
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of α-alkenyl α-amino acids via N–H insertion reactions†
Jun-Xia
Guo
a,
Ting
Zhou
a,
Bin
Xu
 a,
Shou-Fei
Zhu
*ab and
Qi-Lin
Zhou
ab
a,
Shou-Fei
Zhu
*ab and
Qi-Lin
Zhou
ab
aState Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China. E-mail: sfzhu@nankai.edu.cn; Fax: +86-22-2350-4087
First published on 28th October 2015
Abstract
A new highly enantioselective route to α-alkenyl α-amino acid derivatives, which are important naturally occurring compounds with attractive bioactivity and synthetic utility, was developed using a N–H insertion reaction of vinyldiazoacetates and tert-butyl carbamate cooperatively catalyzed by achiral dirhodium(II) carboxylates and chiral spiro phosphoric acids under mild, neutral conditions. This reaction has a broad substrate scope, a fast reaction rate (turnover frequency > 6000 h−1), a high yield (61–99%), and excellent enantioselectivity (83–98% ee). The chiral spiro phosphoric acid, which is proposed to realize the enantioselectivity of the insertion reaction by promoting the proton transfer of a ylide intermediate by acting as a chiral proton shuttle catalyst, can suppress several usual side reactions of vinyldiazoacetates and broaden the applications of these versatile carbene precursors in organic synthesis. To our knowledge, it is the first highly enantioselective carbene insertion reaction of vinyldiazoacetates with heteroatom–hydrogen bonds in which the heteroatom has lone-pair electrons.
α-Amino acids are vital building blocks of peptides, proteins, and many other bioactive compounds, and the development of highly efficient and enantioselective methods for the synthesis of diverse α-amino acids has been a long-standing goal of synthetic chemists. Over the past several decades, many catalytic methods have been established for the synthesis of α-alkyl and α-aryl substituted α-amino acids.1 However, even though chiral α-alkenyl α-amino acids are important naturally occurring compounds2 with attractive bioactivity3 and synthetic utility (Fig. 1 and Scheme 1),4 few enantioselective catalytic methods for their synthesis have been reported.5 Moreover, only two types of chiral α-alkenyl α-amino acids (γ-mono-substituted vinylglycines5a,b and β-carbonyl vinylglycines5c,d) can be prepared via these reported methods. Therefore, general enantioselective catalytic methods for preparing optically active α-alkenyl α-amino acids and their derivatives are highly desired. The challenge in the enantioselective synthesis of chiral α-alkenyl α-amino acids lies in the lability of the products toward racemization and undesirable isomerization of the double bond.
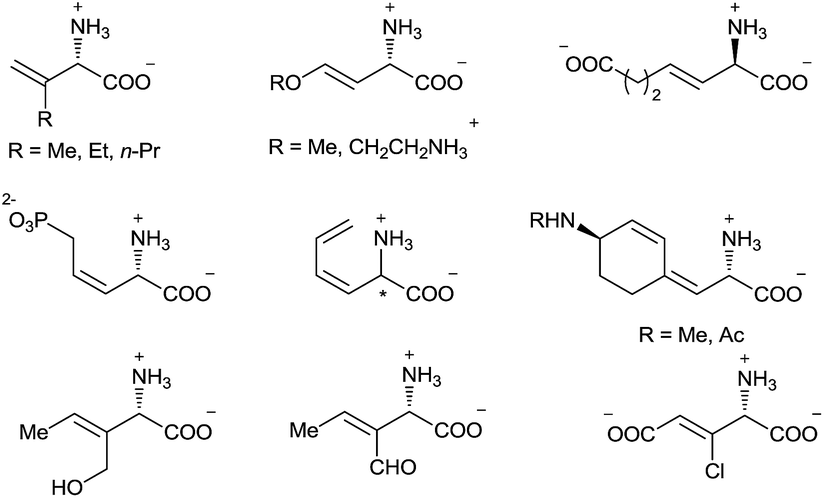 | ||
| Fig. 1 Selected naturally occurring α-alkenyl α-amino acids. | ||
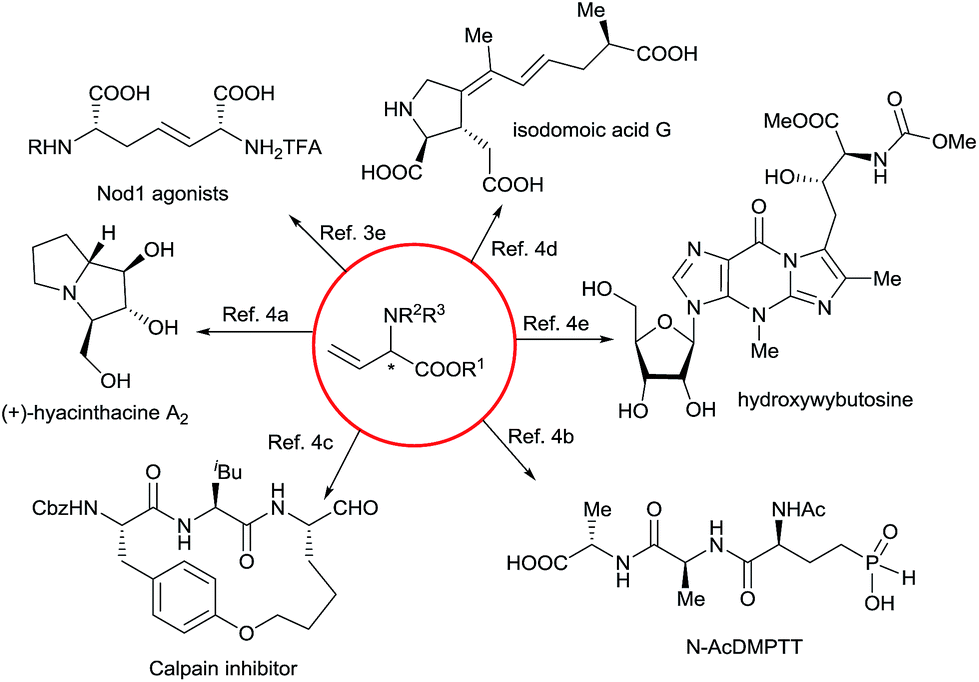 | ||
| Scheme 1 Synthetic utilities of vinylglycines. | ||
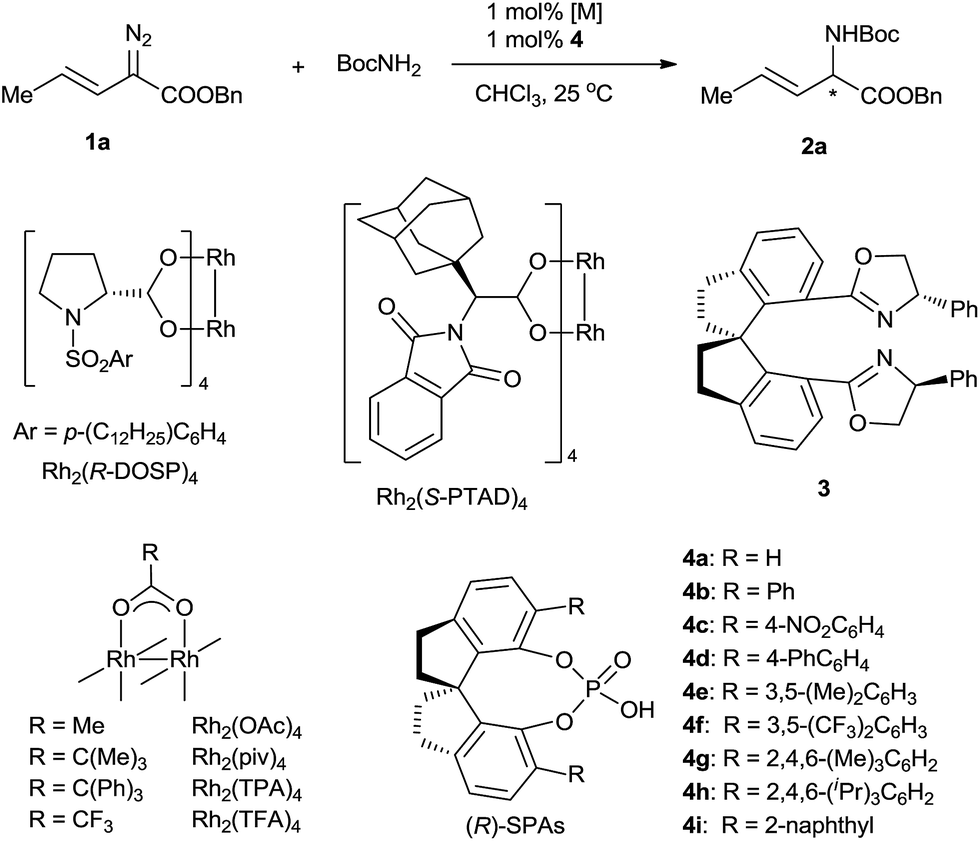
Transition-metal-catalyzed carbenoid insertion into N–H bonds is one of the most efficient methods for constructing C–N bonds, and remarkable progress in asymmetric N–H insertion reactions has been achieved in recent years.6 However, asymmetric N–H insertion reactions of vinyldiazoacetates, which could be used to produce chiral α-alkenyl α-amino acid derivatives, remain an unresolved problem.7 Such reactions can be expected to be challenging because the highly reactive olefin moiety of the vinyldiazoacetates might undergo migration, rearrangement, or cyclopropanation in the presence of traditional metal-complex catalysts.8 For instance, Doyle and co-workers9 studied the reaction of 3-(trialkylsiloxy)-2-diazo-3-butenoate with aldehyde-derived hydrazones using chiral dirhodium catalysts but found that the reaction occurred at the vinyl terminus (referred to as vinylogous N–H insertion), instead of at the α position, to generate α,β-unsaturated γ-amino acid derivatives. Fu and co-workers6f described an asymmetric N–H insertion of 2-diazo-4-phenylbut-3-enoate with good enantioselectivity (87% ee) but very low yield (25%) in a footnote (experimental data not given). Herein we report that vinyldiazoacetates and tert-butyl carbamate undergo a highly enantioselective N–H insertion reaction cooperatively catalyzed by achiral dirhodium(II) carboxylates and chiral spiro phosphoric acids (SPAs) under mild, neutral conditions. This reaction, which constitutes a new route to α-alkenyl α-amino acid derivatives, has a broad substrate scope, a fast reaction rate (turnover frequency > 6000 h−1), a high yield (61–99% yields), and excellent enantioselectivity (83–98% ee). The SPA is proposed to promote the proton transfer of a ylide intermediate by acting as a chiral proton shuttle catalyst, consequently achieving the enantioselectivity of the insertion reaction. Moreover, the SPA suppresses several usual side reactions of vinyldiazoacetates and broadens the applications of these versatile carbene precursors in organic synthesis. To our knowledge, this is the first highly enantioselective carbene insertion reaction of vinyldiazoacetates with heteroatom–hydrogen bonds in which the heteroatom has lone-pair electrons.10
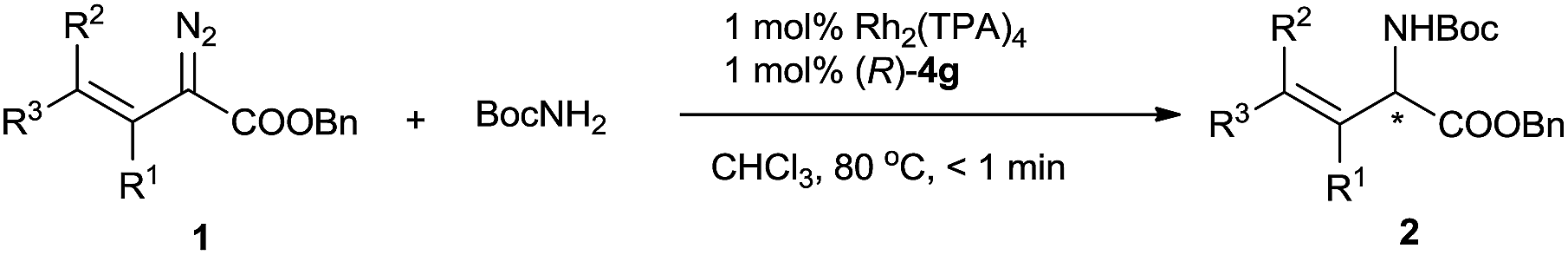
To evaluate various chiral catalysts, we carried out the insertion reaction of (E)-benzyl 2-diazopent-3-enoate (1a) with tert-butyl carbamate in CHCl3 at 25 °C (Table 1). The traditional chiral metal-complex catalysts for carbene insertion reactions, including copper and palladium complexes with chiral spirobisoxazoline ligand 3,11 Rh2(R-DOSP)4,12 and Rh2(S-PTAD)4,13 exhibited only modest yields and low enantioselectivities (entries 1–4). We next turned to cooperative catalysts composed of achiral dirhodium complexes and SPAs 4, which may accelerate the proton shift step of the insertion reaction by acting as chiral proton shuttles (entries 5–13).14 The use of the SPAs significantly increased the yields of the desired N–H insertion products and suppressed double bond rearrangement and carbene dimerization. The SPA (R)-4g, which bears 6,6′-di[2,4,6-(Me)3C6H2] substituents, exhibited the best performance (78% yield, 61% ee; entry 11). The investigation of various achiral dirhodium complexes revealed that the steric characteristics of the complexes strongly affected the enantioselectivity of the N–H insertion reaction (entries 14–16). With dirhodium complex Rh2(TPA)4, which has bulky carboxylate ligands, the reaction was complete in <1 min and afforded a high yield (74%) of the desired product with excellent enantioselectivity (96% ee) (entry 16). Considering their significant effects on the enantioselectivity of the reaction, the rhodium catalysts are most likely involved in the proton transfer step.6i Chlorinated solvents dichloromethane and dichloroethane were suitable for the N–H insertion reaction, whereas the use of THF or toluene dramatically lowered the enantioselectivity (to 3% ee and 6% ee, respectively; Table S1, ESI†). Increasing the reaction temperature increased the yield of the reaction (entries 17–19). The unexpected high enantioselectivity at 80 °C (entry 19) implies that the SPAs can effectively promote the proton shift of the unstable ylide intermediate in this insertion reaction even under harsh conditions.6i,14h
|
|
|||||
|---|---|---|---|---|---|
| Entry | [M] | SPA | Time | Yieldb (%) | eec (%) |
a Reaction conditions: [Rh]/4/1a/BocNH2 = 0.002![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.002:0.2:0.2 (mmol) in 3 mL CHCl3, 25 °C.
b Isolated yield.
c Determined using HPLC using a Chiralcel OD-H column.
d Using 5 mol% catalyst.
e Performed at 0 °C.
f Performed at 60 °C.
g Performed at 80 °C. :0.002:0.2:0.2 (mmol) in 3 mL CHCl3, 25 °C.
b Isolated yield.
c Determined using HPLC using a Chiralcel OD-H column.
d Using 5 mol% catalyst.
e Performed at 0 °C.
f Performed at 60 °C.
g Performed at 80 °C.
|
|||||
| 1d | Pd(PhCN)2Cl2 and 3 | None | 12 h | <5 | — |
| 2d | Cu(MeCN)4PF6 and 3 | None | 12 h | 37 | 11 |
| 3 | Rh2(R-DOSP)4 | None | 3 min | 23 | 12 |
| 4 | Rh2(S-PTAD)4 | None | 1 min | 41 | 12 |
| 5 | Rh2(OAc)4 | (R)-4a | 2 min | 66 | 4 |
| 6 | Rh2(OAc)4 | (S)-4b | 3 min | 66 | −37 |
| 7 | Rh2(OAc)4 | (R)-4c | 4 min | 64 | 11 |
| 8 | Rh2(OAc)4 | (R)-4d | 2 min | 60 | 57 |
| 9 | Rh2(OAc)4 | (R)-4e | 4 min | 76 | 19 |
| 10 | Rh2(OAc)4 | (R)-4f | 2 min | 53 | 31 |
| 11 | Rh2(OAc)4 | (R)-4g | 2 min | 78 | 61 |
| 12 | Rh2(OAc)4 | (S)-4h | 3 min | 58 | −2 |
| 13 | Rh2(OAc)4 | (R)-4i | 3 min | 64 | 34 |
| 14 | Rh2(piv)4 | (R)-4g | <1 min | 37 | 71 |
| 15 | Rh2(TFA)4 | (R)-4g | 3 h | 35 | 59 |
| 16 | Rh2(TPA)4 | (R)-4g | <1 min | 74 | 96 |
| 17e | Rh2(TPA)4 | (R)-4g | <1 min | 66 | 95 |
| 18f | Rh2(TPA)4 | (R)-4g | <1 min | 86 | 97 |
| 19g | Rh2(TPA)4 | (R)-4g | <1 min | 93 | 96 |
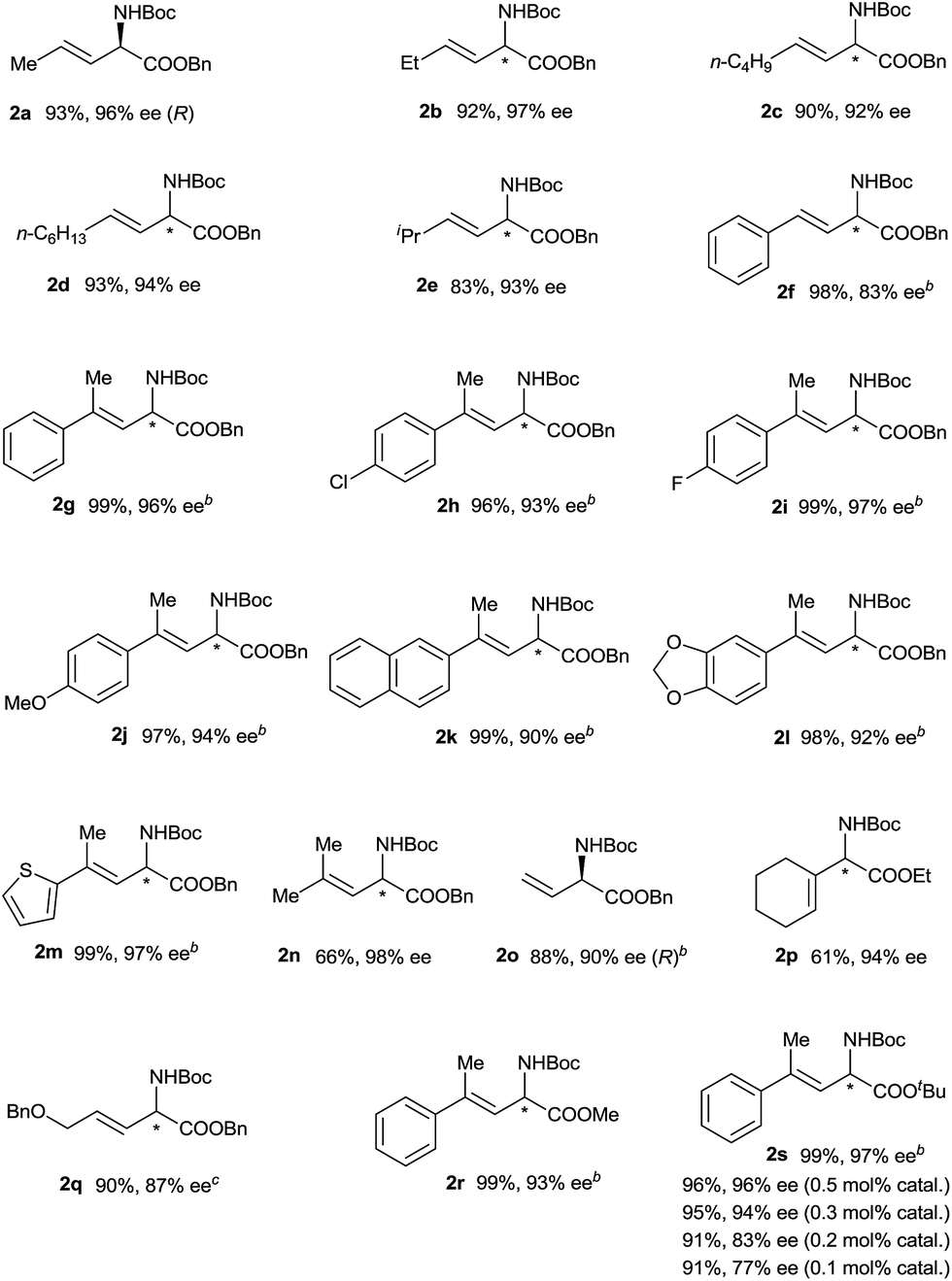
Using the optimized reaction conditions, we carried out reactions of vinyldiazoacetate substrates 1 bearing various substituents on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (Table 2).15 Impressively, all the reactions were complete within 1 min (turnover frequency > 6000 h−1). The length of the γ-alkyl group (R3) had a negligible effect on the yield and enantioselectivity of the reaction (1a–1d). The reaction of γ-isopropyl-substituted substrate 1e proceeded in a slightly lower yield, and this result may be attributable in part to increased steric hindrance due to the branched alkyl group. (E)-Benzyl 2-diazo-4-phenylbut-3-enoate (1f), which has a γ-phenyl group, also underwent the insertion reaction, but the enantioselectivity was lower than that for vinyldiazoacetates with a γ-alkyl group. The N–H insertion reactions of γ,γ-disubstituted vinyldiazoesters 1g–1n also exhibited excellent enantioselectivities. Substrates with substituted phenyl groups (1h–1j), fused rings (1k and 1l), and a thiophenyl moiety (1m) were tolerated. The reaction of benzyl 2-diazo-4-methylpent-3-enoate (1n), which has γ,γ-dimethyl groups, afforded excellent enantioselectivity (98% ee) albeit with a relatively low yield (66%). Benzyl 2-diazobut-3-enoate (1o), which bears a terminal olefin, was also a suitable substrate for the N–H insertion reaction, which afforded the ester form of vinylglycine in high yield with excellent enantioselectivity. A vinyldiazoacetate with a cyclic olefin moiety (1p) also afforded the desired product (61% yield, 94% ee). The benzyloxy functional group in the side chain of vinyldiazoacetate 1q was also tolerated. The size of the ester moiety slightly affected the enantioselectivity; bulkier esters gave higher enantioselectivities (compare substrates 1g, 1r, and 1s). The cooperative catalytic system is highly active. The catalyst loading could be reduced to 0.5 mol% or even 0.3 mol% catalyst without significantly affecting the outcome of the N–H insertion reaction of diazoester 1s. The further reduction in the catalyst loading to 0.2 mol% and 0.1 mol%, leads to lower enantioselectivity (83% ee and 77% ee, respectively). The reaction could be easily performed at a gram scale (Scheme 2), which demonstrates its potential for practical applications.
C bond (Table 2).15 Impressively, all the reactions were complete within 1 min (turnover frequency > 6000 h−1). The length of the γ-alkyl group (R3) had a negligible effect on the yield and enantioselectivity of the reaction (1a–1d). The reaction of γ-isopropyl-substituted substrate 1e proceeded in a slightly lower yield, and this result may be attributable in part to increased steric hindrance due to the branched alkyl group. (E)-Benzyl 2-diazo-4-phenylbut-3-enoate (1f), which has a γ-phenyl group, also underwent the insertion reaction, but the enantioselectivity was lower than that for vinyldiazoacetates with a γ-alkyl group. The N–H insertion reactions of γ,γ-disubstituted vinyldiazoesters 1g–1n also exhibited excellent enantioselectivities. Substrates with substituted phenyl groups (1h–1j), fused rings (1k and 1l), and a thiophenyl moiety (1m) were tolerated. The reaction of benzyl 2-diazo-4-methylpent-3-enoate (1n), which has γ,γ-dimethyl groups, afforded excellent enantioselectivity (98% ee) albeit with a relatively low yield (66%). Benzyl 2-diazobut-3-enoate (1o), which bears a terminal olefin, was also a suitable substrate for the N–H insertion reaction, which afforded the ester form of vinylglycine in high yield with excellent enantioselectivity. A vinyldiazoacetate with a cyclic olefin moiety (1p) also afforded the desired product (61% yield, 94% ee). The benzyloxy functional group in the side chain of vinyldiazoacetate 1q was also tolerated. The size of the ester moiety slightly affected the enantioselectivity; bulkier esters gave higher enantioselectivities (compare substrates 1g, 1r, and 1s). The cooperative catalytic system is highly active. The catalyst loading could be reduced to 0.5 mol% or even 0.3 mol% catalyst without significantly affecting the outcome of the N–H insertion reaction of diazoester 1s. The further reduction in the catalyst loading to 0.2 mol% and 0.1 mol%, leads to lower enantioselectivity (83% ee and 77% ee, respectively). The reaction could be easily performed at a gram scale (Scheme 2), which demonstrates its potential for practical applications.
 | ||
| Scheme 2 The gram-scale experiment. | ||
This N–H insertion reaction of vinyldiazoacetates provides a new route to chiral α-amino acids. For example, L-vinylglycine was easily prepared in good yield by the hydrolysis of insertion product (S)-2o (Scheme 3, eqn (1)).16 Hydrogenation of (R)-2b over a Pd/C catalyst and subsequent acidic hydrolysis gave α-alkyl-α-amino acid 6 (94% yield, eqn (2)).16 The olefin moieties in the products can be expected to undergo transformations such as dihydroxylation, cyclopropanation, and epoxidation,4,16 making this reaction potentially useful for synthesizing various optically active α-amino acid derivatives.
 | ||
| Scheme 3 Synthesis of optically active α-amino acids. | ||
In summary, a highly efficient asymmetric synthesis of α-alkenyl α-amino acids was realized by means of the N–H insertion reaction of vinyldiazoacetates with tert-butyl carbamate cooperatively catalyzed by achiral dirhodium(II) carboxylates and chiral SPAs. The wide substrate scope, good yield, high enantioselectivity, fast reaction rate, and mild, neutral conditions make this N–H insertion reaction widely applicable for the preparation of chiral α-amino acid derivatives. The combination of SPAs and dirhodium(II) carboxylates exhibits a special advantage in the transformation of highly functionalized vinyldiazoacetates by minimizing the side-reactions, and has potential applications in other enantioselective transformations involving vinyldiazoacetates.
Acknowledgements
We thank the National Natural Science Foundation of China, the National Basic Research Program of China (2012CB821600), the “111” project (B06005) of the Ministry of Education of China, the National Program for Support of Top-notch Young Professionals, and the Fundamental Research Funds for the Central Universities for financial support.Notes and references
- For reviews, see: (a) R. M. Williams, in Synthesis of Optically Active α-Amino Acids, ed. J. E. Baldwin, Organic Chemistry Series, Pergamon Press, Oxford, 1989 Search PubMed; (b) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef PubMed; (c) J. Michaux, G. Niel and J.-M. Campagne, Chem. Soc. Rev., 2009, 38, 2093 RSC; for some elegant examples, see: (d) S. J. Zuend, M. P. Coughlin, M. P. Lalonde and E. N. Jacobsen, Nature, 2009, 461, 968 CrossRef CAS PubMed; (e) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed; (f) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed.
- For a review, see: D. B. Berkowitz, B. D. Charette, K. R. Karukurichi and J. M. McFadden, Tetrahedron: Asymmetry, 2006, 17, 869 CrossRef CAS.
- For recent bioactivity studies of α-alkenyl α-amino acids, see: (a) K. L. McPhail, D. J. Armstrong, M. D. Azevedo, G. M. Banowetz and D. I. Mills, J. Nat. Prod., 2010, 73, 1853 CrossRef CAS PubMed; (b) X. Lee, Á. Fox, J. Sufrin, H. Henry, P. Majcherczyk, D. Haas and C. Reimmann, J. Bacteriol., 2010, 192, 4251 CrossRef CAS PubMed; (c) A. Halgren, M. Azevedo, D. Mills, D. Armstrong, M. Thimmaiah, K. McPhail and G. Banowetz, J. Appl. Microbiol., 2011, 111, 949 CrossRef CAS PubMed; (d) X. Lee, M. D. Azevedo, D. J. Armstrong, G. M. Banowetz and C. Reimmann, Environ. Microbiol. Rep., 2013, 5, 83 CrossRef CAS PubMed; (e) Ž. Jakopin, M. Gobec, J. Kodela, T. Hazdovac, I. Mlinarič-Raščan and M. S. Dolenc, Eur. J. Med. Chem., 2013, 69, 232 CrossRef PubMed.
- For recent synthetic applications of optically active vinylglycine, see: (a) P. Dewi-Wülfing and S. Blechert, Eur. J. Org. Chem., 2006, 1852 CrossRef; (b) Y. Xiao, K. Lee and P. Liu, Org. Lett., 2008, 10, 5521 CrossRef CAS PubMed; (c) A. D. Abell, M. A. Jones, J. M. Coxon, J. D. Morton, S. G. Aitken, S. B. McNabb, H. Y.-Y. Lee, J. M. Mehrtens, N. A. Alexander, B. G. Stuart, A. T. Neffe and R. Bickerstaffe, Angew. Chem., Int. Ed., 2009, 48, 1455 CrossRef CAS PubMed; (d) S. E. Denmark, J. H.-C. Liu and J. M. Muhuhi, J. Am. Chem. Soc., 2009, 131, 14188 CrossRef CAS PubMed; (e) A. Hienzsch, C. Deiml, V. Reiter and T. Carell, Chem.–Eur. J., 2013, 19, 4244 CrossRef CAS PubMed; (f) M. D. Lebar, T. J. Lupoli, H. Tsukamoto, J. M. May, S. Walker and D. Kahne, J. Am. Chem. Soc., 2013, 135, 4632 CrossRef CAS PubMed.
- (a) J.-X. Ji, J. Wu and A. S. C. Chan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11196 CrossRef CAS PubMed; (b) Q. Kang, Z.-A. Zhao and S.-L. You, Org. Lett., 2008, 10, 2031 CrossRef CAS PubMed; (c) T. Akiyama, K. Daidouji and K. Fuchibe, Org. Lett., 2003, 5, 3691 CrossRef CAS PubMed; (d) M. Shi, G.-N. Ma and J. Gao, J. Org. Chem., 2007, 72, 9779 CrossRef CAS PubMed.
- For reviews, see: (a) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998, ch. 8 Search PubMed; (b) C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 9148 CrossRef CAS PubMed; (c) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS; (d) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; for selected examples, see: (e) B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834 CrossRef CAS PubMed; (f) E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 12066 CrossRef CAS PubMed; (g) Z. Hou, J. Wang, P. He, J. Wang, B. Qin, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 4763 CrossRef CAS PubMed; (h) H. Saito, T. Uchiyama, M. Miyake, M. Anada, S. Hashimoto, T. Takabatake and S. Miyairi, Heterocycles, 2010, 81, 1149 CrossRef CAS; (i) B. Xu, S.-F. Zhu, X.-L. Xie, J.-J. Shen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 11483 CrossRef CAS PubMed; (j) Y. Zhu, X. Liu, S. Dong, Y. Zhou, W. Li, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2014, 53, 1636 CrossRef CAS PubMed; (k) B. Xu, S.-F. Zhu, X.-D. Zuo, Z.-C. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913 CrossRef CAS PubMed.
- For selected non-asymmetric N–H insertion of vinyldiazoacetates, see: (a) J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256 CrossRef CAS PubMed; (b) Y. Yue, Y. Wang and W. Hu, Tetrahedron Lett., 2007, 48, 3975 CrossRef CAS; (c) O. Pavlyuk, H. Teller and M. C. McMills, Tetrahedron Lett., 2009, 50, 2716 CrossRef CAS; (d) J. H. Hansen and H. M. L. Davies, Chem. Sci., 2011, 2, 457 RSC.
- For a review on enantioselective carbene transfer reactions with vinyldiazoacetates, see: (a) H. M. L. Davies, Aldrichimica Acta, 1997, 30, 107 CAS; for selected examples, see: C–H insertion/Cope rearrangement, (b) H. M. L. Davies and Q. Jin, J. Am. Chem. Soc., 2004, 126, 10862 CrossRef CAS PubMed; (c) H. M. L. Davies, X. Dai and M. S. Long, J. Am. Chem. Soc., 2006, 128, 2485 CrossRef CAS PubMed; Si–H insertion, (d) H. M. L. Davies, T. Hansen, J. Rutberg and P. R. Bruzinski, Tetrahedron Lett., 1997, 38, 1741 CrossRef CAS; (e) R. Sambasivan and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 9289 CrossRef CAS PubMed; cyclopropanation, (f) H. M. L. Davies, P. R. Bruzinski, D. H. Lake, N. Kong and M. J. Fall, J. Am. Chem. Soc., 1996, 118, 6897 CrossRef CAS; (g) H. M. L. Davies, D. G. Stafford, B. D. Doan and J. H. Houser, J. Am. Chem. Soc., 1998, 120, 3326 CrossRef CAS; cyclopropenation, (h) J. F. Briones, J. Hansen, K. I. Hardcastle, J. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 17211 CrossRef CAS PubMed.
- X. Xu, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2012, 51, 9829 CrossRef CAS PubMed.
- Landais and co-workers reported an enantioselective O–H insertion of ethyl (E)-2-diazo-4-phenylbut-3-enoate with water catalyzed by a chiral dirhodium catalyst, but only obtained 8% ee. P. Bulugahapitiya, Y. Landais, L. Parra-Rapado, D. Planchenault and V. Weber, J. Org. Chem., 1997, 62, 1630 CrossRef CAS.
- (a) S.-F. Zhu, B. Xu, G.-P. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2012, 134, 436 CrossRef CAS PubMed; (b) X.-G. Song, S.-F. Zhu, X.-L. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 2555 CrossRef CAS PubMed; (c) Q.-Q. Cheng, S.-F. Zhu, Y.-Z. Zhang, X.-L. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2013, 135, 14094 CrossRef CAS PubMed; (d) X.-L. Xie, S.-F. Zhu, J.-X. Guo, Y. Cai and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 2978 CrossRef CAS PubMed; (e) Q.-Q. Cheng, H. Xu, S.-F. Zhu and Q.-L. Zhou, Acta Chim. Sin., 2015, 73, 326 CrossRef CAS ; see also ref. 6d.
- H. M. L. Davies, T. Hansen and M. R. Churchill, J. Am. Chem. Soc., 2000, 122, 3063 CrossRef CAS.
- R. P. Reddy, G. H. Lee and H. M. L. Davies, Org. Lett., 2006, 8, 3437 CrossRef CAS PubMed.
- For recent reviews on cooperative catalysis, see: (a) J. Zhou, Multicatalyst System in Asymmetric Catalysis, Wiley, Hoboken, 2015 Search PubMed; (b) A. E. Allen and D. W. C. McMillan, Chem. Sci., 2012, 3, 633 RSC; (c) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed; (d) Z.-P. Yang, W. Zhang and S.-L. You, J. Org. Chem., 2014, 79, 7785 CrossRef CAS PubMed; (e) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365 CrossRef CAS PubMed; for the initial investigation into the enantioselective insertion reactions by using chiral proton-shuttle catalysts, see ref. 6i; for further applications, see: (f) H. Saito, D. Morita, T. Uchiyama, M. Miyake and S. Miyairi, Tetrahedron Lett., 2012, 53, 6662 CrossRef CAS; (g) H. Qiu, D. Zhang, S. Liu, L. Qiu, J. Zhou, Y. Qian, C. Zhai and W. Hu, Acta Chim. Sin., 2012, 70, 2484 CrossRef CAS; (h) B. Xu, S.-F. Zhu, Z.-C. Zhang, Z.-X. Yu, Y. Ma and Q.-L. Zhou, Chem. Sci., 2014, 5, 1442 RSC; (i) Y. Ni, X. Guo, W. Hu and S. Liu, Chin. J. Org. Chem., 2014, 34, 107 CrossRef CAS; (j) B. Xu, M.-L. Li, X.-D. Zuo, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2015, 137, 8700 CrossRef CAS PubMed; for mechanistic studies, see: (k) Y. Liang, H. Zhou and Z.-X. Yu, J. Am. Chem. Soc., 2009, 131, 17783 CrossRef CAS PubMed; (l) H. K. Kisan and R. B. Sunoj, Chem. Commun., 2014, 50, 14639 RSC; (m) X.-C. Wang, X.-S. Song, L.-P. Guo, D. Qu, Z.-Z. Xie, F. Verpoort and J. Cao, Organometallics, 2014, 33, 4042 CrossRef CAS; (n) H. K. Kisan and R. B. Sunoj, J. Org. Chem., 2015, 80, 2192 CrossRef CAS PubMed.
- For the preparation of vinyldiazoacetates, see: (a) M. P. Doyle, M. Yan, W. Hu and L. S. Gronenberg, J. Am. Chem. Soc., 2003, 125, 4692 CrossRef CAS PubMed; (b) B. D. Schwartz, J. R. Denton, Y. Lian, H. M. L. Davies and C. M. Williams, J. Am. Chem. Soc., 2009, 131, 8329 CrossRef CAS PubMed ; see also ref. 10.
- K. O. Hallinan, D. H. G. Crout and W. Errington, J. Chem. Soc., Perkin Trans. 1, 1994, 3537 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; spectral data for all new compounds; HPLC or SFC charts for all insertion products. See DOI: 10.1039/c5sc03558a |
| This journal is © The Royal Society of Chemistry 2016 |