift of ca. 900 cm−1. Neither 5a nor 9b exhibited detectable phosphorescence in frozen dichloromethane at 77 K.
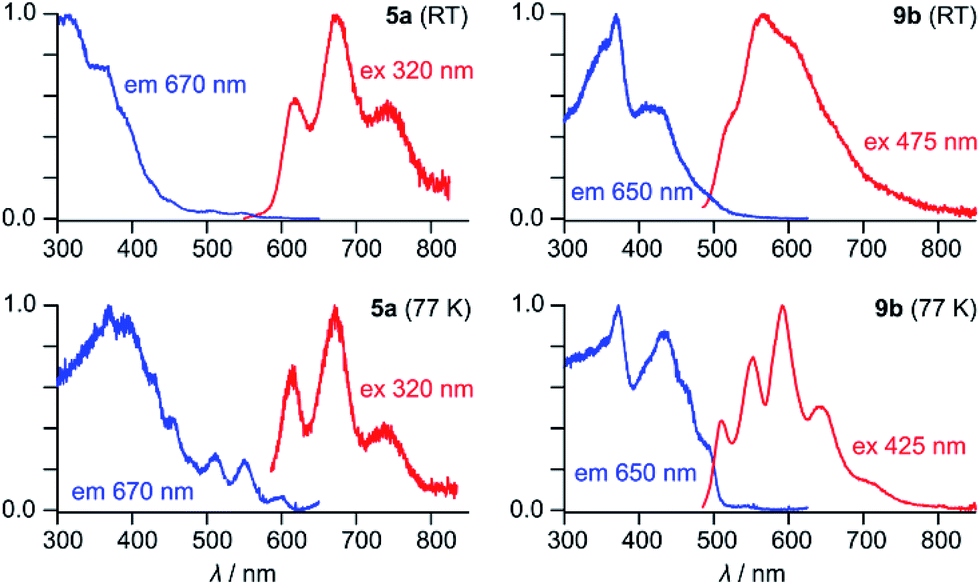 | ||
| Fig. 2 Fluorescence emission and excitation spectra of 5a and 9b recorded in dichloromethane. | ||
The 1H NMR spectra of 5a and 5a2+ (Fig. 3) each contain a large number of signals, consistent with the low effective molecular symmetry (Cs) of the nanographene. Because of the severe overlap of the aryl signals, only partial spectral assignment could be achieved with the use of 1H COSY and ROESY spectra. The differences of the chemical shifts reveal a drastic change in the magnetic properties of 5a attendant upon oxidation, indicative of a considerable increase of the aromatic character of the core. In the spectrum of neutral 5a, the indole signals 4-H, 5-H, and 6-H were found at 6.69, 6.65, and 6.49 ppm, respectively, obscured by the numerous p-chlorophenyl peaks clustered in the 7.2–6.2 ppm range. With the exception of the 9- and 26-Ar groups adjacent to the indole, the aryl signals of 5a are shifted slightly upfield relative to their positions in 8a, an effect caused by steric crowding of the substituents. In 5a2+, the indole signals are shifted by up to 3.3 ppm towards the lower field relative to 5a, whereas smaller relocations are observed for the Me group (+1.6 ppm) and aryl signals (up to +0.8 ppm). Such a distance dependence of the deshielding effect is consistent with a marked increase of the diatropic ring current in the oxidized nanographene core. This diatropic effect is clearly reproduced by GIAO-DFT calculations of proton shifts, performed for 5a2+ and 5a (Fig. S26–S28†). The molecular structure of 5a2+ requires 75 carbon signals (including 51 quaternary sp2 centers), which were identified in the 13C NMR spectrum, and assigned into chemically distinct groups using correlation spectroscopy and GIAO-DFT calculations (Fig. S9–S10†). In particular, the six non-equivalent carbons of the inner ring A were identified in a markedly upfield region (98.7–109.7 ppm). 1H and 13C shifts obtained from the spectroscopic analyses correlate very well with the GIAO predictions.
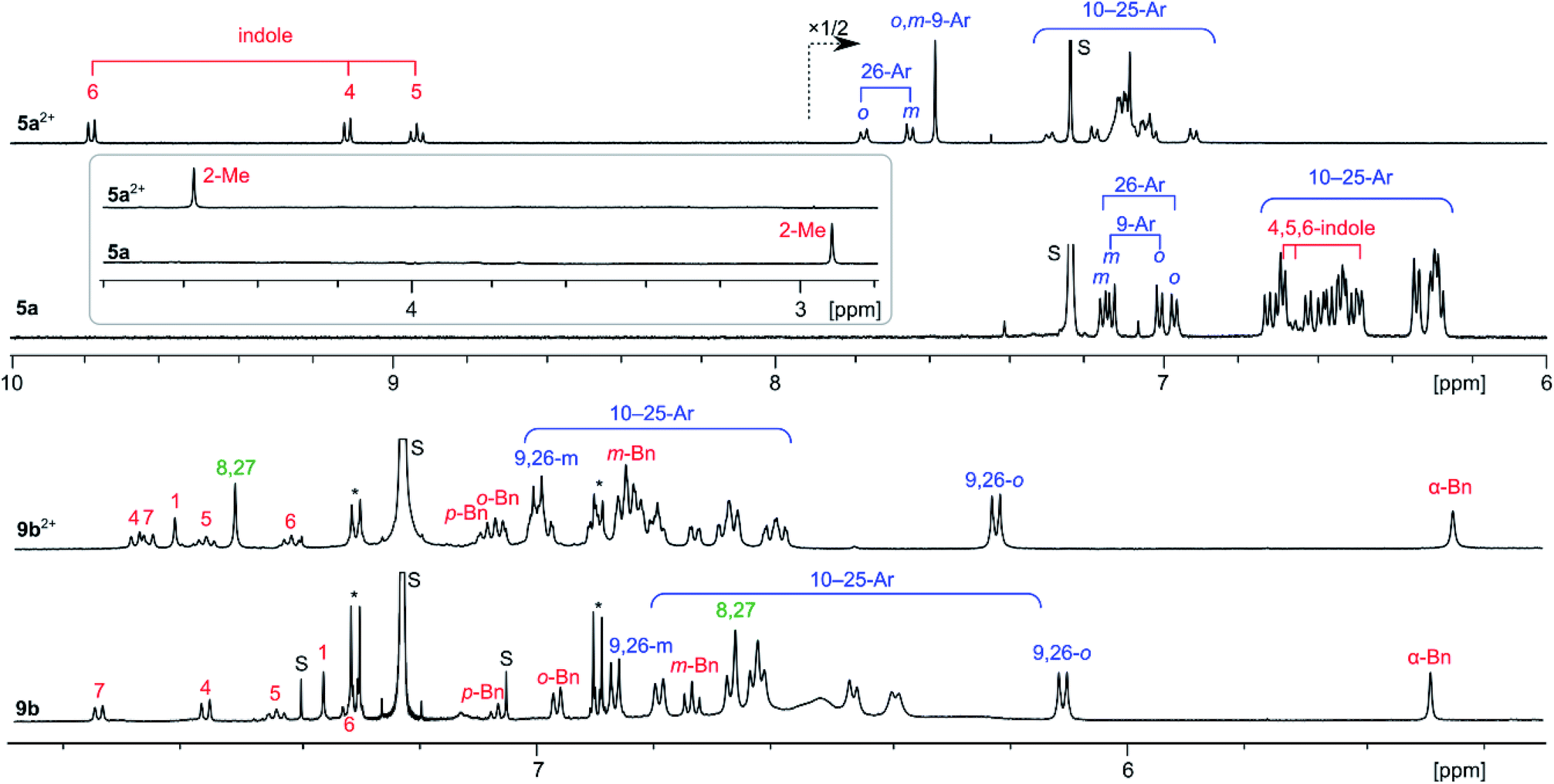 | ||
| Fig. 3 1H NMR spectra (CDCl3, 600 MHz) recorded for [5a][SbCl6]2, 5a (300 K), [9b][SbCl6]2, and 9b (260 K). Signal assignments are based on correlation spectroscopy (see the ESI†). | ||
The 1H NMR spectra of 9b2+ and 9b reveal the chemical equivalence of 9-Ar and 26-Ar substituents, which is only possible in the case of a non-fused indole unit. In both species, signals corresponding to the intact 3-(1-benzyl)-indolyl group and the unique signal of pyrrole 8,27-α protons were identified using correlation spectroscopy (Fig. 3). These features and the absence of other α-pyrrole signals clearly confirm that the oxidative coupling occurred only between adjacent pyrrole units. In contrast to the 5a/5a2+ pair, the 1H shifts of 9b and 9b2+ are similar, indicating that the oxidation of the fused core does not lead to any significant increase of overall diatropicity.
DFT-optimized geometries of the indole-containing nanographene are similar in all oxidation states (5a, 5a+ and 5a2+). As shown in Fig. 4 for the dication 5a2+, the fused core is relatively planar, with slight out-of-plane distortions along the periphery. In contrast to substituted HPHAC's, in which the rotation axes of aryl groups are coplanar with the core, in 5a2+, the substituents are bent away from the central plane and tilted, to reduce steric interactions. Out of eight distinct conformers differing in the relative arrangement of aryl groups, the majority have relative energies within a range of only 1 kcal mol−1. The lowest-energy structure is characterized by an alternating up-and-down arrangement of consecutive pyrrole units. The inter-subunit C–C bonds in 5a2+ (those formed in the course of oxidative coupling) are in the range 1.418 to 1.450 Å and are systematically shorter in comparison to the corresponding distances in 5a (1.436 to 1.470 Å). In contrast to the parent HPHAC system 1, which was predicted by DFT to adopt a bowl shape,3 the DFT-optimized structures of the unsubstituted 5, 5+, and 52+ are completely planar, indicating that the internal strain of the fused framework is partly relieved by the introduction of the 7-membered ring.
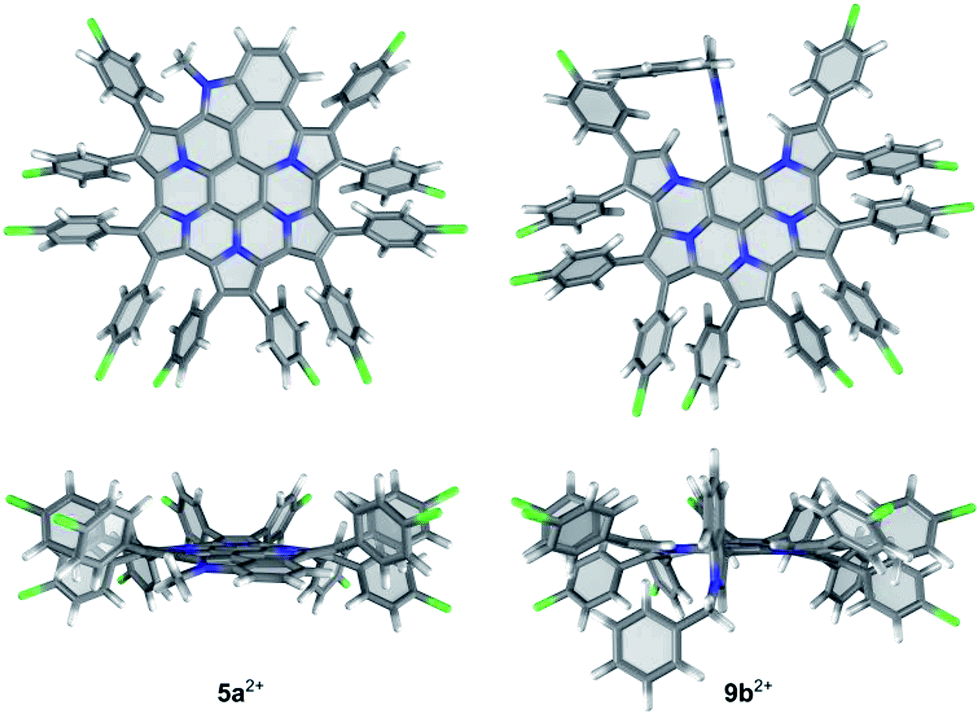 | ||
| Fig. 4 DFT-optimized geometries of 5a2+ and 9b2+. The lowest-energy conformer found for 5a2+ is shown. | ||
The optimized structure of 9b2+ shows that the indole moiety is perpendicular to the fused nanographene unit and is confined between the two adjacent pyrrolic α-hydrogens. In 5b2+, the presence of the benzyl substituent has negligible influence on the conformation of the aromatic core, in comparison with 5a2+.
5a, obtained by in situ reduction of [5a][SbCl6]2 with zinc amalgam, was subjected to a spectrophotometric titration with BAHA (Fig. 1). This experiment revealed the intermediate formation of a light brown radical cation 5a+, characterized by extended NIR absorption, reaching up to 2400 nm. Subsequent addition of BAHA to the solution led to the recovery of 5a2+, however, no further oxidation could be induced with either BAHA or the more strongly oxidizing (NO)[SbF6]. The two observable oxidation events are sufficiently well separated to produce approximate isosbestic points. Alternatively, 5a+ could be generated by comproportionation of 5a2+ and 5a combined in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in a dichloromethane solution. The radical nature of 5a+ was confirmed using ESR spectroscopy, which yielded a signal at g = 2.0026 with no resolvable hyperfine structure (Fig. S15†). Trace amounts of the 5a+ radical form spontaneously in solutions of both 5a and 5a2+, leading to a gradual broadening of the 1H NMR signals. This effect was suppressed by the addition of a small amount of BAHA or zinc amalgam to the solutions of 5a2+ and 5a, respectively, to enable the recording of well-resolved NMR spectra. In the case of 5a2+ the differential broadening of the indole signals (4,5,6-H) caused by the radical admixture was found to correlate well with the calculated spin density distribution (Fig. S24 and S25†).
:1 molar ratio in a dichloromethane solution. The radical nature of 5a+ was confirmed using ESR spectroscopy, which yielded a signal at g = 2.0026 with no resolvable hyperfine structure (Fig. S15†). Trace amounts of the 5a+ radical form spontaneously in solutions of both 5a and 5a2+, leading to a gradual broadening of the 1H NMR signals. This effect was suppressed by the addition of a small amount of BAHA or zinc amalgam to the solutions of 5a2+ and 5a, respectively, to enable the recording of well-resolved NMR spectra. In the case of 5a2+ the differential broadening of the indole signals (4,5,6-H) caused by the radical admixture was found to correlate well with the calculated spin density distribution (Fig. S24 and S25†).
The chemical oxidation of 8a with BAHA, followed spectrophotometrically in dilute DCM solutions, revealed that, even with a substoichiometric amount of the oxidant, the reaction leads directly to 5a2+, without the formation of the radical cation 5a+. This observation, which contrasts with the behavior of 5a, indicates that either (a) 5a2+ is the direct product of the coupling or (b) the small amount of 5a, produced progressively during the reaction, is the first species to consume any excess of the introduced oxidant. The analogous oxidation of 8b revealed the initial formation of 9b2+, which was gradually replaced with 5b2+ as the titration progressed. It seems that, because of the steric hindrance introduced by the benzyl group, the conversion of 9b2+ into 5b2+ is slowed down sufficiently to enable the isolation of the former species by precipitation of its SbCl6 salt. However, when 9b2+ is not precipitated from the solution, it is oxidized to 5b2+ with observable kinetics.
Voltammetric experiments performed on [5a][SbCl6]2 revealed two electrochemically reversible redox events at −0.17 and −0.02 V (vs. Fc/Fc+), corresponding respectively to the 5a0/5a+ and 5a+/5a2+ couples (Fig. S16†). The observed potential difference corresponds to a comproportionation constant Kc of ca. 3.5 × 102, sufficiently high to make the radical cation an observable species in solution. At 0.80 and 1.04 V, two additional oxidations were observed, likely associated with the formation of higher cationic species. The large gap between the second and third oxidation (0.82 V) is thought to result from the aromatic stabilization of 5a2+. Interestingly, when precursor 8a was electrolyzed in tetrabutylammonium perchlorate/dichloromethane at a constant potential of 1.4 V vs. Fc/Fc+, the formation of 5a2+ was observed spectrophotometrically (Fig. S17–S18†). The 5a2+ thus formed yielded an electrochemical signature matching that of the chemically generated dication.
TD-DFT calculations performed for the differently oxidized states of 5a and 9b yielded results in very good agreement with the experiment (Table 2 and S2–S7†). In particular, an excellent linear correlation was found between the experimental and theoretical HLGs (Fig. S29†). At the PCM(CH2Cl2)/TD-B3LYP/6-31G(d,p) level of theory, a number of partially allowed transitions (f < 0.09) are predicted for 5a (400–570 nm), engaging excitations between three highest occupied and four lowest unoccupied MO levels. Similarly weak absorptions in the visible region are found in the TD spectrum of 9a. In contrast, excitations to levels higher than LUMO provide no significant contributions to the calculated vis-NIR transitions of 5a2+ and other cationic species. 5a2+, 9b+, and 9b2+ are shown to have very similar optical band-gaps with a lower oscillator strength predicted for the latter species, as experimentally observed. Additionally, a significantly smaller HLG is correctly predicted for 5a+.
| Absorptiona | Emissiona | |||||
|---|---|---|---|---|---|---|
| λ max (ε) nm (103 M−1 cm−1) | HLGb eV | λ 0–0 nm | Stokes shift cm−1 | Φ F | τ F ns | |
| a In dichloromethane solutions. b Optical HOMO–LUMO gap determined from the lowest energy band. c Fluorescence quantum yield. d Fluorescence decay time. e Shoulder. f Determined from emission and excitation spectra at 77 K. | ||||||
| 5a | 595 (0.9), 550 (2.4), 520 (2.7), 450e, 430 (11.8) | 2.08 | 630 (615) | 678 | 0.005 | 4.1 |
| 5a+ | 2050 (5.9), 1560 (6.0), 1285 (9.3), 1120 (8.8), 805 (7.9), 658 (6.8), 516 (10.8), 484 (12.5) | 0.60 | ||||
| 5a2+ | 1525 (10.3), 1270 (10.3), 1080 (18.3), 656 (12.8), 566 (15.3), 520 (20.0), 498 (20.5) | 0.81 | ||||
| 9b | 475e | 2.61 | ∼520e (510) | 926f | 0.006 | 5.9 |
| 9b+ | 1540, 1275, 1090, 712, 647, 586 | 0.81 | ||||
| 9b2+ | 1530 (1.5), 1140 (24.0), 1020 (17.4), 705 (13.6), 572 (10.6), 439 (22.8) | 0.81 | ||||
| Speciesa/parameter | Charge | |||
|---|---|---|---|---|
| n = 0 | n = 1 | n = 2 | ||
| a PCM(CHCl3)/B3LYP/6-31G(d,p) geometries. b PCM(DCM)/TD-B3LYP/6-31G(d,p). c HOMO–LUMO gap. d Oscillator strength. e GIAO-B3LYP/6-31G(d,p). | ||||
| 5an+ | ||||
| Distance [Å] | Cα–Cβ | 1.387–1.402 | 1.393–1.418 | 1.405–1.435 |
| Cβ–Cβ | 1.426–1.445 | 1.418–1.428 | 1.407–1.417 | |
| Cα–N | 1.376–1.416 | 1.372–1.416 | 1.368–1.415 | |
| Cα–Cα | 1.441–1.456 | 1.432–1.438 | 1.418–1.423 | |
| HLG,b,c eV (nm) | 2.19 (567) | 0.71 (1748) | 0.93 (1332) | |
| f , | 0.009 | 0.106 | 0.156 | |
|
||||
| 5n+ | ||||
| NICS(1)e [ppm] | A, B | −8.5, 6.2 | −19.7, −10.1 | |
| C–G | −0.9 to 5.5 | −12.6 to −14.0 | ||
| H, I | −9.9, −8.3 | −10.2, −21.1 | ||
| J–N | −10.2 to −6.8 | −14.7 to −11.1 | ||
| HLG,b,c eV (nm) | 2.20 (564) | 0.78 (1590) | 1.00 (1242) | |
| f , | 0.007 | 0.061 | 0.092 | |
|
||||
| 9bn+ | ||||
| HLG,b,c eV (nm) | 2.62 (472) | 0.92 (1348) | 0.99 (1255) | |
| f , | 0.006 | 0.282 | 0.019 | |
|
||||
| 9n+ | ||||
| HLG,b,c eV (nm) | 2.63 (471) | 1.04 (1194) | 1.05 (1180) | |
| f , | 0.004 | 0.156 | 0.017 | |
|
||||
| 1n+ | ||||
| HLG,b,c eV (nm) | 2.41 (515) | 1.05 (1175) | 1.35 (918) | |
| f , | 0.000 | 0.083 | 0.123 | |
Because of the different substitution patterns found in 5a and the reported derivatives of 1,3,4 the influence of 5-6-7 fusion and peripheral expansion on the optical bandgaps of pyrrole-based nanographenes cannot be precisely separated from substituent effects. The TD-DFT band gap calculated for the unsubstituted 52+ (1.00 eV) is smaller by 0.35 eV than in 12+ (Table 2). This decrease is noteworthy given that it is caused by the addition of only four centers to a 36-atom π-conjugated system. The HLG differences are progressively smaller between the lower oxidation levels of 5 and 1 (0.27 eV for the radical cation, and 0.21 eV for the neutral state).
Molecular orbital diagrams obtained for 9b2+ reveal that the highest occupied levels, HOMO and H−1, are very close in energy (Fig. 5). The HOMO orbital is localized on the nanographene core and 9,26-Ar groups, whereas H−1 has the highest coefficients on the non-fused indole unit. No such HOMO localization is seen in the contiguously conjugated 5a2+. The lowest energy transition predicted by TD-DFT for 9b2+ is found to consist almost exclusively of the H−1 to LUMO excitation (for a similar case explored by TD-DFT see ref. 58). Because the latter orbital is localized on the nanographene, the transition has a considerable intramolecular charge-transfer (ICT) character,59 leading to the unexpectedly low optical band gap of 9b2+.
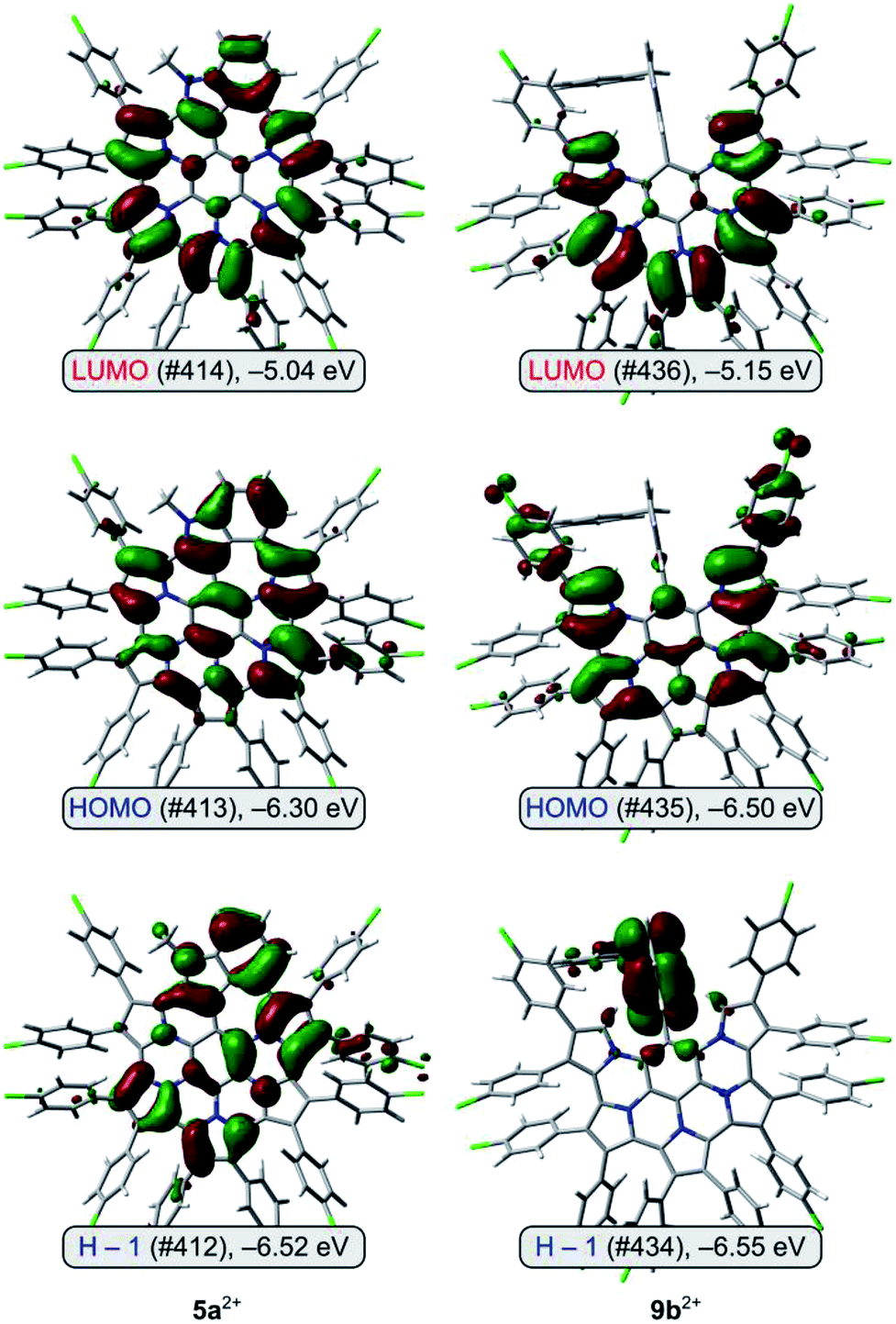 | ||
| Fig. 5 Frontier molecular orbitals for 5a2+ and 9b2+ (PCM(DCM)/TD-B3LYP/6-31G(d,p)). | ||
The effect of oxidation on the magnetic properties of 5 was visualized by probing GIAO shieldings 1 Å above the molecular plane (Fig. 6). The resulting NICS(1) maps60,61 show that in the neutral 5, the overall shielding can be described as a superposition of the ring currents induced in the seven constituent subunits (five pyrroles, one benzene, and one indole). The outer subunits create a deshielding zone in the interior of the nanographene (rings B–G), partly cancelling the shielding effect above the central benzene ring A. In the dication, the NICS(1) scan revealed strongly negative values above the entire ring system, in line with the experimentally observed enhancement of diatropicity. The shielding magnitude is particularly large above the central ring A and the indole ring I, and noticeably weaker above rings B and H. Such a variation of shielding values indicates that the peripheral conjugation in 52+ is dominated by 26-π-electron circuits, represented by the valence structure I-52+ (Fig. 6). These circuits include the indole ring I, while circumventing the peripheral nitrogen (ring H). Alternative circuit types II and III, which respectively exclude and include the entire indole moiety, apparently provide smaller contributions to the outer conjugation in 52+. The conjugation model proposed here is analogous to the typical description of macrocyclic aromaticity in oligopyrroles.62 In analogy to the trends found in the latter family of compounds, it may be proposed that the reduction of optical band gap, observed for 5a2+, is linked to the expansion of the peripheral circuit in 52+ (26 electrons) in comparison with the analogous circuit in 12+ (22 electrons). The closest structural analogy for this change can be found in cyclo[6]- and cyclo[7]pyrrole,63 which contain conjugation pathways isoelectronic with those in 12+ (22 e) and 52+ (26 e), respectively. In these cyclopyrroles, peripheral expansion was observed to reduce the optical band gap from 1.57 to 1.32 eV.
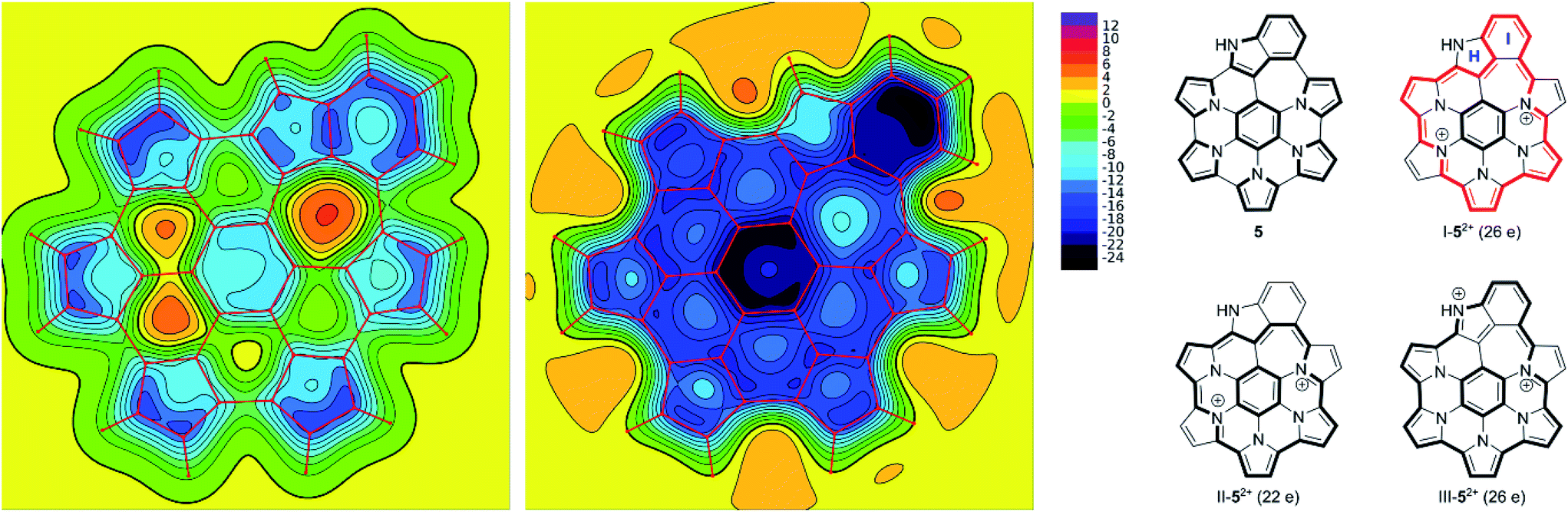 | ||
| Fig. 6 XY NICS(1) maps evaluated for 5 and 52+ (left and right, respectively). GIAO shieldings (shown with negative values) were calculated at the B3LYP/6-31G(d,p) level of theory 1 Å above the molecular plane. | ||
The diatropic ring current associated with conjugation pathway I should produce significant downfield shifts of peripheral protons (notably those attached to ring I), as observed experimentally for 5a2+. It can also be proposed that the upfield 13C shifts of ring-A carbons originate in part from the diatropic shielding contribution. By altering the effective bond orders, peripheral contributions such as I–III are expected to increase pyrrolic Cα–Cβ distances while reducing the Cβ–Cβ, Cα–N and interpyrrolic Cα–Cα bond lengths. Such changes are indeed consistently observed in the DFT-optimized geometries of 5a and 5a2+, with intermediate values of bond lengths observed for 5a+ (Table 1 and Fig. S21–S23†).
Conclusions
The synthesis of the 5-6-7 nanographene presented herein shows that complex heteroaromatic fusion patterns can be accessed by direct oxidative coupling of judiciously designed precursors. The extension of the HPHAC motif, comprising a 7-membered ring and a peripheral benzo unit, results in a considerable red shift of fluorescence emission in the neutral state and an analogous shift of the NIR absorption in the cationic states. The NIR emission of expanded nanographenes, coupled with their redox activity, might become an attractive feature in the context of potential applications, e.g. in the design of fluorescence probes,64,65 provided that these systems can be tuned to provide substantially higher fluorescence quantum yields. The changes of optical properties can be linked to the aromatic conjugation along the edge of the nanographene, which is specifically expanded by the addition of peripheral rings, and which produces a distinct ring current in the dicationic state. These observations clearly indicate that expansion of electron-rich heteroaromatic nanographenes is an efficient strategy for designing low-bandgap chromophores and fluorophores.Acknowledgements
The project was funded by the National Science Centre of Poland (decision DEC-2012/0707/EE/ST5ST5/0078100781). Quantum-chemical calculations were performed in the Centres for Networking and Supercomputing of Poznań and Wrocław.Notes and references
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC
.
-
Q. Ye, Z. Sun, C. Chi and J. Wu, in Graphene Chemistry, ed. D.-E. Jiang and Z. Chen, John Wiley & Sons, Ltd, 2013, pp. 393–424 Search PubMed
- M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 CrossRef CAS PubMed
- M. Takase, T. Narita, W. Fujita, M. S. Asano, T. Nishinaga, H. Benten, K. Yoza and K. Müllen, J. Am. Chem. Soc., 2013, 135, 8031–8040 CrossRef CAS PubMed
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30–31 CrossRef CAS PubMed
- V. V. Diev, K. Hanson, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, Angew. Chem., Int. Ed., 2010, 49, 5523–5526 CrossRef CAS PubMed
- V. V. Diev, C. W. Schlenker, K. Hanson, Q. Zhong, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, J. Org. Chem., 2012, 77, 143–159 CrossRef CAS PubMed
- C. Jiao, N. Zu, K.-W. Huang, P. Wang and J. Wu, Org. Lett., 2011, 13, 3652–3655 CrossRef CAS PubMed
- D. Myśliwiec, B. Donnio, P. J. Chmielewski, B. Heinrich and M. Stępień, J. Am. Chem. Soc., 2012, 134, 4822–4833 CrossRef PubMed
- D. Myśliwiec and M. Stępień, Angew. Chem., Int. Ed., 2013, 52, 1713–1717 CrossRef PubMed
- E. Gońka, P. J. Chmielewski, T. Lis and M. Stępień, J. Am. Chem. Soc., 2014, 136, 16399–16410 CrossRef PubMed
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed
- P.-A. Bouit, A. Escande, R. Szűcs, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524–6527 CrossRef CAS PubMed
- K. Imamura, K. Takimiya, T. Otsubo and Y. Aso, Chem. Commun., 1999, 1859–1860 RSC
- K. Y. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and V. G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367–7370 CrossRef CAS PubMed
- A. A. Gorodetsky, C.-Y. Chiu, T. Schiros, M. Palma, M. Cox, Z. Jia, W. Sattler, I. Kymissis, M. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7909–7912 CrossRef CAS PubMed
- C. J. Martin, B. Gil, S. D. Perera and S. M. Draper, Chem. Commun., 2011, 47, 3616–3618 RSC
- S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed
- L. Chen, S. R. Puniredd, Y.-Z. Tan, M. Baumgarten, U. Zschieschang, V. Enkelmann, W. Pisula, X. Feng, H. Klauk and K. Müllen, J. Am. Chem. Soc., 2012, 134, 17869–17872 CrossRef CAS PubMed
- C.-Y. Chiu, B. Kim, A. A. Gorodetsky, W. Sattler, S. Wei, A. Sattler, M. Steigerwald and C. Nuckolls, Chem. Sci., 2011, 2, 1480–1486 RSC
- Q. Xiao, T. Sakurai, T. Fukino, K. Akaike, Y. Honsho, A. Saeki, S. Seki, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2013, 135, 18268–18271 CrossRef CAS PubMed
- Q. Zhang, H. Peng, G. Zhang, Q. Lu, J. Chang, Y. Dong, X. Shi and J. Wei, J. Am. Chem. Soc., 2014, 136, 5057–5064 CrossRef CAS PubMed
- X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535–538 CrossRef CAS PubMed
- T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS PubMed
- X.-Y. Wang, F.-D. Zhuang, R.-B. Wang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764–3767 CrossRef CAS PubMed
- M. Krieg, F. Reicherter, P. Haiss, M. Ströbele, K. Eichele, M.-J. Treanor, R. Schaub and H. F. Bettinger, Angew. Chem., Int. Ed., 2015, 54, 8284–8286 CrossRef CAS PubMed
- S. M. Draper, D. J. Gregg and R. Madathil, J. Am. Chem. Soc., 2002, 124, 3486–3487 CrossRef CAS PubMed
- Y. Fogel, M. Kastler, Z. Wang, D. Andrienko, G. J. Bodwell and K. Müllen, J. Am. Chem. Soc., 2007, 129, 11743–11749 CrossRef CAS PubMed
- D. Wu, X. Feng, M. Takase, M. C. Haberecht and K. Müllen, Tetrahedron, 2008, 64, 11379–11386 CrossRef CAS
- D. Wu, W. Pisula, M. C. Haberecht, X. Feng and K. Müllen, Org. Lett., 2009, 11, 5686–5689 CrossRef CAS PubMed
- D. Wu, W. Pisula, V. Enkelmann, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 9620–9621 CrossRef CAS PubMed
- J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem., Int. Ed., 2010, 49, 8209–8213 CrossRef CAS PubMed
- Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed
- A. Graczyk, F. A. Murphy, D. Nolan, V. Fernández-Moreira, N. J. Lundin, C. M. Fitchett and S. M. Draper, Dalton Trans., 2012, 41, 7746–7754 RSC
- A. L. Pinardi, J. I. Martínez, A. Jančařík, I. G. Stará, I. Starý, M. F. López, J. Méndez and J. Á. Martín-Gago, Chem. Commun., 2014, 50, 1555–1557 RSC
- L. P. Wijesinghe, B. S. Lankage, G. M. Ó. Máille, S. D. Perera, D. Nolan, L. Wang and S. M. Draper, Chem. Commun., 2014, 50, 10637–10640 RSC
- B. He, A. B. Pun, L. M. Klivansky, A. M. McGough, Y. Ye, J. Zhu, J. Guo, S. J. Teat and Y. Liu, Chem. Mater., 2014, 26, 3920–3927 CrossRef CAS
- B. He, J. Dai, D. Zherebetskyy, T. L. Chen, B. A. Zhang, S. J. Teat, Q. Zhang, L. Wang and Y. Liu, Chem. Sci., 2015, 6, 3180–3186 RSC
- K. T. Kim, J. W. Lee and W. H. Jo, Macromol. Chem. Phys., 2013, 214, 2768–2773 CrossRef CAS
- C. Bronner, S. Stremlau, M. Gille, F. Brauße, A. Haase, S. Hecht and P. Tegeder, Angew. Chem., Int. Ed., 2013, 52, 4422–4425 CrossRef CAS PubMed
- J. Cai, C. A. Pignedoli, L. Talirz, P. Ruffieux, H. Söde, L. Liang, V. Meunier, R. Berger, R. Li, X. Feng, K. Müllen and R. Fasel, Nat. Nanotechnol., 2014, 9, 896–900 CrossRef CAS PubMed
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed
- H. A. M. Biemans, C. Zhang, P. Smith, H. Kooijman, W. J. J. Smeets, A. L. Spek and E. W. Meijer, J. Org. Chem., 1996, 61, 9012–9015 CrossRef CAS PubMed
- M. Lazerges, M. Jouini, P. Hapiot, P. Guiriec and P.-C. Lacaze, J. Phys. Chem. A, 2003, 107, 5042–5048 CrossRef CAS
- K. Yamamoto, T. Harada, M. Nakazaki, T. Naka, Y. Kai, S. Harada and N. Kasai, J. Am. Chem. Soc., 1983, 105, 7171–7172 CrossRef CAS
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed
- A. Pradhan, P. Dechambenoit, H. Bock and F. Durola, J. Org. Chem., 2013, 78, 2266–2274 CrossRef CAS PubMed
- K. Y. Cheung, X. Xu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 3910–3914 CrossRef CAS PubMed
- X. Li, Y. Zhu, J. Shao, L. Chen, S. Zhao, B. Wang, S. Zhang, Y. Shao, H.-L. Zhang and X. Shao, Angew. Chem., Int. Ed., 2015, 54, 267–271 CrossRef CAS PubMed
- C.-Y. He, S. Fan and X. Zhang, J. Am. Chem. Soc., 2010, 132, 12850–12852 CrossRef CAS PubMed
- D. H. R. Barton, J. Kervagoret and S. Z. Zard, Tetrahedron, 1990, 46, 7587–7598 CrossRef CAS
- J. L. Bullington, R. R. Wolff and P. F. Jackson, J. Org. Chem., 2002, 67, 9439–9442 CrossRef CAS PubMed
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed
- F. A. Bell, A. Ledwith and D. C. Sherrington, J. Chem. Soc. C, 1969, 2719–2720 RSC
- B. Evans and K. M. Smith, Tetrahedron Lett., 1977, 18, 3079–3082 CrossRef
- T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC
- Y. Yamanoi, K. Takahashi, T. Hamada, N. Ohshima, M. Kurashina, Y. Hattori, T. Kusamoto, R. Sakamoto, M. Miyachi and H. Nishihara, J. Mater. Chem. C, 2015, 3, 4316–4320 RSC
- X.-H. Duan, X.-Y. Li, R.-X. He and X.-M. Cheng, J. Chem. Phys., 2005, 122, 084314 CrossRef PubMed
- Q. T. Zhang and J. M. Tour, J. Am. Chem. Soc., 1998, 120, 5355–5362 CrossRef CAS
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed
- A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed
- M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288–4340 CrossRef PubMed
- T. Köhler, D. Seidel, V. Lynch, F. O. Arp, Z. Ou, K. M. Kadish and J. L. Sessler, J. Am. Chem. Soc., 2003, 125, 6872–6873 CrossRef PubMed
-
X. Zhang, S. Bloch, W. Akers and S. Achilefu, in Current Protocols in Cytometry, John Wiley & Sons, Inc., 2001 Search PubMed
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectra of new compounds, and details of computational studies. See DOI: 10.1039/c5sc03280f |
| This journal is © The Royal Society of Chemistry 2016 |