
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective fluorination of α-branched aldehydes and subsequent conversion to α-hydroxyacetals via stereospecific C–F bond cleavage†
Kazutaka
Shibatomi
*,
Kazumasa
Kitahara
,
Takuya
Okimi
,
Yoshiyuki
Abe
and
Seiji
Iwasa
Department of Environmental and Life Sciences, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku-cho, Toyohashi 441-8580, Japan. E-mail: shiba@ens.tut.ac.jp
First published on 16th November 2015
Abstract
The highly enantioselective fluorination of α-branched aldehydes was achieved using newly developed chiral primary amine catalyst 1. Furthermore, the C–F bond cleavage of the resulting α-fluoroaldehydes proceeded smoothly under alcoholic alkaline conditions to yield the corresponding α-hydroxyacetals in a stereospecific manner. Accordingly, the one-pot conversion of α-branched aldehydes into α-hydroxyacetals was achieved for the first time in high enantioselectivity.
Enantioselective construction of fluorinated chiral stereogenic centers is synthetically important, because the resulting fluorides are expected to be useful intermediates for fluorinated drugs and agricultural agents.1 Despite the extraordinary interest in practical synthetic methodologies towards chiral tertiary fluorides, until very recently, catalytic enantioselective methods capable of introducing fluorine atoms onto a tertiary carbon center have been primarily limited to the fluorination of active methine compounds.2–4 The chiral secondary amine-catalyzed electrophilic fluorination of aldehydes is a highly useful method for the construction of fluorinated stereogenic centers.5 Although this method yields α-fluoroaldehydes with high enantioselectivity when α-monosubstituted aldehydes are used as substrates, fluorination of α-branched aldehydes with secondary amine catalysts generally exhibits low enantioselectivity.5a,5b To the best of our knowledge, there are only three reports on the enantioselective fluorination of α-branched aldehydes yielding tertiary fluorides with acceptable enantiopurity.6–8 Notably, Jørgensen and co-workers reported the asymmetric fluorination of α-alkyl-α-aryl aldehydes achieving high enantioselectivity (up to 90% ee) with a new primary amine catalyst with non-biaryl atropisomeric chirality.6 However, the isolated yields of the fluorinated products were not satisfactory for some reasons. Although we also reported the asymmetric fluorination of α-chloroaldehydes via the kinetic resolution mechanism, affording α-chloro-α-fluoroaldehydes with high enantioselectivities, moderate enantioselectivities were observed when α,α-dialkylaldehydes were employed.7 Here, we report the organocatalytic fluorination of α-branched aldehydes, using a newly developed chiral primary amine catalyst 1; this approach affords the corresponding α-fluoroaldehydes in high chemical yields and enantioselectivities (Scheme 1). We also found that the resulting α-fluoroaldehydes could be converted into α-hydroxyacetals, bearing chiral tertiary alcohol moieties, and their optical purity could be maintained, which suggested that the reaction proceeded via a stereospecific C–F bond cleavage. These results shed new light on C–F bond activation,9 and will be useful because the resulting chiral tertiary alcohols may be valuable intermediates in the synthesis of biologically active compounds.
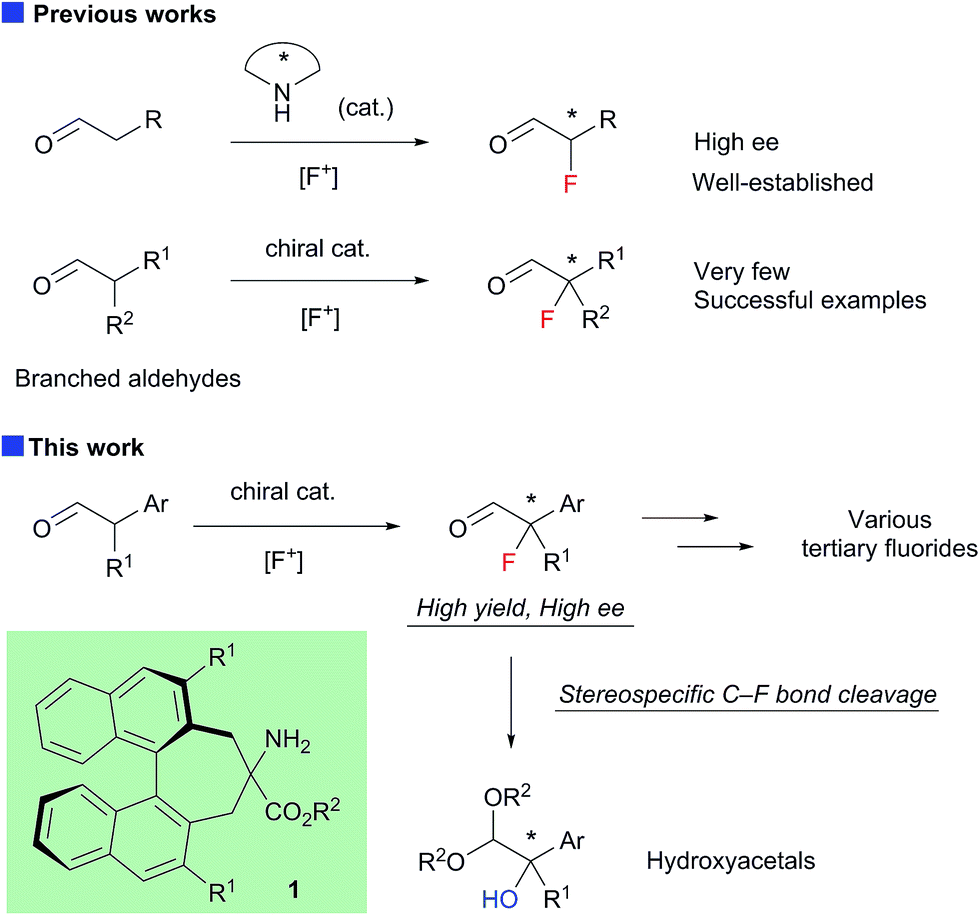 | ||
| Scheme 1 Asymmetric α-fluorination of aldehydes. | ||
The structure of the new chiral primary amine catalyst 1 is shown in Scheme 2.10 An ester moiety and substituents at the 3,3′-positions on the binaphthyl backbone are expected to influence the chirality of the resulting products. Catalyst 1 was synthesized according to the procedure shown in Scheme 2. First, (R)-3,3′-diaryl-2,2′-bis(bromomethyl)-1,1′-binaphthyl (2) was prepared from commercially available (R)-BINOL via a reported procedure.11 Compound 2 was then converted into the desired amino ester 1via alkylative cyclization with ethyl isocyanoacetate and subsequent acid hydrolysis of the isocyano group.
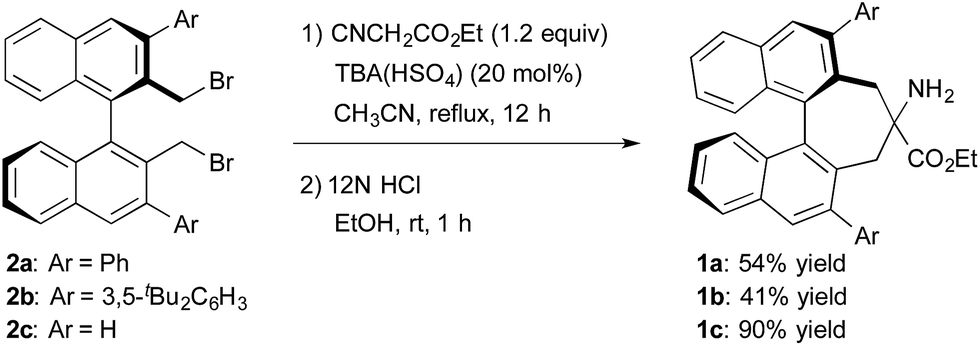 | ||
| Scheme 2 Synthesis of primary amine catalysts. | ||
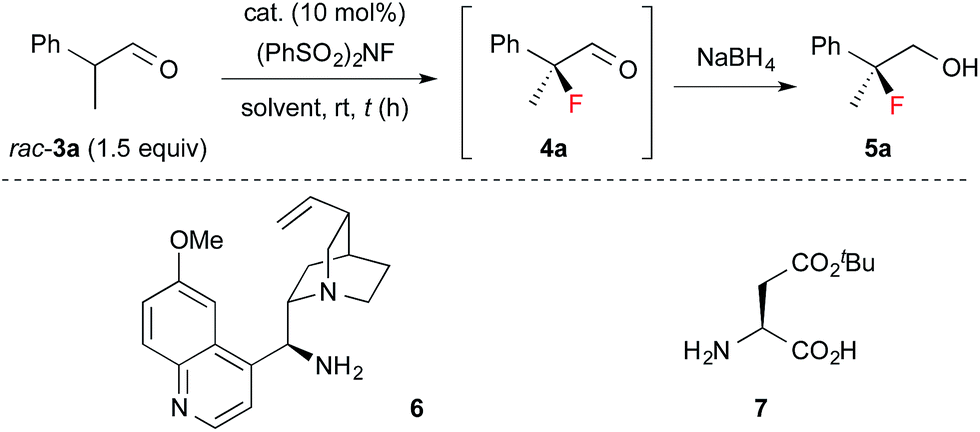
Next, 1 was applied in the enantioselective fluorination of α-branched aldehydes (Table 1). Fluorination of 2-phenylpropanal (3a) was carried out with N-fluorobenzenesulfonimide (NFSI) in the presence of 10 mol% 1a to yield 2-fluoro-2-phenylpropanal (4a) in a high conversion. The fluorinated product was isolated after reduction to primary alcohol 5a, due to difficulties in the purification of 4a. Thus, 5a was isolated in a sufficiently high chemical yield, but with poor enantioselectivity (entry 1). To our delight, the enantioselectivity of the fluorination dramatically improved to 90% ee by employing catalyst 1b, which has bulky aryl substituents at the 3,3′-positions (entry 2). As expected, the use of catalyst 1c without aryl substituents in the 3,3′-positions yielded a nearly racemic product (entry 3). The optimal solvent for the reaction was found to be toluene (entries 4–7). The enantioselectivity and reaction rate were slightly increased by adding 10 mol% 3,5-dinitrobenzoic acid as a co-catalyst (entry 10). We also confirmed that chiral primary amines 6 and 7, which were reported to induce high enantioselectivity in the amination of α-branched aldehydes,12 were ineffective in the fluorination of 3a (entries 11 and 12). The absolute configuration of 5a was determined to be S, by comparison of its optical rotation with that of the reported value.6
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time (h) | Yieldb (%) | eec (%) |
| a Reactions were carried out with 1.5 equiv. of rac-3a based on NFSI in the presence of 10 mol% 1 unless otherwise noted. b Isolated yield of 5a. c Absolute configuration of the major enantiomer is specified in parenthesis. d 1.5 equiv. of NFSI was used based on rac-3a. e At 0 °C. f 10 mol% 3,5-(NO2)2C6H3CO2H was used as a co-catalyst. g 5 mol% catalyst was used with 15 mol% TFA. h 20 mol% catalyst. | |||||
| 1 | 1a | Toluene | 24 | 79 | 51 (S) |
| 2 | 1b | Toluene | 2 | 97 | 90 (S) |
| 3 | 1c | Toluene | 24 | 71 | 3 |
| 4 | 1b | CH2Cl2 | 18 | 86 | 74 (S) |
| 5 | 1b | EtOAc | 4 | 99 | 82 (S) |
| 6 | 1b | t BuOMe | 3 | 97 | 86 (S) |
| 7 | 1b | MeOH | 48 | <10 | n.d. |
| 8d | 1b | Toluene | 6 | 82 | 88 (S) |
| 9e | 1b | Toluene | 48 | 73 | 93 (S) |
| 10e,f | 1b | Toluene | 48 | 86 | 95 (S) |
| 11g | 6 | CHCl3 | 24 | 76 | 13 (R) |
| 12h | 7 | THF | 2 | 98 | 13 (S) |
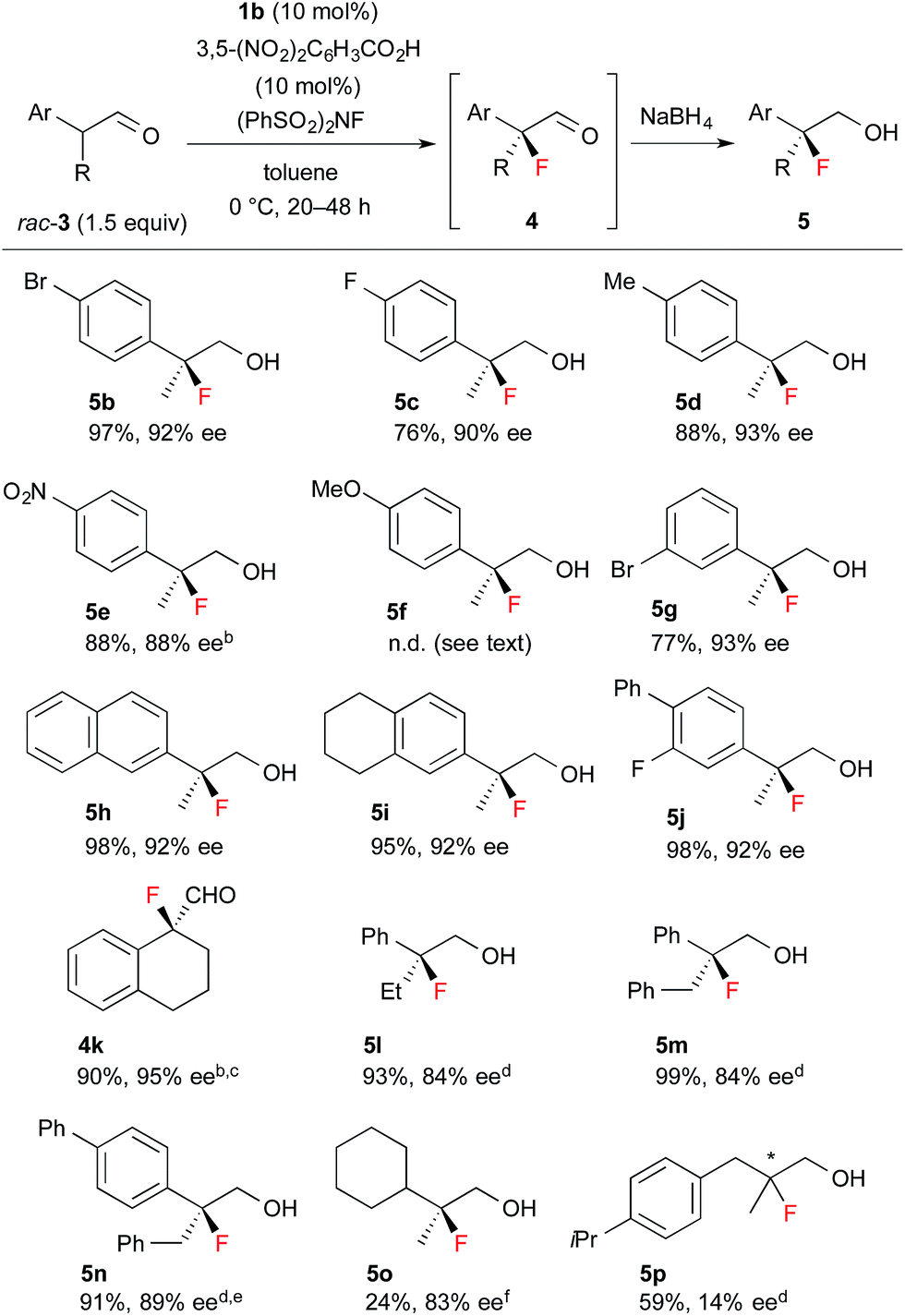
Encouraged by the results obtained with amine catalyst 1b, we attempted to expand the substrate scope of the fluorination reaction. As summarized in Table 2, various α-alkyl-α-aryl aldehydes were successfully fluorinated to afford the corresponding α-fluoroaldehydes in high yields with high enantioselectivities. On the other hand, the reaction with α,α-dialkyl aldehyde 3o yielded the product with good enantioselectivity but in poor yield, while the reaction with 3p showed disappointingly low enantioselectivity. Although it was observed that the reaction with 3f yield the corresponding fluoroaldehyde 4f in good conversion by NMR measurement of the reaction mixture, reduction of 4f to 5f gave a complicated mixture, thus we could not determine those enantiopurity.
| a Reactions were carried out with 1.5 equiv. of rac-3 based on NFSI in the presence of 10 mol% 1b and 3,5-(NO2)2C6H3CO2H. Isolated yield of 5 are described, except for 4k. b Purified product contained ca. 5% of an inseparable by-product. c At rt. for 2 h. d At rt. for 12–24 h. e 20 mol% catalyst. f 30 mol% catalyst. |
|---|
|
The resulting fluorides can be converted into a variety of other tertiary fluorides (Scheme 3). First, allyl fluorides 8 were synthesized by Horner–Wadsworth–Emmons reaction of α-fluoroaldehydes 4 in good yield. Next, fluorohydrine 5j was oxidized to carboxylic acid 9,13 which is a fluorinated analogue of a non-steroidal anti-inflammatory agent, flurbiprofen.
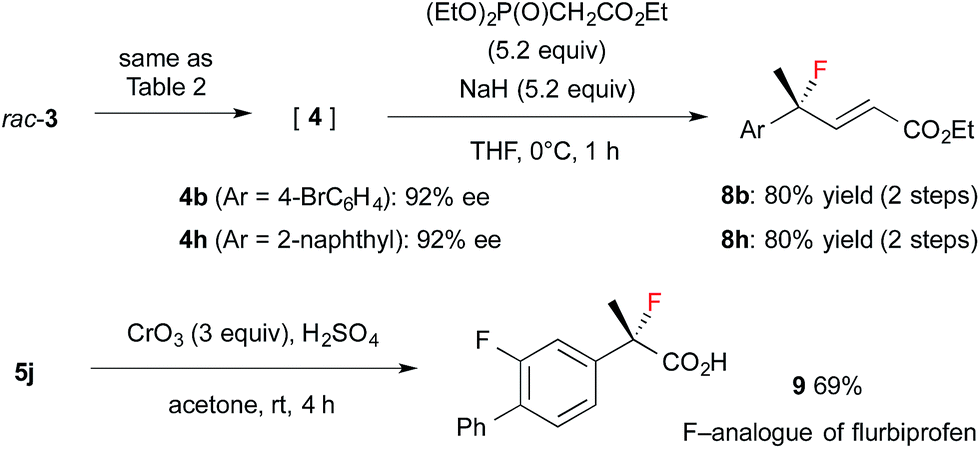 | ||
| Scheme 3 Synthesis applications of α-fluoroaldehydes. | ||
We further investigated the synthetic utility of α-fluoroaldehydes 4. Although, in general, the cleavage of carbon–fluorine bonds is not facile due to the strength of the bond, methods for C–F bond activation have recently garnered significant interest.9 The SN2-type nucleophilic substitution of sp3-alkylfluorides is known to be a challenging reaction; in particular, there are very few examples of the substitution of tertiary alkylfluorides.14 We recently reported that the SN2 reaction of α-chloro-α-keto esters with sodium azide and alkylthiols proceeds smoothly, despite the fact that the reaction occurs at a tertiary carbon.15 This finding encouraged us to examine the nucleophilic substitution of α-fluoroaldehydes 4. First, typical nucleophiles such as sodium azide and alkylthiols were surveyed, but the desired product was not obtained. Eventually, we found that treatment of 4a with NaOMe in methonal yielded the corresponding α-hydroxyacetal 10a in a good conversion (Table 3).16 Due to the difficulties in purifying 4a, enantioselective fluorination of 3a and subsequent hydroxyacetalization were performed in a one-pot fashion. Notably, the enantiopurity of 10a was nearly the same as that of 4a. This result indicated that the C–F bond cleavage occurred in a stereospecific manner. As summarized in Table 3, various α-hydroxyacetals 10 were synthesized in good yields with high enantioselectivities via the sequential fluorination–alkaline treatment. When the second step was carried out with NaH in ethylene glycol, the corresponding α-hydroxy cyclic acetal 12 was obtained. The present method would be a good alternative to direct oxidation of α-branched aldehydes.8,17 Our method does not require the use of any explosive oxidant and simultaneously protects the carbonyl group. The resulting 10a could be easily converted into α-hydroxy ester 13 without loss of enantiopurity (Scheme 4). The absolute configuration of 13 was determined to be R, by comparison of reported optical rotation values;18 these results confirmed that this transformation involved the Walden inversion.
| a Isolated yields of 10–12 from 3 are described. b The first step was carried out at rt. c es = (ee of 10–12)/(ee of 4). d The second step was carried out at rt. under reflux conditions. Purified product contained ca. 10% of an inseparable by-product. e The second step was carried out with NaH in ethylene glycol instead of NaOR2/R2OH. |
|---|
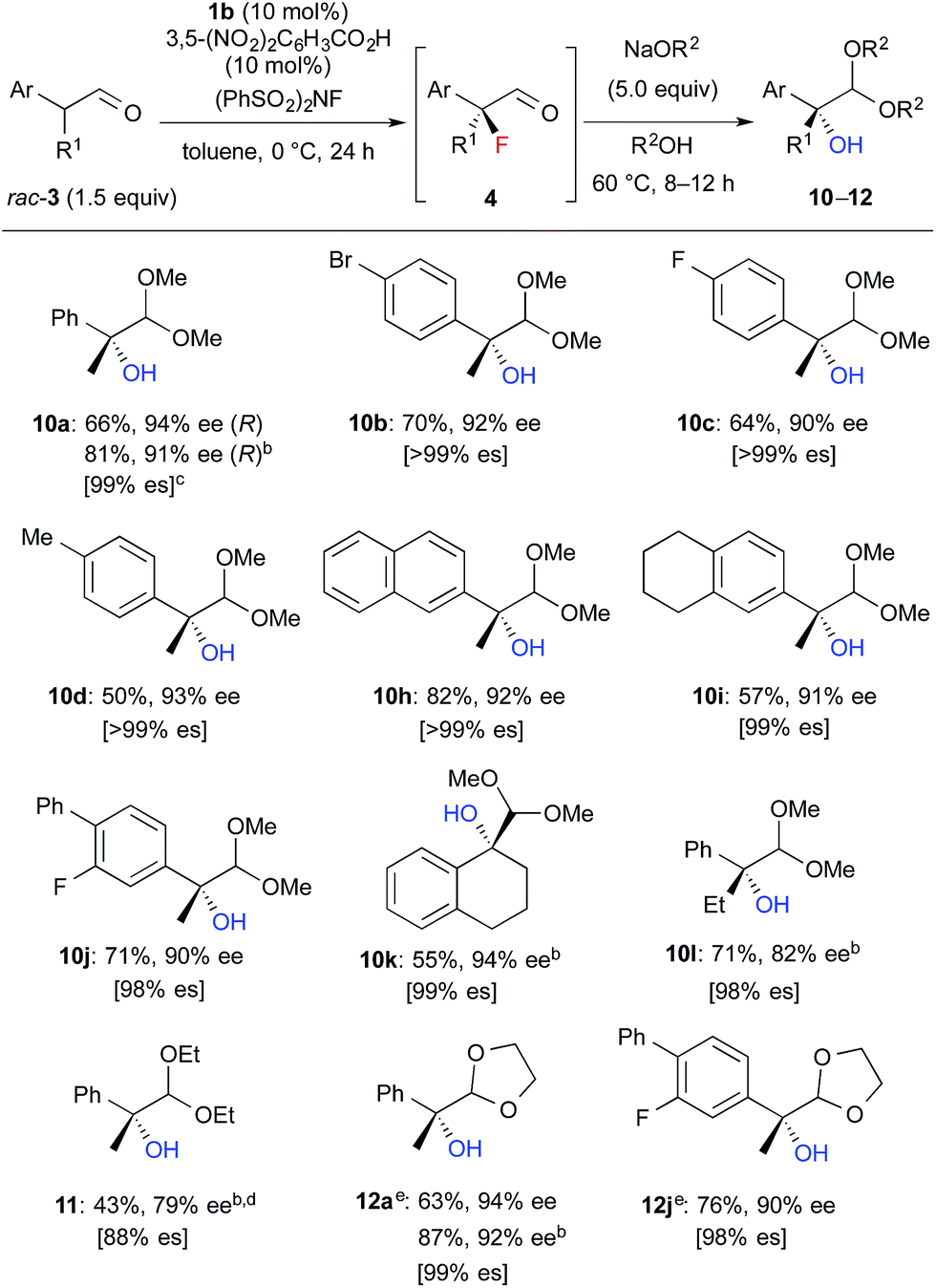
|
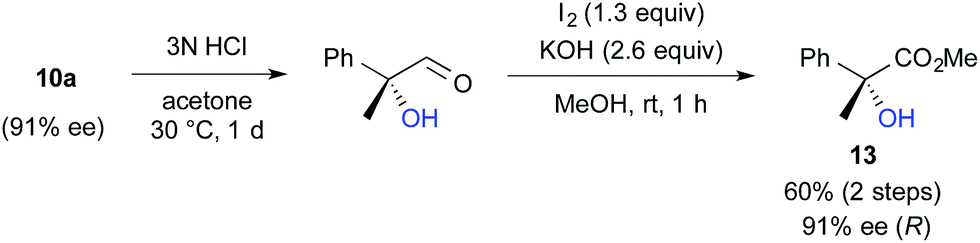 | ||
| Scheme 4 Synthesis of α-hydroxyesters. | ||
The proposed reaction mechanism for the formation of hydroxyacetals 10 is shown in Scheme 5. 1H NMR studies revealed that α-fluoroaldehyde 4 is in equilibrium with hemiacetal I in d4-methanol. Upon treatment with NaOMe, epoxide II is formed via intramolecular SN2 displacement, which involves the stereospecific cleavage of C–F bond. Then, regeneration of the carbonyl moiety and subsequent acetalization or direct SN2-type ring opening of II with methoxide affords hydroxyacetal 10.
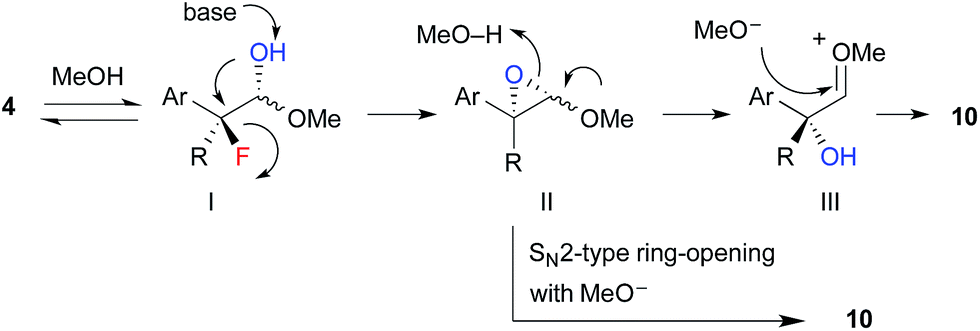 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we developed a new class of chiral primary amine catalysts and successfully applied them in the enantioselective fluorination of α-branched aldehydes. Further, we found that the resulting fluoroaldehydes could be converted into the corresponding α-hydroxyacetals via stereospecific C–F bond cleavage.Acknowledgements
This study was supported by Daiichi Sankyo Co., Ltd. and a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” (26105728) from MEXT, Japan. We thank the Nippon Synthetic Chemical Industry Co., Ltd. for supplying ethyl isocyanoacetate, which was used in the synthesis of the chiral amine catalyst.Notes and references
- (a) V. Gouverneur and K. Müller, Fluorine in Pharmaceutical and Medicinal Chemistry, Imperial College Press, London, 2012 Search PubMed; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons, Chichester, 2009 Search PubMed.
- For pioneering studies on enantioselective fluorination of active methine compounds, see: (a) L. Hintermann and A. Togni, Angew. Chem., Int. Ed., 2000, 39, 4359 CrossRef CAS; (b) Y. Hamashima, K. Yagi, H. Takano, L. Tamás and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 14530 CrossRef CAS PubMed; (c) Y. Hamashima, T. Suzuki, H. Takano, Y. Shimura and M. Sodeoka, J. Am. Chem. Soc., 2005, 127, 10164 CrossRef CAS PubMed; (d) N. Shibata, J. Kohno, K. Takai, T. Ishimaru, S. Nakamura, T. Toru and S. Kanemasa, Angew. Chem., Int. Ed., 2005, 44, 4204 CrossRef CAS PubMed ; see also a very recent review on asymmetric fluorination: ; (e) V. Bizet, T. Besset, J.-A. Ma and D. Cahard, Curr. Top. Med. Chem., 2014, 14, 901 CrossRef CAS PubMed.
- For recent examples of enantioselective fluorination of α,α-disubstituted carbonyl compounds, see: (a) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376 CrossRef CAS PubMed; (b) R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268 CrossRef CAS PubMed; (c) X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225 CrossRef CAS PubMed; (d) S. Y. Lee, S. Neufeind and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 8899 CrossRef CAS PubMed.
- For examples of other approach, see: (a) É. Bélanger, K. Cantin, O. Messe, M. Tremblay and J.-F. Paquin, J. Am. Chem. Soc., 2007, 129, 1034 CrossRef PubMed; (b) H. P. Shunatona, N. Früh, Y.-M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 7724 CrossRef CAS PubMed; (c) J. Wu, Y.-M. Wang, A. Drljevic, V. Rauniyar, R. J. Phipps and F. D. Toste, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 13729 CrossRef CAS PubMed; (d) Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 5520 CrossRef CAS PubMed.
- For pioneering studies, see: (a) M. Marigo, D. Fielenbach, A. Braunton, A. Kjœrsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 3703 CrossRef CAS PubMed; (b) D. D. Steiner, N. Mase and C. F. Barbas III, Angew. Chem., Int. Ed., 2005, 44, 3706 CrossRef CAS PubMed; (c) T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826 CrossRef CAS PubMed; (d) D. Enders and M. R. M. Hüttle, Synlett, 2005, 991 CrossRef CAS.
- S. Brandes, B. Niess, M. Bella, A. Prieto, J. Overgaard and K. A. Jørgensen, Chem. –Eur. J., 2006, 12, 6039 CrossRef CAS PubMed.
- (a) K. Shibatomi and H. Yamamoto, Angew. Chem., Int. Ed., 2008, 47, 5796 CrossRef CAS PubMed; (b) K. Shibatomi, T. Okimi, Y. Abe, A. Narayama, N. Nakamura and S. Iwasa, Beilstein J. Org. Chem., 2014, 10, 323 CrossRef PubMed.
- Very recently, new primary amine-catalyzed enantioselective fluorination of α-alkyl-α-aryl aldehydes achieving up to 86% ee was published, see: M. R. Witten and E. N. Jacobsen, Org. Lett., 2015, 17, 2772 CrossRef CAS PubMed.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed; (b) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS PubMed.
- Although the synthesis of 1c was reported previously, there is no report on its use in organocatalysis. For the synthetic method, see: (a) J.-P. Mazaleyrat, A. Gaucher, M. Wakselman, L. Tchertanov and J. Guilhem, Tetrahedron Lett., 1996, 37, 2971 CrossRef CAS; (b) K. Shibatomi, T. Muto, Y. Sumikawa, A. Narayama and S. Iwasa, Synlett, 2009, 241 CrossRef CAS.
- (a) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 1999, 121, 6519 CrossRef CAS; (b) T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 5139 CrossRef CAS PubMed.
- (a) C. Liu, Q. Zhu, K.-W. Huang and Y. Lu, Org. Lett., 2011, 13, 2638 CrossRef CAS PubMed; (b) A. Desmarchelier, H. Yalgin, V. Coeffard, X. Moreau and C. Greck, Tetrahedron Lett., 2011, 52, 4430 CrossRef CAS; (c) A. Theodorou, G. N. Papadopoulos and C. G. Kokotos, Tetrahedron, 2013, 69, 5438 CrossRef CAS.
- (a) H. Fujisawa, T. Fujiwara, Y. Takeuchi and K. Omata, Chem. Pharm. Bull., 2005, 53, 524 CrossRef CAS PubMed; (b) Y. Yamauchi, T. Fukuhara, S. Hara and H. Senboku, Synlett, 2008, 438 CAS; (c) I. T. Schiefer, S. Abdul-Hay, H. Wang, M. Vanni, Z. Qin and G. R. J. Thatcher, J. Med. Chem., 2011, 54, 2293 CrossRef CAS PubMed.
- For examples of SN2-type substitution of C–F bonds at a chiral carbon center, see: (a) L. Zhang, W. Zhang, J. Liu and J. Hu, J. Org. Chem., 2009, 74, 2850 CrossRef CAS PubMed; (b) A. M. Träff, M. Janjetovic, L. Ta and G. Hilmersson, Angew. Chem., Int. Ed., 2013, 52, 12073 CrossRef PubMed; (c) P. A. Champagne, J. Pomarole, M.-E. Thérien, Y. Benhassine, S. Beaulieu, C. Y. Legault and J.-F. Paquin, Org. Lett., 2013, 15, 2210 CrossRef CAS PubMed.
- (a) K. Shibatomi, A. Narayama, Y. Soga, T. Muto and S. Iwasa, Org. Lett., 2011, 13, 2944 CrossRef CAS PubMed; (b) K. Shibatomi, Y. Soga, A. Narayama, I. Fujisawa and S. Iwasa, J. Am. Chem. Soc., 2012, 134, 9836 CrossRef CAS PubMed; (c) K. Shibatomi, M. Kotozaki, N. Sasaki, I. Fujisawa and S. Iwasa, Chem. –Eur. J., 2015, 21, 14095 CrossRef CAS PubMed.
- For the formation of α-hydroxyacetals with α-chloroaldehydes, see: H. Arasaki, M. Iwata, D. Nishimura, A. Itoh and Y. Masaki, Synlett, 2004, 546 CAS.
- (a) N. Demoulin, O. Lifchits and B. List, Tetrahedron, 2012, 68, 7568 CrossRef CAS; (b) O. Lifchits, N. Demoulin and B. List, Angew. Chem., Int. Ed., 2011, 50, 9680 CrossRef CAS PubMed.
- L. C. Wieland, H. Deng, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 15453 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including characterization date, copies of 1H, 13C, 19F NMR and HPLC traces. See DOI: 10.1039/c5sc03486h |
| This journal is © The Royal Society of Chemistry 2016 |