DOI:
10.1039/C5SC03612G
(Edge Article)
Chem. Sci., 2016,
7, 2821-2826
Design and synthesis of biphenyl and biphenyl ether inhibitors of sulfatases†
Received
23rd September 2015
, Accepted 10th January 2016
First published on 11th January 2016
Abstract
Inhibitors of sulfatase-2 are putative anticancer agents, but the discovery of potent small molecules targeting this enzyme has proved challenging. Based on molecular modelling, two series of sulfatase-2 inhibitors have been developed with biphenyl and biphenyl ether scaffolds judiciously substituted with sulfamate, carboxylate and other polar groups (e.g. amino). Inhibition of aryl sulfatase A and B was also determined. The biphenyl ether derivatives were less selective for sulfatase-2 over aryl sulfatase B than the biphenyl series. All biphenyl ether derivatives inhibited aryl sulfatase A, whereas only amino derivatives inhibited aryl sulfatase B significantly. In the biphenyl series few derivatives exhibited activity against aryl sulfatase B. The trichloroethylsulfamate group was identified as a new pharmacophore enabling potent inhibition of all of the sulfatases studied.
Introduction
Sulfatase-1 (Sulf-1) and sulfatase-2 (Sulf-2) catalyse the hydrolysis of the 6-O-sulfate group attached to glucosamine residues in heparan sulfate proteoglycans (HSPGs).1 The HSPG substrates consist of a protein core linked to heparan sulfate polysaccharide chains derived from repeating disaccharide units comprising a hexuronic acid (glucuronic acid or iduronic acid) and glucosamine. Removal of the 6-O-sulfate affects the mobilisation of heparan bound growth factors and cytokines,2 including fibroblast growth factor (FGF) and wingless-related integration site (wnt).3 The role of FGF in cell proliferation, invasion, migration and angiogenesis,4 and of wnt in cell growth and proliferation,5 implicate the sulfatases as potential targets for therapeutic intervention in certain diseases including pulmonary fibrosis6 and prostate,7 colorectal,8 ovarian, and breast cancers.9 Very few small molecule inhibitors of Sulf-1 and Sulf-2 have been reported.10,11 On re-synthesis, the purported activity against Sulf-2 of a group of monosaccharide sulfamate-based inhibitors could not be replicated.12 The nitrone ‘OKN-007’ is a suboptimal Sulf-2 inhibitor tool compound, having several proposed potential chemical and biological mechanisms of action in addition to Sulf-2 inhibition, including suppression of NO production, S-nitrosylation of critical proteins and inhibition of NF-κB activation.13 The difficulty in identifying small molecule inhibitors of Sulf-2 has led to the investigation of indirect methods to affect Sulf-2 through inhibition of the proteosomal machinery.14 However, in this paper we report new saccharide mimics that inhibit Sulf-2 directly. We have also identified the trichloroethylsulfamate group as a new pharmacophore for Sulf-2 inhibition. Among the compounds described are the first relatively potent small molecule inhibitors of Sulf-2 that will aid further biological studies and assist elucidation of the role of Sulf-2 in cancer.
De novo inhibitor design
There is no crystal structure for Sulf-2 and a lack of either small molecule inhibitors or biological assay suitable for a high-throughput screening campaign. Given these constraints, the structure of heparan sulfate proteoglycans, which are the endogenous ligands of Sulf-2, was used as a template for the design of potential Sulf-2 inhibitors. With no solved structures of a HSPG, the principal structural motifs of HSPG reported to be substrates for Sulf-2 are the highly sulfated regions, which closely resemble heparin in structure.15 However, structures of heparin in solution have been solved by NMR (pdb code 1hpn).16 These were compared with structures of heparin bound to protein partners such as the NK1 growth factor (pdb code 1gmn), 3-O-sulfotransferase (pdb code 1t8u) and heparin lyase (pdb code 3ina). The bound form was found to resemble the solution α-helical form, with interactions to protein binding partners being dominated by contacts between the sulfate and carboxylate functionalities on the surface of the heparin helix. Negatively charged sulfate and carboxylate groups cover most of the accessible surface of the heparin polymer, and have been shown to regulate the binding of heparin to the enzyme 3-O-sulfotransferase.16,17 X-ray crystal structures of FGF-1 and FGF-2 in complex with heparin oligosaccharides also revealed the importance of the sulfate and carboxylate groups for binding.18 These studies demonstrated that interactions of the charged sulfate and carboxylate groups on the heparin oligosaccharide with basic amino acids on protein binding partners are the main contributors to tight and specific binding. Mimicking these ionic interactions was a key consideration for de novo design of potential non-saccharide based Sulf-2 inhibitors.
Potential sulfamate inhibitors were designed with the aim of identifying hit compounds with improved Sulf-2 inhibition and physicochemical properties compared to the monosaccharides.12 The arylsulfamate group has been shown to be a privileged structure for the inhibition of sulfatases,19 although much of this work was focused on steroid sulfatase (STS). Potential Sulf-2 inhibitors were designed initially by overlaying a primary phenylsulfamate (A-ring) with the 6-O sulfate of glucosamine of a representative trisaccharide taken from the reducing end of the heparin solution structure (1hpn). Vectors where the phenyl ring could be substituted to allow incorporation of a second polar group that could overlay a surface polar group on the heparin helix were explored. Compounds from both biphenyl (Fig. 1) and biphenyl ether sulfamate (Fig. 2) series were energy minimised and found to enable such a superposition of polar functionality. A primary sulfamate group at the 3-position of the (A-ring) in the biphenyl series enabled substituents on the second phenyl ring to overlay the N-sulfate of glucosamine or the O-sulfate of iduronic acid in the template structure. In addition to these rationally designed derivatives, the biphenyl core was used to probe additional polar and ionic interactions through incorporation of polar functional groups at the ortho, meta and para positions of the B-ring. In the biphenyl ether sulfamate scaffold the oxygen linker allows different conformations with alternative positioning of groups on the B-ring relative to the A-ring.
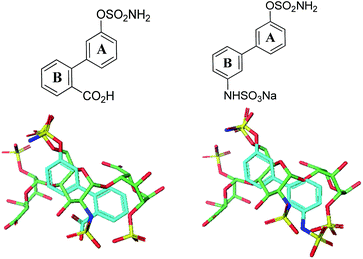 |
| | Fig. 1 Representative overlays of biphenyl targets (cyan) with a heparin-derived trisaccharide template (green) created using the proprietary structural visualisation program MoViT (Pfizer). | |
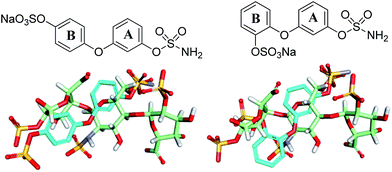 |
| | Fig. 2 Representative overlays of biphenyl ethers (cyan) with a heparin-derived trisaccharide template (green). | |
Synthesis of inhibitors
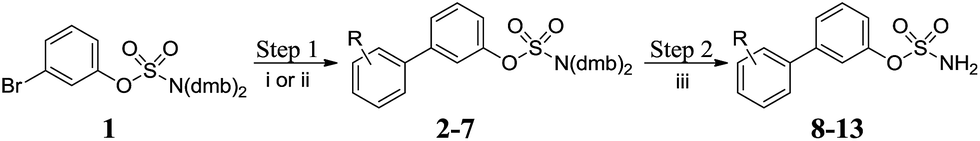
A number of highly efficient synthetic routes were implemented that should have wide applicability for sulfamate synthesis. Key to the synthesis of the desired templates was our previously reported sulfamate protecting group methodology using the N-2,4-dimethoxybenzyl (dmb) group,20 which was applied to the synthesis of libraries of biphenyl- and biaryl ether-sulfamates. Microwave-assisted Suzuki–Miyaura cross-coupling of 1 in the presence of potassium carbonate and Pd(PPh3)4 afforded 2 in 75% yield (method i, Scheme 1 and Table 1). Alternative Suzuki conditions (method ii, Scheme 1 and Table 1) were required for the preparation of 3–7. All deprotections proceeded at room temperature in dilute TFA, resulting in high yields of the desired primary sulfamates 8–13.
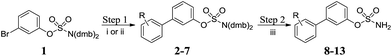 |
| | Scheme 1 Synthesis of biphenyl sulfamates 8–13. Reagents and conditions: (i) K2CO3, RC6H4B(OH)2, Pd(PPh3)4, MeCN, 120 °C, μW, 20 min; (ii) RC6H4B(pin), 2 M aq. Na2CO3, Pd(dppf)Cl2, dioxane, 80 °C, μW, 20 min; (iii) 10% TFA/DCM, RT, 2 h. | |
Table 1 Summary of yields for synthesis of biphenyl sulfamates
| R |
Step 1 |
Step 2 |
| Method |
Yield |
Yield |
| 2-CO2Me |
i |
2 75% |
8 88% |
| 3-CO2H |
ii |
3 79% |
9 82% |
| 4-CO2H |
ii |
4 77% |
10 90% |
| 2-NH2 |
ii |
5 82% |
11 83% |
| 3-NH2 |
ii |
6 77% |
12 82% |
| 4-NH2 |
ii |
7 72% |
13 82% |
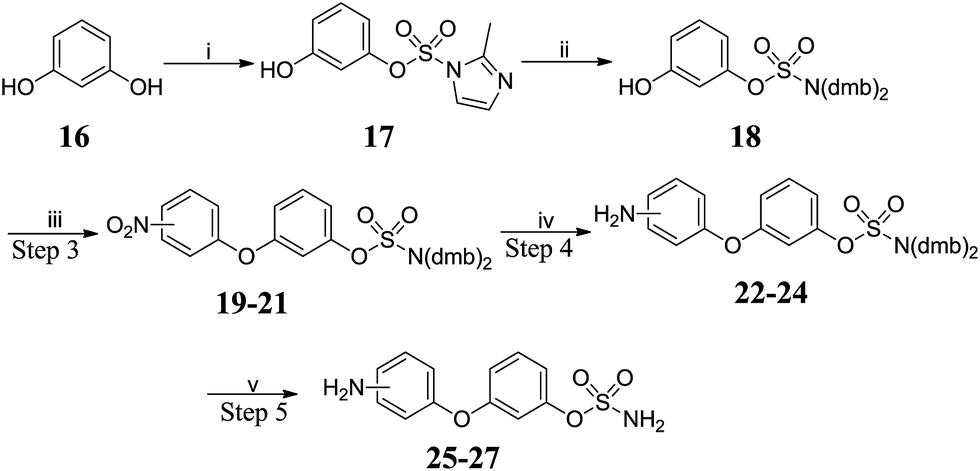
The preparation of amino-substituted biaryl ether-sulfamates 25–27 started from resorcinol 16 (Scheme 2 and Table 2). The use of high dilution (0.08 M) and low temperature prevented side-reactions during the methylation/imidazolium displacement step. SNAr reaction of fluoronitrobenzenes with 18 gave biphenyl ether intermediates 19–21, which were reduced via palladium-catalysed flow hydrogenation. Deprotection of 22–24 gave high yields of the desired primary sulfamates 25–27.
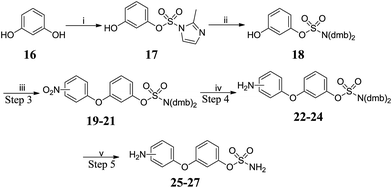 |
| | Scheme 2 Synthesis of biaryl ether-sulfamates 25–27. Reagents and conditions: (i) 1,1′-sulfonylbis(2-methyl-1H-imidazole), Cs2CO3, MeCN, 120 °C, μW, 15 min, 80%; (ii) (a) Me3O·BF4, DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (8:1), 0 °C to RT, 9 h; (b) bis(2,4-dimethoxy benzyl)amine, MeCN, 42 °C, 24 h, 60%; (iii) fluoronitrobenzene, K2CO3, DMF, 150 °C, μW, 20 min; (iv) H2, 10% Pd/C, MeOH:THF (3:1), RT, 24 h; (v) 10% TFA/DCM, RT, 2 h. :THF (8:1), 0 °C to RT, 9 h; (b) bis(2,4-dimethoxy benzyl)amine, MeCN, 42 °C, 24 h, 60%; (iii) fluoronitrobenzene, K2CO3, DMF, 150 °C, μW, 20 min; (iv) H2, 10% Pd/C, MeOH:THF (3:1), RT, 24 h; (v) 10% TFA/DCM, RT, 2 h. | |
Table 2 Summary of yields for synthesis of biaryl ether-sulfamates
| Position |
Step 3 |
Step 4 |
Step 5 |
| 2′- |
19 80% |
22 85% |
25 90% |
| 3′- |
20 75% |
23 87% |
26 82% |
| 4′- |
21 85% |
24 86% |
27 65% |
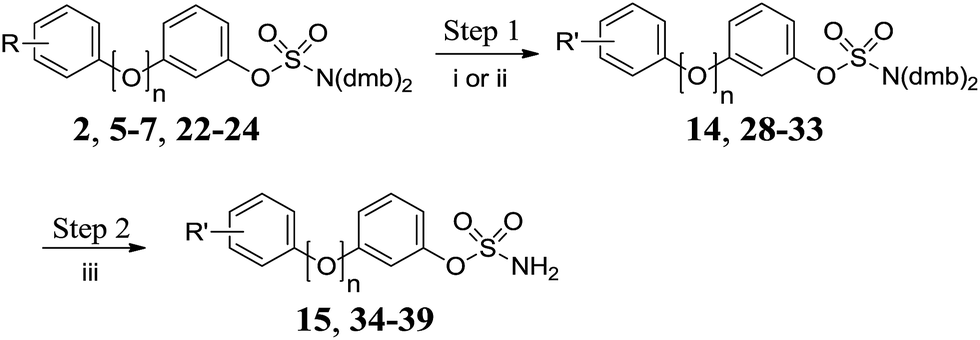
Methyl ester 2 was hydrolysed under basic conditions and the resulting carboxylic acid 14 was deprotected to give 15 (Scheme 3 and Table 3). Acetylation of aniline derivatives 5–7 and 22–24 afforded acetamidobiphenyls 28–33, which were deprotected to give 34–39.
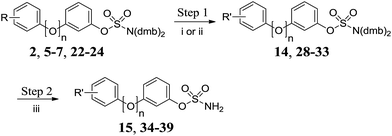 |
| | Scheme 3 Functionalisation/deprotection of protected biphenyl sulfamates. Reagents and conditions: (i) LiOH, H2O/THF, 60 °C, 24 h, 80%; (ii) Ac2O, NEt3, DCM, RT, 24 h; (iii) 10% TFA/DCM, RT, 2 h. | |
Table 3 Summary of yields for functionalisation/deprotection of protected biphenyl sulfamates
| R |
R′ |
n
|
Step 1 |
Step 2 |
| Method |
Yield |
Yield |
| 2-CO2Me |
2-CO2H |
0 |
i |
14 80% |
15 89% |
| 2-NH2 |
2-NHAc |
0 |
ii |
28 87% |
34 86% |
| 3-NH2 |
3-NHAc |
0 |
ii |
29 89% |
35 91% |
| 4-NH2 |
4-NHAc |
0 |
ii |
30 88% |
36 84% |
| 2-NH2 |
2-NHAc |
1 |
ii |
31 87% |
37 85% |
| 3-NH2 |
3-NHAc |
1 |
ii |
32 88% |
38 94% |
| 4-NH2 |
4-NHAc |
1 |
ii |
33 86% |
39 92% |
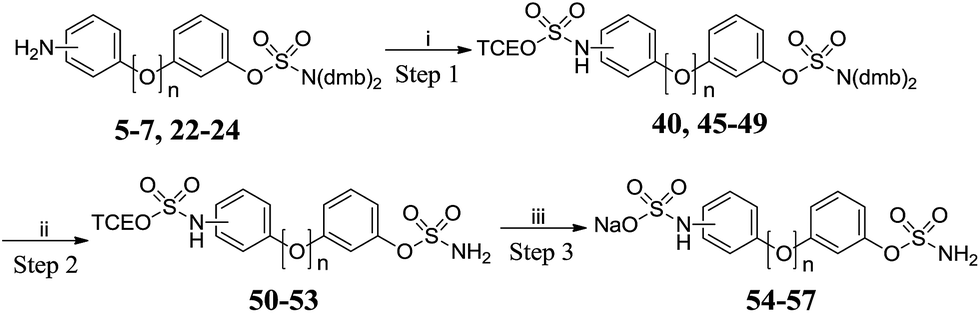
Attempted preparation of the 2′-sulfamic acid derivative by reaction of aniline 5 with sulfur trioxide-pyridine complex was unsuccessful. Taylor et al. reported the 2,2,2-trichloroethyl (TCE) group as an effective protecting group for arylsulfate esters21 and this methodology was adapted to the synthesis of protected sulfamic acids. TCE chlorosulfate 42 was obtained by reacting sulfuryl chloride with one equivalent of 2,2,2-trichloroethanol 41 (Scheme 4). Reaction of 42 with 2-methylimidazole gave 2-methylimidazole-1-sulfonate 43, which was methylated with Meerwein's salt to give 44.
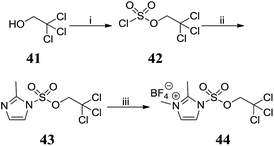 |
| | Scheme 4 Synthesis of 2,3-dimethyl-1-((2,2,2-trichloroethoxy)-sulfonyl)-1H-imidazol-3-ium tetrafluoroborate 44. Reagents and conditions: (i) SO2Cl2, pyridine, Et2O, −78 °C, 4 h, 83%; (ii) 2-methylimidazole, THF, 0 °C to RT, 16 h, 95%; (iii) Me3O·BF4, DCM, 0 °C to RT, 20 h, 86%. | |
Reacting the appropriate anilines with 3 molar equivalents of 44 under microwave irradiation in acetonitrile at 120 °C for 20 min afforded the meta- (45 and 48) and para- (46 and 49) derivatives, but poor conversion was observed with ortho-anilines 5 and 22 (Scheme 5 and Table 4). The trichloroethyl group was stable under the acidic conditions used for sulfamate deprotection. Cleavage of the trichloroethyl group was achieved using zinc in a mixture of methanol and acetate buffer with no desulfamoylation. The resulting sulfamic acids were converted to sodium salts using ion exchange chromatography, affording targets 54–57 in high yields.
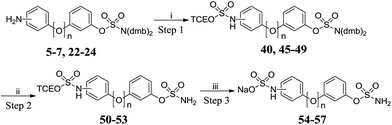 |
| | Scheme 5 Synthesis of amino-sulfate derivatives 54–57. Reagents and conditions: (i) 44, MeCN, 120 °C, μW, 20 min; (ii) 10% TFA/DCM, RT, 2 h; (iii) (a) Zn powder, MeOH, acetate buffer pH 4.65, 60 °C, 2 h; (b) Dowex 50W8X2 Na+ form, H2O. | |
Table 4 Summary of yields for the synthesis of amino-sulfate derivatives
| Position |
n
|
Step 1 |
Step 2 |
Step 3 |
| 2′- |
0 |
40 0% |
— |
— |
| 3′- |
0 |
45 75% |
50 86% |
54 84% |
| 4′- |
0 |
46 82% |
51 91% |
55 80% |
| 2′- |
1 |
47 15% |
— |
— |
| 3′- |
1 |
48 78% |
52 86% |
56 75% |
| 4′- |
1 |
49 90% |
53 89% |
57 70% |
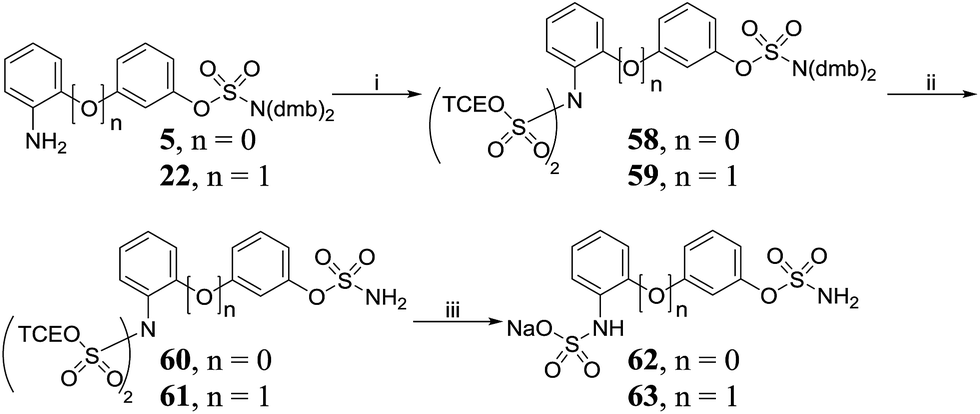
Protection of 2-aminobiphenyls 5 and 22 with TCE chlorosulfate 44 produced bis-N,N-sulfated aminobiphenyls 58 and 59, respectively (Scheme 6), which were deprotected to afford 60 and 61. A one-pot deprotection/mono-sulfate hydrolysis of 60 and 61 led to the isolation of 62 and 63.
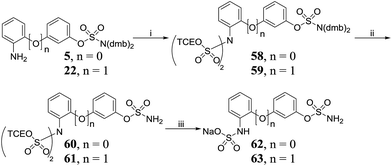 |
| | Scheme 6 Synthesis of amino-sulfate derivatives 62, 63. Reagents and conditions: (i) 44, NEt3, DMAP, THF, 0 °C to RT, 24 h, n = 0 70%, n = 1 55%; (ii) 10% TFA/DCM, RT, 2 h, n = 0 90%, n = 1 86%; (iii) Zn powder, MeOH, acetate buffer pH 4.65, AcOH, RT, 24 h; Dowex 50W8X2 Na+ form, H2O, n = 0 50%, n = 1 33%. | |
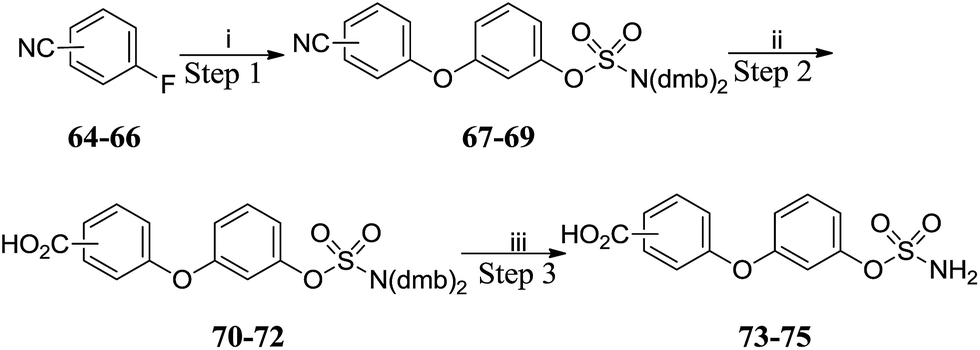
SNAr reactions with fluorobenzonitriles 64–66 gave cyanobiphenyl ethers 67–69 which were hydrolysed to the corresponding benzoic acids 70–72 (Scheme 7 and Table 5). Acidic deprotection of the sulfamate moiety afforded targets 73–75. Thus, sets of substituted biphenyl and biphenyl ether sulfamates were prepared. Compounds 76–82 (Table 6) bearing a single substituent on the aromatic core were also prepared by analogous routes (see ESI data†).
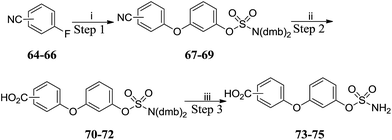 |
| | Scheme 7 Synthesis of benzoic acid derivatives 73–75. Reagents and conditions: (i) 18, K2CO3, DMF, 150 °C, μW, 20 min; (ii) 2 M aq. NaOH, dioxane, 130 °C, μW, 2 h; (iii) 10% TFA/DCM, RT, 2 h. | |
Table 5 Summary of yields for synthesis of benzoic acid derivatives
| Position |
Step 1 |
Step 2 |
Step 3 |
| 2′- |
67 81% |
70 45% |
73 90% |
| 3′- |
68 61% |
71 80% |
74 94% |
| 4′- |
69 72% |
72 65% |
75 92% |
Table 6 Monosubstituted phenyl and biphenyl derivatives
|
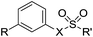
|
| Cmpd |
R |
X |
R′ |
|
76
|
Ph |
O |
NH2 |
|
77
|
Ph |
O |
NMe2 |
|
78
|
OPh |
O |
NH2 |
|
79
|
Ph |
NH |
OCH2CCl3 |
|
80
|
OPh |
NH |
OCH2CCl3 |
|
81
|
Ph |
O |
OCH2CCl3 |
|
82
|
Ph |
NH |
OCH2CF3 |
|
83
|
H |
NH |
OCH2CCl3 |
Biological evaluation
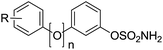
Compounds were assayed for their ability to inhibit the Sulf-2-catalysed desulfation of 4-methylumbelliferyl sulfate (4-MUS) to the fluorescent 4-methylumbelliferone (MU). To determine their sulfatase selectivity, inhibition of aryl sulfatases A (ARSA) and B (ARSB) was also assessed. To date no suitable benchmark inhibitors of these sulfatase have been reported. In the biphenyl series (Table 7A), only the trichloroethylsulfamates (50 and 51) exhibited high Sulf-2 inhibition giving almost complete inhibition of sulfatase activity at 1 mM concentration. The position and nature of the substituent on the B ring had a pronounced effect on the sulfatase inhibition. In the majority of cases the presence of a substituent on the B-ring was detrimental to Sulf-2 inhibition when compared with unsubstituted biphenyl sulfamate 76. Aminosulfates 54 and 62 and carboxylic acid 15 retained potency. Surprisingly, the synthetic trichloroethylsulfamate intermediates 50 and 51 exhibited potent inhibition. Compounds 8, 10, 15, 50 and 51 were also relatively potent ARSA inhibitors. The tertiary dimethylsulfamate 77 exhibited no Sulf-2 inhibition (0% inh @ 1 mM). Similar SAR was observed with the 3-phenoxyphenyl template (Table 7B) with the trichloroethylsulfamates 52 and 53 again proving to be superior inhibitors against all sulfatases tested. Aminosulfates 56 and 63 were the best Sulf-2 inhibitors lacking a trichloroethylsulfamate group. In summary, the design strategy of introducing polar groups to mimic polar groups of the endogenous substrate did not give the anticipated improvement in Sulf-2 inhibition, whereas the presence of a relatively large, lipophilic trichloroethylsulfamate group on the B-ring in both templates provided superior inhibitors.
Table 7 Sulfatase inhibition data for biphenylsulfamates. Data are means of two or more determinations. n.d. = not determined
|
|
| Cpd |
R |
Sulf-2% inh @ 1 mM |
ARSA% inh @ 1 mM |
ARSB% inh @ 1 mM |
|
A: n = 0
|
|
76
|
H |
38 |
14 |
65 |
|
8
|
2′-CO2Me |
20 |
86 |
0 |
|
15
|
2′-CO2H |
31 |
98 |
0 |
|
9
|
3′-CO2H |
12 |
59 |
0 |
|
10
|
4′-CO2H |
22 |
85 |
0 |
|
11
|
2′-NH2 |
21 |
0 |
0 |
|
12
|
3′-NH2 |
20 |
0 |
0 |
|
13
|
4′-NH2 |
6 |
61 |
0 |
|
34
|
2′-NHAc |
4 |
33 |
0 |
|
35
|
3′-NHAc |
8 |
52 |
33 |
|
36
|
4′-NHAc |
3 |
0 |
0 |
|
62
|
2′-NHSO3Na |
37 |
26 |
0 |
|
54
|
3′-NHSO3Na |
31 |
0 |
0 |
|
55
|
4′-NHSO3Na |
4 |
0 |
0 |
|
60
|
2′-N(SO3CH2CCl3)2 |
10 |
63 |
0 |
|
50
|
3′-NHSO3CH2CCl3 |
99 |
92 |
67 |
|
51
|
4′-NHSO3CH2CCl3 |
96 |
99 |
68 |
|
|
B: n = 1
|
|
78
|
H |
32 |
n.d. |
n.d. |
|
73
|
2′-CO2H |
10 |
44 |
0 |
|
74
|
3′-CO2H |
30 |
49 |
0 |
|
75
|
4′-CO2H |
23 |
33 |
0 |
|
25
|
2′-NH2 |
28 |
75 |
17 |
|
26
|
3′-NH2 |
31 |
75 |
0 |
|
27
|
4′-NH2 |
14 |
75 |
0 |
|
37
|
2′-NHAc |
11 |
29 |
48 |
|
38
|
3′-NHAc |
10 |
57 |
0 |
|
39
|
4′-NHAc |
14 |
45 |
0 |
|
63
|
2′-NHSO3Na |
56 |
54 |
93 |
|
56
|
3′-NHSO3Na |
58 |
95 |
85 |
|
57
|
4′-NHSO3Na |
13 |
34 |
77 |
|
61
|
2′-N(SO3CH2CCl3)2 |
18 |
82 |
40 |
|
52
|
3′-NHSO3CH2CCl3 |
100 |
100 |
96 |
|
53
|
4′-NHSO3CH2CCl3 |
98 |
96 |
86 |
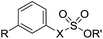
To understand the origin of sulfatase inhibition by compounds 50–53, compounds 79 and 80, lacking a primary sulfamate, were prepared and found to be also potent Sulf-2 inhibitors (Table 8), indicating that the primary sulfamate group is not essential for sulfatase inhibition with these templates. Compound 79 exhibited some selectivity over ARSA and ARSB, whereas compound 80 was a good inhibitor for all of the sulfatases. Compound 83, the simple phenyl trichloroethylsulfamate derivative did not retain Sulf-2 inhibitory activity, suggesting that the trichloroethylsulfamate group is not sufficient per se to impart sulfatase inhibition and is not conferring pan-sulfatase activity through a non-specific mechanism of action. Compound 83 did, however, possess inhibitory activity against both ARSA and ARSB. Trichloroethylsulfate 81 and trifluoroethylsulfamate 82 exhibited no Sulf-2 inhibition, but retained activity against ARSA and ARSB. Thus, it appears that the trichloroethylsulfamate group is a new motif for Sulf-2 inhibition, and that, in combination with lipophilic groups, inhibitors of Sulf-2 lacking a primary sulfamate moiety can be developed.
Table 8 Sulfatase inhibition data for analogues lacking a primary sulfamate group. Data are means of two or more determinations
|
|
| Cpd |
R |
X |
R′ |
Sulf-2% inh @ 1 mM |
ARSA% inh @ 1 mM |
ARSB% inh @ 1 mM |
|
83
|
H |
NH |
CH2CCl3 |
3 |
63 |
68 |
|
79
|
Ph |
NH |
CH2CCl3 |
100 |
0 |
61 |
|
80
|
OPh |
NH |
CH2CCl3 |
99 |
74 |
99 |
|
81
|
Ph |
O |
CH2CCl3 |
1 |
70 |
85 |
|
82
|
Ph |
NH |
CH2CF3 |
5 |
78 |
92 |
Sulf-2 IC50 values were determined for the most potent inhibitors (Table 9). Compound 50 had the highest Sulf-2 inhibitory activity with an IC50 value of 167 μM. The ARSA and ARSB inhibitory activity of all compounds tested proved to be higher than their Sulf-2 inhibitory activity, providing the first reported inhibitors of ARSA and ARSB with IC50 < 1 mM. Following screening at 1 mM, ARSA inhibition by 80 was evaluated over the concentration range 0.025–1 mM. Concentration-dependent inhibition was not observed and hence an IC50 value could not be determined. Aryl sulfamate-based inhibitors of steroid sulfatase have demonstrated irreversible inhibition profiles.15 Further studies will be required to determine if the aryl sulfamates such as 50, and trichloroethylsulfamates such as 80 behave as irreversible sulfatase inhibitors.
Table 9 Sulfatase IC50 data for inhibitors 50–51, 53, 79–80. IC50 values were calculated using non-linear regression analysis for 7 different inhibitor concentrations each performed in duplicate to give the mean ± SEM
| Compound |
Sulf-2 IC50 (μM) |
ARSA IC50 (μM) |
ARSB IC50 (μM) |
|
50
|
167 ± 5 |
55 ± 11 |
130 ± 6 |
|
51
|
566 ± 28 |
91 ± 4 |
101 ± 8 |
|
53
|
390 ± 11 |
108 ± 2 |
116 ± 10 |
|
79
|
254 ± 3 |
No inhibition |
30 ± 11 |
|
80
|
298 ± 3 |
nd |
34 ± 9 |
Conclusions
Two new series of sulfatase inhibitors have been developed based on biphenyl and biphenyl ether templates, with highly effective synthetic procedures providing access to a variety of sulfamates and aminosulfates. The trichloroethylsulfamates 50–53, 79 and 80 exhibited favourable Sulf-2 inhibition. The trichloroethylsulfamate group has therefore been identified as a new pharmacophore for sulfatase inhibition. Further studies will be required to determine the origin of sulfatase inhibition by aryl trichloroethylsulfamates. The most potent Sulf-2 inhibitors lacking a trichloroethylsulfamate group were biphenyl ether aminosulfates 56 and 63. The diphenyl ether derivatives also proved to be moderate to good inhibitors of ARSA, whereas significant inhibition of ARSB was only observed with substituted amino derivatives. This trend was not retained in the biphenyl series with few derivatives exhibiting activity against ARSB. However, the first inhibitors of ARSA and ARSB with IC50 < 1 mM are reported. Additional SAR studies are underway to improve Sulf-2 potency and to delineate the factors affecting selectivity.
Author contributions
DCM, BTG and RJG designed the inhibitors; BTG, DCM and DRN wrote the manuscript; TR, AB (Bertoli) and AB (Brennan) performed the chemical synthesis supervised by CC, BTG, RJG and DCM; SFA and GSB performed the biological assays supervised by DRN and HLR.
Acknowledgements
The authors thank: Cancer Research UK and Pfizer Global Research and Development for financial support, the EPSRC National Mass Spectrometry Service at the University of Wales (Swansea) for mass spectrometric determinations, Professor S. Rosen and Dr M. Singer, University of California San Francisco, for invaluable advice on establishing the Sulf-2 assay and Dr I. R. Hardcastle for helpful discussions.
Notes and references
- M. Morimoto-Tomita, K. Uchimura, Z. Werb, S. Hemmerich and S. D. Rosen, J. Biol. Chem., 2002, 277, 49175 CrossRef CAS PubMed.
- K. Uchimura, M. Morimoto-Tomita, A. Bistrup, J. Li, M. Lyon, J. Gallagher, Z. Werb and S. D. Rosen, BMC Biochem., 2006, 7, 2 CrossRef PubMed.
- R. Nawroth, A. van Zante, S. Cervantes, M. McManus, M. Hebrok and S. D. Rosen, PLoS One, 2007, 2, e392 Search PubMed.
- N. Turner and R. Grose, Nat. Rev. Cancer, 2010, 10, 116 CrossRef CAS PubMed.
- A. Klaus and W. Birchmeier, Nat. Rev. Cancer, 2008, 8, 387 CrossRef CAS PubMed.
- X. Yue, J. Lu, L. Auduong, M. D. Sides and J. A. Lasky, Glycobiology, 2013, 23, 709 CrossRef CAS PubMed.
- C. M. Vicente, M. A. Lima, H. B. Nader and L. Toma, J. Exp. Clin. Cancer Res., 2015, 34, 25 CrossRef PubMed.
- C. M. Vicente, M. A. Lima, E. A. Yates, H. B. Nader and L. Toma, Mol. Cancer Res., 2015, 13, 510 CrossRef CAS PubMed.
- A. Khurana, D. Beleford, X. He, J. Chien and V. Shridhar, Am. J. Cancer Res., 2013, 3, 34 CAS.
- M. Schelwies, D. Brinson, S. Otsuki, Y.-H. Hong, M. K. Lotz, C.-H. Wong and S. R. Hanson, ChemBioChem, 2010, 11, 2393 CrossRef CAS PubMed.
- X. Zheng, X. Gai, S. Han, C. D. Moser, C. Hu, A. M. Shire, R. A. Floyd and L. R. Roberts, Genes, Chromosomes Cancer, 2013, 52, 225 CrossRef CAS PubMed.
- D. C. Miller, B. Carbain, G. S. Beale, S. F. Alhasan, H. L. Reeves, U. Baisch, D. R. Newell, B. T. Golding and R. J. Griffin, Org. Biomol. Chem., 2015, 13, 5279 CAS.
- R. A. Floyd, H. K. Chandru, T. He and R. Towner, Anti-Cancer Agents Med. Chem., 2011, 11, 373 CrossRef CAS PubMed.
- A. Khurana, D. Jung-beom, X. He, S. Kim, R. C. Busby, L. Lorenzon and V. Shridhar, Clin. Exp. Metastasis, 2013, 30, 407 CrossRef CAS PubMed.
- O. M. Saad, H. Ebel, K. Uchimura, S. D. Rosen, C. R. Bertozzi and J. A. Leary, Glycobiology, 2005, 15, 818 CrossRef CAS PubMed.
- B. Mulloy and M. J. Forster, Glycobiology, 2000, 10, 1147 CrossRef CAS PubMed.
- R. T. Moon, A. D. Kohn, G. V. D. Ferrari and A. Kaykas, Nat. Rev. Genet., 2004, 5, 691 CrossRef CAS PubMed.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 390 CrossRef CAS.
- M. P. Thomas and B. V. L. Potter, J. Med. Chem., 2015, 58, 7364 CrossRef PubMed.
- T. Reuillon, A. Bertoli, R. J. Griffin, D. C. Miller and B. T. Golding, Org. Biomol. Chem., 2012, 10, 7610 CAS.
- Y. Liu, I. F. F. Lien, S. Ruttgaizer, P. Dove and S. D. Taylor, Org. Lett., 2004, 6, 209 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc03612g |
| ‡ Deceased 24 September 2014. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a