
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zwitterionic and biradicaloid heteroatomic cyclopentane derivatives containing different group 15 elements†
Alexander
Hinz
a,
Axel
Schulz
*ab and
Alexander
Villinger
a
aInstitut für Chemie, Universität Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany. E-mail: axel.schulz@uni-rostock.de
bAbteilung Materialdesign, Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 27th October 2015
Abstract
The formal cyclopentane-1,3-diyl derivatives [E1(μ-NTer)2({E2C} = NDmp)] (Ter = 2,6-dimesityl-phenyl, Dmp = 2,6-dimethylphenyl) were prepared by 1,1-insertion of CNDmp into the N–E2 bond of [E1(μ-NTer)2E2] (E1 = N, P; E2 = P, As). The insertion does not occur for E1 = E2 = As or E2 = Sb. Dependent on the choice of formal radical centres E, either a biradicaloid or a zwitterion was obtained. The biradicaloid features a P and an As radical center and its biradical character was established by computations as well as characteristic reactivity with respect to the formation of a housane derivative and the activation of molecules bearing multiple bonds, which was demonstrated using the example of PCtBu. In contrast, the formally N,As- and N,P-centered biradicaloids are better regarded as zwitterionic species in accord with computations and diminished reactivity, as neither housane formation nor activation of multiple bonds could be observed.
Introduction
Biradicals and biradicaloids are highly reactive species that can occur in the processes of bond formation and bond breaking. They were discussed as intermediates even in Diels–Alder reactions by M. Dewar et al.1 Hence, the study of biradicaloids is of general importance. Excellent reviews on this topic were recently published by F. Breher and M. Abe.2,3 While for cyclopentane-1,3-diyl (Scheme 1, species A) several stable main group derivatives are known,4–12 especially cyclopentane-1,3-diyls are elusive. The parent cyclopentane-1,3-diyl was first observed in 1975 by Buchwalter and Closs, and since then targeted repeatedly by theoretical and in situ spectroscopic studies. To date, several heteroatom-substituted derivatives of cyclopentanediyl bearing different substituents are known (selected examples: Scheme 1, species B–D).13–26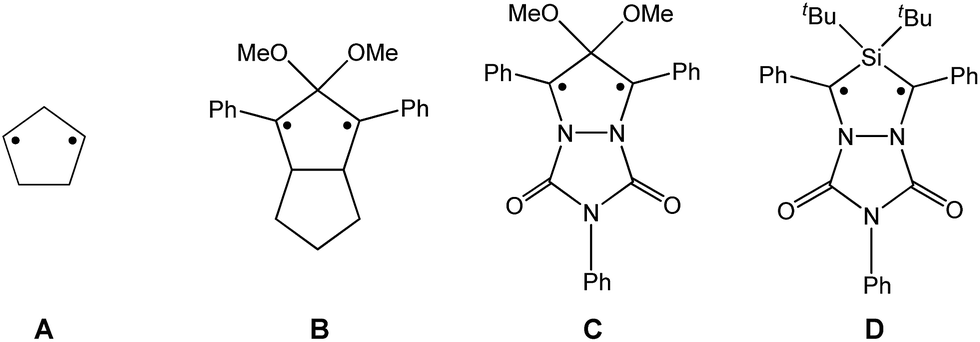 | ||
| Scheme 1 Selected known cyclopentane-1,3-diyl derivatives (A–D). | ||
Until recently, all known cyclobutane-1,3-diyl derivatives incorporated equivalent radical centres, even though several examples investigated by the groups of Power and Yoshifuji are known featuring differing bridging moieties.27–31 A synthetic protocol was devised by our group, enabling the synthesis of the formal group-15-substituted cyclobutanediyls [As(μ-NTer)2P] and [E1(μ-NTer)2E2] (E1 = N with E2 = P, As, Sb; 1E1E2 in Scheme 2).32,33 The reactivity of singlet biradicaloids was mainly studied using the examples of diboradiphosphonio-cyclobutanediyls [iPr2P(μ-BtBu)]2,35,36 diphosphacyclobutane-diyls [ClC(μ-PMes*)]2 (Mes* = 2,4,6-tri-tertbutylphenyl),37,38 diphosphadiazanediyls [P(μ-NTer)]2 (Ter = 2,6-bis(2,4,6-trimethylphenyl)phenyl),39,40 and digermynes R2Ge2 (R = 2,6-bis(2,6-diisopropylphenyl)phenyl).31,41–43 For the triazenide-derived species [E2(μ-NTer)2N], only diminished reactivity was observed, hence these are better regarded as zwitterionic compounds than as biradicaloids in agreement with computational studies. In the case of [Sb(μ-NTer)2P], the biradicaloid was found to be a transient intermediate, whose existence could be proven by trapping experiments.34
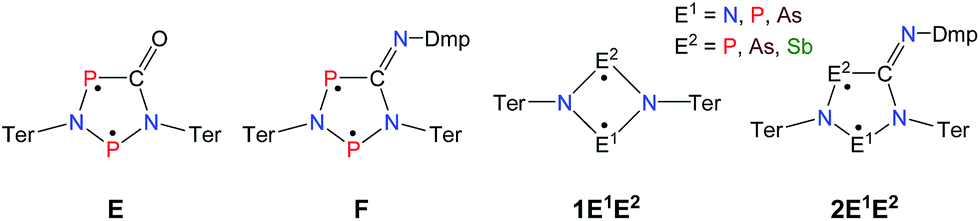 | ||
| Scheme 2 Stable cyclopentane-1,3-diyl derivatives (E and F), group-15-substituted cyclobutanediyls (1) and the targeted cyclopentane-1,3-diyls (2). | ||
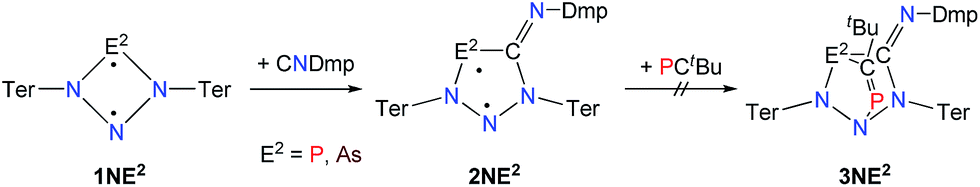
A viable access to a stable singlet derivative of formal heteroatomic cyclopentane-1,3-diyls was found in the 1,1-insertion of carbon monoxide, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O, into cyclodiphospha diazanediyl, [P(μ-NTer)]2 (1PP), which afforded species E (Scheme 2).44 Subsequent systematic investigations targeted the activation of isonitriles, CN–R (R = tBu, Dmp, N(SiMe3)2, Ter; Ter = 2,6-dimesityl-phenyl, Dmp = 2,6-dimethylphenyl), with diphosphadiazanediyl 1PP. By variation of the organic substituent, steric and electronic properties of the isonitrile could be varied. These could be adjusted to cleanly afford the cyclopentane-1,3-diyl derivative, when 2,6-dimethylphenyl-isonitrile was utilized (species F, Scheme 2).45 In this contribution, we report on the formation of cyclopentane-1,3-diyls bearing different group 15 radical centres (2E1E2) by reaction of the available group 15 cyclobutanediyl derivatives (1E1E2) with a selected isonitrile, CN–Dmp (Schemes 3–5).
O, into cyclodiphospha diazanediyl, [P(μ-NTer)]2 (1PP), which afforded species E (Scheme 2).44 Subsequent systematic investigations targeted the activation of isonitriles, CN–R (R = tBu, Dmp, N(SiMe3)2, Ter; Ter = 2,6-dimesityl-phenyl, Dmp = 2,6-dimethylphenyl), with diphosphadiazanediyl 1PP. By variation of the organic substituent, steric and electronic properties of the isonitrile could be varied. These could be adjusted to cleanly afford the cyclopentane-1,3-diyl derivative, when 2,6-dimethylphenyl-isonitrile was utilized (species F, Scheme 2).45 In this contribution, we report on the formation of cyclopentane-1,3-diyls bearing different group 15 radical centres (2E1E2) by reaction of the available group 15 cyclobutanediyl derivatives (1E1E2) with a selected isonitrile, CN–Dmp (Schemes 3–5).
 | ||
| Scheme 3 Not accessible cyclopentane-1,3-diyl derivatives (2AsAs, 2NSb, 2PSb). | ||
 | ||
| Scheme 4 Formation of 2NP and 2NAs. | ||
 | ||
| Scheme 5 Synthesis and reactivity of P,As-centered cyclopentane-1,3-diyl derivative 2PAs, housane formation on irradiation (365 nm) and addition reaction to 3PAs. | ||
Results and discussion
Synthesis
Cyclobutanediyl derivatives 1E1E2 are strongly coloured compounds.32,33,46 Thus, the reactions can often easily be followed visually, apart from reaction monitoring by NMR spectroscopy. While the diphosphadiazanediyl 1PP readily reacted with 2,6-dimethylphenyl-isonitrile to give 2PP,45 no such transformation was observed when the heavier homologue diarsadiazanediyl 1AsAs was utilized (Scheme 3). Also, neither the antimony-containing species, stable 1NSb, nor in situ generated 1PSb, reacted with CNDmp at all.On the contrary, the ring expansion reaction of the triazenide-derived cyclobutanediyl derivatives 1NP and 1NAs proceeded smoothly (Scheme 4) when a solution of CNDmp in benzene was added slowly at ambient temperature to a solution of 1NP and 1NAs, respectively, in benzene. Since the starting material 1NP could not be isolated due to the recurring formation of the triazenide Ter2N3H as an impurity,33 the insertion of the isonitrile and subsequently, the attempted conversion with PCtBu, was investigated by means of spectroscopy. The 31P NMR spectrum of 1NP displays a singlet resonance at +342.4 ppm, that shifts to +167.3 ppm upon addition of the isonitrile, indicating the formation of 2NP in good agreement with 1,2,3,4-triazaphospholes prepared by Müller et al. and Jones et al. utilizing “click reaction” of azides with phosphaalkynes (e.g. C5NH4–N3PC–tBu 167.5 ppm).47–50 It should be noted that various attempts of crystallization only afforded the triazenide Ter2N3H and the product 2NP could not be isolated. Upon insertion of the isonitrile, the colour of the solution changed from yellow (1NP) to red (2NP: λmax = 490, calc. 476 nm).51,52 The attempted addition of PCtBu did not alter any of these characteristics, indicating that no reaction with 2NP occurred in accord with a rather small biradical character (see below).
The reaction of 1NAs with CNDmp similarly resulted in a change of colour from initially yellow (λmax = 379 nm) to red, indicating the presence of 2NAs (λmax = 523, calc. 518 nm). Similar to 2NP, 2NAs features a ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) vibration at 1612 in the Raman and at 1610 cm−1 in the IR spectrum which is significantly different from the ν(CN) vibration of pure CNDmp exhibiting a CN triple bond (2123 cm−1). Crystals suitable for single X-ray studies were obtained after concentration at 4 °C in good yields (83%). Red needle-shaped crystals of 2NAs decompose above 141 °C and are moisture and air sensitive. Like 2NP, 2NAs does not react with PCtBu also displaying diminished biradical character. The molecular structure of 2NAs (Fig. 1) features a planar five-membered N3CAs heterocycle. The As–N bond of 1.875(3) Å is considerably longer than in the known tetrazarsole galliumtrichloride adduct Mes*N4As·GaCl3 (1.784(2), 1.805(2) Å; cf. Σrcov(As–N) = 1.91 Å) indicating single bond character.53 The same holds true for the As–C bond with 1.902(4) Å (Σrcov(As–C) = 1.97 Å). The N–N distances in 2NAs of 1.316(4) and 1.349(4) Å are between the sum of covalent radii for a double and a single bond (1.20, 1.42 Å), indicating delocalized double bond character, while the C49–N3 bond length (1.428(5) Å) corresponds to a single bond (Σrcov(C–N) = 1.46 Å) contrary to the exocyclic C49–N4 bond (1.293(5) Å) which is in the typical range of a CN double bond.54
N) vibration at 1612 in the Raman and at 1610 cm−1 in the IR spectrum which is significantly different from the ν(CN) vibration of pure CNDmp exhibiting a CN triple bond (2123 cm−1). Crystals suitable for single X-ray studies were obtained after concentration at 4 °C in good yields (83%). Red needle-shaped crystals of 2NAs decompose above 141 °C and are moisture and air sensitive. Like 2NP, 2NAs does not react with PCtBu also displaying diminished biradical character. The molecular structure of 2NAs (Fig. 1) features a planar five-membered N3CAs heterocycle. The As–N bond of 1.875(3) Å is considerably longer than in the known tetrazarsole galliumtrichloride adduct Mes*N4As·GaCl3 (1.784(2), 1.805(2) Å; cf. Σrcov(As–N) = 1.91 Å) indicating single bond character.53 The same holds true for the As–C bond with 1.902(4) Å (Σrcov(As–C) = 1.97 Å). The N–N distances in 2NAs of 1.316(4) and 1.349(4) Å are between the sum of covalent radii for a double and a single bond (1.20, 1.42 Å), indicating delocalized double bond character, while the C49–N3 bond length (1.428(5) Å) corresponds to a single bond (Σrcov(C–N) = 1.46 Å) contrary to the exocyclic C49–N4 bond (1.293(5) Å) which is in the typical range of a CN double bond.54
 | ||
| Fig. 1 Molecular structure of 2NAs. Thermal ellipsoids are drawn at 50% probability (173 K). Selected bond lengths [Å] and angles [°]: 2NAs: As1–N1 1.875(3), As1–C49 1.902(4), N1–N2 1.316(4), N2–N3 1.349(4), N3–C49 1.428(5), N4–C49 1.293(5); N1–As1–C49 82.71(16), N2–N1–As1 119.4(2), N1–N2–N3 109.8(3), N2–N3–C49 119.4(3). | ||
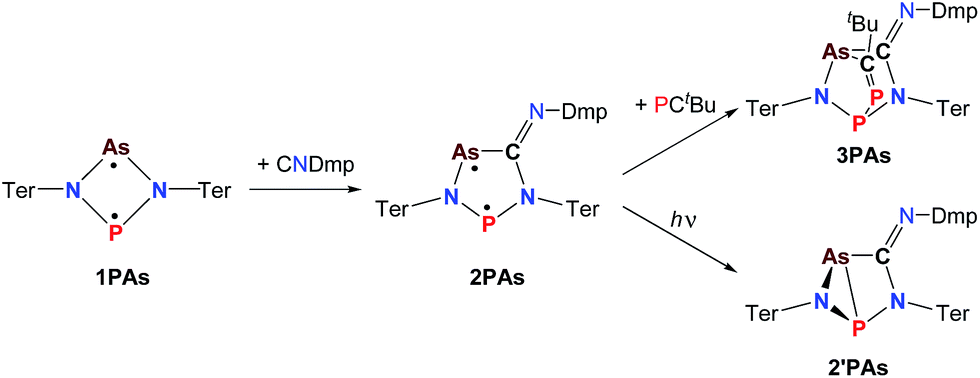
In a next series of experiments we treated a solution of 1PAs in benzene with CNDmp. Within 10 minutes the insertion of CNDmp into the dark purple 1PAs (λmax = 550 nm)32 led in good yields (68%) to a singlet biradicaloid cyclopentanediyl derivative, 2PAs, which is of green colour (λmax = 431, 684, calc. 454, 674 nm, Scheme 5). Interestingly, at the beginning of the reaction the reaction mixture appeared dark grey due to the presence of both the starting material and the reaction product. Astonishingly, the 31P NMR shift merely changed from 268.8 to 269.0 ppm. However, as discussed before, upon incorporation of the isonitrile into the four-membered biradicaloid, the ν(CN) vibration is dramatically shifted from 2123 to 1633 cm−1, as expected for the transition from a C–N triple to double bond. Crystals of 2PAs decompose above 122 °C.
The connectivity is furthermore corroborated by the 13C NMR data, in which the former isonitrile carbon atom gives rise to a resonance at 184.98 ppm, which appears as doublet with a small JCP = 9.9 Hz, indicating a 2J coupling. X-ray diffraction experiments on single crystals of 2PAs revealed a secondary irradiation-induced isomerization to the housane isomer 2′PAs (Scheme 5, Fig. 2, see below), as it was observed for cyclopentanediyl derivatives such as 2PP indicating substantial biradical character.44,45 However contrary to 2PP, for 2PAs this isomerization did not cause the crystals to completely decompose. Two data sets were collected from a single crystal: the first data set without irradiation prior to measurement and the second after 12 hours of X-ray irradiation on the diffractometer. In both cases, the structural model features disordered P and As atoms. While in the first data set, the planar five-membered species is dominant (86% occupation, Fig. 2 top), in the second data set, which was collected after 12 hours of X-ray irradiation, 95% occupation are found for the housane species (Fig. 2 top). In solution, all attempts to generate 2′PAs by UV irradiation of 2PAs led to decomposition, thus no NMR data for 2′PAs could be obtained.
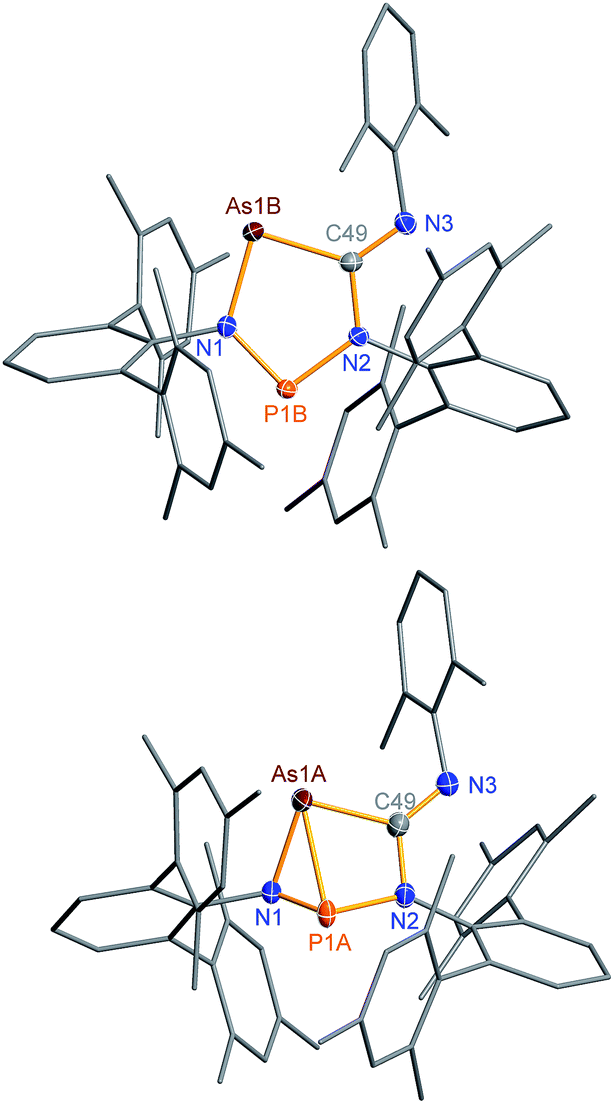 | ||
| Fig. 2 Molecular structure of 2PAs (top) and 2′PAs (bottom). Thermal ellipsoids are drawn at 50% probability (123 K). Selected bond lengths [Å] and angles [°]: 2PAs: As1B–N1 1.874(2), As1B–C49 1.937(2), P1B–N1 1.636(2), P1B–N2 1.691(2), As1B–P1B 3.049(2), P1B–N1–As1B 120.5(1); 2′PAs: As1A–P1A 2.2920(7), As1A–C49 2.011(2), As1A–N1 1.970(2), P1A–N1 1.692(2), P1A–N2 1.801(2), N1–P1A–N2 95.12(7), P1A–N1–As1A 77.10(6). | ||
The biradical character of 2PAs invokes high reactivity, which could be demonstrated in the activation of molecules such as phosphaalkynes, PCtBu, bearing a PC triple bond. The initially green solution of 2PAs in benzene quickly turned yellow upon addition of the phosphaalkyne and formation of 3PAs was observed in good yields (78%, Scheme 5, Fig. 3). The 31P NMR spectrum exhibited an AB spin system (331.8, 156.8 ppm), indicating that exclusively one isomer was formed. The strong JPP coupling of 260 Hz is characteristic for a 1JPP coupling constant. Single crystal X-ray structure elucidation unequivocally revealed the exclusive formation of one isomer (P–P and C–As bonded species, Fig. 3) therefore featuring complete regioselectivity. This regioselectivity is most probably caused by steric hindrance, as the formation of the putative C–P and P–As bonded isomer would considerably distort the “pocket” formed by the two terphenyls, which is already opened to one side due to insertion of the isonitrile (difference between both isomers: ΔE = 48 kJ mol−1). It is interesting to note that for the cyclobutanediyl derivative [As(μ-NTer)2P], the regioselectivity was opposite and only the C–P and P–As bonded isomer was observed due to thermodynamic preference of a C–P over C–As bond.32
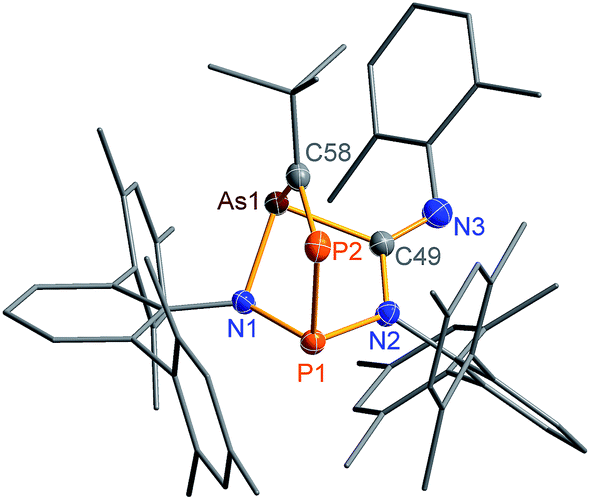 | ||
| Fig. 3 Molecular structure of 3PAs. Thermal ellipsoids are drawn at 50% probability (173 K). Selected bond lengths [Å] and angles [°]: 3PAs: As1–N1 1.895(2), As1–C58 1.996(2), As1–C49 2.007(2), P1–N1 1.7378(18), P1–N2 1.756(2), P1–P2 2.2822(8), P2–C58 1.673(2), N3–C49 1.270(3), N1–As1–C58 93.47(9), C58–P2–P1 96.42(8), P1–P2–C58–As1–2.50(13). | ||
Single crystals of 2PAs/2′PAs and 3PAs suitable for structure analysis were obtained from benzene solutions. The most prominent structural feature of 2PAs represents the almost planar five-membered heterocycle with rather short P–N bond lengths (P1B–N1 1.636(2), P1B–N2 1.691(2) Å) in the range of PN double bonds (Σrcov(PN) = 1.62 Å), while the As–N (As1B–N1 1.874(2) Å) and As–C bonds (As1B–C49 1.937(2) Å, Fig. 2) are in the range of single bonds (see also 2NAs, Fig. 1).53,55,56
The structure changes dramatically upon irradiation and formation of the housane 2′PAs. The transannular P–As distance is shortened from 3.049(2) to 2.2920(7) Å clearly indicating the presence of a transannular P–As single bond (Σrcov(P–As) = 2.32 Å). Additionally, the P–N–As angle strongly decreases from 120.5(1) to 77.10(6)°. The three-membered As–N–P ring is almost perpendicular condensed to the four-membered As–P–N–C ring. These experimental structural parameters are in good agreement with those of DFT computations (see below and ESI†).
The phosphaalkyne addition product 3PAs shows a puckered five-membered ring with a transannular P–As distance of 2.918(2) Å. The P–C bridging bond length amounts to 1.673(2) Å in accord with a PC double bond.
Computations – bonding and biradical character
To shed some light into the bonding and biradical character, MO (Fig. 4), NBO (Scheme 6) and CASSCF computations have been carried out. MO and NBO computations show formal 6π electronic 2E1E2 five-membered heterocycles (Table 1). A common electronic feature of the heterocycles 2NP, 2NAs, and 2PAs is the weak aromaticity as indicated by NICS values (Table 1).57 The frontier orbitals feature a p-type transannular antibonding π-HOMO and transannular bonding π*-LUMO between the radical centres, in accord with other group 15 singlet biradicaloids (Fig. 4).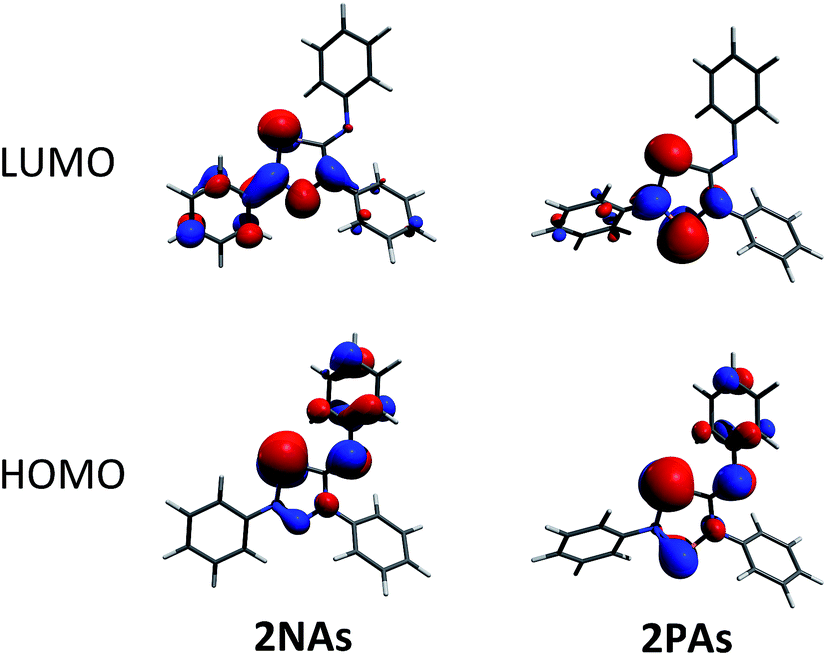 | ||
| Fig. 4 Frontier orbitals of 2NAs (left) and 2PAs (right). For the orbital representations phenyl-substituted model compounds are used for clarity. | ||
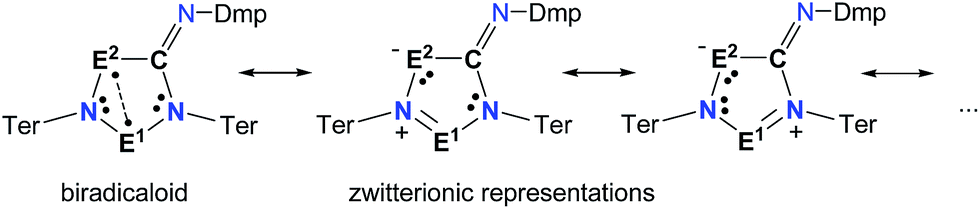 | ||
| Scheme 6 Selected Lewis representations according to NBO analysis. | ||
| 2NP | 2NAs | 2NSba | 2 PAs | 2PSba | 2AsAsa | |
|---|---|---|---|---|---|---|
| a Species not isolated. b Calculated in [kJ mol−1]: ΔEform = E(2) − [E(1) + E(CNDmp)]. c β = 2c22/(c12 + c22), the two main contributions according to CASSCF(6,6) computations were used. d π electrons in cpd = cyclopentanediyl, occupation of pz orbitals in the five-membered heterocycle according to NBO analysis. e In ppm. f Wiberg bond index. | ||||||
| λ maxcalc. | 476 | 518 | 542 | 674 | 715 | 750 |
| ΔEformb | −148.0 | −114.6 | −55.6 | −94.8 | −51.4 | −91.2 |
| S–T gapb | −148.9 | −132.7 | −118.8 | −75.4 | −72.7 | −44.4 |
| β | 13% | 11% | 4% | 24% | 7% | 38% |
| c 1 | 0.946 | 0.956 | 0.961 | 0.916 | 0.982 | 0.889 |
| c 2 | −0.250 | −0.229 | −0.132 | −0.339 | −0.185 | −0.433 |
| BO(E–E)f | 0.313 | 0.288 | 0.262 | 0.389 | 0.343 | 0.434 |
| π e cpdd | 6.51 | 6.51 | 6.55 | 6.49 | 6.53 | 6.50 |
| NICS(0)e | −8.32 | −7.45 | −6.40 | −5.62 | −4.84 | −4.66 |
| NICS(1)e | −6.65 | −6.10 | −5.51 | −3.24 | −3.18 | −2.90 |
To investigate the biradicaloid character of 2E1E2, the singlet–triplet energy gap (ΔES–T) was computed for 2E1E2 and CASSCF(6,6) computations were carried out (CASSCF = complete active space self-consistent field). Experimentally, biradicaloids 2E1E2 show no EPR signal and 1H, 13C, and 31P NMR signals. All 2E1E2 compounds have a singlet ground state in accord with rather large ΔES–T values (Table 1) significantly decreasing the heavier the group 15 elements E1 and E2. CASSCF(6,6) computations confirmed the biradicaloid nature of 2E1E2. The dominant contributions to the CI wave function arise from the HOMO/LUMO exchange. The biradicaloid character can be estimated by using the formula: β = 2c22/(c12 + c22).58 Hence, upon insertion of the isonitrile into the four-membered ring of 1 the biradical character is preserved compared to the starting material 1E1E2. Therefore, as illustrated in Scheme 6 and Table 1, the zwitterionic character increases (biradical character decrease) along E1 = As < P < N and E2 = Sb < As < P. For example, a biradical character β of only 13% was computed for 2NP and 11% for 2NAs, respectively (CASSCF(6,6), coefficients of main contributions 0.946, −0.250 for 1NP and 0.956, −0.229 for 1NAs).58 However, 2PAs features substantial biradical character of β = 24% (CASSCF(6,6), c1 = 0.916 and c2 = −0.339), in agreement with the experimental fact that this species is capable of activating molecules containing triple bonds (vide supra) contrary to 2NP or 2NAs. Moreover, the larger zwitterionic character of 2NAs compared to 2PAs is also manifested by the HOMO of 2NAs featuring very large coefficients at As and very small ones at N, while for 2PAs the contributions are distributed almost evenly.
The computational data show a correlation between biradical character β and Wiberg bond index (WBI) between the two radical centers (Table 1). The WBI(E1–E2) of all considered species 2E1E2 ranges from 0.262 to 0.434, which originates from partial occupation of the transannularly bonding LUMO. This reflects strong antiferromagnetic coupling between the radical centers E1 and E2. NICS values (between −2.9 and −8.3, Table 1, cf. benzene −11.5 ppm, azulene −21.5 [5 ring] and −8.3 [7 ring] ppm) decrease when heavier elements are incorporated in the five-membered ring, indicating less stabilization by electron delocalization within the heterocycle, which is due to diminished orbital overlap between E and the adjacent N or C atoms.
In analogy to the activation of CO with 1PP and the reaction of 1PP with different isonitriles, we suggest a mechanism involving the formation of a [1.1.1]bicyclic intermediate, which subsequently rearranges to give the cyclopentanediyl derivative (Scheme 7).44,45 The formation of the [1.1.1]bicyclic species is endothermic for all species 2E1E2 with E1 or E2 being N, increasing in the order As (62.8) < P (82.0) < Sb (126.8 kJ mol−1) for E. For the heavier homologues with E1 being P, it is exothermic and the reaction energy increases in the same order: E2 = As (−68.8) < P (−49.9) < Sb (−28.9 kJ mol−1). The second reaction step, the rearrangement from the [1.1.1]bicycle to the planar five-membered ring, is exothermic in every case. In this case, there is a tendency of the reaction becoming less exothermic as the pnictogen E2 becomes heavier (ΔRE: N > P > As > Sb; e.g.2PAs −66.5, 2PAs −30.9, 2PSb −7.0; Table S3†), with the exception of E1 = N and E2 = Sb, which is slightly more exothermic than for E2 = As. In all combinations of E1 and E2 being N, P, As, or Sb, the overall insertion reaction is exothermic (e.g.2NP −148.0, 2NAs −114.6, 2NSb −55.6 kJ mol−1; Tables S2 and S3†). Interestingly, all cyclopentanediyl derivatives 1 in which E1 is heavier than E2 are thermodynamically more stable than the observed species, in which E2 is heavier than E1. This means, that the observed products are formed owing to kinetic reaction control. This is plausible, since the heavier E–N bonds are weaker and hence more readily activated. This can be corroborated with computed transition states for the rearrangement from [1.1.1]bicycle to planar five-membered ring for the example of a phenyl-substituted model compound of 2PAs, in which the insertion into the N–As bond requires 16 kJ mol−1 less activation energy than into the N–P bond.
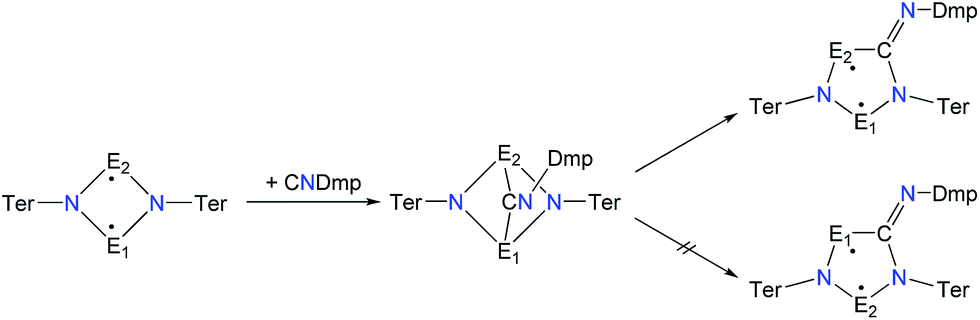 | ||
| Scheme 7 Proposed reaction mechanism for isonitrile activation with 1E1E2 (E1 is lighter than E2). | ||
Finally, we want to address the issue of housane formation. Computational studies indicate, that 2PAs is more favorable than the housane isomer 2′PAs by 94 kJ mol−1. The computed activation barrier for the formation of the P–As bond amounts to 167 kJ mol−1 and breaking the bond 73 kJ mol−1. These values are higher than computed for the previously investigated housanes (42 kJ mol−1 difference in energy, activation barrier of 83 kJ mol−1).45 This provides an explanation for the slower decomposition in the X-ray beam of the diffractometer, which allowed the structure determination of 2PAs as well as 2′PAs. However, upon UV irradiation, decomposition occurred, preventing the isolation of the housane species 2′PAs.
The electronic situation of both isomers clearly differs, as the housane features a bent bond between the former radical centres, while in the biradicaloid there is no direct interaction between P and As. This is apparent from the maximum in the ELF (electron localization function) aside the P–As axis of 2′PAs, which also features a localized double bond (Fig. 5).
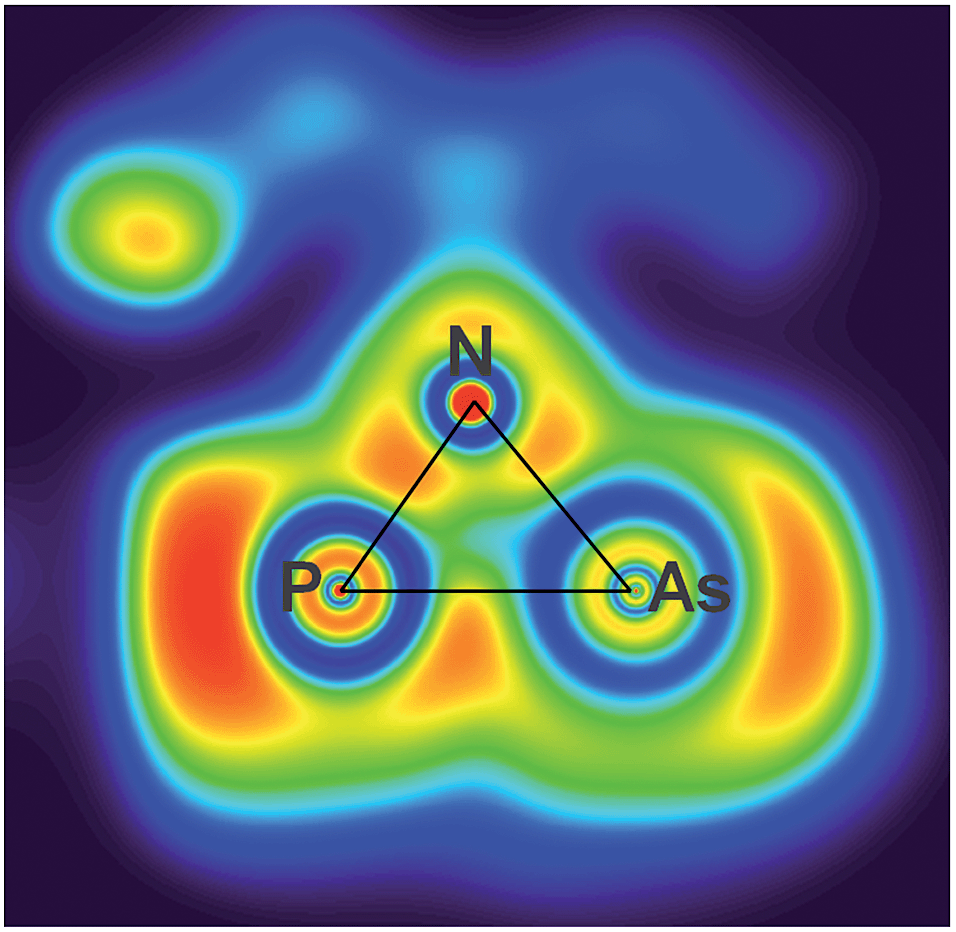 | ||
| Fig. 5 ELF representation of 2P′As utilizing a phenyl substituted model compound for clarity. A section of the N–P–As plane is shown. The maximum is located aside the axis between As (left) and P (right). | ||
Conclusions
In summary, the ring expansion reaction of cyclobutanediyls with isonitriles enabled the synthesis of three new group 15 derivatives of cyclopentane-1,3-diyl featuring N/P, N/As, or P/As atoms as radical centres within the five-membered heterocycle. However, there are limitations to this insertion reaction, and the preparation of cyclopentane-1,3-diyls bearing N/Sb, As/As, or P/Sb radical centres remains a challenge. Two reasons can be accountable for this: (i) in the zwitterionic border case, due to strong polarization the valence electron density distributed far from equally between the formal radical centres, so the biradical reactivity is diminished, and (ii) by incorporating heavier elements in the heterocycle (e.g. within the homologous series 2NP, 2NAs, 2NSb), the distance between the radical centres is large and hence the orbital overlap is small, thereby reducing the stability of the heavier cyclopentane-1,3-diyl species. This is reflected in the decreasing relative stability of the singlet ground state compared to the lowest lying triplet state.The new cyclopentane-1,3-diyl derivatives containing an N3 moiety (E1 = N) have strongly polarized N–E2 bonds, a rather small biradical character and therefore are better referred to as zwitterions, which is also manifested by their inability to activate molecules bearing multiple bonds. In contrast, the P/As centered biradicaloid 2PAs exhibits a considerable biradical character, higher reactivity and can be isomerized to the short-bond species 2′PAs or be utilized in small molecule activation.
Acknowledgements
The authors thank the DFG (SCHU 1170/11-1) for financial support. M.Sc. Jonas Bresien is gratefully acknowledged for setting up and maintaining Gaussian and NBO software on the cluster computer. A. H. thanks the GDCh for financial support.Notes and references
- M. J. S. Dewar, S. Olivella and J. J. Stewart, J. Am. Chem. Soc., 1986, 108, 5771–5779 CrossRef CAS PubMed.
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., 1995, 107, 640–642 ( Angew. Chem., Int. Ed. , 1995 , 34 , 555–557 ) CrossRef.
- D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1882 CrossRef CAS PubMed.
- H. Sugiyama, S. Ito and M. Yoshifuji, Angew. Chem., 2003, 115, 3932–3934 ( Angew. Chem., Int. Ed. , 2003 , 42 , 3802–3804 ) CrossRef.
- C. Cui, M. Brynda, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 124, 6510–6511 CrossRef PubMed.
- K. Takeuchi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 12478–12481 CrossRef CAS PubMed.
- C. Cui, M. Brynda, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 6510–6511 CrossRef CAS PubMed.
- P. Henke, T. Pankewitz, W. Klopper, F. Breher and H. Schnöckel, Angew. Chem., 2009, 121, 8285–8290 ( Angew. Chem., Int. Ed. , 2009 , 48 , 8141–8145 ) CrossRef.
- H. Cox, P. B. Hitchcock, M. F. Lappert and L. J.-M. Pierssens, Angew. Chem., 2004, 116, 4600–4604 ( Angew. Chem., Int. Ed. , 2004 , 43 , 4500–4504 ) CrossRef.
- S.-H. Zhang, H.-W. Xi, K. H. Lim, Q. Meng, M.-B. Huang and C.-W. So, Chem.–Eur. J., 2012, 18, 4258–4263 CrossRef CAS PubMed.
- S. L. Buchwalter and G. L. Closs, J. Am. Chem. Soc., 1975, 97, 3857–3858 CrossRef CAS.
- F. Kita, W. Nau, W. Adam and J. Wirz, J. Am. Chem. Soc., 1995, 8670–8671 CrossRef CAS.
- W. R. Roth, F. Bauer and R. Breuckmann, Chem. Ber., 1991, 124, 2041–2046 CrossRef CAS.
- W. Adam, H. García, M. Diefenbach, V. Martí, M. Olivucci and E. Palomares, J. Am. Chem. Soc., 2002, 124, 12192–12199 CrossRef CAS PubMed.
- M. Abe, C. Ishihara, S. Kawanami and A. Masuyama, J. Am. Chem. Soc., 2005, 127, 10–11 CrossRef CAS PubMed.
- A. Maeda, T. Oshita, M. Abe and T. A. Ishibashi, J. Phys. Chem. B, 2014, 118, 3991–3997 CrossRef CAS PubMed.
- W. Adam, M. Diefenbach and V. Martí, Eur. J. Org. Chem., 2003, 592–596 CrossRef CAS.
- T. H. Peterson and B. K. Carpenter, J. Am. Chem. Soc., 1993, 115, 5466–5478 CrossRef CAS.
- M. Abe, S. Kawanami, C. Ishihara and M. Nojima, J. Org. Chem., 2004, 69, 5622–5626 CrossRef CAS PubMed.
- W. Adam, K. Goller, T. Kammel and K. Peters, J. Org. Chem., 1995, 60, 308–316 CrossRef CAS.
- W. Adam, H. Platsch, J. Sendelbach and J. Wirz, J. Org. Chem., 1993, 58, 1477–1482 CrossRef CAS.
- M. Abe, W. Adam and W. M. Nau, J. Am. Chem. Soc., 1998, 120, 11304–11310 CrossRef CAS.
- M. Abe, W. Adam, W. T. Borden, M. Hattori, D. A. Hrovat, M. Nojima, K. Nozaki and J. Wirz, J. Am. Chem. Soc., 2004, 126, 574–582 CrossRef CAS PubMed.
- J. Ye, Y. Fujiwara and M. Abe, Beilstein J. Org. Chem., 2013, 9, 925–933 CrossRef CAS PubMed.
- S. Ito, M. Kikuchi, J. Miura, N. Morita and M. Yoshifuji, J. Phys. Org. Chem., 2012, 25, 733–737 CrossRef CAS.
- S. Ito, M. Kikuchi, M. Yoshifuji, A. J. Arduengo, T. A. Konovalova and L. D. Kispert, Angew. Chem., 2006, 118, 4447–4451 ( Angew. Chem., Int. Ed. , 2006 , 45 , 4341–4345 ) CrossRef.
- H. Sugiyama, S. Ito and M. Yoshifuji, Chem.–Eur. J., 2004, 10, 2700–2706 CrossRef CAS PubMed.
- S. Ito, M. Kikuchi, H. Sugiyama and M. Yoshifuji, J. Organomet. Chem., 2007, 692, 2761–2767 CrossRef CAS.
- X. Wang, Y. Peng, M. M. Olmstead, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2009, 131, 14164–14165 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., 2014, 127, 678–682 ( Angew. Chem., Int. Ed. , 2014 , 54 , 668–672 ) CrossRef.
- A. Hinz, A. Schulz, A. Villinger and J.-M. Wolter, J. Am. Chem. Soc., 2015, 137, 3975–3980 CrossRef CAS PubMed.
- A. Hinz, J. Rothe, A. Schulz and A. Villinger, Dalton Trans., 2016, 2, 2016 Search PubMed.
- H. Amii, L. Vranicar, H. Gornitzka, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 1344–1345 CrossRef CAS PubMed.
- G. Fuks, N. Saffon, L. Maron, G. Bertrand and D. Bourissou, J. Am. Chem. Soc., 2009, 131, 13681–13689 CrossRef CAS PubMed.
- M. Sebastian, A. J. Hoskin, M. Nieger, L. Nyulászi and E. Niecke, Angew. Chem., 2005, 117, 1429–1432 ( Angew. Chem., Int. Ed. , 2005 , 44 , 1405–1408 ) CrossRef.
- M. Sebastian, M. Nieger, D. Szieberth, L. Nyulászi and E. Niecke, Angew. Chem., 2004, 116, 647–651 ( Angew. Chem., Int. Ed. , 2004 , 43 , 637–641 ) CrossRef.
- A. Hinz, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Chem.–Eur. J., 2014, 20, 14659–14673 CrossRef CAS PubMed.
- A. Hinz, A. Schulz and A. Villinger, Chem.–Eur. J., 2014, 20, 3913–3916 CrossRef CAS PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- C. Cui, M. M. Olmstead, J. C. Fettinger, G. H. Spikes and P. P. Power, J. Am. Chem. Soc., 2005, 127, 17530–17541 CrossRef CAS PubMed.
- X. Wang, Y. Peng, Z. Zhu, J. C. Fettinger, P. P. Power, J. Guo and S. Nagase, Angew. Chem., 2010, 122, 4697–4701 ( Angew. Chem., Int. Ed. , 2010 , 49 , 4593–4597 ) CrossRef.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., 2015, 127, 2815–2819 ( Angew. Chem., Int. Ed. , 2015 , 54 , 2776–2779 ) CrossRef.
- A. Hinz, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2015, 137, 9953–9962 CrossRef CAS PubMed.
- S. Demeshko, C. Godemann, R. Kuzora, A. Schulz and A. Villinger, Angew. Chem., 2013, 125, 2159–2162 ( Angew. Chem., Int. Ed. , 2013 , 52 , 2105–2108 ) CrossRef.
- J. A. W. Sklorz, S. Hoof, M. G. Sommer, F. Weißer, M. Weber, J. Wiecko, B. Sarkar and C. Müller, Organometallics, 2014, 33, 511–516 CrossRef CAS.
- J. A. W. Sklorz, S. Hoof, N. Rades, N. de Rycke, L. Könczöl, D. Szieberth, M. Weber, J. Wiecko, L. Nyulászi, M. Hissler and C. Müller, Chem.–Eur. J., 2015, 21, 11096–11109 CrossRef CAS PubMed.
- S. L. Choong, C. Jones and A. Stasch, Dalton Trans., 2010, 39, 5774–5776 RSC.
- S. L. Choong, A. Nafady, A. Stasch, A. M. Bond and C. Jones, Dalton Trans., 2013, 42, 7775–7780 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- All computations were carried out at the PBE1PBE or CASSCF(6,6) level of theory. For Sb, a relativistic pseudopotential was used (ECP46MDF 4 46), while for all other atoms 6-31G(d,p) was employed.
- A. Schulz and A. Villinger, Angew. Chem., 2008, 120, 614–617 ( Angew. Chem., Int. Ed. , 2008 , 47 , 603–606 ) CrossRef.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- M. Kuprat, A. Schulz and A. Villinger, Angew. Chem., 2013, 125, 7266–7270 ( Angew. Chem., Int. Ed. , 2013 , 52 , 7126–7130 ) CrossRef.
- S. Herler, P. Mayer, J. Günne, A. Schulz, A. Villinger, J. J. Weigand and J. Schmedt auf der Günne, Angew. Chem., 2005, 117, 7968–7971 ( Angew. Chem., Int. Ed. , 2005 , 44 , 7790–7793 ) CrossRef.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef.
- E. Miliordos, K. Ruedenberg and S. S. Xantheas, Angew. Chem., 2013, 125, 5848–5851 ( Angew. Chem., Int. Ed. , 2013 , 52 , 5736–5739 ) CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, full characterization of all compounds and computational details. CCDC 1421413–1421416. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03515e |
| This journal is © The Royal Society of Chemistry 2016 |