
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The first intermolecular interrupted imino-Nazarov reaction: expeditious access to carbocyclic nucleoside analogues†
Ronny
William
 ,
Wei Lin
Leng
,
Siming
Wang
and
Xue-Wei
Liu
*
,
Wei Lin
Leng
,
Siming
Wang
and
Xue-Wei
Liu
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: xuewei@ntu.edu.sg
First published on 27th October 2015
Abstract
The analogous imino-variant of the Nazarov reaction suffers from unfavorable energetics associated with the ring closure process, thereby limiting the utility of this class of reactions. In this report, the realization of the first intermolecular interrupted imino-Nazarov reaction utilizing silylated pyrimidine derivatives as nucleophilic trapping partners culminated in an expedient synthetic route to carbocyclic nucleoside analogues. The potential application of silylated nucleobases to intercept the oxyallyl cation in other variants of the Nazarov reaction provides vast opportunities to develop new strategies for the formation of carbocyclic nucleoside analogues.
Carbocyclic nucleosides represent an emerging class of privileged therapeutic compounds which exhibit interesting biological properties.1,2 Substitution of endocyclic oxygen by a methylene unit in a carbocyclic nucleoside derivative imparts greater stability in the C–N pseudoglycosidic bond towards both chemical and enzymatic processes as compared to the parent nucleoside counterpart.2 Some examples of naturally-occurring and synthetic carbocyclic nucleosides with potent pharmacological activity, such as aristeromycin,3 neplanocin A,4 carbovir5 and abacavir,6 are depicted in Fig. 1.
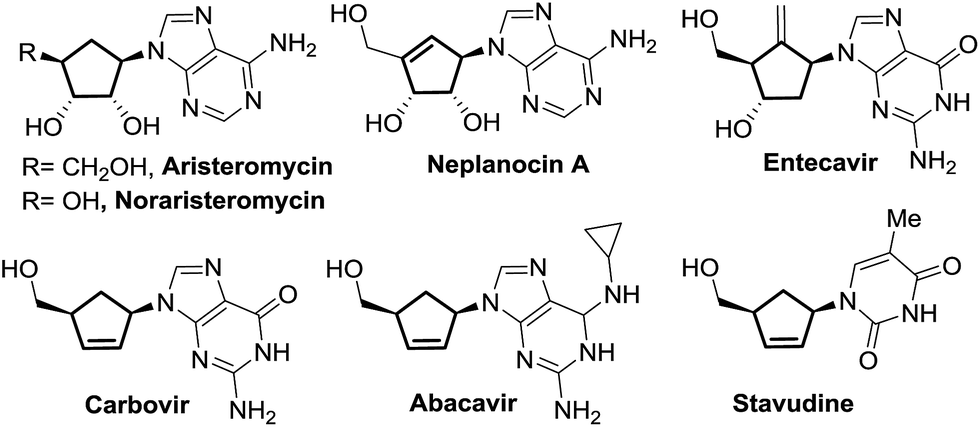 | ||
| Fig. 1 Biologically active carbocyclic nucleoside analogues. | ||
Nazarov cyclization involving 4π electrocyclic ring closure of the pentadienyl cation has long been regarded as a versatile method to construct five-membered carbocycles which are ubiquitous in nature.7 In the classical Nazarov reaction, a variety of nucleophiles have been utilized to ingeniously intercept the cyclopentenyl cation formed following cyclization in inter- or intramolecular fashion, allowing rapid access to more elaborate cyclopentanoid compounds.8 The imino variant of the Nazarov reaction9 and the related intercepted process,9f in contrast, have not been exploited to a great extent due to unfavourable energetics of the ring closure reaction.10
Our previous success in the development of efficient domino processes involving intramolecular imino-Nazarov cyclization of the 1-aminopentadienyl cation prompted us to explore the fundamentally similar intermolecular capture of the transient oxyallyl cation intermediate.9h In particular, we became interested in the prospect of employing a silylated nucleobase as the intermolecular trapping partner to render formation of the nucleobase-containing cyclopentanone, which can be potentially transformed into carbocyclic nucleoside analogues (Scheme 1). It is notable that nitrogen nucleophiles have not been widely used in interrupted Nazarov reactions, presumably a consequence of the incompatibility of basic nitrogen-based nucleophiles with a highly acidic promoter, which is often necessary to effect cyclization. Tius reported a silica gel-/florisil-promoted amine intercepted Nazarov reaction,11 whereas West utilized organoazides as viable N-nucleophiles to trap the oxyallyl cation12 (Scheme 1). The present work described herein represents the first example in which a silylated nucleobase serves as an effective trapping agent for the oxyallyl cation in Nazarov-type cyclization.
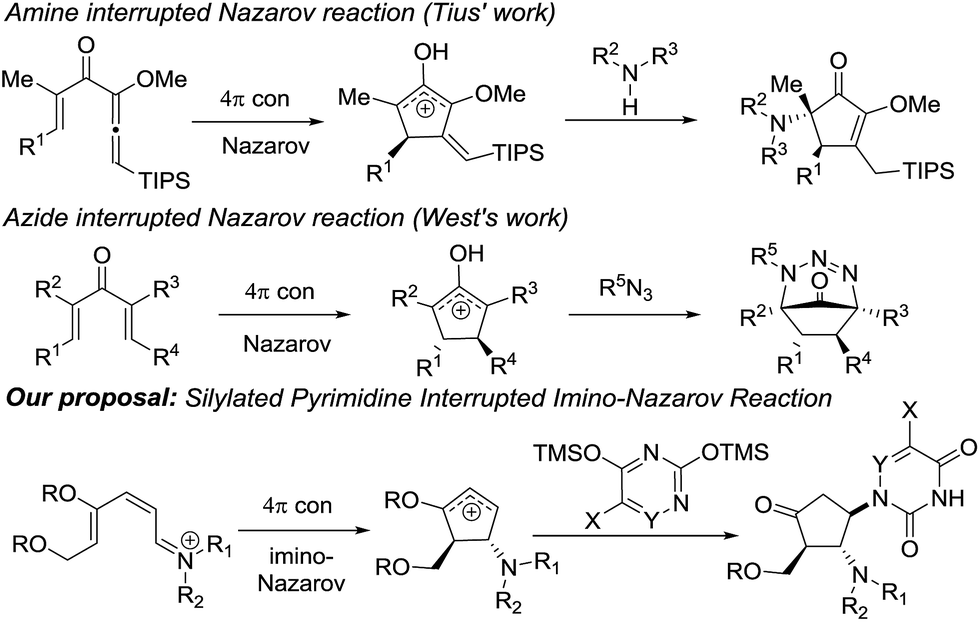 | ||
| Scheme 1 N-nucleophiles in interrupted Nazarov reactions. | ||
Our initial efforts focused on the treatment of 4,6-dimethoxyhexa-2,4-dienal 1a, N-benzyl-4-methoxyaniline 2a and O,O′-bis(trimethylsilyl)thymine 3a in the presence of a Lewis acid catalyst. The requisite 4,6-dimethoxyhexa-2,4-dienal 1a could be obtained readily from 3,4,5-tri-O-methyl-D-glucal as an inseparable mixture of 2Z, 4Z and 2Z, 4E isomers in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio, as described previously.9h We began our investigation with SnCl4 as the choice of Lewis acid catalyst as it is commonly used to activate silylated nucleobases in the Vorbrüggen method for preparation of nucleosides.13 To our delight, when the reaction was carried out in the presence of 30 mol% of SnCl4 in acetonitrile at 25 °C, dienal 1a was fully consumed within an hour. Instead of the expected cyclopentanone product, however, the corresponding highly functionalized cyclic enol ether 4a was isolated in 63% yield (Table 1, entry 1). Structural elucidation and stereochemical determination of compound 4a was accomplished based on extensive 2D NMR (COSY and NOESY) studies. On the basis of the NOESY experiment, the absence of an observed correlation between protons at C-4 and C-5 indicates a trans configuration between the substituents. Additionally, a strong correlation between the protons of benzylic methylene and the proton at C-3 suggests that nucleophilic attack of silylated thymine occurs trans to the amino group at C-4. As compound 4a retained the enol ether functionality, possibly allowing introduction of an electrophile at the α carbon, we decided to pursue formation of this type of carbocyclic nucleoside.14
:4 ratio, as described previously.9h We began our investigation with SnCl4 as the choice of Lewis acid catalyst as it is commonly used to activate silylated nucleobases in the Vorbrüggen method for preparation of nucleosides.13 To our delight, when the reaction was carried out in the presence of 30 mol% of SnCl4 in acetonitrile at 25 °C, dienal 1a was fully consumed within an hour. Instead of the expected cyclopentanone product, however, the corresponding highly functionalized cyclic enol ether 4a was isolated in 63% yield (Table 1, entry 1). Structural elucidation and stereochemical determination of compound 4a was accomplished based on extensive 2D NMR (COSY and NOESY) studies. On the basis of the NOESY experiment, the absence of an observed correlation between protons at C-4 and C-5 indicates a trans configuration between the substituents. Additionally, a strong correlation between the protons of benzylic methylene and the proton at C-3 suggests that nucleophilic attack of silylated thymine occurs trans to the amino group at C-4. As compound 4a retained the enol ether functionality, possibly allowing introduction of an electrophile at the α carbon, we decided to pursue formation of this type of carbocyclic nucleoside.14
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) |
| a Unless otherwise noted, reactions were performed using dienal 1a (0.1 mmol, 1 equiv.), aniline 2a (0.1 mmol, 1 equiv.) and silylated thymine 3a (0.2 mmol, 2 equiv.) with 30 mol% of catalyst in 1 mL of solvent. b NMR yield determined by 1H NMR analysis of the crude reaction against CH2Br2 as an internal standard. c Reaction was carried out in the presence of 20 mol% of InBr3. PMP = p-methoxyphenyl. | |||
| 1 | SnCl4 | CH3CN | 63 |
| 2 | TiCl4 | CH3CN | 57 |
| 3 | Cu(OTf)2 | CH3CN | 52 |
| 4 | FeBr3 | CH3CN | 58 |
| 5 | InBr3 | CH3CN | 93 |
| 6 | In(OTf)3 | CH3CN | 71 |
| 7 | InBr3 | CH2Cl2 | 79 |
| 8 | InBr3 | THF | 65 |
| 9 | InBr3 | DMF | — |
| 10c | InBr3 | CH3CN | 61 |
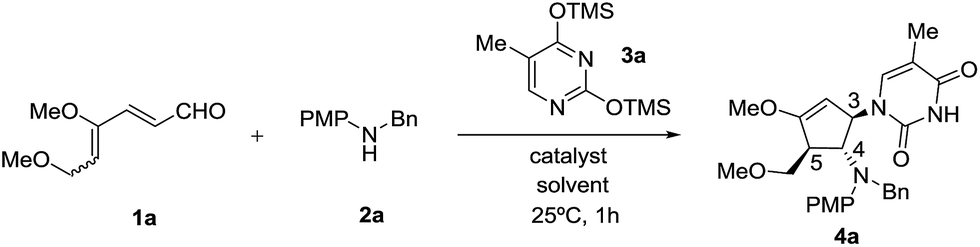
Encouraged by this satisfactory initial result, we directed our attention to the investigation of a series of Lewis acids including TiCl4, Cu(OTf)2, FeBr3, InBr3 and In(OTf)3 in acetonitrile as the solvent. Although all tested Lewis acids were able to promote the desired intermolecular interrupted imino-Nazarov reaction, InBr3 emerged as the best catalyst, affording 4a in 93% yield diastereoselectively (Table 1, entries 2–6). The reaction gave a lower yield when carried out in CH2Cl2 or THF (79% and 65% respectively), whereas no desired product 4a was observed in DMF (Table 1, entries 7–9). An attempt to perform the present domino reaction using a lower loading of InBr3 (20 mol%) led to a diminished yield of 4a (Table 1, entry 10).
With the optimal conditions in hand, we set out to demonstrate the general utility of this transformation by varying the silylated pyrimidine derivatives employed to furnish a library of carbocyclic nucleoside analogues 4b–4k (Scheme 2). Simple unsubstituted silylated uracil furnished the cyclopentenol scaffold bearing a natural uracil group at C-3 4b in 91% yield. In general, substitution at the 5-position of silylated pyrimidine nucleophiles is tolerated in the present intermolecular interrupted imino-Nazarov reaction. Electron-withdrawing halogen substituents did not significantly influence the reactivity of silylated pyrimidine in trapping the oxyallyl cation, affording the desired product 4d–4g in yields ranging from 87–91%. When silylated uracil derivatives with strongly electron-withdrawing groups such as –CF3 and –NO2 at the 5-position were used, the efficiency of the trapping process diminished slightly, giving 79% and 81% yield of 4h and 4i, respectively. Finally, modified uracil derivatives with more distinct variation in the core skeleton, including silylated 2-thiouracil and 5-azauracil, successfully participate in the reactions to provide 4j and 4k in good yields.
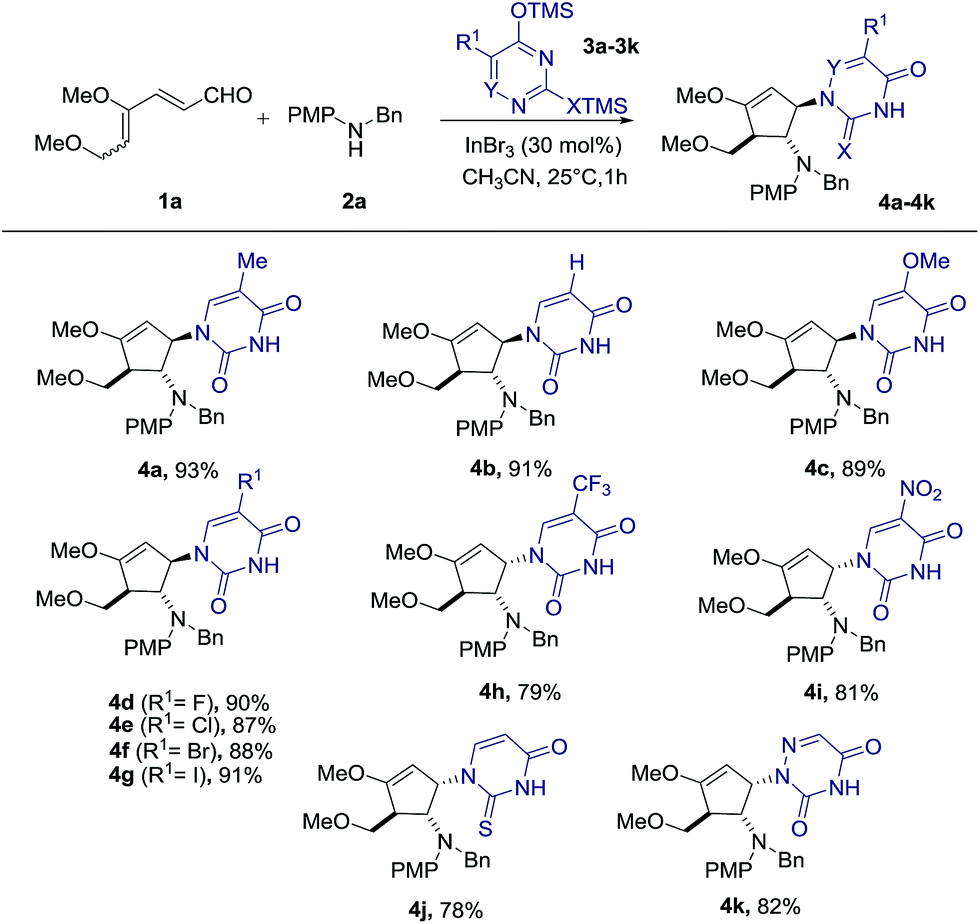 | ||
| Scheme 2 Scope of silylated uracil derivatives. | ||
Having established the scope of silylated pyrimidine, we then directed our attention to examine the applicability of this reaction to a number of secondary anilines (Scheme 3). Various substituents including methyl, allyl, propargyl and 4-methoxybenzyl groups on the aniline nitrogen were well-tolerated, allowing formation of the carbocyclic nucleoside-like compounds 4l–4o. A more electron-deficient aniline with a –Cl group at the para position of the aniline ring resulted in a moderate yield of 56% of the intercepted product 4p.
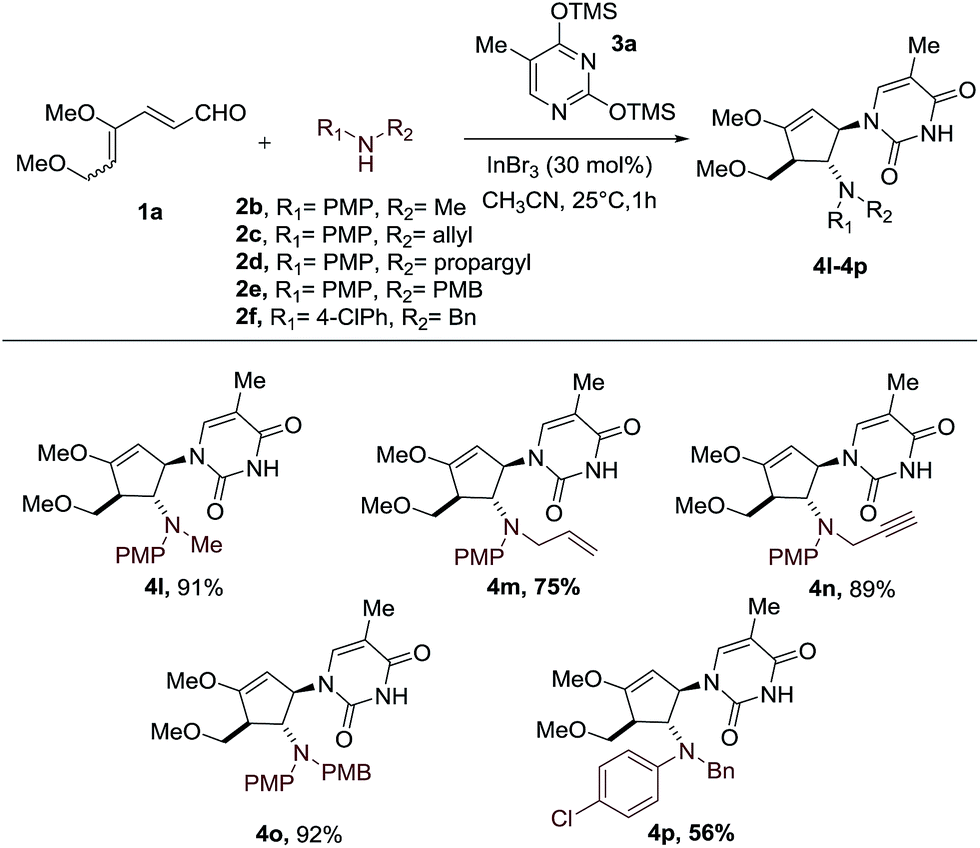 | ||
| Scheme 3 Scope of secondary anilines. | ||
Subsequently, we proceeded to perform the reaction starting with different dienals which can similarly be derived from the corresponding glycals (Scheme 4). During the preparation of benzyl- and MOM-protected dienal 1b and 1c, as well as dienal 1d derived from L-rhamnal, we found that the geometrical isomers (2Z, 4Z and 2Z, 4E) turn out to be separable, in contrast to the methyl protected counterpart 1a. The reactions with these dienals were thus carried out using the pure Z-isomers instead of a mixture of isomers. When the benzyl protected dienal 1b was subjected to the same reaction conditions, the interruption of the imino-Nazarov reaction proceeded smoothly to give an excellent yield of 4q (Scheme 4). In the case of the more labile methoxymethyl protected dienal 1c, the reaction still led to the formation of 4r, albeit in a much lower yield of 52%, along with a substantial amount of 4-aminocyclopentenone (Scheme 4).15 It is notable that the presence of alkoxy (–OMe/–OBn) or methoxymethyl (–OMOM) groups is imperative for the success of this transformation, due to stabilization of the allyl cation intermediate.
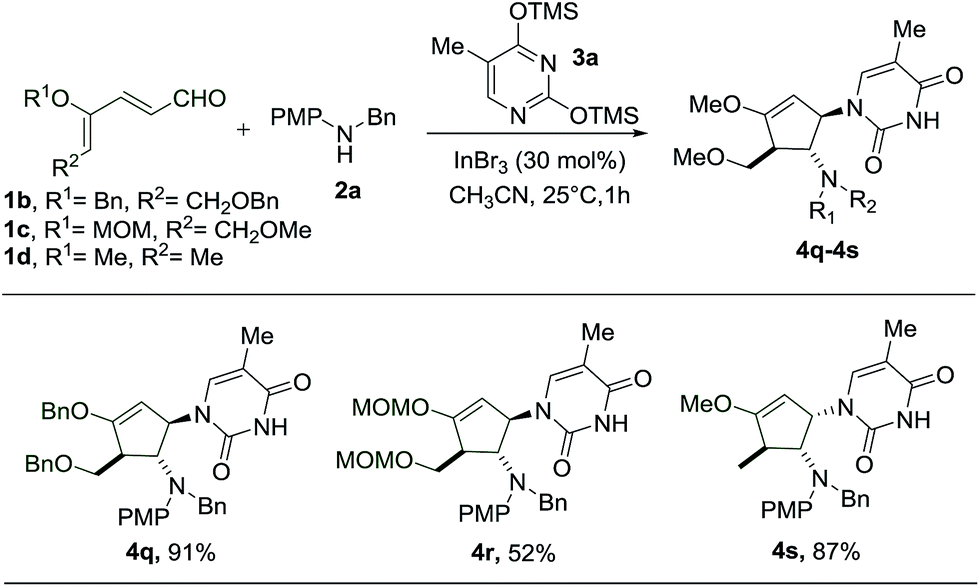 | ||
| Scheme 4 Scope of glycal-derived dienals. | ||
To test our hypothesis that isomerization occurs prior to cyclization, we treated the 4Z isomer 1b and 4E isomer 1b′ separately with aniline 2a and silylated thymine 3a under the same reaction conditions (Scheme 5). As expected, both isomers led to the formation of the same diastereomeric product 4q, indicating that the initial cyclization provided only the trans oxyallyl cation intermediate, a consequence of alkene isomerization.16
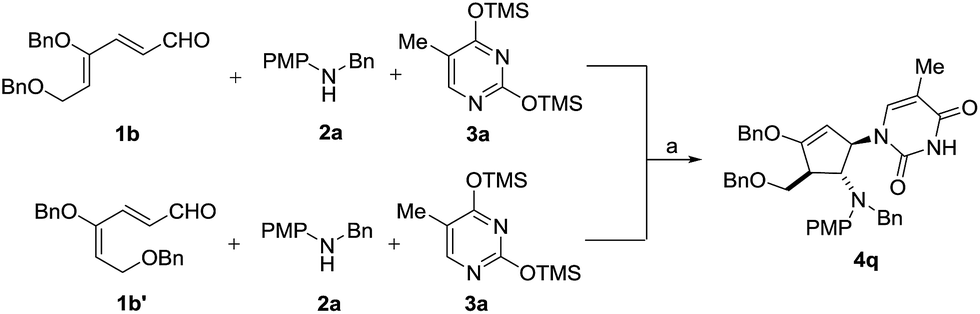 | ||
| Scheme 5 Stereoconvergence cyclization of 1b and 1b′. Reaction conditions: (a) 30 mol% InBr3, CH3CN, 25 °C. Yield of 4q: 91% from 1b and 85% from 1b′. | ||
It is noteworthy that, in all cases, the reactions exhibited excellent diastereoselectivity with regard to the nucleophilic addition of silylated pyrimidine to the oxyallyl cation species. To confirm that the diastereofacial selectivity arises due to steric hindrance, a simple theoretical DFT calculation for the possible trajectory of addition to the oxyallyl cation was conducted (see Fig. S1 in ESI†). The results of our theoretical studies correlate well with the experimental observation of complete diastereoselectivity.
Besides silylated pyrimidine, trimethylsilyl azide proved to be a competent nucleophile in trapping the oxyallyl cation, giving adduct 6 as the only diastereomer in which the azido group was incorporated into the cyclopentanoid framework. The instability of compound 6 in silica gel prevented us from isolating it in its pure form. The ESI mass spectrum of the crude product with molecular ion at m/z (M+ + H) coupled with its 1H NMR analysis provided strong evidence for the formation of 6. Sequential hydrogenation on Pd/C in methanol resulted in the reduction of azide and alkene, along with removal of the benzyl group, rendering conversion of compound 6 to diaminocyclopentane 7 with an overall yield of 84% for 2 steps (Scheme 6). The structure and stereochemical assignment were determined based on a combination of 1D and 2D (COSY and NOESY) NMR studies. To date, there is only a single documented example of azidation of the oxyallyl cation, reported by West, using Me2AlN3 as the azide source.8e
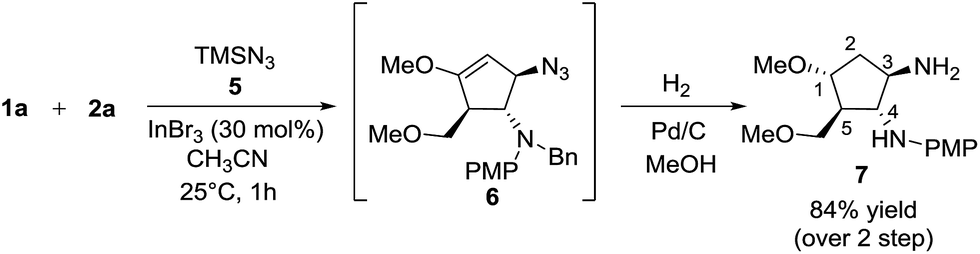 | ||
| Scheme 6 One-pot azide trapping/hydrogenation. | ||
Conclusions
In conclusion, we have developed the first examples of intermolecular interrupted imino-Nazarov reactions using silylated pyrimidine to provide convenient access to carbocyclic nucleoside analogues. The novel utility of using a silylated nucleobase as the nucleophilic trapping partner to intercept the fleeting oxyallyl cation from the imino-Nazarov reaction can potentially be extended to other variants of the Nazarov reaction.Acknowledgements
We gratefully acknowledge Nanyang Technological University (RG6/13 and SUG) and the Ministry of Education, Singapore (MOE 2013-T3-1-002) for the financial support of this research.Notes and references
- (a) S. W. Schnelle, Curr. Top. Med. Chem., 2002, 2, 1087 CrossRef; (b) J. Velcicky, A. Lanver, J. Lex, A. Prokop, T. Wiederm and H.-G. Schmalz, Chem.–Eur. J., 2004, 10, 5087 CrossRef CAS PubMed; (c) W. Hatton, D. Arosio, M. Re, D. Giudici, A. Bernardi and P. Seneci, C. R. Chim., 2010, 13, 1284 CrossRef CAS.
- A. Mieczkowski and L. A. Agrofoglio, Modified Nucleosides: in Biochemistry, Biotechnology and Medicine, ed. P. Herdewijn, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, p. 393 Search PubMed.
- (a) E. W. Dunham and R. J. Vince, J. Pharmacol. Exp. Ther., 1986, 238, 954 CAS; (b) R. J. Parry, in Secondary-Metabolite Biosynthesis and Metabolism, eds. R. Petroski, and S. McCormick, Springer, US, 1992, vol. 44, ch. 77, pp. 89-104 Search PubMed.
- (a) S. Yaginuma, N. Muto, M. Tsujino, Y. Sudate, M. Hayashi and M. Otani, J. Antibiot., 1981, 34, 359 CrossRef CAS PubMed; (b) M. Hayashi, S. Yaginuma, H. Yoshioka and K. Nakatsu, J. Antibiot., 1981, 34, 675 CrossRef CAS PubMed.
- (a) R. Vince, Nucleic Acids Symp. Ser., 1991, 25, 193 CAS; (b) R. Vince and M. Hua, J. Med. Chem., 1990, 33, 17 CrossRef CAS PubMed.
- (a) S. M. Daluge, M. T. Martin, B. R. Sickles and D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 297 CrossRef CAS PubMed; (b) P. S. Hervey and L. M. Perry, Drugs, 2000, 60, 447 CrossRef CAS PubMed.
- For reviews on Nazarov cyclization see: (a) S. E. Denmark, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, New York, 1991, vol. 5, p. 751 Search PubMed; (b) M. A. Tius, Eur. J. Org. Chem., 2005, 2193 CrossRef CAS; (c) H. Pellissier, Tetrahedron, 2005, 61, 6479 CrossRef CAS; (d) A. J. Frontier and C. Collison, Tetrahedron, 2005, 61, 7577 CrossRef CAS; (e) T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531 CrossRef CAS; (f) N. Shimada, C. Stewart and M. A. Tius, Tetrahedron, 2011, 67, 5851 CrossRef CAS PubMed; (g) M. A. Tius, Acc. Chem. Res., 2003, 36, 284 CrossRef CAS PubMed; (h) W. T. Spencer III, T. Vaidya and A. J. Frontier, Eur. J. Org. Chem., 2013, 3621 CrossRef PubMed; (i) M. A. Tius, Chem. Soc. Rev., 2014, 43, 2979 RSC; (j) M. J. D. Grandi, Org. Biomol. Chem., 2014, 12, 5331 RSC; (k) D. R. Wenz and J. Read de Alaniz, Eur. J. Org. Chem., 2015, 23 CrossRef CAS.
- (a) T. N. Grant, C. J. Rieder and F. G. West, Chem. Commun., 2009, 5676 RSC; (b) C. J. Rieder, R. J. Fradette and F. G. West, Heterocycles, 2010, 80, 1413 CrossRef CAS; (c) Y.-K. Wu, R. McDonald and F. G. West, Org. Lett., 2011, 13, 3584 CrossRef CAS PubMed; (d) D. J. Kerr, M. Miletic, J. H. Chaplin, J. M. White and B. L. Flynn, Org. Lett., 2012, 14, 1732 CrossRef CAS PubMed; (e) Y. Kwon, R. McDonald and F. G. West, Angew. Chem., Int. Ed., 2013, 52, 8616 CrossRef CAS PubMed; (f) J. H. Chaplin, K. Jackson, J. M. White and B. L. Flynn, J. Org. Chem., 2014, 79, 3659 CrossRef CAS PubMed; (g) F. M. LeFort, V. Mishra, G. D. Dexter, T. D. R. Morgan and D. J. Burnell, J. Org. Chem., 2015, 80, 5877 CrossRef CAS PubMed.
- (a) M. A. Tius, C. C. Chu and R. Nieves-Colberg, Tetrahedron Lett., 2001, 42, 2419 CrossRef CAS; (b) S. Suárez-Pantiga, E. Rubio, C. Alvarez-Rúa and J. M. González, Org. Lett., 2009, 11, 13 CrossRef PubMed; (c) W. F. Bow, A. K. Basak, A. Jolit, D. A. Vicic and M. A. Tius, Org. Lett., 2010, 12, 440 CrossRef CAS PubMed; (d) N. Shimada, B. O. Ashburn, A. K. Basak, W. F. Bow, D. A. Vicic and M. A. Tius, Chem. Commun., 2010, 46, 3774 RSC; (e) Z.-X. Ma, S. He, W. Song and R. P. Hsung, Org. Lett., 2012, 14, 5736 CrossRef CAS PubMed; (f) S. A. Bonderoff, T. N. Grant, F. G. West and M. Tremblay, Org. Lett., 2013, 15, 2888 CrossRef CAS PubMed; (g) S. Wang, R. William, K. K. G. Seah and X.-W. Liu, Green Chem., 2013, 15, 3180 RSC; (h) R. William, S. Wang, F. Ding, E. N. Arviana and X.-W. Liu, Angew. Chem., Int. Ed., 2014, 53, 10742 CrossRef CAS PubMed.
- D. A. Smith and C. W. Ulmer II, J. Org. Chem., 1997, 62, 5110 CrossRef CAS.
- (a) F. Dhoro and M. A. Tius, J. Am. Chem. Soc., 2005, 127, 12472 CrossRef CAS PubMed; (b) F. Dhoro, T. E. Kristensen, V. Stockmann, G. P. A. Yap and M. A. Tius, J. Am. Chem. Soc., 2007, 129, 7256 CrossRef CAS PubMed.
- (a) A. Rostami, Y. Wang, A. M. Arif, R. McDonald and F. G. West, Org. Lett., 2007, 9, 703 CrossRef CAS PubMed; (b) D. Song, A. Rostami and F. G. West, J. Am. Chem. Soc., 2007, 129, 12019 CrossRef CAS PubMed; (c) O. Scadeng, M. J. Ferguson and F. G. West, Org. Lett., 2011, 13, 114 CrossRef CAS PubMed.
- H. Vorbrüggen and C. Ruh-Pohlenz, Organic Reactions, ed. L. A. Paquette, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, vol. 55, pp. 1–630 Search PubMed.
- (a) J. Nie, H.-W. Zhu, H.-F. Cui, M.-Q. Hua and J.-A. Ma, Org. Lett., 2007, 9, 3053 CrossRef CAS PubMed; (b) M. Fujiwara, M. Kawatsura, S. Hayase, M. Nanjo and T. Itoh, Adv. Synth. Catal., 2009, 351, 123 CrossRef CAS; (c) G. E. Hutson, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 4988 CrossRef CAS PubMed.
- M. A. Tius, D. P. Astrab, A. H. Fauq, J. B. Ousset and S. Trehan, J. Am. Chem. Soc., 1986, 108, 3438 CrossRef CAS.
- For similar observation of isomerization prior to Nazarov cyclization, see: (a) S. E. Denmark, M. A. Wallace and C. B. Walker, J. Org. Chem., 1990, 55, 5543 CrossRef CAS; (b) S. Giese and F. G. West, Tetrahedron Lett., 1998, 39, 8393 CrossRef CAS; (c) S. Giese and F. G. West, Tetrahedron, 2000, 56, 10221 CrossRef CAS; (d) L. Leclerc and M. A. Tius, Org. Lett., 2003, 5, 1171 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03559g |
| This journal is © The Royal Society of Chemistry 2016 |