
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotassium ion-selective fluorescent and pH independent nanosensors based on functionalized polyether macrocycles†
Zdeňka
Jarolímová
a,
Mahesh
Vishe
b,
Jérôme
Lacour
 *b and
Eric
Bakker
*a
*b and
Eric
Bakker
*a
aDepartment of Inorganic and Analytical Chemistry, University of Geneva, Quai Ernest-Ansermet 30, CH-1211 Geneva, Switzerland. E-mail: eric.bakker@unige.ch
bDepartment of Organic Chemistry, University of Geneva, Quai Ernest-Ansermet 30, CH-1211 Geneva, Switzerland. E-mail: jerome.lacour@unige.ch
First published on 8th October 2015
Abstract
We present here a new family of pH insensitive ion-selective optical sensors based on emulsified nanospheres containing densely functionalized 15-, 16-, 18- and 20-membered pyreneamide derivatives. These compounds were successfully synthesized by the reaction of α-diazo-β-ketoesters with cyclic ethers of the desired size in the presence of dirhodium complexes followed by a stereo-selective tandem amidation-transposition process and characterized by 1H-NMR, 13C-NMR, IR, HR-ESI-MS, UV-VIS and fluorescence spectroscopy and potentiometry. Their unique structure consisting of a crown ether ring linked to pyrene moieties through amide groups exhibits on–off switchable behavior upon binding of specific cations and allows one to incorporate these chemosensors as fluorescent ionophores into ion-exchange nanospheres. The nanosphere matrix is composed of bis(2-ethylhexyl)sebacate (DOS), poly(ethylene glycol) (PEG), sodium tetrakis 3,5-bis(trifluoromethyl)phenyl borate and pyreneamide functionalized 18-crown-6 ether (18C6). These optode nanoparticles exhibit a strong affinity to the potassium cation over other metal ions up to the millimolar concentration range in an exhaustive detection mode. The logarithmic complex formation constant was determined using the segmented sandwich membrane method and was found to be 6.5 ± 0.3 (SD) in PVC membrane plasticized with NPOE and 5.3 ± 0.3 (SD) in DOS with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex stoichiometry. The nanosensors were characterized in broad range of pH from 4 to 10 and the same linear calibration curves were obtained in the concentration range from 10−7 M to 10−5 M and thus the pH dependent response was largely overcome. These nanosensors are sufficiently stable, simple to prepare, exhibit a rapid response and their nanoscale size makes them suitable for sensing purposes in samples of limited dimensions.
:1 complex stoichiometry. The nanosensors were characterized in broad range of pH from 4 to 10 and the same linear calibration curves were obtained in the concentration range from 10−7 M to 10−5 M and thus the pH dependent response was largely overcome. These nanosensors are sufficiently stable, simple to prepare, exhibit a rapid response and their nanoscale size makes them suitable for sensing purposes in samples of limited dimensions.
Introduction
Ion-selective sensors play an important role in daily life as they are important research tools in fundamental chemical science as well as in applied work such as clinical diagnostics or environmental analysis.1,2 During the last decades, ionophore-based ion-selective sensors (ISEs) that are interrogated under electrochemical or optical conditions have been developed for various analytes.3–8 In recent years, the exploration and the development of optochemical sensors have been the subjects of growing scientific attention. Today, a number of optical chemical probes, ionophore based ion-selective fiber-optical sensors, sensor layers and micro/nano particles exist for the recognition of ions and molecules.9–19 Typically, the sensing mechanism is based on the optical response of a chromophore due to the interaction with a reference or a target ion.A main interest in chemical sensors has been the development of chromogenic reagents soluble in aqueous media that are suitable and compatible for sensing of biological, clinical and environmental samples. However, many such reagents exhibit practical limitations owing to their poor solubility in water that is caused by their highly conjugated structures.20,21 Therefore, a solubility enhancement in aqueous solution is required prior to their application, which may be achieved either by a chemical modification of the structure by introducing hydrophilic groups or by incorporation the reagent into a hydrophobic polymeric sensing phase known as “bulk optode”.22–24 In this case, the optical receptor (also called chromoionophore or fluoroionophore) is often responsive to a reference ion, typically H+, and should exhibit a sufficiently high fluorescence quantum yield or extinction coefficient, good solubility in the polymeric sensing matrix, high lipophilicity to avoid leaching problems, and a high photostability. In addition to the chromoionophore, the matrix may contain a lipophilic ion-exchanger and a second ionophore that is selective to the target ion.9 The working mechanism of such sensors is based on chemical equilibria where electroneutrality within the bulk sensing phase is maintained either by ion exchange or co-extraction processes.25–27 The degree of protonation and its spectral characteristics of the chromoionophores depend on the activity of both competing ions, H+ and the target ion in the sample solution. Because of this, a major disadvantage of so-called ion-selective bulk optical sensors is the strong dependence of the sensor readout on the sample pH in the entire response range9,10,28,29 and the pH must be known or kept constant for reliable measurements of the target ion.
More recently, a drastic miniaturization of ion selective bulk optodes to the micro and nanoscale was achieved to allow for the sensing of small volumes as in intracellular ion imaging applications.2,28,30–32 In these systems, dissolved ion-exchanger and ionophore are assembled inside the core of functionalized self assembled polymeric spheres made of PVC, PEG or pluronic F-127. These nanospheres may work under equilibrium conditions where the sample is not altered by the sensor, or in an exhaustive mode where the sensor consumes the analyte from the sample.33 Based on this concept, Xie et al. have presented a system where the pH dependence of ion selective optodes may be overcome by using exhaustive ion selective nanoparticles in a limited pH window.34
The main interest of this work has been focused on the development of an optical ion sensor by eliminating the strong pH cross-sensitivity of bulk optodes described above while maintaining compatibility with aqueous solutions. To fulfill these aims, we focused our attention on the synthesis of fluorescent sensors that interact selectively with metal ions and quench or enhance the fluorescence signal upon binding with a specific cation without having to control the sample pH.
Crown ethers are known to complex alkali, alkaline earth cations and transition metals. The cavity dimensions, shape, substituent effects, conformational flexibility and donor atom type (S, O, N) control the metal ion recognition stability and the selectivity towards cations.35–42 Here, crown ethers of the desired size (15-, 16-, 18- and 20-membered rings) were functionalized with pyreneamide derivatives as fluorescent moieties that are known to not only exhibit monomer emission but also excimer emission due to the π–π interaction between two pyrene moieties.37,43 Their specific structure favors a close interaction between amide and crown ether oxygens and the guest metal atom, resulting in binding induced conformational changes with on–off switchable properties. Both monomer and excimer emission signals of pyrene units change effectively and cooperatively upon specific binding.
Their complex hydrophobic structures result in a poor solubility and weak or no fluorescence properties in water, so the compounds were incorporated into the hydrophobic core of emulsified ion-exchange nanospheres containing plasticizer. A detailed characterization of the synthesized macrocyclic compounds was performed in broad range of pH using potentiometry and fluorescence spectroscopy.
Experimental section
Reagents and solutions
Potassium chloride, sodium chloride, lithium chloride, magnesium chloride dihydrate, calcium chloride anhydrous, barium chloride anhydrous, copper(II)chloride, lead(II)nitrate, quinine sulfate, anthracene, tetrahydrofuran (THF), dichloromethane (DCM), ethanol (EtOH), tetrakis(4-chlorophenyl)borate tetradodecylammonium salt (ETH 500), bis(2-ethylhexyl)sebacate (DOS), dodecyl nitrophenyloctyl ether (d-NPOE), poly(vinyl chloride) (PVC, high molecular weight), sodium tetrakis 3,5-bis(trifluoromethyl)phenyl borate (NaTFPB), polyethylene glycol 6000 (PEG), hydrochloric acid, 1-aminopyrene were purchased from Sigma-Aldrich with analytical or spectroscopic grade (used without further purification). Aqueous solutions were prepared by dissolving the appropriate salts in Milli-Q-purified water. Dry 1,4-dioxane is obtained by distillation over Na/benzophenone and conserved over 4 Å molecular sieves. Dry tetrahydrofuran is obtained by filtration through appropriate activated alumina drying column. Tetrahydropyran (THP) is purchased from Acros (purity: 99%) and conserved over 4 Å molecular sieves. Oxetane and potassium tert-butoxide (t-BuOK) are purchased from Aldrich, used without any purification and conserved over 4 Å molecular sieves. However, we have found that the quality of commercial oxetane varies strongly from one chemical provider to the next and, sometimes, from one batch to the other within the same company. It is of utmost importance that the solution takes a blue color upon dissolution of dirhodium catalysts into oxetane and not a red one. Dirhodium catalysts were purchased from Strem Chemical Co. and used as received, methyl 2-diazo-3-oxobutanoate was readily prepared by using p-acetamidobenzene-sulfonyl azide (p-ABSA) as diazo-transfer reagent.44Instrumentation and measurements
NMR spectra were recorded on 300, 400 or 500 MHz Bruker spectrometer at 20 °C unless otherwise stated. 1H-NMR chemical shifts are given in ppm relative to Me4Si with the solvent resonance used as the internal standard (CDCl3δ = 7.26 ppm). 13C-NMR (125 or 101 MHz) chemical shifts were given in ppm relative to Me4Si with the solvent resonance used as the internal standard (CDCl3 = 77.16 ppm). IR spectra were recorded using an ATR sampler and are reported in wave numbers (cm−1). Melting points (Mp) were measured in open capillary tubes and are uncorrected. Electrospray mass spectra (ESI+) were obtained by the Department of Mass Spectrometry of the University of Geneva. All reactions involving air sensitive compounds were carried out under dry N2 or Ar by means of an inert gas/vacuum double manifold line and standard Schlenk techniques. Flash column chromatography was performed with silica gel 40–63 μm or alumina (neutral Brockmann I, 50–200 μm). A double – junction Ag/AgCl/3 M KCl/1 M LiOAc reference electrode was used in potentiometric measurements (Metrohm Autolab, Utrecht, The Netherlands). pH values were determined using a Metrohm 827 pH meter (Metrohm Autolab, Utrecht, The Netherlands). Zero current potentials (electromotive force, EMF) were measured with a 16-channel electrode monitor EMF-16 interface from Lawson Laboratories Inc. All electrode potential measurements were performed at laboratory ambient conditions in magnetically stirred solution. The fluorescent spectra were measured with a fluorescence spectrometer (Fluorolog3, Horiba Jobin Yvon) with excitation at 346 nm and emission spectra were recorded between 360 nm and 650 nm for samples containing various cation concentrations. By adjusting the slit widths, the signal at all wavelengths was kept under 106 CPS to ensure that it was within the linear range of the detector. The maximum fluorescence intensity of the monomer unit at the emission wavelength of 406 nm, without any addition of analyte ions, was used to normalize the fluorescence intensity for the analyte calibration curves.The absorbance was measured with a UV-VIS spectrometer (Specord 250 plus, Analytic Jena, AG, Germany). Disposable poly(methyl methacrylate) cuvettes with path length of 1 cm were used for UV-VIS and fluorescent interrogation and quartz cuvettes with path length of 1 cm were used for quantum yield measurements.
Measurements of quantum yield
The quantum yields were measured in 10 mm path length fluorescence cuvettes. In order to minimize re-absorption effects, absorbance was recorded in the range of 0 to 0.1 A.U. The UV-VIS absorbance and fluorescence spectra of increasing concentration were collected for anthracene, quinine as references and for the samples. Quantum yield of the macrocyclic compounds was determined by the comparative method of Williams et al. and was calculated according to the following equation: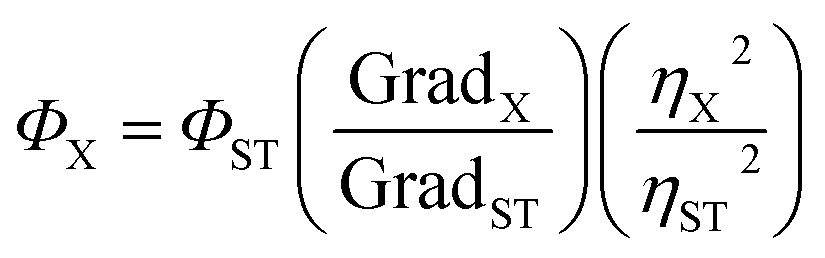 | (1) |
Synthesis of polyether macrocycles and conformational bias
The synthesis of the crown ethers functionalized with pyrene groups was performed in two steps. In the first step, methyl 2-diazo-3-oxobutanoate was allowed to react with cyclic ethers of different size (oxetane, THF, THP, dioxane and oxepane) in the presence of a dirhodium catalyst, usually Rh2(Oct)4, to prepare 15-, 16-, 18- and 20-membered conjugated polyether macrocycles.45 Then, the resulting derivatives were subjected to a stereoselective tandem amidation-transposition process to yield mono or bispyreneamide macrocycles as single racemic stereoisomers.46 More specifically, for this second step, t-BuOK was added as a solid to the suspension of starting macrocycles and 1-aminopyrene dissolved in dry THF at −100 °C (cooling bath of EtOH and liquid N2). After 2 min, the cooling bath was removed and the reaction was allowed to reach room temperature on its own. The suspension was stirred for an additional 2 h. The reaction mixture was purified by column chromatography (SiO2) and preparative TLC without further treatment or work-up. Detailed synthetic procedures and the 1H-NMR, 13C-NMR, IR and HR-ESI-MS characterization of pyreneamide 15-crown-4 ether (15C4), pyreneamide 16-crown-4 ether (16C4), pyreneamide 18-crown-4 ether (18C4), pyreneamide 18-crown-6 ether (18C6) and pyreneamide 20-crown-4 ether (20C4) are shown in the ESI (see Fig. S1–S19†). Importantly, isomerized 18C4 and 18C6 derivatives present well-defined conformations.46 In fact, due to strong allylic 1,3-strain interactions occurring between the exocyclic double bonds and the adjacent stereogenic centers, the amide moieties are disposed in an essentially perpendicular orientation to the mean plane of the macrocycles. This conformational bias induces a spatial proximity between the aromatic residues and hydrogen bonding interactions between the amides N–H and neighboring oxygen atoms or, in the case of the 18C6 macrocycle, with a guest water molecule. As a consequence and in respect to the cavity, the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of the amide groups are turned respectively inwards and outwards. This can be clearly seen in the X-ray diffraction analysis of 18C6 depicted in Fig. 1. Detailed parameters of the X-ray structure are shown in Table S1 and Fig. S20 in the ESI.†
O bonds of the amide groups are turned respectively inwards and outwards. This can be clearly seen in the X-ray diffraction analysis of 18C6 depicted in Fig. 1. Detailed parameters of the X-ray structure are shown in Table S1 and Fig. S20 in the ESI.†
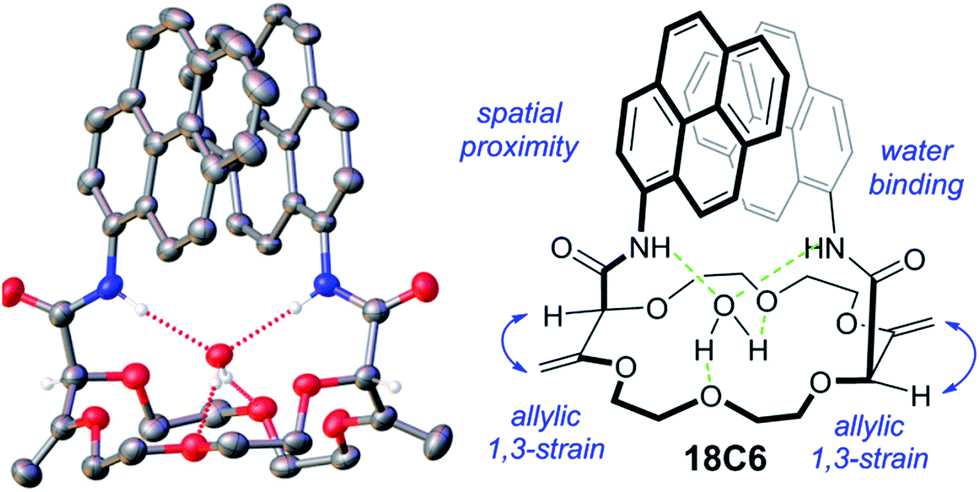 | ||
| Fig. 1 “Diaxial”-like conformation of 18C6. Displacement ellipsoid plot of the crystal structure drawn at 50% probability. Only one of the four superimposable macrocycles present in the asymmetric unit is depicted. A solvent molecule is also omitted for clarity. | ||
Composition of membrane cocktails
The final composition of membrane cocktails for selectivity screening is shown in Table S2.† All components were dissolved in THF. THF was used to enhance the solubility of the solid compounds into the plasticizer and was subsequently removed by evaporation before casting the membranes at ambient conditions. Porous polypropylene (PP) membranes (Celgard, 22.1 mm2 surface area and 25 μm thicknesses, provided by Membrana, Wuppertal, Germany) were used as supporting material for membranes M1–M6. The PP membranes were washed with THF for 5 min to remove contaminants before using. When the membrane was completely dry, an excess volume of 3 μL of the cocktail solution was deposited on it. The membranes (M1–M6) were left in the Petri dish for ∼10 min to ensure a homogeneous and reproducible impregnation of the pores. The pore filling solution composition is assumed to remain identical to the initial THF-free cocktail. Afterwards, the membranes were conditioned in 1 mM solution of KCl for 3 h. Finally, the membrane was mounted in an Ostec electrode body (Oesch Sensor Technology, Sargans, Switzerland). The inner compartment was filled with 10 mM KCl.The membrane cocktail M7 for determination of the stability constant was poured into a glass ring (22 mm in diameter) placed on a slide glass and dried overnight at room temperature in a dust-free environment. Small disks were punched from the cast film and mounted in Ostec electrode bodies. The membranes were placed into 1 mM solution of KCl for several hours. The inner compartment was filled with 1 mM KCl. The sandwich membrane was made by pressing two individual membranes (one without macrocyclic compound and one with the same components and additional macrocylic compound) together after drying with filter paper. The fused membrane was mounted into the Ostec electrode body and immediately measured.
Nanosensor preparation
The final composition of THF cocktails for nanoparticle preparation is shown in Table 1. All components were dissolved in 3 mL of THF to form a homogenous solution. A 0.5 mL aliquot of this cocktail was pipetted and injected into 4.5 mL of deionized water on a vortex with a spinning speed of 1000 rpm. The resulting clear mixture was blown with compressed air on the surface for 20 minutes to remove THF, giving a clear particle suspension. Nanoparticles were freshly prepared before starting the experiments. No preconditioning of the nanoparticles was required. All experiments were performed in unbuffered Milli-Q-purified water, all pH values of the tested solutions were adjusted with HCl or glycine solution to avoid sodium and buffer interferences.| NP1 | NP2 | NP3 | NP4 | NP5 | NP6 | |
|---|---|---|---|---|---|---|
| NaTFPB | 1.80 | 1.80 | 1.80 | 1.80 | 0.85 | 1.80 |
| 15C4 | 2.00 | — | — | — | — | — |
| 16C4 | — | 3.20 | — | — | — | — |
| 18C4 | — | — | 2.60 | — | — | — |
| 20C4 | — | — | — | 3.40 | — | — |
| 18C6 | — | — | — | — | 1.7 | 3.40 |
| DOS | 12 | 12 | 12 | 12 | 12 | 12 |
| PEG | 45 | 45 | 45 | 45 | 45 | 45 |
Results and discussions
The synthesis and characterization of densely functionalized 15-, 16-, 18- and 20-membered macrocyclic derivatives as a neutral carrier are reported here. The fluorescent ionophores were synthesized following the modified procedure published by Vishe et al.46 and the functionalization of the crown ethers with pyrenyl units linked through the amide bridge is depicted in the reaction shown in Fig. S1.† The synthesized compounds shown in Fig. 2 were characterized on the basis of elemental analysis, IR, 1H-NMR, 13C-NMR, IR, HR-ESI-MS, potentiometry, and UV-VIS and fluorescence spectroscopy. Details of the characterization are available in the ESI.† All compounds were found to be highly hydrophobic and therefore only slightly soluble in pure water and completely soluble in aprotic organic solvents such as DCM or THF, apart from 16C4, which is slightly soluble in THF.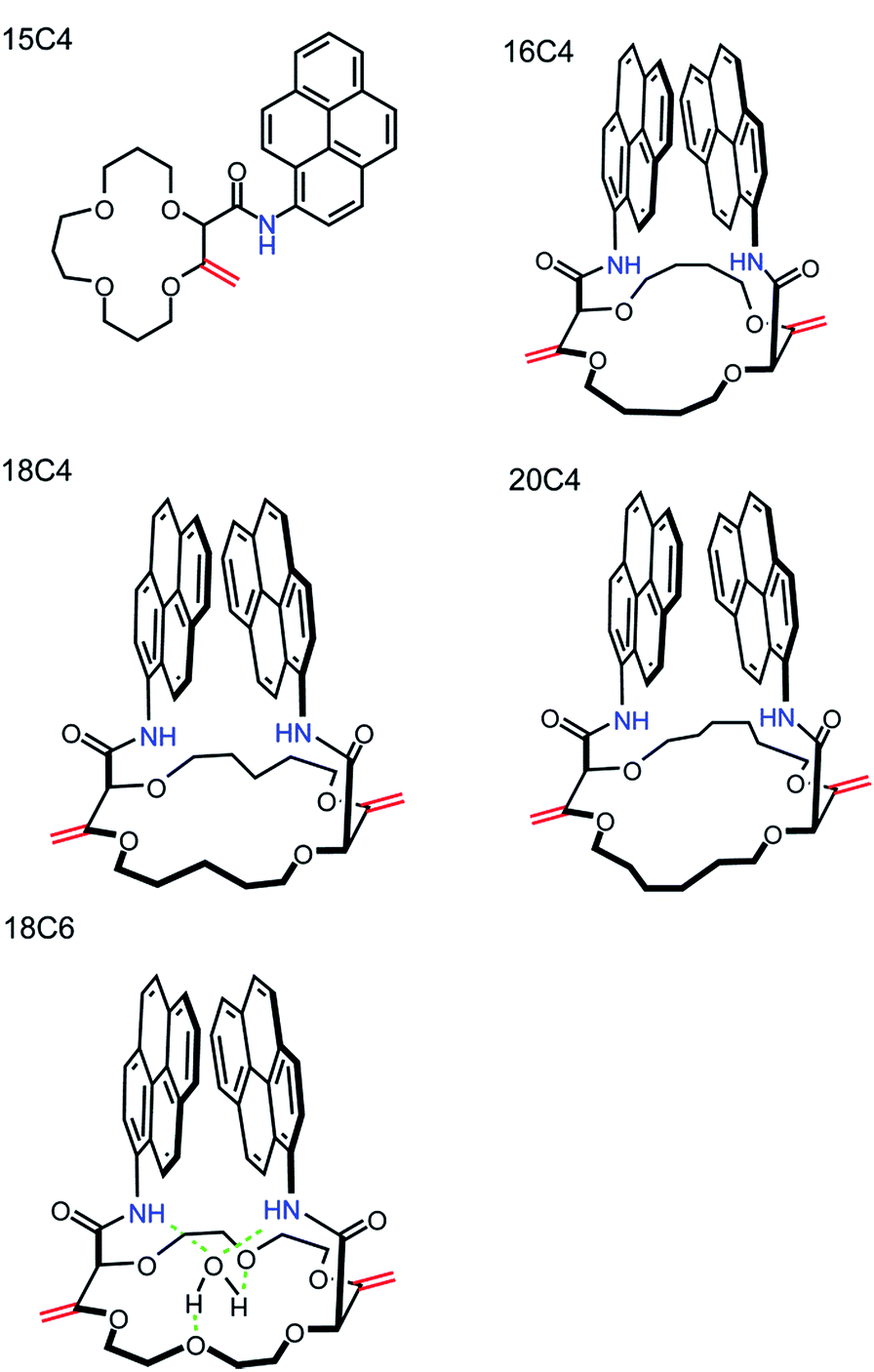 | ||
| Fig. 2 Structures of the studied polyether macrocycles. | ||
The optical properties of pyreneamides 15C4, 16C4, 18C4, 18C6 and 20C4 were characterized by absorbance and fluorescence spectroscopy in THF and DCM solution. UV-VIS spectroscopic analyses of the compounds were carried out in the range 300–600 nm and the typical absorption band for pyrene at 346 nm was obtained. The fluorescence quantum yields was determined in THF and DCM and compared with the reference systems quinine and anthracene. Owing to the presence of pyreneamides groups, the molecules exhibit a sufficiently high fluorescence quantum yield to function as fluoroionophores. The quantum yields of all compounds were similar to each other in THF and most of them display a higher fluorescence quantum yield in DCM. The values were calculated according to eqn (1) and are summarized in Table 2.47 The fluorescence emission spectra of all pyreneamide derivatives with well-defined peaks were collected in the range 360–650 nm and are shown in Fig. S21.† All compounds apart from 15C4 display both monomer and excimer fluorescence when irradiated at 346 nm in THF and DCM. The excimer fluorescence of 15C4 appears only at higher concentrations of 15C4 (see Fig. S22†) that arises from the close interaction between two pyrene rings when the pyrene rings are ∼10 Å from each other.48 With a 346 nm excitation, the monomer peaks appear at 385–435 nm, depending on compound, and the excimer peaks are observed at longer wavelengths ranging from 435 to 650 nm centred around 480 nm (see Table 2).
| Quantum yield | Emission maximum [nm] | |||||
|---|---|---|---|---|---|---|
| THF | DCM | λ 1 | λ 2 | λ 3 | λ 4 | |
| 15C4 | 0.29 | 0.30 | 384 | 406 | 429 | — |
| 16C4 | 0.27 | 0.32 | 384 | 406 | 429 | 495 |
| 18C4 | 0.38 | 0.46 | 384 | 406 | — | 486 |
| 20C4 | 0.32 | 0.36 | 384 | 406 | 426 | 489 |
| 18C6 | 0.37 | 0.46 | 384 | 406 | — | 489 |
To obtain information about the selectivity of these compounds towards alkali, alkaline earth and transition metal ions in the ion-exchange membranes and nanoparticles, preliminary experiments were carried out in polymeric membrane electrodes characterized by zero current potentiometry, by screening in separate 1 mM solutions of the (mostly chloride) salts of Na+, Li+, Ca2+, Mg2+, Ba2+, Cu2+ and Pb2+, see Experimental. All membranes containing the synthesized compounds exhibited a preference for potassium over the other metal ions. A graphical representation of the affinity of the compounds to the metal ions from these potentiometric experiments is shown in Fig. S23.†
The macrocyclic compounds were subsequently incorporated into the hydrophobic core of polymeric nanospheres by a recently introduced precipitation method.34 In addition to the functionalized crown ethers, the nanospheres were doped with lipophilic ion-exchanger dissolved in a nonpolar plasticizer DOS. The principle of the ion-exchange extraction process is shown in Fig. 3. Once the nanoparticles are in the contact with the ion of interest, the counter ion from the ion-exchanger is readily replaced by the target ion and the hydrophobic receptor in the particle core forms a stable and selective complex of defined stoichiometry with the ion of interest, resulting in spectral changes as discussed in more detail below. Under the conditions chosen here, this process works in an exhaustive detection mode where the analyte is completely consumed by the sensor probe and the nanosensors no longer work under typical equilibrium conditions. The selective uptake is driven by the lipophilic ion receptor while the quantity of extractable ion is defined by the ion-exchanger.
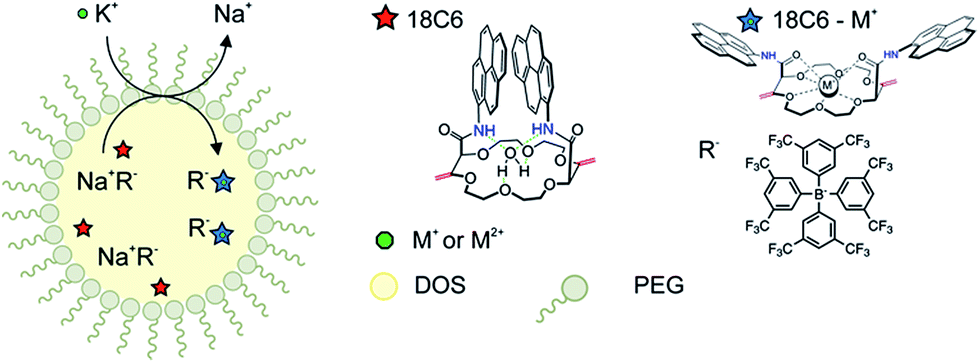 | ||
| Fig. 3 Ion-selective emulsions doped with macrocyclic compound as an ionophore, a plasticizer DOS and the hydrophobic structure of PEG. The sensing mechanism is based on ion-exchange between the target ion K+ in aqueous phase and the counter ion of R− in the organic phase (1) and the specific reaction between 18-crown-6 compound (18C6) and the ion of interest (2). | ||
Based on the above-mentioned findings from the selectivity screening in potentiometry, the selectivity of these compounds for potassium was evaluated in emulsified ion-exchange nanospheres. In comparison with 18C6, the fluorescence intensities of the compounds 15C4, 16C4, 18C4 and 20C4 were hardly affected by the addition of potassium cation and their emission intensities did not change significantly. 18C6 showed the largest signal change in the fluorescent recognition of potassium and was used as the fluoroionophore of choice for further experiments. Fig. 4 represents the normalized fluorescence intensity as a function of potassium concentration for the different macrocyclic compounds and shows that the other compounds exhibited smaller or no fluorescence sensitivity towards potassium. To obtain information about the affinity of the ligand 18C6 towards potassium, its logarithmic complex formation constant was determined using the segmented sandwich method described by Mi et al. as 6.5 ± 0.3 (SD) in polymeric membrane plasticized with a polar o-NPOE plasticizer and 5.3 ± 0.3 for a membrane plasticized with a nonpolar plasticizer DOS (Fig. S24†).49 These values are significantly smaller than those for the well-established potassium-selective ionophore valinomycin (10.0 and 11.0, respectively).49
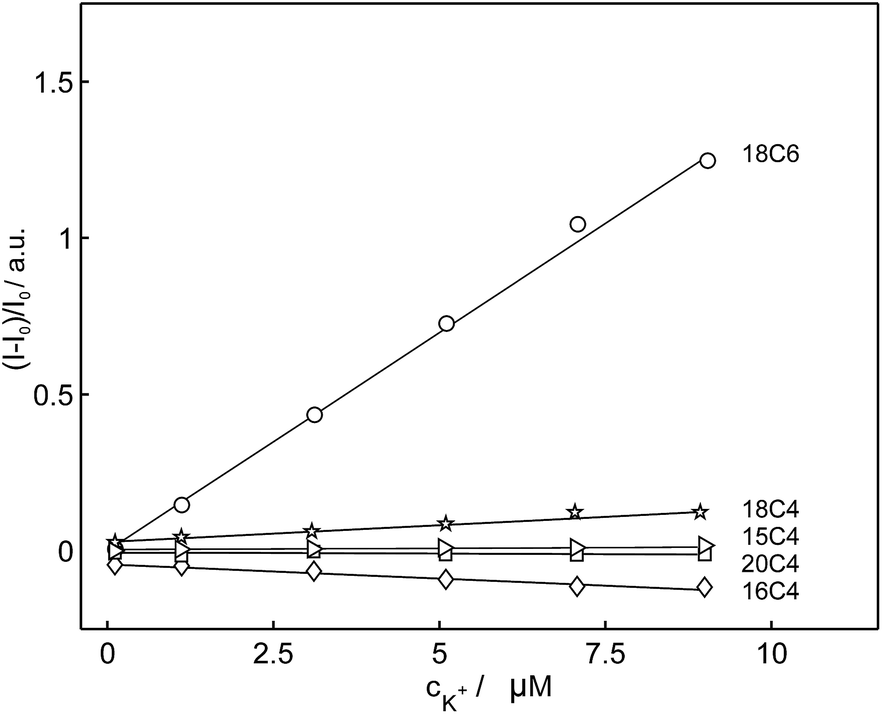 | ||
| Fig. 4 Observed linear calibration curves of normalized fluorescence at 406 nm as a function of potassium concentration for different macrocyclic compounds (15C4, 16C4, 18C4, 18C6, 20C4) in exhaustive fluorescence mode (λexcitation = 346 nm). | ||
To obtain further insight into the fluorescence behavior of 18C6 in the presence of metal ions, the polymeric nanoparticles based on 18C6 were titrated with the cation of interest and fluorescence spectra were collected. As shown in Fig. 5, the optode nanoparticles based on 18C6-membered pyreneamide derivative exhibit a clear affinity to the potassium cation over other metal ions. The addition of potassium induces a change of the fluorescence spectrum in which monomer emission is strongly enhanced and the excimer emission is quenched with no wavelength shift.
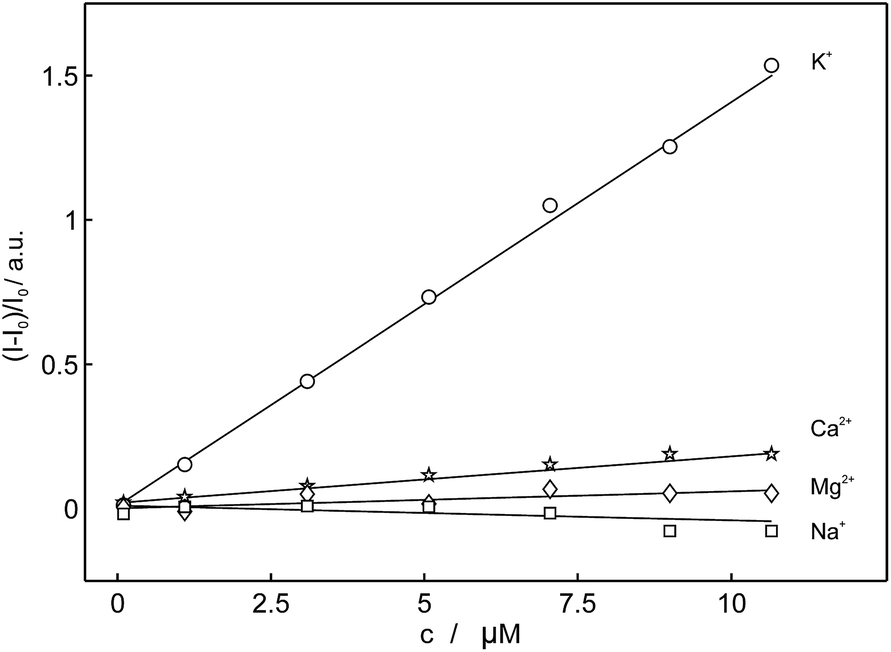 | ||
| Fig. 5 Observed linear calibration curves of normalized fluorescence at 406 nm as a function of concentration of different cations for 18C6 (λexcitation = 346 nm). | ||
The complexation mechanism between 18C6 and potassium is completely different from the interaction between 18C6 and sodium, lithium, calcium, magnesium and lead. From the fluorescence behavior shown in Fig. 6 it can be concluded that the potassium cation interacts with the amide and crown ether oxygens and that this interaction results in a conformational change and switching of the amide groups upon binding. We assume that potassium coordinates in the macrocyclic compound to the amide oxygens that are turned towards the crown ether cycle to form and stabilize the complex with potassium, thereby spatially separating the two pyreneamide arms, resulting in the enhancement of monomer emission and quenching of excimer emission. In this conformation, parallel stacking of the two pyrene moieties is no longer possible and these conformational changes manifest themselves in the fluorescence spectra of Fig. 6a. The proposed mechanism of trapping potassium in 18C6 is shown in Fig. 7.
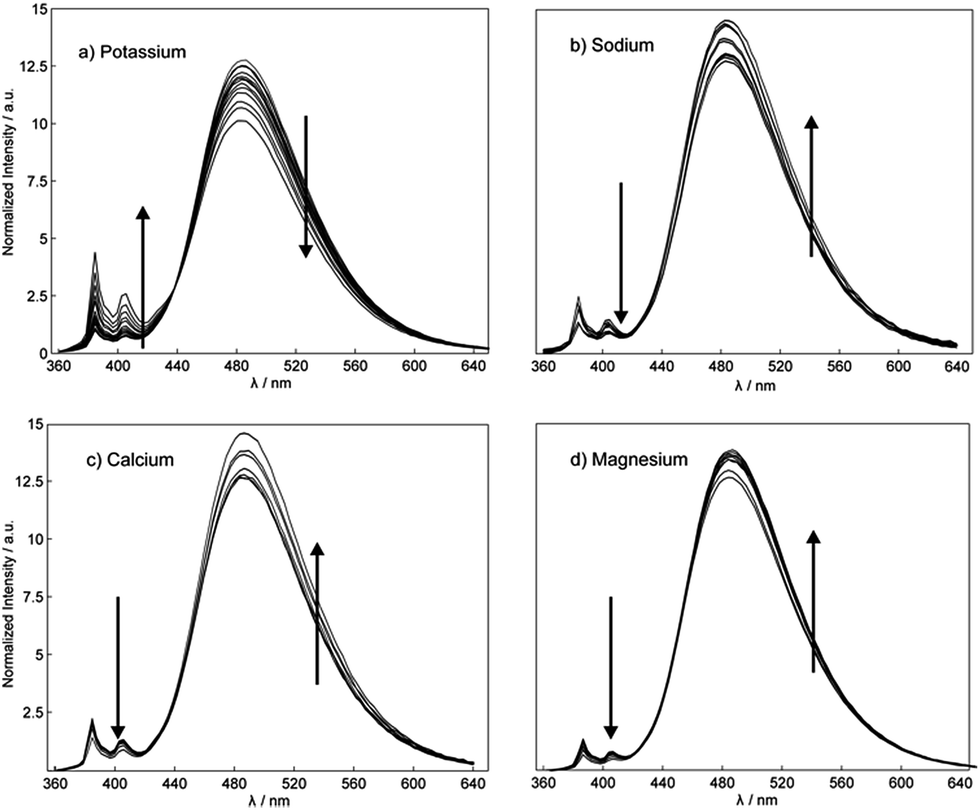 | ||
| Fig. 6 Effect of the addition of (a) potassium, (b) sodium, (c) calcium and (d) magnesium on the fluorescence emission behavior of K+ selective nanospheres (λexcitation = 346 nm) in the concentration range 10−6 to 10−1 M. | ||
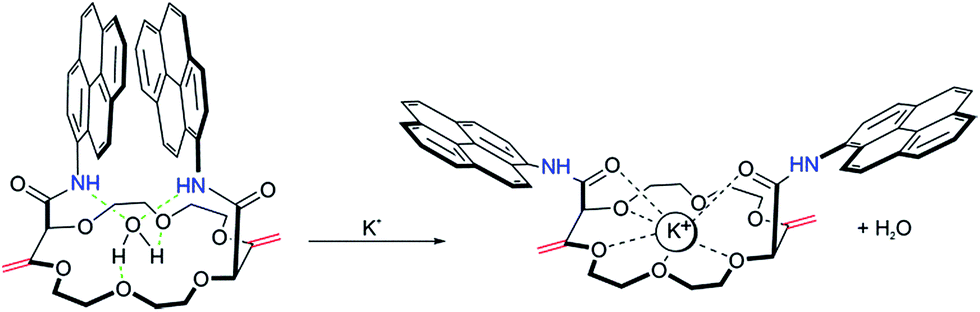 | ||
| Fig. 7 Proposed sensing mechanism of binding of potassium to 18C6, resulting in a conformational change. | ||
As reported in previous studies37,43 the binding mechanism varies with the nature of the investigated cation, and in contrast to potassium, an opposite fluorescence patterns may be produced by a different mode of binding. Other cations such as sodium, calcium and magnesium cause a marked fluorescence quenching in monomer emission and an increase of the excimer emission fluorescence intensity (see Fig. 6b–d).37,43 In this case, the complexation takes place between cation and the amide groups of each fluorophore, resulting in a quenching of the monomer emission and an enhancement of the excimer emission that is attributed to the facing pyrene moieties in a parallel intramolecular π-stacking interaction. Since the intensity ratio of the excimer to the monomer emission Iexcimer/Imonomer is sensitive to conformational changes of the pyreneamide derivatives, changes in the ratio upon metal ion complexation can be an informative parameter. The relative ratios of excimer to monomer bands for 18C6 in the presence of the cations K+, Na+, Ca2+ and Mg2+ were found to be 0.7, 1.1, 2.1 and 1.1, respectively. The larger ratio for sodium, calcium, and magnesium results from the greater conformational rigidity compared to potassium. To confirm the conformational changes of 18C6 related to the metal ion complexation, an X-ray structure elucidation on crystals of 18C6 with various cation salts was attempted, but successful crystals were unfortunately not obtained due to the jellification of the system.
A linear response of the sensor based on 18C6 to potassium was obtained in the concentration range of 0 to 10−5 M as shown in Fig. 4. At higher potassium concentration the sensor response started to deviate from linearity due to limited binding capacity. To achieve complete consumption of the potassium with sensors and to avoid the deviation at higher concentration, the nanospheres must be present in sufficient quantity to extract all available potassium cation in the sample. An advantage of the exhaustive detection is that the response range can be tuned by adjusting the nanosphere loading. To evaluate the influence of the ionophore concentration on the sensor response, the nanoparticles with the same concentration of anionic sites, PEG and the plasticizer, but different concentration of ionophore were used. The concentration was varied from 10−7 to 10−5 M and Fig. S25† exhibits several calibration curves performed at varying concentrations of the ligand 18C6. As expected, the higher the ionophore concentration, the more extended the linear range of the calibration curve.
To verify the selectivity of 18C6 for potassium in the presence of interfering cations and to evaluate their influence on the linear range, a competitive titration experiment was also conducted. Potassium was titrated into a nanoparticles suspension (see Sensor Preparation in Experimental) containing different sodium background levels. The results shown in Fig. 8 clearly demonstrate that the sensor 18C6 exhibits a poorer selectivity towards potassium with respect to conventional K+-selective membrane electrodes and optodes,49 with 1 mM sodium already starting to influence the potassium dosage curve. This suggests that physiological and environmental levels of sodium cannot be tolerated in this assay. While sample dilution will partially alleviate this limitation, the binding selectivity is not yet sufficiently attractive for general practical use.
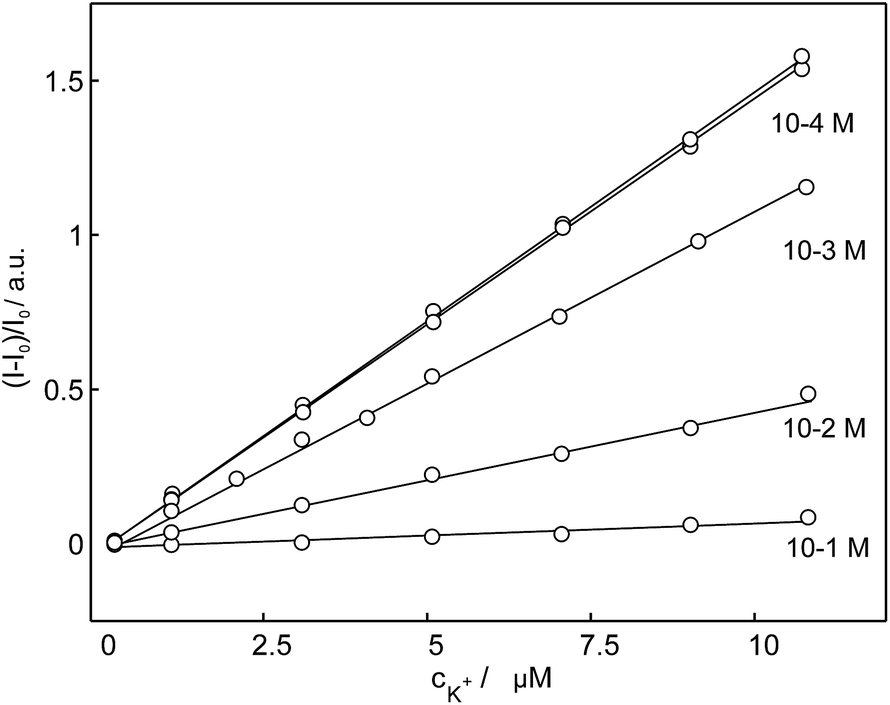 | ||
| Fig. 8 Observed linear calibration curves of normalized fluorescence response of nanospheres based on 18C6 to the potassium cation at different sodium background levels. | ||
As mentioned in the introduction, the pH dependence is the major drawback of conventional ion selective optodes containing H+-chromoionophores and limits the usefulness of this technique. To partially overcome this pH cross-sensitivity, Xie et al. introduced ion-selective exhaustive nanosensors that work well at neutral pH.33 The reported nanosensors exhibited identical response behavior in the pH range 7.4 ± 0.4 and even in unbuffered H2O where the pH was not well-controlled. However, under more acidic conditions the sensor response becomes strongly pH dependent again because the underlying equilibrium still relies on a competitive exchange with hydrogen ions. The compounds synthesized here are stable in a wide pH range and do not alter their spectral properties with pH in comparison to the conventional optodes where the spectral properties depends on the activity of both competing ions. To evaluate the influence of pH, the K+ response of the K+-selective membrane electrodes were characterized in 1 mM KCl in a broad pH range from 2.60 to 10.15 (the pH was adjusted by addition of 1 M HCl and 1 M NaOH), see Fig. 9a. The EMF does not change in the pH range from 3.80 to 10.15 while a pH below 3.80 causes a drift in the potentiometric response. These data are consistent with those obtained in fluorescence experiments. Fig. 9b compares the normalized fluorescence intensities as a function of potassium concentration at different pH. In the presence of potassium the spectra did not show significant variation above pH 4 and the same linear calibration curves for changing K+ concentrations were obtained. At pH below 4, changes are observed from the decomposition of 18C6 as visualized by a color change from colorless to yellowish. The optode pH cross-response was shown to be therefore largely overcome using pyreneamide 18C6 as a model sensor.
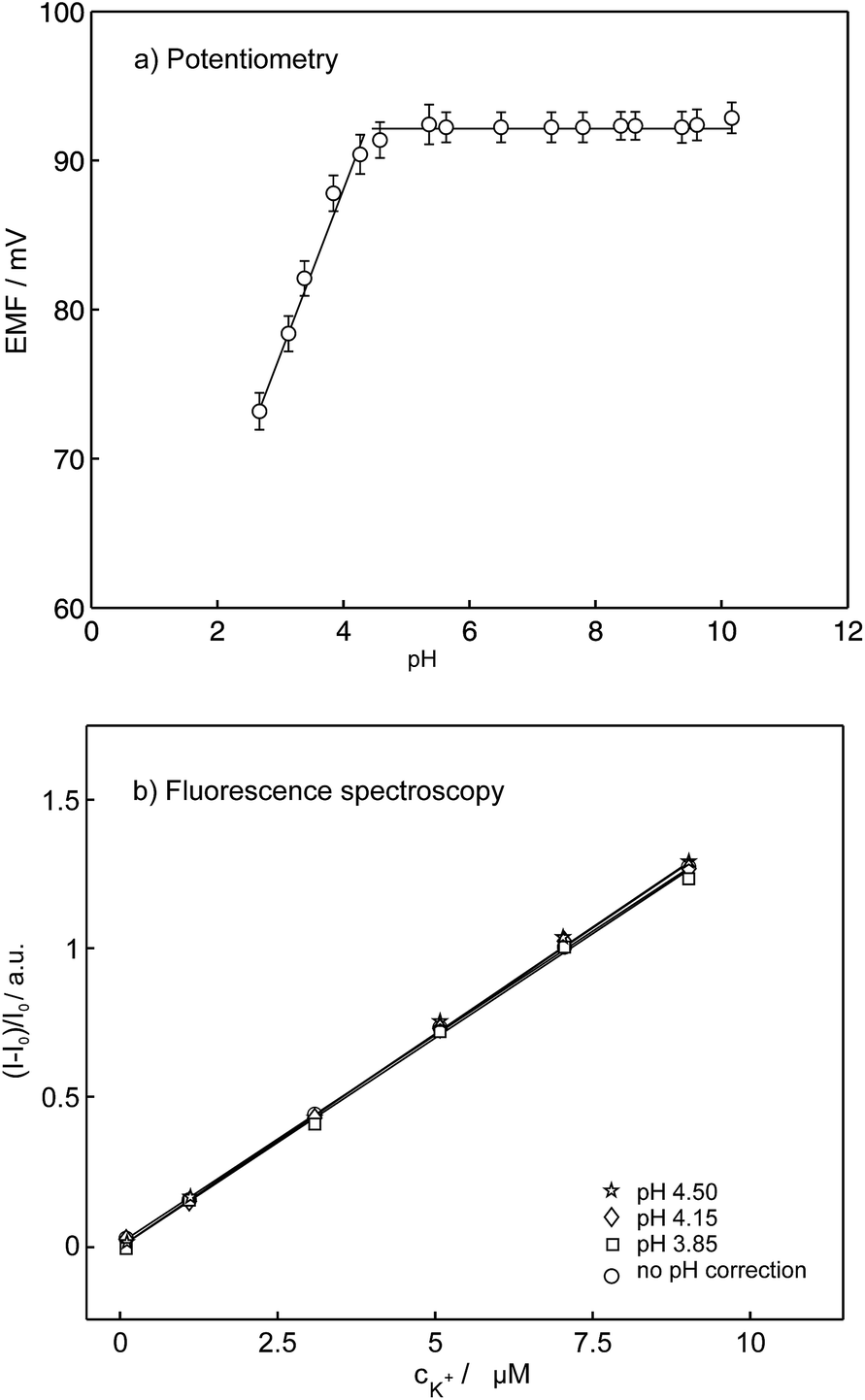 | ||
| Fig. 9 (a) Potentiometric response of the potassium selective sensor to the changes in concentration of H+ at fixed concentration of potassium 1 mM and (b) observed linear calibration curves from the normalized fluorescence spectra as a function of potassium concentration at different pH. | ||
Ionophore-based nanoparticles based on the compounds introduced here are advantageous compared to classical bulk optodes for practical applications since the pH does not have to be controlled. To minimize any matrix effect, the standard addition method was used for the determination of potassium concentration in two mineral water samples, St-Yorre and Volvic. The determined concentrations (152 mg L−1 for St-Yorre and 7.8 mg L−1 for Volvic with a relative standard deviation of less than 3%) agreed quantitatively with the expected potassium levels of 156 mg L−1 and 7.9 mg L−1, respectively. The results also indicate that the selectivity of the potassium nanospheres is sufficient for the application in samples without pH control and that there are no significant interferences from sodium, calcium and magnesium in diluted samples.
Conclusions
We synthesized and characterized a series of novel crown ethers functionalized with pyreneamide derivatives from 1-aminopyrene and crown ethers of desired size in a two-step reaction sequence. A new family of fluorescence ion-selective sensors based on pyreneamide functionalized crown ethers with a reduced sensitivity to pH was introduced. We developed and successfully incorporated the 18C6 derivative that appeared to be the best candidate for potassium recognition into ion-selective nanospheres that operate in an exhaustive detection mode. Pyreneamide 18C6 can be used as an attractive alternative to the H+-chromoionophores that are known to be pH dependent and may provide potential for the design of more selective optical sensors in the future. The presented compound 18C6 is shown to exhibit a dual function in the emulsified nanospheres: it acts as the potassium ionophore and the fluorophore. The utility of the sensor was illustrated by monitoring the concentration of potassium at different pH and the sensor does not exhibit any pH interferences within the pH range from 4 to 10 in potentiometric as well in fluorescence mode. The observed linear range of the sensor was about 10−7 to 10−5 M, which is attractive in view of practical applications such as mineral water sample analysis.Acknowledgements
The authors thank the Swiss National Foundation and the University of Geneva for supporting this research. We also acknowledge Dr Laure Guénée and Dr Céline Besnard for the help with X-ray diffraction.References
- T. Jokic, S. M. Borisov, R. Saf, D. A. Nielsen, M. Kühl and I. Klimant, Anal. Chem., 2012, 84, 6723 CrossRef CAS PubMed.
- M. Telting-Diaz and E. Bakker, Anal. Chem., 2002, 74, 5251–5256 CrossRef CAS.
- Y. Ge, J. Zhu, W. Zhao and Y. Qin, Sens. Actuators, B, 2012, 166, 480–484 CrossRef PubMed.
- V. K. Gupta, R. N. Goyal and R. A. Sharma, Electrochim. Acta, 2009, 54, 4216–4222 CrossRef CAS PubMed.
- V. K. Johns, P. K. Patel, S. Hassett, P. Calvo-Marzal, Y. Qin and K. Y. Chumbimuni-Torres, Anal. Chem., 2014, 86, 6184–6187 CrossRef CAS PubMed.
- A. Kisiel, K. Klucinska, M. Gniadek, K. Maksymiuk and A. Michalska, Talanta, 2015, 144, 398–403 CrossRef CAS PubMed.
- G. Mistlberger, X. Xie, M. Pawlak, G. A. Crespo and E. Bakker, Anal. Chem., 2013, 85, 2983–2990 CrossRef CAS PubMed.
- X. Xie, G. Mistlberger and E. Bakker, J. Am. Chem. Soc., 2012, 134, 16929–16932 CrossRef CAS PubMed.
- E. Bakker, P. Buhlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083–3132 CrossRef CAS PubMed.
- S. L. R. Barker, M. R. Shortreed and R. Kopelman, Anal. Chem., 1997, 69, 990 CrossRef CAS.
- G. J. Mohr, I. Murkovic, F. Lehmann, O. S. Wolfbeis and C. Haider, Sens. Actuators, B, 1997, 39, 239–245 CrossRef CAS.
- E. Park, K. Reid, W. Tang, R. Kennedy and R. Kopelman, J. Mater. Chem., 2005, 15, 2913–2919 RSC.
- O. S. Wolfbeis, Anal. Chem., 2008, 80, 4269–4283 CrossRef CAS PubMed.
- M. Brasuel, R. Kopelman, T. J. Miller, R. Tjalkens and M. A. Philbert, Anal. Chem., 2001, 73, 2221–2228 CrossRef CAS.
- S. J. M. Koskela, T. M. Fyles and T. D. James, Chem. Commun., 2005, 7, 945–947 RSC.
- J. P. Malval, I. Leray and B. Valeur, New J. Chem., 2005, 29, 1089–1094 RSC.
- L. Xie, Y. Qin and H.-Y. Chen, Anal. Chem., 2013, 85, 2617–2622 CrossRef CAS PubMed.
- X. Xie, G. A. Crespo, J. Zhai, I. Szilagyi and E. Bakker, Chem. Commun., 2014, 50, 4592–4595 RSC.
- S. Nagatoishi, T. Nojima, B. Juskowiak and S. Takenaka, Angew. Chem., 2005, 117, 5195–5198 CrossRef PubMed.
- G. McMahon, S. O'Malley, K. Nolan and D. Diamond, ARKIVOC, 2003, 23–31 Search PubMed.
- B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- J. Chen, I. R. Corbin, H. Li, W. Cao, J. D. Glickson and G. Zheng, J. Am. Chem. Soc., 2007, 129, 5798–5799 CrossRef CAS PubMed.
- Z. Cheng, Z. Wu, Z. Xiong, S. S. Gambhir and X. Chen, Bioconjugate Chem., 2005, 16, 1433–1441 CrossRef CAS PubMed.
- K. Kiyose, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2006, 128, 6548–6549 CrossRef CAS PubMed.
- C. Krause, T. Werner, C. Huber and O. S. Wolfbeis, Anal. Chem., 1999, 71, 1544–1548 CrossRef CAS.
- N. Stromberg and S. Hulth, Anal. Chim. Acta, 2001, 443, 215–225 CrossRef CAS.
- H. Sumiyoshi and K. Nakahara, Talanta, 1977, 763 CrossRef CAS.
- I. H. A. Badr, R. D. Johnson, M. Diaz and M. F. Hawthorne, Anal. Chem., 2000, 72, 4249 CrossRef CAS.
- J. M. Dubach, D. I. Harjes and H. A. Clark, J. Am. Chem. Soc., 2007, 129, 8418–8419 CrossRef CAS PubMed.
- H. A. Clark, M. Hoyer, M. A. Philbert and K. R. Kopelman, Anal. Chem., 1999, 71, 4831–4836 CrossRef CAS.
- I. Tsagkatakis, S. Peper and E. Bakker, Anal. Chem., 2001, 73, 315–320 CrossRef CAS.
- K. Wygladacz and E. Bakker, Anal. Chim. Acta, 2005, 532, 61–69 CrossRef CAS PubMed.
- X. Xie, J. Zhai and E. Bakker, Anal. Chem., 2014, 86, 2853–2856 CrossRef CAS PubMed.
- X. Xie, G. Mistlberger and E. Bakker, Anal. Chem., 2013, 85, 9932–9938 CrossRef CAS PubMed.
- M. M. G. Antonisse and D. N. Reinhoudt, Chem. Commun., 1998, 443–448 RSC.
- J. S. Kim, O. J. Shon, J. A. Rim, S. K. Kim and J. Yoon, J. Org. Chem., 2002, 67, 2348–2351 CrossRef CAS PubMed.
- S. K. Kim, S. H. Lee, J. Y. Lee, J. Y. Lee, R. A. Bartsch and J. S. Kim, J. Am. Chem. Soc., 2004, 126, 16499–16506 CrossRef CAS PubMed.
- K. Kimura, H. Oishi, T. Miura and T. Shono, Anal. Chem., 1987, 59, 2331–2334 CrossRef CAS.
- C. J. Pedersen and H. K. Frensdorff, Angew. Chem., Int. Ed., 1972, 11, 16–25 CrossRef CAS PubMed.
- R. Sevcikova, P. Lubal, M. P. C. Campello and I. Santos, Polyhedron, 2013, 62, 268–273 CrossRef CAS PubMed.
- S.-R. Sheen and J.-S. Shih, Analyst, 1992, 117, 1691–1695 RSC.
- B. Whittle, S. R. Batten, J. C. Jeffery, L. H. Rees and M. D. Ward, J. Chem. Soc., Dalton Trans., 1996, 4249–4255 RSC.
- M. Kumar, A. Dhir and V. Bhalla, Eur. J. Org. Chem., 2009, 4534–4540 CrossRef CAS PubMed.
- J. S. Baum, A. Shook, H. M. L. Davies and H. D. Smith, Synth. Commun., 1987, 17, 1709–1716 CrossRef CAS PubMed.
- W. Zeghida, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2010, 49, 7253–7256 CrossRef CAS PubMed.
- M. Vishe, R. Hrdina, A. I. Poblador-Bahamonde, C. Besnard, L. Guenee, T. Buergi and J. Lacour, Chem. Sci., 2015, 6, 4923–4928 RSC.
- A. T. R. Williams, S. A. Wingfield and J. N. Miller, Analyst, 1983, 108, 1067 RSC.
- G. K. Bains, S. H. Kim, E. J. Sorin and V. Narayanaswami, Biochemistry, 2012, 51, 6207–6219 CrossRef CAS PubMed.
- Y. M. Mi and E. Bakker, Anal. Chem., 1999, 71, 5279–5287 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1045592. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03301b |
| This journal is © The Royal Society of Chemistry 2016 |