
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnesium-catalysed nitrile hydroboration†
Catherine
Weetman
,
Mathew D.
Anker
,
Merle
Arrowsmith
,
Michael S.
Hill
*,
Gabriele
Kociok-Köhn
,
David J.
Liptrot
and
Mary F.
Mahon
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@Bath.ac.uk
First published on 20th October 2015
Abstract
A β-diketiminato n-butylmagnesium complex is presented as a selective precatalyst for the reductive hydroboration of organic nitriles with pinacolborane (HBpin). Stoichiometric reactivity studies indicate that catalytic turnover ensues through the generation of magnesium aldimido, aldimidoborate and borylamido intermediates, which are formed in a sequence of intramolecular nitrile insertion and inter- and intramolecular B–H metathesis events. Kinetic studies highlight variations in mechanism for the catalytic dihydroboration of alkyl nitriles, aryl nitriles bearing electron withdrawing (Ar(EWG)CN) and aryl nitriles bearing electron donating (Ar(EDG)CN) substitution patterns. Kinetic isotope effects (KIEs) for catalysis performed with DBpin indicate that B–H bond breaking and C–H bond forming reactions are involved in the rate determining processes during the dihydroboration of alkyl nitriles and Ar(EDG)CN substrates, which display divergent first and second order rate dependences on [HBpin] respectively. In contrast, the hydroboration of Ar(EWG)CN substrates provides no KIE and HBpin is not implicated in the rate determining process during catalysis. Irrespective of these differences, a common mechanism is proposed in which the rate determining steps are deduced to vary through the establishment of several pre-equilibria, the relative positions of which are determined by the respective stabilities of the dimeric and monomeric magnesium aldimide and magnesium aldimidoborate intermediates as a result of adjustments to the basicity of the nitrile substrate. More generally, these observations indicate that homogeneous processes performed under heavier alkaline earth catalysis are likely to demonstrate previously unappreciated mechanistic diversity.
Introduction
The reduction of organic nitriles to primary amines is an essential component of many industrial processes (e.g. the production of dyes, polyesters, agrochemicals and as precursors for pharmaceutical compounds).1 Catalytic nitrile hydrogenation may be achieved under heterogeneous conditions. These latter processes, however, are typically poorly selective and also result in the unwanted formation of imine and secondary amine side products at the high temperatures required.2 While their reliance on the use of poorly abundant and/or toxic precious metals is a further indicator of the unsustainability of these heterogeneous systems, it is notable that the development of well-defined solution-phase catalysis is limited to a handful of reports which deploy similarly expensive species derived from heavy precious metals.3,4 Although the reduction of nitriles may also be achieved through the use of stoichiometric quantities of main group reducing agents such as LiAlH4 and NaBH4,5 the flammable nature of these reagents, coupled with large amounts of inorganic waste by-products, again renders them unattractive. More recent reports have shown that reduction may be accomplished using amine borane reagents,6 whilst other novel hydrogenation methods have exploited the use of catalytic amounts of ‘frustrated’ Lewis pairs to provide the first metal-free systems to reduce nitriles, albeit under rather energetic (120 °C) reaction conditions.7Whilst nitrile hydrogenation provides a direct route to the desired amine product, reductive hydrosilylation or hydroboration can be advantageous in their provision of further functionality to the resultant amine.8 Although the catalytic hydroboration of a wide range of multiply-bonded substrates has been achieved, only a handful of nitrile hydroboration reactions have been devised and all but one of these previous reports described non-catalysed H–B addition and required the use of more activated and less discriminating borane reagents.9 A unique case of a catalysed addition, therefore, has been provided by Nikonov's report of the catalytic hydroboration of nitriles using 5 mol% of the Mo(IV) imido–hydrido complex (I) with catecholborane (HBcat).10 With this system acetonitrile and benzonitrile were reduced to the 1,1-bis(boryl)amine products, which were themselves shown to undergo chemoselective coupling with aldehydes, R′C(O)H, to afford imines RCH2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)R′.
C(H)R′.
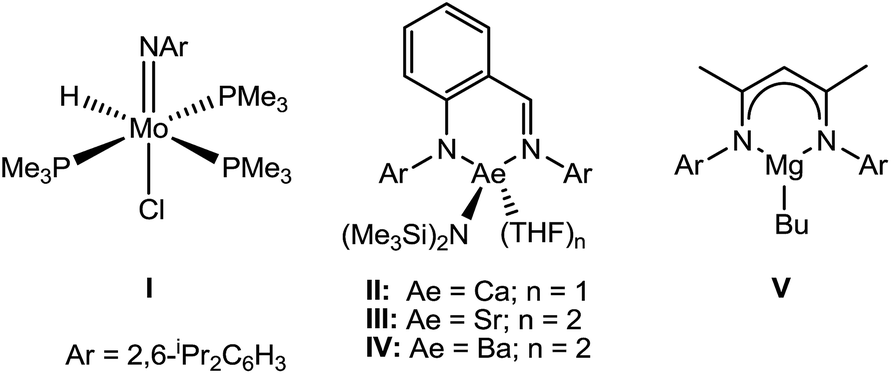
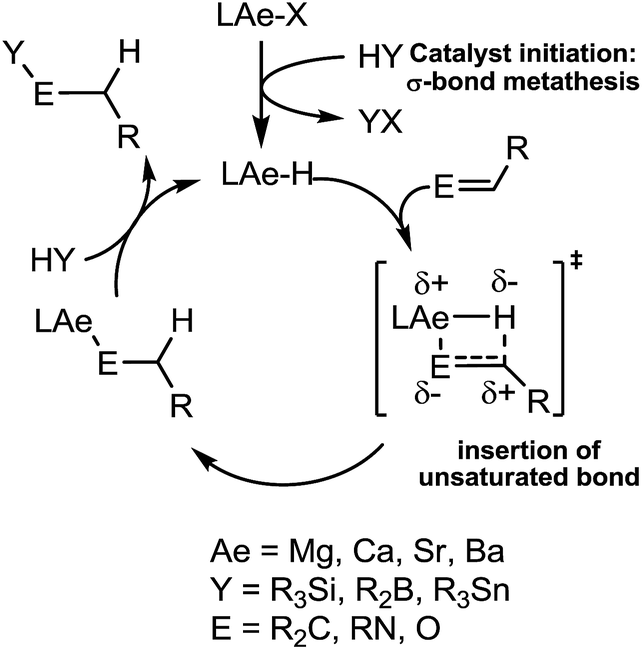
Our own research has focussed on the development of a homogeneous catalytic chemistry for complexes, LAeX (Ae = Mg, Ca, Sr and Ba; L = unreactive spectator ligand; X = reactive substituent), derived from the heavier alkaline-earth elements.11 The negligible toxicity and high natural abundance of calcium and magnesium (the fourth and sixth most abundant lithospheric elements respectively) in particular designate species of this type as environmentally benign and sustainable. Although these are primary motivating factors for the development of this chemistry, an additional major concern is a deconvolution of basic reactivity patterns for this relatively understudied family of elements. Based upon an immutable +2 oxidation state and effectively ionic ligand and substrate binding under catalytic conditions, a level of ‘lanthanide mimetic’ behaviour was initially assumed. Consequently, a wide variety of heterofunctionalisation catalyses, predicated on sequences of regioselective and polarisation-dependent sigma-bond metathesis and insertion events have now been described (Schemes 1 and 2).11 A majority of detailed studies have focussed on the intramolecular hydroamination/cyclisation of aminoalkenes as an appropriate baseline reaction that is well-precedented in homogeneous 4f-element centred catalysis.12,13 Within this one reaction type alone, distinct variations which occur with changing group 2 atomic weight have been rationalised as a consequence of the substantial adjustments to ionic radius and cation charge density on descending the group. While an expectation of some rate variation with alkaline earth identity may appear retrospectively obvious, it is also apparent that a generalised framework accounting for the intrinsic or relative reactivity of the individual elements is yet to emerge. Recent work by Sarazin and Carpentier,14 for example, employing the anilido-imine species II–IV has indicated a general increase in catalytic hydroamination capability with increasing group 2 atomic weight (i.e. Ca < Sr < Ba) whereas our own studies of the homoleptic bis(trimethylsilyl)amides [Ae{N(SiMe3)2}2]2 [Ae = Mg, Ca, Sr, Ba] have evidenced more subtle kinetic dependences upon the identity and characteristics of the Ae2+ cation.11 In such cases we have reasoned that the facility for reaction is determined by an intricate interplay of factors of which complex nuclearity and access to the group 2 centre, in concert with the basicity of the pre-catalytic reagent and an ability to polarise in-coming substrate molecules, are essential to successfully traverse the various transition states involved in the catalytic process.
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
We have previously reported the use of the magnesium alkyl precatalyst (V) for the hydroboration of aldehydes and ketones,15 as well as pyridines,16 aldimines, ketimines17 and CO2 (ref. 18a) with HBpin. Most recently we have described a unique case of isonitrile dihydroboration through use of the same borane and magnesium reagents, which provides access to 1,2-diborylated amines.18b In all cases this reactivity was deduced to be derived from the rapid and unobservable generation of magnesium hydride species through Mg–C/B–H σ-bond metathesis and a sequence of polarised insertion and further metathesis steps (cf.Scheme 2). In common with a recent report of magnesium-catalysed ester reduction with HBpin,19a these reactions are also observed to occur through the assembly of alkoxy- or amido-hydrido borate derivatives, in which the hydride reducing equivalent is already present in the proposed catalyst resting state. During the course of these studies we have previously noted that cyanopyridines underwent double hydroboration of the nitrile C–N triple bond to form N,N-(diboryl)aminopyridines.16 The reaction sequence depicted in Scheme 3 may, thus, be envisaged as a generalised means to effect a similar dihydroboration of organic nitriles. In this case the formation of the 1,1-diborylated amine product (F) requires the consumption of two molecules of HBpin and the intermediacy of an initial magnesium-bound imide (A), a borylated imine (B) as the product of initial B–H/Mg–N metathesis and an amide species (D) formed by formal Mg–H insertion of the borylimine (B). The additional complexity introduced by the requirement for two-fold nitrogen functionalisation, thus, raises a number of issues regarding validity of such a stepwise process and the identity of other potential intermediates such as the borate species shown as C and E shown in Scheme 3. (vide infra).
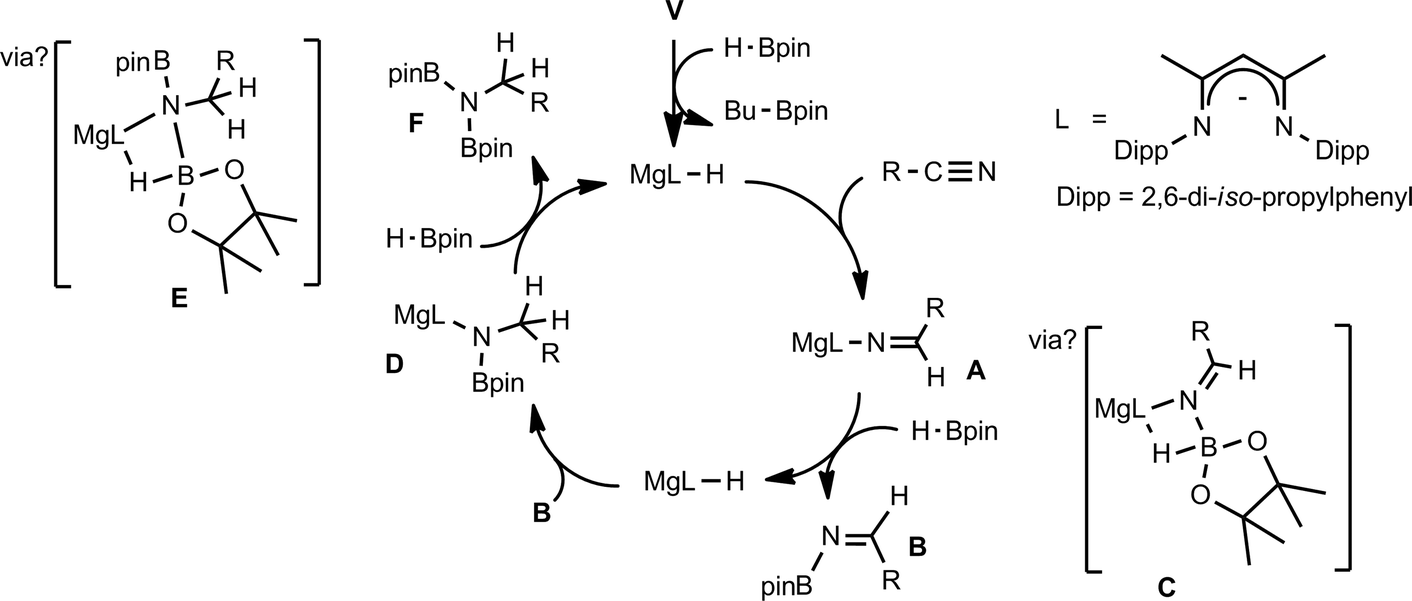 | ||
| Scheme 3 Prototype mechanism for dihydroboration of nitriles. | ||
With these broader considerations in mind we now describe a magnesium-centred protocol for the hydroboration of organic nitriles with the unactivated borane reagent pinacolborane (HBpin). Our selection of a single β-diketiminato magnesium precatalyst (V), which is stable to both Schlenk-type redistribution equilibria and to chemical degradation under conditions of the catalysis, allows for an assessment of the effects of gradual substrate adjustment which suggests that every reaction must be treated on its merits.
Results
Catalytic reaction scope
An initial catalytic reaction using 10 mol% of V with respect to one equivalent of propionitrile with 2 molar equivalents of HBpin at room temperature evidenced slow consumption of the reagents while heating of an identical reaction mixture at 60 °C provided quantitative conversion to the 1,1-bis(boryl)amine product within 30 minutes. The clean formation of this new species was clearly apparent through the emergence of a (2H) triplet methylene resonance in the 1H NMR spectrum at δ 3.42 ppm synchronous with a new singlet signal in the 11B NMR spectrum at δ 29.5 ppm. In contrast an analogous reaction performed without the addition of V evidenced less than 5% consumption of the propionitrile substrate when heated at 60 °C for 16 hours. Encouraged by this result the conditions of the catalytic study were extended to the successful di-hydroboration of the range of alkyl and aryl nitriles summarised in Table 1.|
|
||||
|---|---|---|---|---|
| Entry | R | Time (h) | NMR yielda (%) | Isolated yielda (%) |
| a All NMR reactions were carried out in C6D6 whilst scale-up reactions were carried out in toluene then isolated from hexanes. | ||||
| 1 | Et | 0.5 | >99 | 70 |
| 2 | i-Pr | 1 | 93 | 96 |
| 3 | t-Bu | 5.5 | 97 | 54 |
| 4 | Cy | 1 | 94 | 75 |
| 5 | C6H5 | 12 | 92 | 56 |
| 6 | o-(Me)C6H4 | 15 | 86 | 73 |
| 7 | m-(Me)C6H4 | 15 | 87 | 81 |
| 8 | m-(F)C6H4 | 14 | 88 | 57 |
| 9 | m-(MeO)C6H4 | 15 | 90 | 60 |
| 10 | p-(Me)C6H4 | 13 | 91 | 72 |
| 11 | p-(F)C6H4 | 12 | 94 | 61 |
| 12 | p-(Cl)C6H4 | 12 | 93 | 59 |
| 13 | p-(CF3)C6H4 | 12.5 | 89 | 76 |
| 14 | p-(MeO)C6H4 | 13.5 | 88 | 58 |
| 15 | Ph2CH | 30 | 75 | 43 |
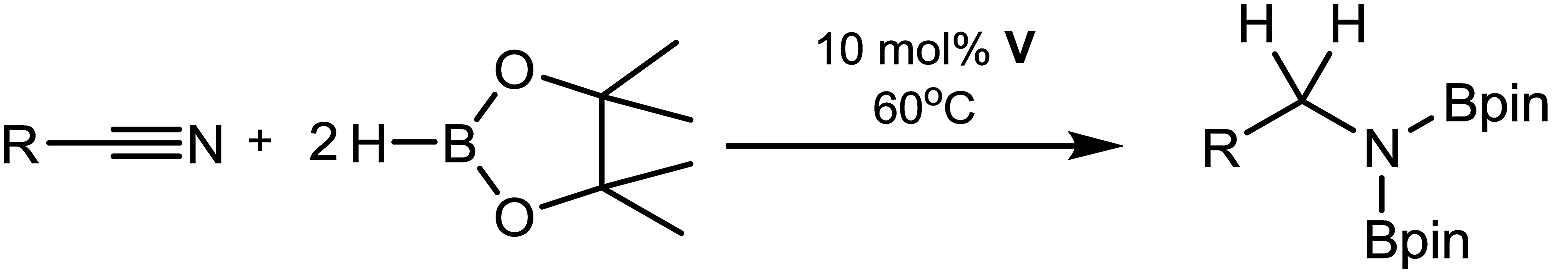
In common with our earlier reports of the magnesium-catalysed hydroboration of aldehydes, ketones, imines and isonitriles,15–17,18b any increase in the overall steric demands of the substrate resulted in a longer reaction time. This is most readily appreciated through consideration of the effects of increasing steric demands upon substitution of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-bound carbon centre of aliphatic nitriles wherein a 10-fold increase in reaction time is observed across the transition from propionitrile (entry 1) to iso-propylnitrile (entry 2) to tert-butylnitrile (entry 3). Aryl nitriles (entries 5–15) also provided clean reactions with no side products, but with markedly longer reaction times. Notably varied reaction times were also required with changing aryl substituent patterns. Although in the case of o-tolunitrile (entry 6) the slower reaction may again be attributed to an increase in substituent steric hindrance proximal to the reaction centre, any minor variability across the range of meta-and para-substituted arylnitriles is more realistically attributed to electronic adjustments across the pi-conjugated substrate or intermediate structure (vide infra). The identities of the products from this catalysis were confirmed by 1H, 11B and 13C NMR spectroscopy and a single crystal X-ray diffraction analysis performed upon the product (compound 1) isolated from the catalytic dihydroboration of propionitrile. The outcome of this latter experiment is shown in Fig. 1, while selected bond lengths and angles and details of the X-ray analysis are provided in Tables 2–4 respectively. The N1–C1 bond length of 1.497(2) Å is consistent with the expected twofold reduction of the nitrile while the N1–C1–C2 angle of 109.1° is a clear indication of sp3 hybridisation at the C1 carbon atom.
C-bound carbon centre of aliphatic nitriles wherein a 10-fold increase in reaction time is observed across the transition from propionitrile (entry 1) to iso-propylnitrile (entry 2) to tert-butylnitrile (entry 3). Aryl nitriles (entries 5–15) also provided clean reactions with no side products, but with markedly longer reaction times. Notably varied reaction times were also required with changing aryl substituent patterns. Although in the case of o-tolunitrile (entry 6) the slower reaction may again be attributed to an increase in substituent steric hindrance proximal to the reaction centre, any minor variability across the range of meta-and para-substituted arylnitriles is more realistically attributed to electronic adjustments across the pi-conjugated substrate or intermediate structure (vide infra). The identities of the products from this catalysis were confirmed by 1H, 11B and 13C NMR spectroscopy and a single crystal X-ray diffraction analysis performed upon the product (compound 1) isolated from the catalytic dihydroboration of propionitrile. The outcome of this latter experiment is shown in Fig. 1, while selected bond lengths and angles and details of the X-ray analysis are provided in Tables 2–4 respectively. The N1–C1 bond length of 1.497(2) Å is consistent with the expected twofold reduction of the nitrile while the N1–C1–C2 angle of 109.1° is a clear indication of sp3 hybridisation at the C1 carbon atom.
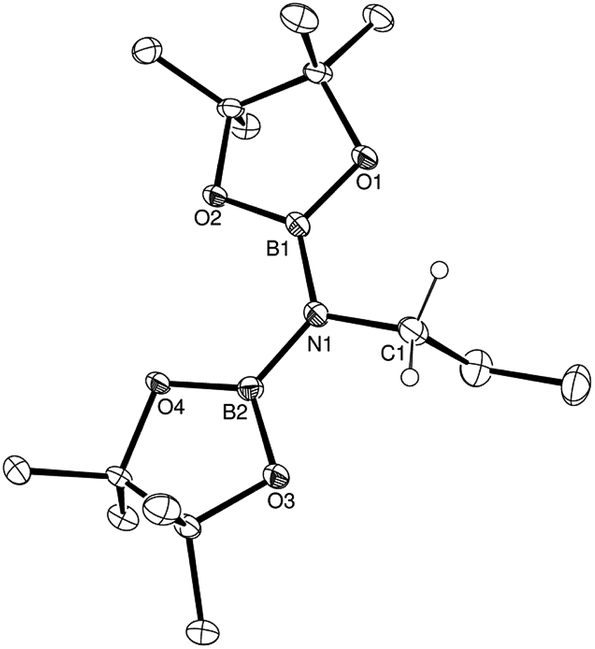 | ||
| Fig. 1 ORTEP representation of the structure of compound 1 (30% probability ellipsoids). Hydrogen atoms, except for those attached to the C1 methylene carbon centre, are omitted for clarity. | ||
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| a N3–B1. b Mg2–N4 2.046(4). c Mg2–N4 2.0467(4). d Mg1–N5. e Mg2–O2 1.953(4). f N(5)–C(30). g N6–C79 1.114(7). | |||||
| N1–B1 | 1.423(2) | — | — | 1.572(2)a | — |
| N1–B2 | 1.430(2) | — | — | — | — |
| N1–C1 | 1.497(2) | — | — | — | — |
| Mg1–N1 | — | 2.0957(16) | 2.1003(16) | 2.0334(13) | 2.033(5)b |
| Mg1–N2 | — | 2.0931(17) | 2.0840(16) | 2.0361(13) | 2.057(4)c |
| Mg1–N3 | — | 2.1062(16) | 2.0951(16) | 2.0825(13) | 1.996(6)d |
| Mg1–O1 | — | — | — | 1.9714(11) | 1.974(4)e |
| N3–C30 | — | 1.263(2) | 1.271(2) | 1.266(2) | 1.188(8)f,g |
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| a B1–N3–C30. b N3–Mg2–N4 93.44(18). c N1–Mg1–N5. d N3–Mg2–N6 103.53(19). e N2–Mg1–N5. f N4–Mg2–N6 105.5(2). g O1–Mg1–N5. h O2–Mg2–N6 97.01(13). | |||||
| B1–N1–B2 | 126.15(15) | — | — | — | — |
| B1–N1–C1 | 117.15(14) | — | — | — | — |
| B2–N1–C1 | 116.41(14) | — | — | 120.10(13)a | — |
| N2–Mg1–N1 | — | 90.19(6) | 90.88(6) | 95.26(5) | 93.76(18)b |
| N1–Mg1–N3 | — | 119.63(6) | 124.69(7) | 132.20(6) | 105.7(2)c,d |
| N2–Mg1–N3 | — | 122.98(7) | 117.31(6) | 115.45(5) | 107.3(2)e,f |
| C30–N3–Mg1 | — | 136.56(13) | 137.55(13) | 150.55(12) | — |
| O1–Mg1–N3 | — | — | — | 71.91(5) | 103.31(19)g,h |
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Empirical formula | C15H31B2NO4 | C37H49MgN3O | C74H98Mg2N6O2 | C40H64MgBN3O2 | C102H132BMg2N6O2 |
| FW (g mol−1) | 311.03 | 1152.20 | 1152.20 | 654.06 | 1533.57 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | P21/n | P21/c | Cc |
| a (Å) | 6.3072(1) | 12.7410(2) | 22.7470(2) | 13.7058(2) | 12.3279(5) |
| b (Å) | 12.4643(3) | 12.9480(2) | 13.2640(1) | 13.1659(2) | 30.1877(9) |
| c (Å) | 12.5184(3) | 20.7600(3) | 24.9170(2) | 22.9509(3) | 26.5175(11) |
| α (°) | 101.1406(10) | 90 | 90 | 90 | 90 |
| β (°) | 100.1735(10) | 92.575(1) | 115.512(1) | 96.1770(10) | 102.622(2) |
| γ (°) | 103.8021(12) | 90 | 90 | 90 | 90 |
| Density (Mg m−3) | 1.133 | 1.118 | 1.128 | 1.055 | 1.058 |
| Z | 2 | 4 | 4 | 4 | 4 |
| μ (Mo Kα) (mm−1) | 0.078 | 0.083 | 0.084 | 0.077 | 0.074 |
| Reflections collected | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 086 086 |
58058 |
113992 |
76184 |
35060 |
| Independent reflections | 3190 | 7818 | 15492 |
9431 | 14727 |
| R int | 0.0465 | 0.0658 | 0.0685 | 0.0814 | 0.1401 |
| R 1, wR2 [I > 2σ(I)] | 0.0495, 0.1087 | 0.0569, 0.1247 | 0.0561, 0.1192 | 0.0498, 0.1103 | 0.0771, 0.1602 |
| R Indices (all data) | 0.0613, 0.1137 | 0.0837, 0.1347 | 0.1025, 0.1405 | 0.0894, 0.1273 | 0.1605, 0.1978 |
Stoichiometric reactions
To provide further insight into the course of these successful catalytic reactions and the viability of intermediate species such as those shown as A–E in Scheme 3, a series of stoichiometric reactions were undertaken. Monitoring of NMR scale reactions performed with t-BuCN was particularly informative due to the ready discrimination of the diagnostic singlet tert-butyl resonances in the 1H NMR spectra. We have earlier reported that V and HBpin react to form a magnesium hydride/borohydride species along with BuBpin within minutes at room temperature.16 Repetition of this initial reaction with subsequent addition of an equimolar equivalent of t-BuCN provided complete CN insertion into the magnesium hydride bond, which was clearly apparent after heating for 12 hours at 60 °C through the formation of a single new compound designated as an aldimide derivative analogous to the species A depicted in Scheme 3. The production of this species was clearly evidenced by the emergence of a downfield singlet (1H by integration) resonance at δ 7.84 ppm assigned as the aldimide methine signal and a set of resonances associated with a single new β-diketiminate ligand environment in the 1H NMR spectrum (see Scheme S1†). Although attempts to isolate this aliphatic aldimide species were unsuccessful, the veracity of this deduction was supported by subsequent analogous reactions performed with m- and p-MeOC6H4CN. While both of the resultant compounds (2 and 3) again displayed characteristic 1H aldimide methine singlet resonances (2, δ 8.63; 3δ 8.58 ppm) in their respective 1H NMR spectra, in these latter cases crystallisation from benzene solutions also provided samples of both compounds suitable for single crystal X-ray diffraction analysis. The results of these analyses are presented in Fig. 2a and b while selected bond lengths and angles and details of the X-ray analyses are provided in Tables 2–4 respectively. Although compounds 2 and 3 are the first magnesium aldimide complexes to be characterised in the solid state, their structures are otherwise analogous to a previously described calcium benzaldimide derivative supported by the identical β-diketiminate ligand.20 As was the case for this heavier congener, the structures of compounds 2 and 3 are dimeric with bridging Mg–N–Mg interactions provided by the aldimide ligands. Whereas the asymmetric unit of compound 2 comprises half of a dimer which straddles a crystallographic inversion centre, each half of the dimeric unit of compound 3 is unique. The gross features of both structures are, however, very similar wherein the magnesium centres are bridged by unsymmetrical Mg–N–Mg interactions. The magnesium to aldimide nitrogen bond lengths in 2 and 3 [2: 2.1062(16), 2.0918(16); 3: 2.0840(16), 2.0882(16), 2.0951(16), 2.0974(16) Å] are shorter than the magnesium to amide contacts observed within topologically related dimeric magnesium benzylamide and pyrollidide derivatives [2.1251(16) and 2.117(2) Å respectively], both of which contain four-coordinate magnesium centres supported by the identical β-diketiminate ligand.21 The transition from formal sp3 to sp2 hybridisation at the bridging nitrogen centres in comparison to these previously described compounds is also reflected by the more obtuse Mg–N–Mg bond angles [90.73(6) and 91.58(9)° versus (2) 95.51(6)°, (3) 95.26(6) and 95.27(6)°] within compounds 2 and 3. As previously highlighted in Harder and co-workers' discussion of their calcium benzaldimide species,20 the planes described by the methoxyphenyl rings of both compounds 2 and 3 are virtually co-planar with the planes formed by the magnesium and the aldimide nitrogen and sp2 carbon centres. A similar in-plane conformation is also a common feature of dimeric diorganoaluminum benzaldimide species, [R2AlNC(Ph)H]2,22 and has been calculated for the model lithium compound, [LiNCH2]2, to lie some 17 kcal mol−1 lower in energy than the alternative conformation in which the methylene unit is perpendicular to the Li2N2 plane.23 In the current case, the angles subtended by the aryl rings and the various Mg–N–C(Ar)H planes within compounds 2 and 3 (2: 0.7°; 3: 11.3, 10.9°) indicate that this stabilisation is further enhanced by delocalisation across the aryl substituents. Although such data should be treated with caution due to the possibility of solid-state crystal packing and dispersion effects, we ascribe the greater co-planarity of 2 in comparison to 3 to the mesomeric influence of the respectively stabilizing 3-methoxy- and destabilising 4-methoxyphenyl substituents. While only providing a minor solid-state effect, we suggest that modulation of aldimide resonance stabilisation is a significant factor during the magnesium-catalysed hydroboration of the range of aryl nitriles listed in Table 1 (entries 5–14) (vide infra).
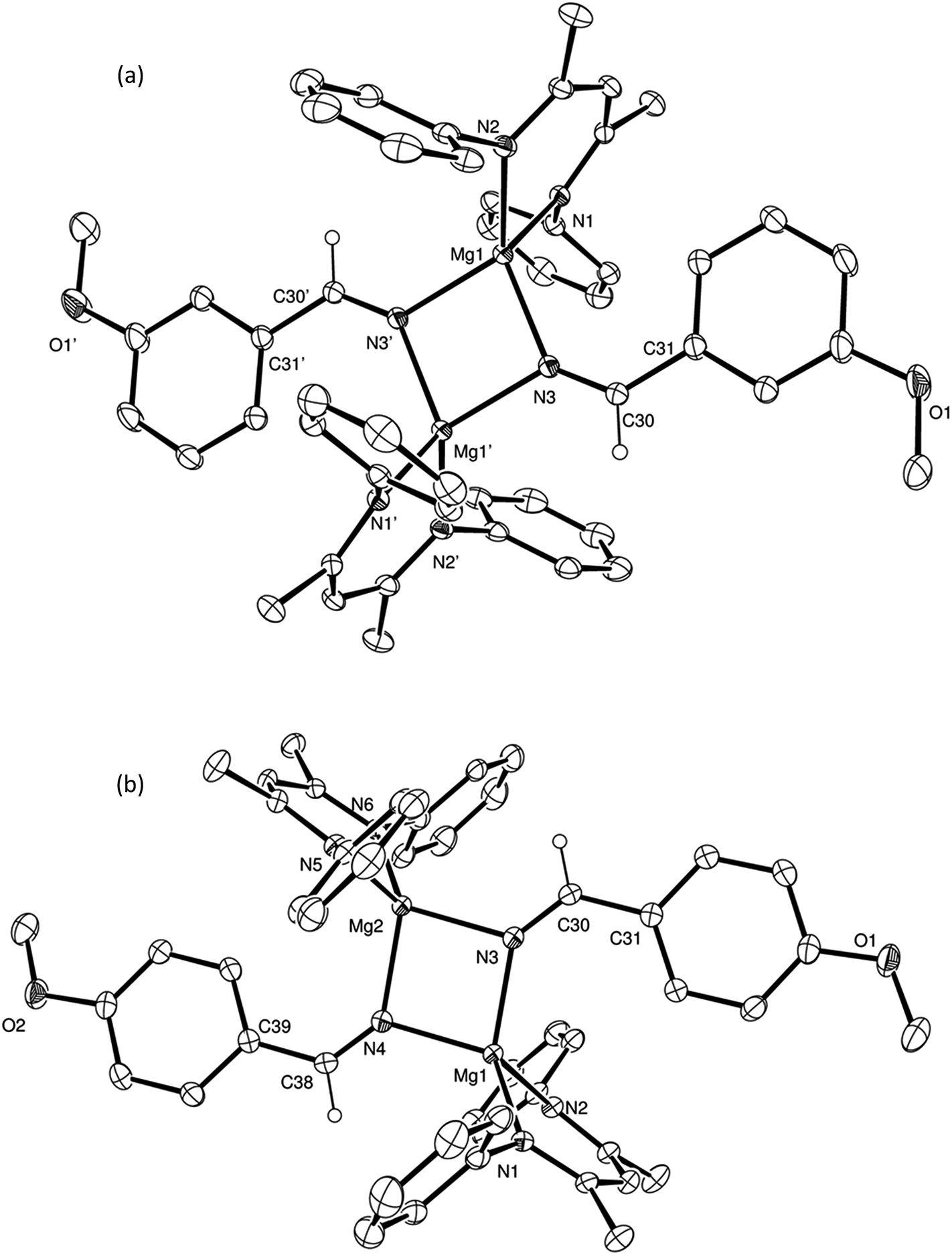 | ||
| Fig. 2 ORTEP representations of (a) compound 2 and (b) compound 3 (30% probability ellipsoids). Hydrogen atoms, except for those attached to the C(30) and, for 3, the C(38) methine carbon centres, and iso-propyl groups are omitted for clarity. Symmetry transformations used to generate equivalent (′) atoms in (b) −x + 1, −y + 1, −z + 1. | ||
Counter to the expectation illustrated in Scheme 3, addition of a further equivalent of HBpin to the imide product generated in the reaction with t-BuCN did not provide any initial evidence of B–H/Mg–N metathesis and production of a borylated imine derivative (shown as B in Scheme 3). Rather, this procedure yielded a single new species, assigned as a magnesium aldimidoborohydride (C in Scheme 3), characterised by a resonance at δ 9.27 ppm in the 11B NMR spectrum in which the integrity of the B–H bond was confirmed by a 1JBH coupling of 105 Hz; a value that is significantly reduced from the three-coordinate HBpin reagent (1JBH = 175 Hz) and more typical of four-coordinate boron.24 Scale up of this reaction in hexane and storage at −30 °C to avoid conversion to the boryl amide species (vide infra) allowed the isolation of the aldimidoborohydride derivative, compound 4, which was fully characterised by multinuclear NMR spectroscopy and a further single crystal X-ray experiment. The results of this latter analysis are presented in Fig. 3 and selected bond lengths and angles and details of the X-ray analysis are provided in Tables 2–4 respectively. The monomeric structure displays a distorted tetrahedral geometry about the magnesium atom with coordination from the aldimidoborohydride ligand provided by interactions with the imide nitrogen centre and one of the oxygen atoms of the pinacolate unit of the anion. Although the formation of borohydride intermediates has been previously implied from our NMR studies of both carbonyl and imine hydroboration15,17 and Sadow's ester reduction catalysis,19a the structure of compound 4 provides the first crystallographic evidence for the intermediacy of borohydride intermediates during any magnesium-mediated hydroboration catalysis. The C(3)–N(3) distance (1.226(2) Å) of the formal CN double bond, although shorter than the aldimide CN distances, comprising 2-coordinate nitrogen, within compounds 2 and 3 (2: 1.263(2); 3: 1.262(2), 1.271(2) Å), is closely comparable to previously reported, albeit exclusively transition metal-coordinated, imine-derived borate anions.25
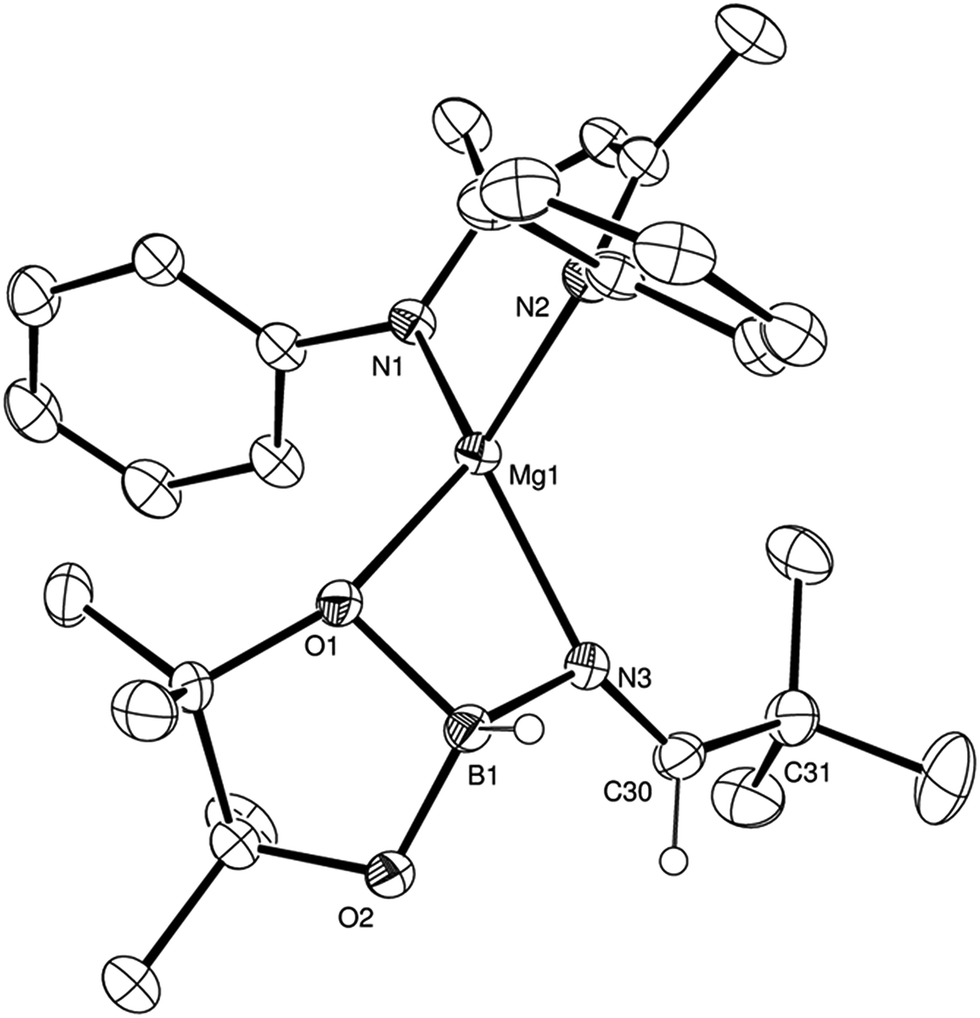 | ||
| Fig. 3 ORTEP representation of compound 4 (30% probability ellipsoids). Hydrogen atoms apart from those attached to B1 and C30 and iso-propyl groups are omitted for clarity. | ||
Samples of compound 4 in C6D6 were observed to undergo continued reaction on standing at room temperature over a period of 12 hours; clearly observed by monitoring of the time-resolved 1H NMR spectra through the disappearance of the downfield 1H aldimido proton signal and the simultaneous appearance of a new singlet resonance at δ 3.35 ppm with an ultimate 2H intensity by integration. The corresponding 11B NMR spectra over this period evidenced a small downfield shift to δ 6.3 ppm with loss of the doublet signal multiplicity. We suggest that these observations are a consequence of an intramolecular hydride shift resulting in reduction of the aldimide residue and production of a magnesium borylamide derivative directly analogous to species D shown in Scheme 3. Addition of a final equivalent of HBpin yielded the desired dihydroboration product, depicted as species F in Scheme 3. Conversion to this latter product, with regeneration of the initial magnesium hydride species, however, required heating at 60 °C obviating any observation of borohydride intermediates akin to species E in Scheme 3.
Notably, analogous addition of one equivalent of HBpin to solutions of compounds 2 and 3 provided no evidence for similar borate formation and provided resonances consistent with the persistence of the unreacted starting compounds. DOSY NMR analysis of these reactions also evidenced no change in the solution diffusion coefficient attributed to the dimeric species even with increasing temperature and the addition of further equivalents of HBpin. We attribute this observation to the additional conjugative stability provided to the dimeric unit by the co-planarity of the 3- and 4-methoxyphenyl substituents with the C–N–Mg linkage.
Isolated samples of compounds 2–4 could themselves be utilised as precatalysts for the hydroboration of m- and p-MeOC6H4CN and t-BuCN respectively. These reactions provided conversions and reaction times that were broadly commensurate with the reactions initiated by V (Table 1; entries 9, 14 and 3), indicating the possible intermediacy of these aldimide and aldimidoborohydride species during the course of each catalysis. Further stoichiometric reactions of alkyl nitriles bearing less sterically demanding substituents than tert-butyl were found to take place with insufficient discrimination to provide useful mechanistic information. A similar reaction of compound V with Ph2CHCN and HBpin, however, highlighted the potential for more complex behaviour during magnesium-catalysed nitrile hydroboration. Upon immediate addition of the reagents, bubbles of gas were observed and single crystals of a new compound (5) were isolated from the unstirred reaction mixture. Although this new species was insufficiently soluble to allow characterisation by solution-state NMR spectroscopy, a single crystal X-ray diffraction analysis revealed compound 5 as the unusual dinuclear magnesium complex shown in Fig. 4. Selected bond length and angle data and details of the X-ray analysis are again provided in Tables 2–4 respectively. The alkaline earth centres of compound 5 are connected through bridging interactions provided by both oxygen atoms of a [H2Bpin]− anion, in a manner reminiscent of that observed in both a previously reported trimeric calcium species, [LCa(H2Bpin)]3 (where L is as defined in Scheme 3),26 and a μ-Mg–H–Mg and O–B–O bridged β-diketiminato magnesium compound the connectivity of which was established through a low resolution (R1 = 10.31%) X-ray diffraction analysis.16 Although each of the magnesium atoms displays a similar pseudo-tetrahedral N3O-coordination sphere, with ligation provided by the bridging borate and a single β-diketiminate anion, the nature of the fourth nitrogen-centred ligand differs across the two Mg centres of the molecule. Whereas Mg(1) is coordinated by a molecule of diphenylacetonitrile, the coordination sphere of Mg(2) is completed by a diphenylketeniminate anion, generated by deprotonation of the nitrile starting material. Magnesium and barium derivatives of the identical diphenylketeniminate anion are precedented by complexes in which the alkaline earth centres were further coordinated by sterically demanding bis(imino)acenaphthene (Dipp-BIAN) radical anions, in which case the magnesium complex was prepared by deprotonation of diphenylacetonitrile by [(Dipp-BIAN)MgMe].27 We suggest that the diphenylketeniminate derivative 5 is formed in a similar manner, in a process which is detrimentally competitive with the desired hydroboration reactivity under catalytic conditions (Table 1, entry 15).
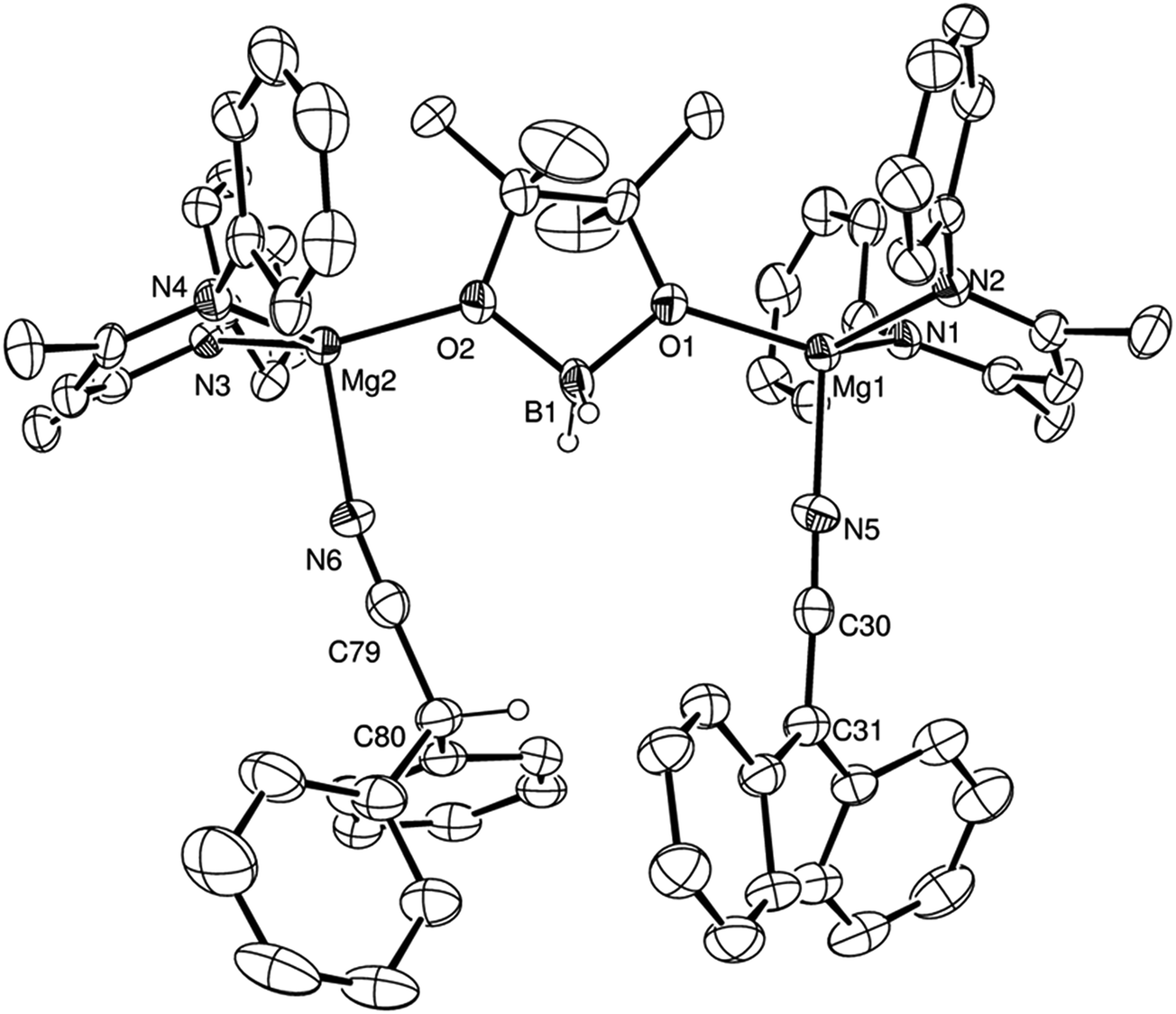 | ||
| Fig. 4 ORTEP representation of compound 5 (30% probability ellipsoids). Iso-propyl groups and hydrogen atoms other than those attached to B1 and C80 have been removed for clarity. | ||
Kinetic studies
To further assess the mechanistic implications of these observations, a kinetic investigation of the hydroboration catalysis was undertaken. Propionitrile was selected as the representative alkyl nitrile substrate due to its provision of opportune reaction rates and clearly observed starting material and product resonances in the 1H NMR spectra. Initial reactions were carried out at 323 K and were monitored by 1H NMR spectroscopy to three half-lives (80% product conversion), using the standard catalytic reaction of 10 mol% of the precatalyst V in conjunction with a 1:2.1 ratio of propionitrile (0.40 M) to HBpin (0.82 M). While the reactions displayed apparent pseudo-zero order kinetic behaviour (Fig. 5a), a first order dependence on [V] was deduced through variation of the catalyst loading with maintenance of the same concentrations of propionitrile and HBpin (Fig. 5b). This latter observation is consistent with other magnesium- and calcium-catalysed reactions in which the alkaline earth centre is coordinated by a β-diketiminate ligand and,12 we suggest, is indicative of the involvement of an isolated magnesium centre during the rate determining process of the catalysis. Experiments under pseudo-first order conditions employing a large excess of propionitrile (8.0 M, 20 equivalents) signified a first order dependence on [HBpin] (Fig. S5–S8†). Although reactions performed with a twentyfold excess of HBpin and with variation of [EtCN] were also indicative of a first order dependence of the reaction rate with changing nitrile concentration (Fig. S9–S12†), further studies highlighted that the data acquired under these conditions may not be truly reflective of the stoichiometric catalytic process. A series of reactions performed with [HBpin] = 8.0 M and [EtCN] = 0.4 M, for example, undertaken with catalyst starting concentrations adjusted between 0.012 M and 0.068 M provided a series of kobs which varied with the third power of [catalyst] (Fig. S13–S18†). In contrast further pseudo-first order experiments employing a large excess of EtCN provided the expected first order variation in rate with respect to changing [catalyst] (Fig. S19–S22†). We, thus, suggest that these latter experiments indicate a possible change in mechanism under pseudo-first order conditions in HBpin. Variation of both [EtCN] and [HBpin] under a concentration regime more representative of the 2:1 HBpin:EtCN ratio required for the dihydroboration (Fig. S23–S30†) also indicated a marked dependence on the reaction stoichiometry and maximum (saturating) substrate concentration limits reminiscent of enzymatic Michaelis–Menten kinetics.
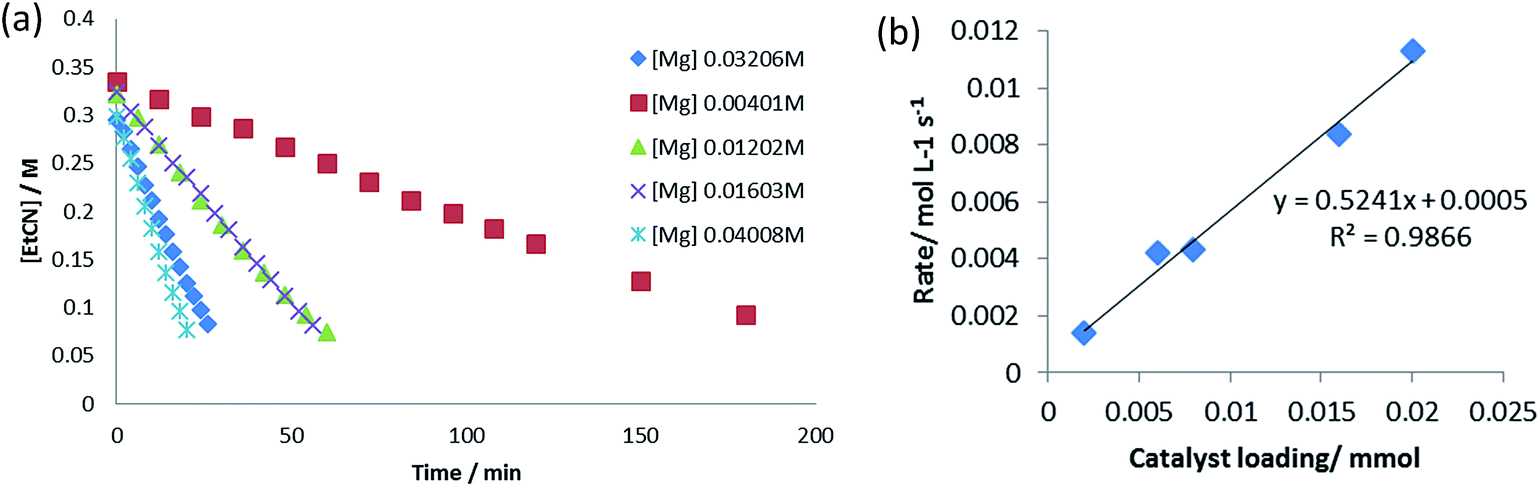 | ||
| Fig. 5 (a) Pseudo-zero order kinetics of propionitrile dihydroboration with varying [V]; (b) propionitrile dihydroboration reaction rate as a function of [V]. | ||
We interpret the suppression of reaction rate at high substrate concentrations on the ability of both reagents to coordinate to the metal centre, either as a neutral nitrile donor or through the likely formation of borohydride species. We suggest that this deduction is further supported by the first order dependence of the reaction rate on [HBpin] in the presence of excess EtCN wherein increasing concentration will favour the displacement of the neutral nitrile from the magnesium coordination sphere allowing the onward formation of intermediate aldimidoborohydride species akin to compound 4 and resultant catalytic turnover. A comparison of the pseudo-zero order rate constants for hydroboration of propionitrile in which HBpin was replaced by DBpin provided a kinetic isotope effect (KIE, kH/kD) of 2.79. Although relatively small, this value would constitute a very large secondary effect and is significantly in excess of comparable results (kH/kD = 1.62) reported by Hartwig et al. during studies of catecholborane metathesis at ruthenium(II) alkyl centres, in which B–H bond breaking processes are integral to the progress of the reaction.28 We, thus, deduce that species analogous to compound 4 do not persist under catalytic conditions but are consumed by intramolecular hydride transfer from HBpin to the metal-bound aldimide fragment.
In contrast to the pseudo-zero order kinetics displayed during the hydroboration of propionitrile, experiments performed to interrogate the rates of reactions displayed by the array of aryl nitriles employed in the study (Table 1) evidenced significantly divergent characteristics. In all cases, reactions performed with the 1:2 nitrile/HBpin ratio required by the reaction stoichiometry conformed to unambiguous first order kinetic behaviour (Fig. S37†), the observed rate constants of which varied internally across the range of aryl substitution patterns employed. These data were employed to construct the chevron-shaped Hammett plot shown in Fig. 6, which clearly indicates a change in reaction mechanism at the transition from overall electron donating [denoted as Ar(EDG)CN] to electron withdrawing [denoted as Ar(EWG)CN] aryl nitrile substitution.29 Although the resultant ρ values [ρ(EDG) = −1.46; ρ(EWG) = +0.68] are relatively small and must be interpreted with caution,30 the change in sign is indicative of minor, yet mechanistically significant, adjustments to the redistribution of electronic charge during the traversal of the rate determining transition state(s).
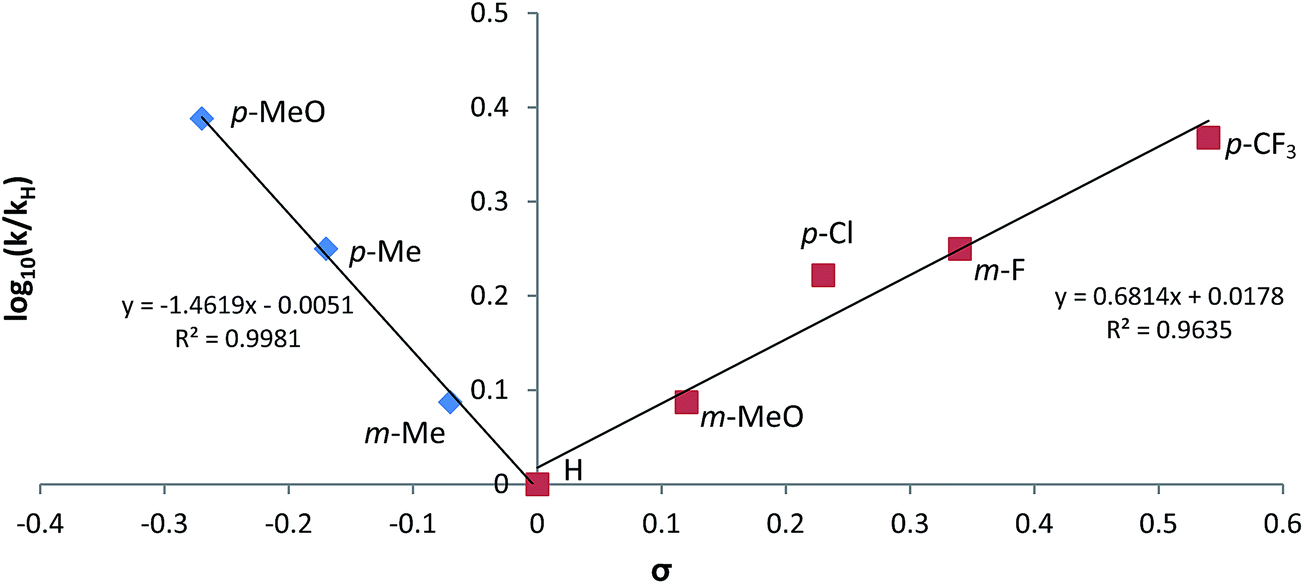 | ||
| Fig. 6 Hammett plot for dihydroboration of variously substituted aryl nitriles catalysed by V. | ||
This apparent change in mechanism was interrogated through kinetic studies of the hydroboration of nitrile substrates bearing representative electron donating (p-MeOC6H4CN) and electron withdrawing (m-MeOC6H4CN) aryl substitution. For the hydroboration of p-MeOC6H4CN a first order dependence on the concentration of [catalyst] (Fig. S41 and S43†) was again consistent with catalysis mediated by a single magnesium centre. A series of reactions conducted at 323 K with 10 mol% of V under pseudo-first order conditions employing 20 molar equivalents of the nitrile substrate, however, indicated a second order dependence on [HBpin] for p-MeOC6H4CN (Fig. S57†). An inverse dependence on [Ar(EDG)CN] again denoted nitrile inhibition as a key influence on the observed reaction rate (Fig. S50†). We suggest that the experimental reaction order in [HBpin] does not reflect the molecularity of the rate determining process. Rather, the second order dependence on [HBpin] is likely the result of two sequential reactions between an intermediate magnesium species and the borane substrate (vide infra).
Despite similar global first order rate behaviour, analogous kinetic experiments for the hydroboration of m-MeOC6H4CN evidenced dissimilar dependences on the concentrations of the reaction components. In contrast to the expectation provided by the kinetic analyses of the EtCN- and p-MeOC6H4CN-based reactions, these latter experiments highlighted a second order dependence on the precatalyst concentration [V] (Fig. S80†). Furthermore, whereas reactions performed with DBpin and p-MeOC6H4CN provided kH/kD = 2.44 commensurate with that deduced for propionitrile hydroboration, analogous experiments performed with m-MeOC6H4CN did not provide any retardation or acceleration of the reaction rate and kH/kD = 1.00. These latter data confirm that the HBpin substrate plays an influential role in the reduction of aryl nitriles with electron donating substitution patterns but has no influence on the turnover limiting process during the magnesium-catalysed hydroboration of more electron poor nitrile substrates.
In a similar manner to that noted for kinetic experiments performed with EtCN, the rates of dihydroboration reactions performed on both aryl nitriles studied in the presence of either a large excess of HBpin or the nitrile substrate were seen to display variable reaction orders in [catalyst] which were dependent on the concentration regime employed. Whilst we consider it imprudent to speculate on the origin of these adjustments, it is clear that the reactions again undergo a change in mechanism in the presence of a saturating concentration of one or the other substrate. With this proviso in place, however, it is clearly apparent that a change in the disposition of the nitrile substrate yields a decisive influence over the course of the catalysis.
Further insight into the nature of the mechanistic discontinuities was, thus, obtained from variable temperature kinetic studies of all three reaction types. The macro- and microscopic activation parameters for the hydroboration of propionitrile, p-MeOC6H4CN and m-MeOC6H4CN mediated by 10 mol% V were determined through Arrhenius and Eyring analyses for reactions performed at four temperatures with standard errors calculated using the least squares method for linear regression. The data presented in Table 5 again evidence considerable variability and are strongly reflective of the dissimilar turnover limiting and bond breaking and forming processes across the three substrate classes. Whilst the hydroboration of propionitrile is enthalpically disadvantaged, the catalysis benefits from a markedly less negative (effectively zero) activation entropy than that deduced for either aryl nitrile substrate. This observation, in conjunction with the significant KIE, further implicates a combination of borane and nitrile substrates which are pre-assembled at the catalytic magnesium centre prior to B–H bond rupture. We thus suggest that the structure of compound 4 (Fig. 3) provides a viable solid-state model for the assembled catalytic intermediate during the hydroboration of alkyl nitriles prior to an intramolecular B–N bond forming and B–H bond breaking process to form a magnesium borylamido species which is in turn rapidly consumed by facile Mg–N/B–H metathesis with a further equivalent of HBpin.
| Nitrile | E a (kJ mol−1) | ΔH≠ (kJ mol−1) | ΔS≠ (J K−1 mol−1) |
|---|---|---|---|
| EtCN | 106.5 (±3.0) | 103.8 (±3.0) | −4.1 (±9.4) |
| p-MeOC6H4CN | 53.1 (±5.8) | 50.4 (±5.8) | −174.8 (±18.2) |
| m-MeOC6H4CN | 46.2 (±3.1) | 43.5 (±3.1) | −197.6 (±9.7) |
In contrast, the ΔH≠ and ΔS≠ values for reactions encompassing both electron donating and electron withdrawing aryl substituents are notably influenced by entropic rather than enthalpic considerations. While the enthalpic contribution to the microscopic activation free energies for both aryl nitrile-based catalyses are ca. 50% of that deduced for EtCN, a significantly negative activation entropy of ca. −190 J K−1 mol−1 is, in both cases, indicative of the associative formation of activated complexes through the assembly of two or more reaction components and must be accounted for in any mechanistic rationale.
Discussion
The kinetic data and the demarcation between electron donating and electron withdrawing aryl nitrile substitution illustrated by the Hammett analysis (Fig. 6) are suggestive of significant variability across the catalytic reactions. We suggest, however, that each reaction conforms to a common mechanism and that the differing kinetic profiles for the three substrate classes are indicative of a series of substrate-dependent pre-equilibria established during the multi-step catalytic reaction.Scheme 4 provides a catalytic mechanism that, although common to each substrate, allows the discrimination of divergent turnover limiting behaviour for (a) alkyl nitriles (RCN), (b) aryl nitriles with appended electron donating groups [Ar(EDG)CN] and (c) aryl nitriles with electron withdrawing groups [Ar(EWG)CN]. Entrance to the catalytic manifold presents a common initiation step involving the transformation of the magnesium precatalyst (V) into a hydride intermediate (G) through metathesis of the magnesium–butyl bond with HBpin (designated as the circled reaction 1 in Scheme 4). Reaction of the nitrile substrate with magnesium hydride and the formation of aldimide derivatives (H and I) exemplified by the isolation of compounds 2 and 3 appears facile, irrespective of nitrile substrate identity (reaction 2). Whilst the activation of organonitriles toward different nucleophiles (e.g. water, alcohols, amines,31 mercaptans,32 phosphines,33 oximes34) by coordination to transition metal centres is well precedented and has been rationalised by an increase of the effective positive charge induced at the nitrile sp-carbon centre,35 this process appears to have little impact on the overall facility for catalytic turnover. We, thus, propose that the rate determining processes of the reactions diverge under the influence of a series of pre-equilibria within the catalytic manifold, which are themselves dictated by modulating nitrile basicity and magnesium aldimide stability. Although only minor variations in experimental and calculated gas-phase basicities and proton affinities have been reported across a wide variety of organic nitriles,36 even slight changes in basicity induced by the relative stabilizing influence of the N-alkyl or N-aryl residue will affect the access of HBpin to magnesium and the consequent formation of borate species akin to compound 4 necessary for successful catalytic turnover. From this perspective, the variation in kinetic behaviour across the three substrate classes (a)–(c) may be discriminated.
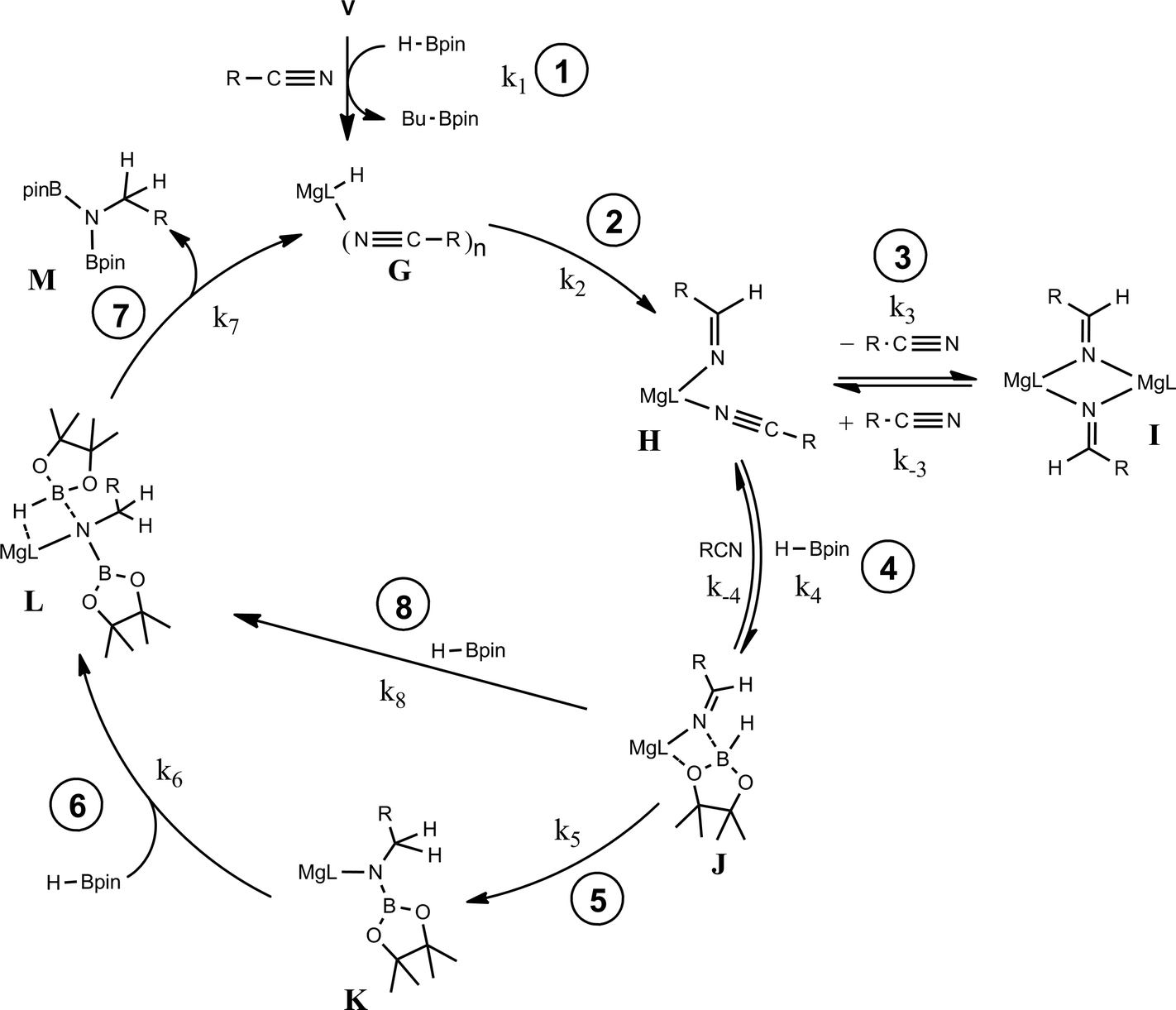 | ||
| Scheme 4 | ||
Case (a): alkyl nitriles (RCN)
The more basic character of alkyl nitriles ensures that the monomer/dimer equilibrium depicted as reaction 3 in Scheme 4 favours the monomeric species, H (i.e. k−3 ≫ k3). The assembly of aldimidohydridoborate anions (J) such as that confirmed by the crystallographic characterisation of compound 4 requires the displacement of pre-coordinated nitrile by the HBpin substrate (reaction 4 depicted in Scheme 4), an equilibration process that is reflected in the inhibition of catalysis at higher propionitrile concentrations. Once formed, the facility of subsequent imine reduction by intramolecular boron-to-carbon hydride transfer to form the borylamide intermediate K will be dictated by the relative stability of the aldimide fragment. Subsequent Mg–N metathesis with a second equivalent of HBpin will then provide the ultimate bis(boryl)amine product (M) via the assembly of a further borate intermediate (L) (reactions 6 and 7, Scheme 4). For alkyl nitrile substrates we suggest that this process will be relatively facile under catalytic conditions with k5 > k−4 (Scheme 5) yielding the negligible entropic component (ΔS≠ = −4.1 (±9.4) J K−1 mol−1) of the free energy of activation for the alkyl nitrile hydroboration catalysis.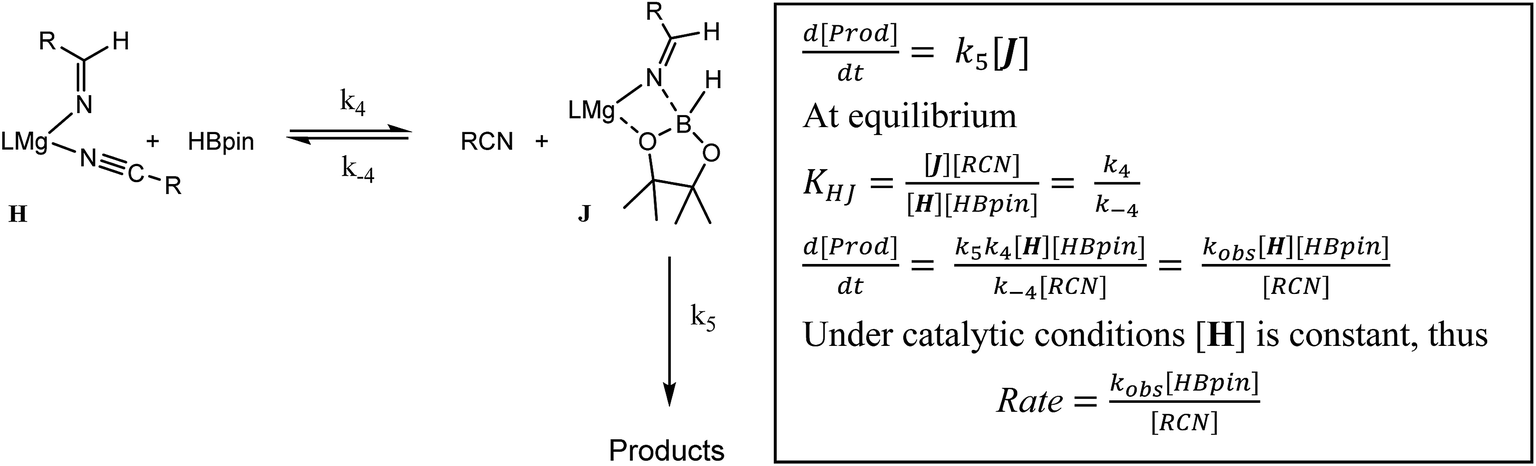 | ||
| Scheme 5 | ||
The catalytic hydroboration of alkyl nitriles is consequently determined by the pre-equilibration of H to J and its subsequent consumption through B–H transfer to the coordinated aldimide fragment. The observed rate of catalysis for alkyl nitrile hydroboration is, thus, dictated by not only the ability of HBpin to replace nitrile in the magnesium coordination sphere but also the consequent ease of intramolecular CN hydride reduction.
Case (b): aryl nitriles with appended electron donating groups (Ar(EDG)CN)
As highlighted by the isolation of compounds 2 and 3, magnesium aldimide derivatives bearing N-aryl substitution will benefit from considerably enhanced conjugative stability in comparison to those bearing alkyl residues of comparable steric demands. We suggest, therefore, that the kinetic profile observed for the hydroboration of p-MeOC6H4CN reflects the resistance to intramolecular hydride transfer of aldimidoborate species analogous to compound 4 (species J in Scheme 4). Although the ease of formation of such species is again dictated by the equilibrium shown as reaction 4 in Scheme 4, the conjugative stability of the resultant aldimidoborate toward intramolecular CN reduction suggests that the sequence of reactions 5–7 in Scheme 4 is inoperable for these intermediates. In such cases, we suggest that hydride transfer and consequent catalytic turnover occurs through reaction of species analogous to J with a further molecule of HBpin (shown as reaction 8 in Scheme 4). The KIE (2.44) associated with this process is comparable to that observed for alkyl nitrile hydroboration, which leads us to deduce that this most likely occurs as a sequence of elementary processes rather than an alternative pathway involving simultaneous aldimide reduction and Mg–N/B–H metathesis. The notably negative activation entropy deduced for this reaction (ΔS≠ = −174.8 (±18.2) J K−1 mol−1) is also congruent with pre-equilibration of Ar(EDG)CN and HBpin and the onward reaction with a second molecule of HBpin. Consideration of the entire process illustrated by Scheme 6 predicts a rate law which correctly encapsulates the experimentally determined second order dependence on [HBpin] while also accounting for the inhibitory effects of increasing nitrile concentration and the first order reliance on changing precatalyst concentration.
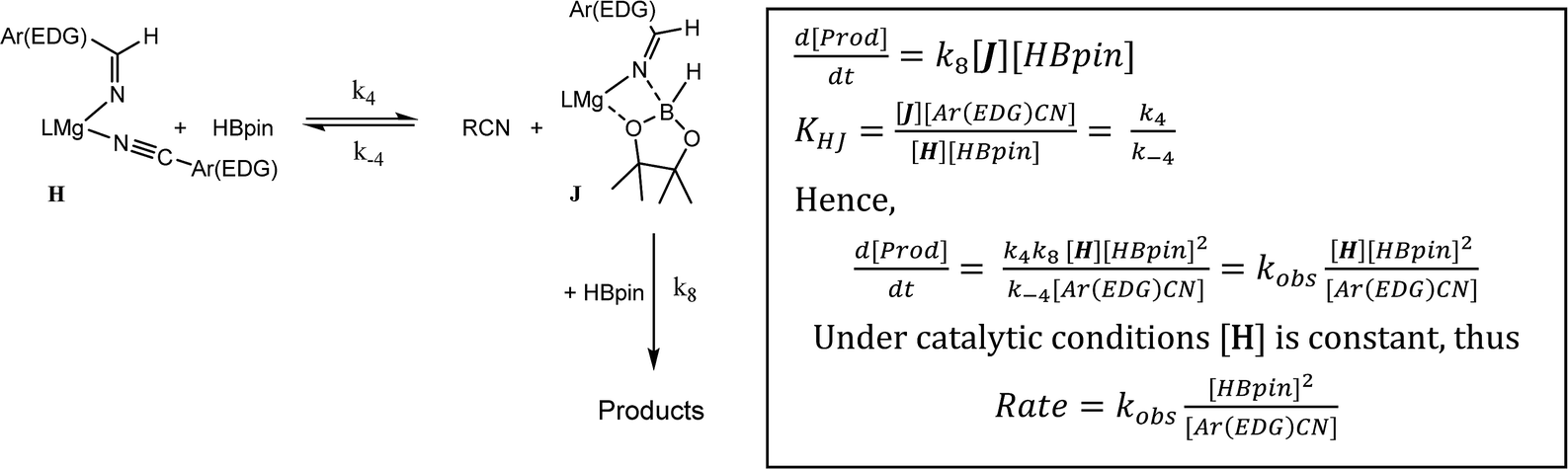 | ||
| Scheme 6 | ||
Case (c): aryl nitriles with appended electron withdrawing groups (Ar(EWG)CN)
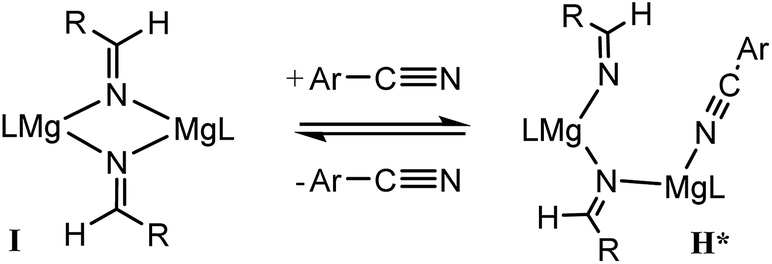
For 3-methoxybenzonitrile, Mg–H insertion will provide a dimeric aldimide (I) with a solution structure similar to that depicted for 2 in Fig. 2a. The apparent independence of the reaction rate on [HBpin] and the absence of a KIE resulting from its replacement in the reaction with DBpin indicates that HBpin is uninvolved in the rupture of this dimeric aldimide. The pre-equilibrium shown in Scheme 7 is, thus, entirely dependent on the ability of the weakly basic nitrile to coordinate to magnesium with rupture of the dimeric resting state to form an intermediate denoted as H* (vide infra). Use of the steady state approximation allows the derivation of a rate law that is dependent on [I] and may be identified as the rate determining process in a hydroboration catalysis in which k3 ≫ k−3 ≪ k4. | ||
| Scheme 7 | ||
We suggest that this process, therefore, occurs with only partial rupture of the dimeric unit as illustrated in Scheme 8, whereupon subsequent reaction with HBpin can only take place at the terminal aldimide to magnesium bond of the unsymmetrical dimeric intermediate (H*). Under this regime the role of the nitrile substrate is encapsulated by the experimentally deduced second order dependence on initial precatalyst concentration, [V], and is simply reflective of the involvement of two magnesium centres in the formation of H*.
 | ||
| Scheme 8 | ||
Conclusion
In conclusion, the β-diketiminato magnesium species V has been demonstrated as an active precatalyst for the HBpin-derived hydroboration of a range of alkyl and aryl nitriles to form bis(boryl)amines. Catalysis proceeds under mild conditions, with reasonable catalyst loadings and is proposed to occur through a sequence of magnesium-mediated B–H insertion and metathesis steps that are crucially dependent on a variety of pre-equilibration steps that are dictated by minor variations in substrate basicity and the stability of mono- and dimeric intermediates. These observations indicate that, somewhat counter to historical prejudice, there is likely to be considerable variation across even superficially identical reactions when catalysed by alkaline earth reagents.Acknowledgements
We thank the EPSRC (UK) (EP/I014519/1) and the University of Bath for funding.References
- (a) The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) L. A. Oro, D. Carmona and J. M. Fraile, in Metal-Catalysis in Industrial Organic Processes, ed. G. P. Chiusoli and P. M. Maitlis, RSC Publishing, London, 2006; pp. 79–113 Search PubMed.
- (a) J. Z. Saavedra, A. Resendez, A. Rovira, S. Eagon, D. Haddenham and B. Singaram, J. Org. Chem., 2012, 77, 221 CrossRef CAS PubMed; (b) L. Hegedús and T. Máthe, Appl. Catal., A, 2005, 296, 209 CrossRef PubMed; (c) L. Hegedús, T. Máthe and T. Kárpáti, Appl. Catal., A, 2008, 349, 40 CrossRef PubMed; (d) S. Maki, M. Okawa, T. Makii, T. Hirano and H. Niwa, Tetrahedron Lett., 2003, 44, 3717 CrossRef CAS; (e) S. P. Bawane and S. B. Sawant, Chem. Eng. J., 2004, 103, 13 CrossRef CAS PubMed; (f) C. de Bellefon and P. Fouilloux, Catal. Rev.: Sci. Eng., 1994, 36, 459 CrossRef CAS PubMed.
- (a) R. A. Grey, G. P. Pez and A. Wallo, J. Am. Chem. Soc., 1981, 103, 7536 CrossRef CAS; (b) T. Suarez and B. Fontal, J. Mol. Catal., 1988, 45, 335 CrossRef CAS; (c) A. M. Joshi, K. S. MacFarlane, B. R. James and P. Frediani, in Progress in Catalysis, ed. K. J. Smith and E. C. Sanford, Elsevier, New York, 1992, pp. 143–146 Search PubMed; (d) D. K. Mukherjee, B. K. Palit and C. R. Saha, J. Mol. Catal., 1994, 88, 57 CrossRef CAS; (e) T. Yoshida, T. Okano and S. Otsuka, J. Chem. Soc., Chem. Commun., 1979, 19, 870 RSC; (f) C. S. Chin and B. Lee, Catal. Today, 1992, 14, 135 CAS; (g) S. Takemoto, H. Kawamura, Y. Yamada, T. Okada, A. Ono, E. Yoshikawa, Y. Mizobe and M. Hidai, Organometallics, 2002, 21, 3897 CrossRef CAS; (h) M. Vilches-Herrera, S. Werkmeister, K. Junge, A. Börner and M. Beller, Catal. Sci. Technol., 2014, 4, 629 RSC.
- For a recent review, see: (a) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289 CrossRef CAS; (b) S. Enthaler, D. Addis, K. Junge, G. Erre and M. Beller, Chem.–Eur. J., 2008, 14, 9491 CrossRef CAS PubMed; (c) S. Enthaler, K. Junge, D. Addis, G. Erre and M. Beller, ChemSusChem, 2008, 1, 1006 CrossRef CAS PubMed; (d) R. Reguillo, M. Grellier, N. Vautravers, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2010, 132, 7854 CrossRef CAS PubMed; (e) K. Rajesh, B. Dudle, O. Blaque and H. Berke, Adv. Synth. Catal., 2011, 353, 1479 CrossRef CAS PubMed; (f) C. Gunanathan, M. Hoelscher and W. Leitner, Eur. J. Inorg. Chem., 2011, 3381 CrossRef CAS PubMed; (g) I. Cabrita and A. C. Fernandes, Tetrahedron, 2011, 67, 8183 CrossRef CAS PubMed; (h) X. Miao, J. Bidange, P. H. Dixneuf, C. Fischmeister, C. Bruneau, J.-L. Dubois and J.-L. Couturier, ChemCatChem, 2012, 4, 1911 CrossRef CAS PubMed; (i) S. Werkmeister, C. Bornschein, K. Junge and M. Beller, Chem.–Eur. J., 2013, 19, 4437 CrossRef CAS PubMed; (j) Z. Lu and T. J. Williams, Chem. Commun., 2014, 50, 5391 RSC; (k) S. Werkmeister, K. Junge, B. Wendt, A. Spannenberg, H. Jiao, C. Bornschein and M. Beller, Chem.–Eur. J., 2014, 20, 4227 CrossRef CAS PubMed.
- J. Seyden-Penne, Reductions by Alumino and Borohydrides in Organic Synthesis, Wiley-VCH, New York, 2nd edn, 1997 Search PubMed.
- T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Tetrahedron Lett., 2011, 52, 6652 CrossRef CAS PubMed.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050 CrossRef CAS PubMed.
- See, for example: (a) S. Das, S. Zhou, D. Addis, S. Enthaler, K. Junge and M. Beller, Top. Catal., 2010, 53, 979 CrossRef CAS; (b) S. Das, B. Wendt, M. Möller, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 1662 CrossRef CAS PubMed; (c) S. Laval, W. Dayoub, A. Favre-Reguillon, M. Berthod, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron Lett., 2009, 50, 7005 CrossRef CAS PubMed; (d) A. M. Caporusso, N. Panziera, P. Pertici, E. Pitzalis, P. Salvadori, G. Vitulli and G. Martra, J. Mol. Catal. A: Chem., 1999, 150, 275 CrossRef CAS; (e) D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004 CrossRef CAS PubMed; (f) R. J. P. Corriu, J. J. E. Moreau and M. Pataud-Sat, J. Organomet. Chem., 1982, 228, 301 CrossRef CAS; (g) D. V. Gutsulyak and G. I. Nikonov, Angew. Chem., Int. Ed., 2010, 49, 7553 CrossRef CAS PubMed; (h) T. Murai, T. Sakane and S. Kato, J. Org. Chem., 1990, 55, 449 CrossRef CAS.
- A. Togni and H. Grützmacher, Catalytic Heterofunctionalisation, Wiley-VCH, 2001 Search PubMed.
- A. Y. Khalimon, P. F. Lyudmila, G. Kuzmina and G. I. Nikonov, Chem. Commun., 2012, 48, 455 RSC.
- (a) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. London, Ser. A, 2010, 466, 927 CrossRef CAS; (b) S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS PubMed; (c) M. Arrowsmith and M. S. Hill, Alkaline Earth Chemistry: Applications in Catalysis, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, vol. 1, p. 1189 Search PubMed; (d) M. R. Crimmin and M. S. Hill, Homogeneous Catalysis with Organometallic Complexes of Group 2, in Topics in Organometallic Chemistry, 2013, vol. 45, p. 191 Search PubMed.
- See, for example: (a) M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042 CrossRef CAS PubMed; (b) M. Arrowsmith, M. R. Crimmin, A. G. M. Barrett, M. S. Hill, G. Kociok-Köhn and P. A. Procopiou, Organometallics, 2011, 30, 1493 CrossRef CAS; (c) M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS PubMed.
- (a) G. A. Molander and J. A. C. Romero, Chem. Rev., 2002, 102, 2161 CrossRef CAS PubMed, and references therein: (b) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed.
- (a) B. Liu, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2012, 51, 4943 CrossRef CAS PubMed; (b) B. Liu, T. Roisnel, J.-F. Carpentier and J.-F. Sarazin, Chem.–Eur. J., 2013, 19, 2784 CrossRef CAS PubMed; (c) S. Tobisch, Chem.–Eur. J., 2011, 17, 14974 CrossRef CAS PubMed; (d) S. Tobisch, Chem.–Eur. J., 2015, 18, 6765 CrossRef PubMed.
- M. Arrowsmith, T. Hadlington, M. S. Hill and G. Kociok-Köhn, Chem. Commun., 2012, 48, 4567 RSC.
- M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556 CrossRef CAS.
- M. Arrowsmith, M. S. Hill, T. Hadlington and G. Kociok-Köhn, Chem.–Eur. J., 2013, 19, 2776 CrossRef CAS PubMed.
- (a) M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826 RSC; (b) C. Weetman, M. S. Hill and M. F. Mahon, Chem. Commun., 2015, 51, 14477 RSC.
- (a) D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959 RSC; see also: (b) N. L. Lampland, M. Hovey, D. Mukherjee and A. D. Sadow, ACS Catal., 2015, 5, 4219 CrossRef CAS.
- J. Spielmann and S. Harder, Chem.–Eur. J., 2007, 13, 8928 CrossRef CAS PubMed.
- A. G. M. Barrett, I. J. Casely, M. R. Crimmin, M. S. Hill, J. R. Lachs, M. F. Mahon and P. A. Procopiou, Inorg. Chem., 2009, 48, 4445 CrossRef CAS PubMed.
- W. Uhl and M. Matar, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1214 CAS.
- J. Kaneti, P. V. R. Schleyer, T. Clark, A. J. Kos, G. W. Spitmagel, J. G. Andrade and J. B. Moffats, J. Am. Chem. Soc., 1986, 108, 1481 CrossRef CAS.
- D. Reed, Chem. Soc. Rev., 1993, 22, 109 RSC.
- See, for example: (a) P. Binger, F. Sandmeyer and C. Kruger, Organometallics, 1995, 14, 2969 CrossRef CAS; (b) A. C. Hillier, T. Fox, H. W. Schmalle and H. Berke, J. Organomet. Chem., 2003, 669, 14 CrossRef CAS; (c) J. R. Galsworthy, C. E. Housecroft, J. S. Humphrey, X. Song, A. J. Edwards and A. J. Rheingold, J. Chem. Soc., Dalton Trans., 1994, 3273 RSC.
- S. Harder and J. Spielmann, J. Organomet. Chem., 2012, 698, 7 CrossRef CAS PubMed.
- (a) I. L. Fedushkin, A. G. Morozov, V. A. Chudakova, G. K. Fukin and V. K. Cherkasov, Eur. J. Inorg. Chem., 2009, 4995 CrossRef CAS PubMed; (b) I. L. Fedushkin, A. G. Morozov, O. V. Rassadin and G. K. Fukin, Chem.–Eur. J., 2005, 11, 5749 CrossRef CAS PubMed.
- J. F. Hartwig, S. Bhandari and P. R. Rablen, J. Am. Chem. Soc., 1994, 116, 1839 CrossRef CAS.
- For related deductions during the Rh-catalysed hydroboration of vinyl arenes, see: D. R. Edwards, Y. B. Hleba, C. J. Lata, L. A. Calhoun and C. M. Crudden, Angew. Chem., Int. Ed., 2007, 46, 7799 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, 2006, University Science Books, Sausalito, California Search PubMed.
- (a) R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299 CrossRef CAS; (b) R. Cini, P. A. Caputo, F. P. Intini and G. Natile, Inorg. Chem., 1995, 34, 1130 CrossRef CAS; (c) N. A. Bokach, V. Y. Kukushkin, M. L. Kuznetsov, D. A. Garnovskii, G. Natile and A. J. L. Pombeiro, Inorg. Chem., 2002, 41, 2041 CrossRef CAS PubMed; (d) K. V. Luzyanin, M. Haukka, N. A. Bokach, M. L. Kuznetsov, V. Y. Kukushkin and A. J. L. Pombeiro, J. Chem. Soc., Dalton Trans., 2002, 1882 RSC.
- R. Ros, J. Renaud and R. Roulet, J. Organomet. Chem., 1976, 104, 271 CrossRef CAS.
- (a) F. A. Cotton, L. M. Daniels, C. A. Murillo and X. Wang, Polyhedron, 1998, 17, 2781 CrossRef CAS; (b) F. A. Cotton and F. E. Kühn, J. Am. Chem. Soc., 1996, 118, 5826 CrossRef CAS.
- (a) V. Y. Kukushkin, I. V. Ilichev, G. Wagner, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, J. Chem. Soc., Dalton Trans., 1999, 3047 RSC; (b) V. Y. Kukushkin, I. V. Ilichev, M. A. Zhdanova, G. Wagner and A. J. L. Pombeiro, J. Chem. Soc., Dalton Trans., 2000, 1567 RSC.
- (a) M. L. Kuznetsov, J. Mol. Struct.: THEOCHEM, 2004, 674, 33 CrossRef CAS PubMed; (b) R. M. Oballa, H.-C. Truchon, C. I. Bayly, N. Chauret, S. Day, S. Crane and C. Berthelette, Bioorg. Med. Chem. Lett., 2007, 17, 998 CrossRef CAS PubMed.
- (a) O. Castafio, R. Notario, K. Hori and J.-L. M. Abboud, Struct. Chem., 1996, 7, 321 CrossRef; (b) O. Exner, S. Böhm, M. Decouzon, J.-F. Gal and P. C. Maria, J. Chem. Soc., Perkin Trans. 2, 2002, 2, 168 Search PubMed; (c) R. H. Staley, J. E. Kleckner and J. L. Beauchamp, J. Am. Chem. Soc., 1976, 98, 2081 CrossRef CAS; (d) C. Colominas, M. Orozco, F. J. Luque, J. I. Borrell and J. Teixido, J. Org. Chem., 1998, 63, 4947 CrossRef CAS; (e) E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 41 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full characterisation data, details of the X-ray analyses of compounds 1–5, protocols and data associated with the kinetic analyses and NMR spectra. CCDC 1018705–1018709. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03114a |
| This journal is © The Royal Society of Chemistry 2016 |