
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rationally designed metal-binding helical peptoid for selective recognition processes†
Maria
Baskin
and
Galia
Maayan
*
Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Technion City, Hailfa 32000, Israel. E-mail: gm92@tx.technion.ac.il
First published on 8th January 2016
Abstract
Metal-binding biopolymers play a significant role in processes, such as regulation, recognition and catalysis, due to their high affinity towards specific metal ions, which they bind selectively from the cellular pool. Many enzymes can bind two or more metal ions, each at a specific binding site, to enable efficient cooperative function. Imitating these recognition abilities might lead to the production of biomimetic materials such as unique chelators and catalysts. Herein, we report a rationally designed helical peptoid bearing two distinct metal binding ligands at positions i and i + 3 (Helix HQT i + 3), which enables the selective recognition of one or two metal ions depending on its environment. Using various spectroscopic techniques, we describe (1) the selective intramolecular binding of Cu2+ and its extraction from a mixture of neighboring metal ions in high concentrations, and (2) the selective intermolecular binding of two different metal ions, including the pair Cu2+ and Zn2+, one at each binding site, for the generation of hetero-bimetallic peptoid duplexes. Thorough analysis and comparison between the spectroscopic data and association constants of the metal complexes formed by Helix HQT i + 3 and those formed by non-helical peptoids, or helical peptoids in which the two metal binding ligands are not pre-organized, revealed that the unique recognition processes performed by Helix HQT i + 3 are controlled by both the sequence and the structure of the peptoid.
Introduction
Metal ions are key elements in the structure and function of natural biopolymers, being employed in tasks spanning from signal transduction to electron transfer and catalysis. Biopolymers capable of binding metal ions exhibit high affinity and especially high selectivity towards specific metal ions that are required for their utility. Many metalloproteins found in nature have multiple coordination sites suitable for binding of at least two different metal ions, each at a distinct site, thus enabling cooperative and challenging catalytic tasks. Therefore, an important goal in the designing of metal-binding proteins, peptides and peptide mimics is to understand how to generate structures that selectively bind different metal ions in distinct sites.1 We note here that recognition processes for the metal ions Cu, Zn and Fe are especially interesting because these metals, being the three most abundant trace elements in biological and ecological systems, play central roles in the structure and function of proteins and all are essential components in many enzymes. Moreover, because there are only a few metal-binding side chains available for natural biopolymers, the high selectivity towards specific metal ion(s) is achieved by their 3-D structure (the folds), which controls intramolecular vs. intermolecular binding as well as steric and electronic effects. Mimicking such hetero bimetallic motifs by synthetic oligomers that can also fold might lead to unique cooperative catalysts in which each metal ion has a distinct role in the overall process towards high efficiency and selectivity.In recent years, chemists started to explore possibilities to imitate these unique recognition properties, mostly by developing biomimetic foldamers2 capable of binding metal ions.3–5 Peptoids, N-substituted glycine oligomers, are peptidomimetics capable of forming stable helical structures that resemble the polyproline type helices if chiral bulky side groups are incorporated within their backbone.6 Peptoids can be easily synthesized on solid support using the “submonomer” method,7 which employs primary amines and thus does not require protection and de-protection steps and which enables the incorporation of innumerable functional groups at specified N-positions along their spine. Moreover, peptoids are biocompatible8 and both their sequences and secondary structures exhibit high stability.9 These features, specifically the great versatility of the peptoid backbone, which can be easily modified, thus have the potential to include various ligands for selective coordination of different metal ions and/or for the creation of different types of complexes, together with their ability to form secondary structures as an additional tool for controlling recognition, make them excellent candidates to carry out selective recognition processes.
Although peptoids research is generally well developed, its extension to metal coordination is still very limited. Currently, there are only a few examples of metallopeptoids, including, among others, a peptoid that mimics the zinc finger motif,5a peptoid chelators selective for Cr6+ or Cd2+,5g,l and peptoid catalysts bearing a phenanthroline ligand for the binding of Cu+.10 These examples demonstrate that metal-binding peptoids are not only excellent candidates for mimicking biological structural motifs, but they also exhibit a great potential for applications such as selective extraction of metal ions from various media and biomimetic cooperative catalysis. Despite these advances, the number of such examples is still very insufficient, and they are all currently limited to the binding of one metal ion per peptoid chelator. Moreover, the relationship between the peptoids sequence and their metal coordination properties has been scarcely explored.5a Herein, we describe for the first time, the rational design of a peptoid oligomer that can mimic the recognition ability of biopolymers by (i) selectively binding Cu2+ from a mixture of various neighboring metal ions in higher concentrations and (ii) binding two different metal ions selectively and simultaneously in two distinct coordination sites. We also demonstrate that these unique recognition capabilities are controlled by both the structure and sequence of the peptoid.
Results and discussion
Peptoid design for selective Cu2+, Cu2+/Zn2+ and Cu2+/Fe3+ recognition processes
It was previously shown that the pre-organization of two 8-hydroxyquinoline (HQ) ligands at positions i and i + 3 of a helical peptoid hexamer (H26), such that they face the same side of the helix, led to the exclusive intramolecular coordination of Cu2+ with very high affinity (K > 1014 mol−1 in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 4:1 (ref. 5a) and 1.43 ± 0.46 × 1012 mol−1 in MeOH:H2O 1:5 (ref. 11)). However, the intermolecular binding of two metal ions to form the peptoid duplex was not possible, probably because this product is less thermodynamically favored than the intramolecular complex, especially when the coordinative ligands are both identical and pre-organized. To control and study the recognition properties of metal-binding helical peptoids, we sought to start by replacing one HQ ligand in H26 with a different ligand targeting a peptoid that includes two distinct metal binding sites at positions i and i + 3, enabling metal coordination in both intramolecular and intermolecular modes. For this purpose, we chose 2,2′:6′,2′′-terpyridine (Terpy) as the second ligand, resulting in the design of peptoid Helix HQT i + 3 (Scheme 1). We assumed that this peptoid would have high affinity and high selectivity for Cu2+via intramolecular binding to both HQ and Terpy because such coordination can lead to a square pyramid geometry, which can be stabilized by Cu2+ but not by neighboring metal ions, such as Zn2+, Fe3+, Co2+, Ni2+ and Mn2+, that can also bind these ligands.12 Moreover, as this peptoid includes two distinct binding sites, we anticipated that intermolecular coordination of two different metal ions would be also possible, probably under modified reaction conditions. Such binding will lead to the production of unique hetero-bimetallic peptoid duplexes in which Cu2+ is bound to two HQ ligands, stabilizing a tetragonal geometry, and a second metal ion, e.g. Zn2+, Fe3+ or Co2+, is bound to two Terpy ligands, stabilizing an octahedral geometry (see Scheme 1).12c,d
:H2O 4:1 (ref. 5a) and 1.43 ± 0.46 × 1012 mol−1 in MeOH:H2O 1:5 (ref. 11)). However, the intermolecular binding of two metal ions to form the peptoid duplex was not possible, probably because this product is less thermodynamically favored than the intramolecular complex, especially when the coordinative ligands are both identical and pre-organized. To control and study the recognition properties of metal-binding helical peptoids, we sought to start by replacing one HQ ligand in H26 with a different ligand targeting a peptoid that includes two distinct metal binding sites at positions i and i + 3, enabling metal coordination in both intramolecular and intermolecular modes. For this purpose, we chose 2,2′:6′,2′′-terpyridine (Terpy) as the second ligand, resulting in the design of peptoid Helix HQT i + 3 (Scheme 1). We assumed that this peptoid would have high affinity and high selectivity for Cu2+via intramolecular binding to both HQ and Terpy because such coordination can lead to a square pyramid geometry, which can be stabilized by Cu2+ but not by neighboring metal ions, such as Zn2+, Fe3+, Co2+, Ni2+ and Mn2+, that can also bind these ligands.12 Moreover, as this peptoid includes two distinct binding sites, we anticipated that intermolecular coordination of two different metal ions would be also possible, probably under modified reaction conditions. Such binding will lead to the production of unique hetero-bimetallic peptoid duplexes in which Cu2+ is bound to two HQ ligands, stabilizing a tetragonal geometry, and a second metal ion, e.g. Zn2+, Fe3+ or Co2+, is bound to two Terpy ligands, stabilizing an octahedral geometry (see Scheme 1).12c,d
 | ||
| Scheme 1 A rational design of a helical peptoid oligomer capable of intramolecular binding of one metal ion or intermolecular binding of two different metal ions in a selective manner. | ||
Synthesis and characterization of the peptoid Helix HQT i + 3 and its Cu2+ complex
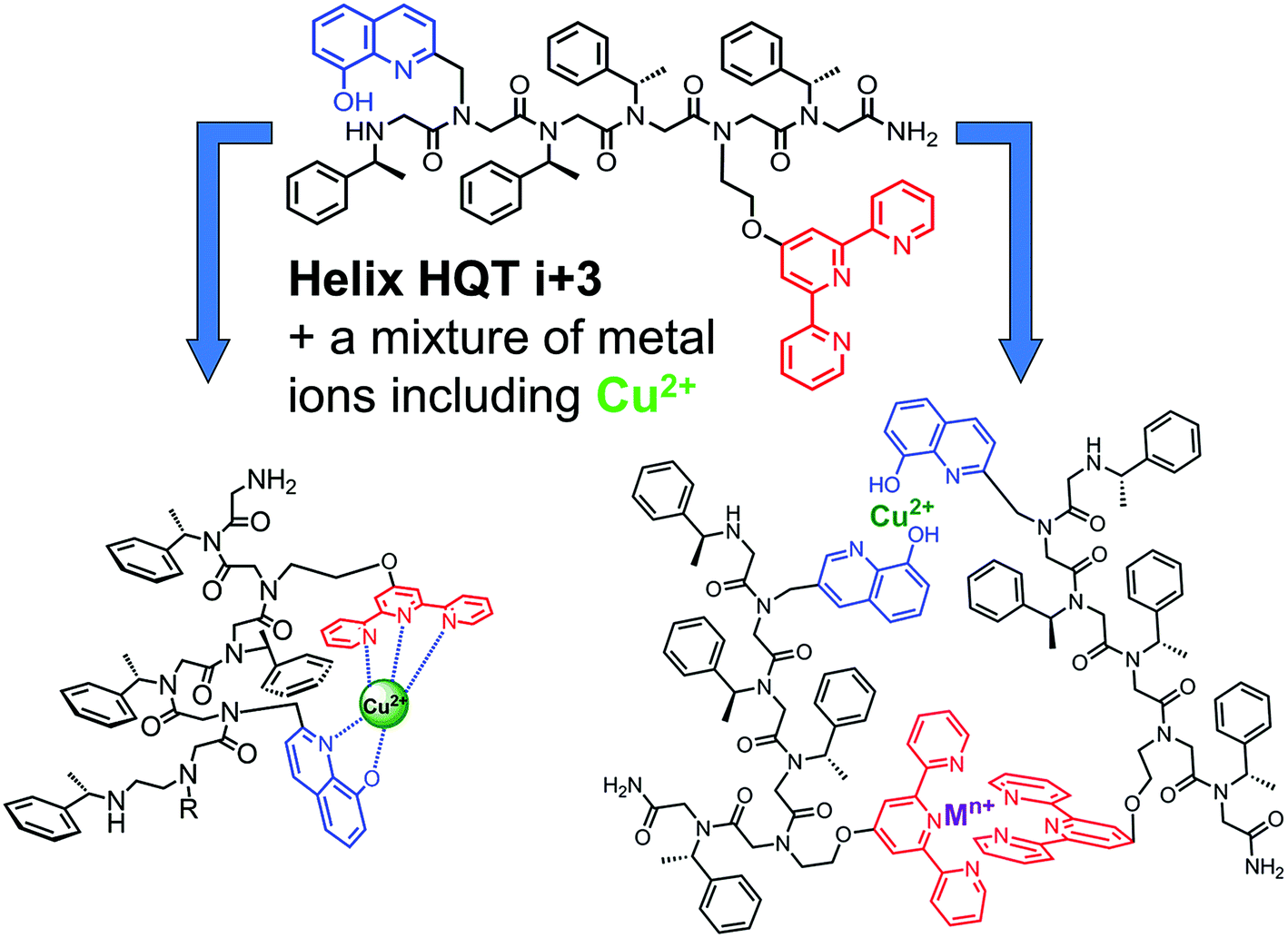
The peptoid Helix HQT i + 3 was synthesized on Rink amide resin using a previously reported variation of the peptoid submonomer protocol,13 cleaved from the solid support and purified by HPLC (>99% purity). The molecular weight measured by electrospray ionization mass spectrometry (ESI MS) was consistent with the expected mass. Metal-free peptoid Helix HQT i + 3 exhibits absorption bands near λ = 245 and 278 nm, in MeOH:H2O 4:1, arising from the ligands HQ and Terpy, respectively. Upon addition of copper acetate, these two bands diminished simultaneously and new absorption bands near λ = 259, 316 and 328 nm were produced (Fig. 1A). A peptoid-to-metal ratio plot constructed from these UV-Vis titrations was consistent with a 1:1 peptoid:Cu ratio, demonstrating the formation of the intramolecular complex (Helix HQT i + 3)Cu (Fig. 1A, inset). The peptoid-to-metal ratio was verified by ESI MS and the isotopic analysis showed no evidence for the formation of higher order complexes (e.g. 2:2 complexes, see ESI†).
 | ||
| Fig. 1 (A) UV-Vis spectra and peptoid-to-metal ratio plot for the titration of Helix HQT i + 3 with Cu2+. The peptoid (17 μM) in MeOH:H2O 4:1 solution was titrated with 2 μL aliquots of a metal ion (5 mM in H2O) in multiple steps (red = free ligand, black = metal complex). (B) Non-linear regression fit and UV-Vis spectra for the titration of Helix HQT i + 3 with Cu2+. The peptoid (7 μM) in MeOH:H2O 1:5 solution was titrated with 2 μL aliquots of a metal ion (2 mM in H2O) in multiple steps (red = free ligand, black = metal complex). (C) CD spectra of Helix HQT i + 3 (100 μM in MeOH:H2O 4:1) before (red) and after (green) the addition of 1 equivalent Cu2+. | ||
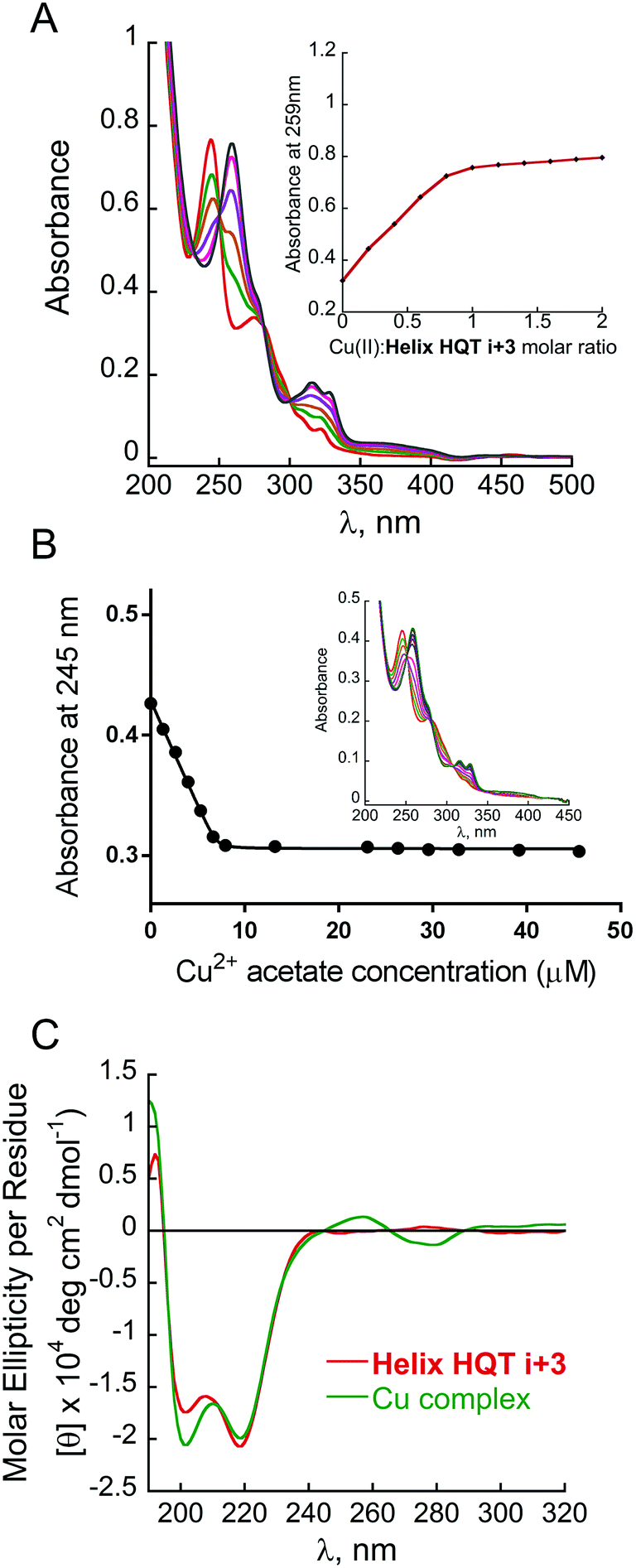
The association constant of (Helix HQT i + 3)Cu was calculated by a nonlinear regression curve fitting obtained from UV titration experiments at lower concentrations in MeOH:H2O 1:5 (Fig. 1B). The value for the formation of (Helix HQT i + 3)Cu is K = 1.03 ± 0.49 × 1013 M−1. This value reflects a strong binding affinity, which is higher by one order of magnitude than that of H26Cu in the same titration conditions, thus supporting our design principles. The circular dichroism (CD) spectrum of Helix HQT i + 3 in aqueous methanol showed double minima near λ = 202 and 220 nm, which is characteristic of a peptoid helix6c,d (Fig. 1C, red line). Adding 1 equivalent of Cu2+ to the CD cuvette produced exciton couplet CD peaks between 240 and 300 nm, the region corresponding to the 8-hydroxyquinoline π–π* transition, with a maximum at 257 nm and a minimum at 279 nm, crossing ε = 0 near 265 nm.5b,k EPR measurements of the solid (Helix HQT i + 3)Cu complex indicated the presence of Cu2+ (Fig. 2) and the Hamiltonian parameters obtained from the simulated spectra were g∥ = 2.23, g⊥ = 2.070 and A∥ = 175 G, which are consistent with a square pyramidal coordination geometry.14
 | ||
| Fig. 2 X-band EPR spectrum of (Helix HQT i + 3)Cu (blue line) and its corresponding simulated spectrum (red line). The measurements were performed in the solid state at rt., with TEMPO as a reference (g = 2.0058). | ||
Selective recognition of Cu2+ by Helix HQT i + 3
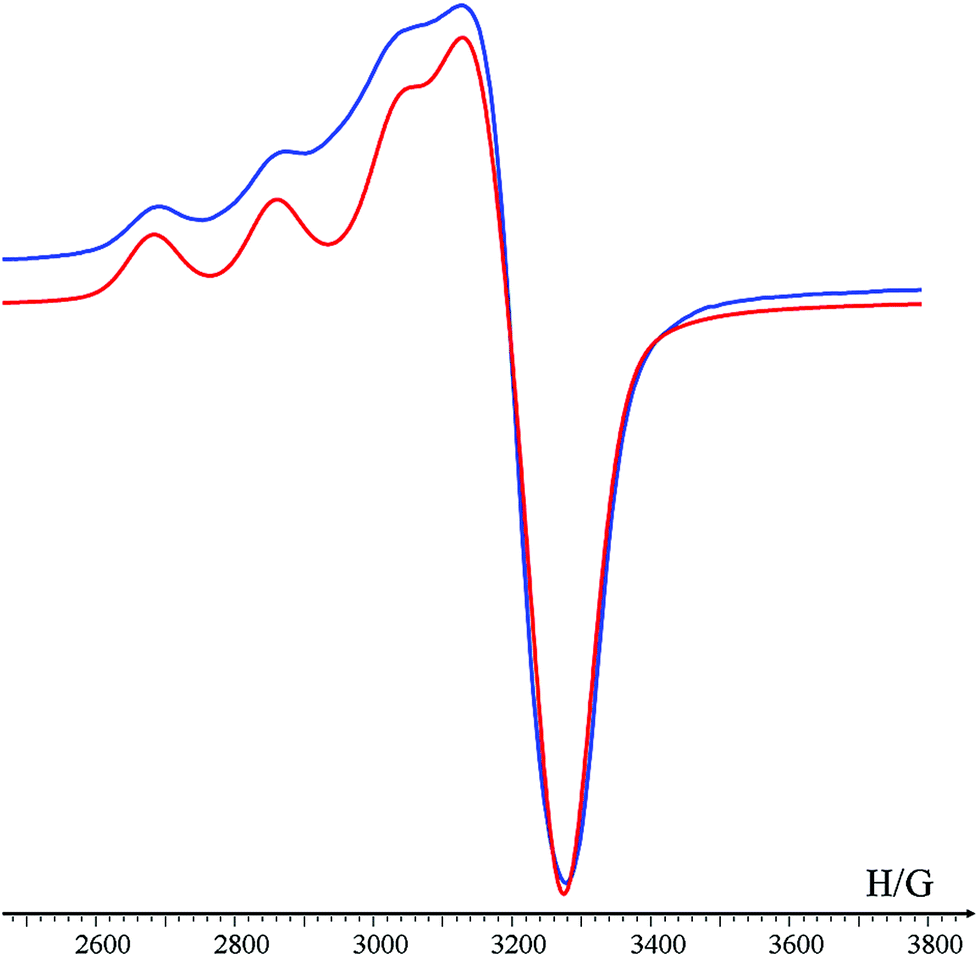
The initial selectivity assessment of Helix HQT i + 3 towards Cu2+ was conducted by treating the peptoid with a 1 equivalent mixture solution containing the metal ions Cu2+, Co2+, Zn2+, Fe3+, Mn2+ and Ni2+ in MeOH:H2O 4:1. Interestingly, the UV spectrum of this solution was identical to the UV spectrum of (Helix HQT i + 3)Cu, suggesting that this peptoid exhibits high selectivity towards Cu2+ from the tested mixture. ESI MS analysis of this mixture solution displayed only the mass of the Cu complex, supporting the selective binding of Cu2+ from the mixture solution (Fig. S77†). For comparison, the formation of (Helix HQT i + 3)M (M = Co2+, Zn2+, Fe3+, Mn2+ or Ni2+) was characterized by UV-Vis titrations and by ESI MS under the same reaction conditions. The peptoid-to-metal ratio plots revealed 1:1 intramolecular binding with all the metal ions except with Fe3+, in which the binding was intermolecular, with distinct absorption bands for each metal complex; the ESI MS analysis reflected the mass of each complex (see ESI†). These results support the observation that the UV-Vis and ESI MS spectra corresponding to the reaction of Helix HQT i + 3 with the mixture solution produced only the Cu2+ complex. To further evaluate the selectivity of Helix HQT i + 3 towards Cu2+, we tested its binding in mixtures containing higher concentrations of the different metal ions relative to Cu2+, i.e. 1 equivalent Cu2+ and up to 20 equivalents of Co2+, Zn2+, Fe3+, Mn2+ and Ni2+ mixture, using UV-Vis and ESI-MS. The obtained UV-Vis spectra were identical to the spectrum of (Helix HQT i + 3)Cu in all the examined ratios (see Fig. 3A for the ratio 1:10) and the ESI MS spectra demonstrated exclusively the mass of (Helix HQT i + 3)Cu.
 | ||
| Fig. 3 Selective binding of Cu2+ in aqueous methanol: (A) UV-Vis spectra of Helix HQT i + 3 (red line), its Cu complex (green line) and the complex formed from a mixture solution of 1:10 Cu2+:other metal ions (blue line). (B) Association constants for the formation of (Helix HQT i + 3)M complexes. | ||
To quantify these results, we calculated the association constants of Helix HQT i + 3 with each metal ion (Fig. 3B).15 The values for the formation of (Helix HQT i + 3)M (M = Zn2+, Co2+, Ni2+ and Mn2+) were K = 3.60 ± 0.46 × 1011, 2.53 ± 0.35 × 1011, 1.52 ± 0.09 × 1010 and 1.13 ± 0.06 × 1010 M−1, respectively. These results clearly demonstrate that Cu2+ coordination is at least one order of magnitude higher than that of the other metal ions, consistent with the high selectivity observed when binding from the mixture solutions. According to these data, selective binding of Cu2+ can occur from solutions containing about 27, 40, 670 and 885 times higher concentrations of Zn2+, Co2+, Ni2+ and Mn2+, respectively. To validate these findings, we obtained the UV spectra of solutions containing a mixture of 1 equivalent of Cu2+ and 22–27 equivalent of Zn2+, as well as solutions containing a mixture of 1 equivalent Cu2+ and 35–40 equivalent of Co2+ (see ESI†). The results indicated that selectivity for Cu2+ is retained in solutions having 25 equivalent of Zn2+ and 35 equivalent of Co2+, which in both cases is consistent with the calculated binding constants.
Finally, we were interested in investigating whether the selectivity for Cu2+ is thermodynamically or kinetically driven. Thus, we decided to examine the ability of Cu2+ to replace any other metal ion pre-bound to Helix HQT i + 3. For this purpose, the complexes (Helix HQT i + 3)M (M = Co2+, Zn2+, Mn2+ or Ni2+) in a UV cuvette were treated with 1 equivalent of Cu2+ in aqueous methanol and the UV spectra were obtained before and after the addition of Cu2+. The final UV-Vis spectra revealed that Cu2+ was able to replace the metal ions Zn2+ and Mn2+, forming the thermodynamically stable complex (Helix HQT i + 3)Cu2+, whereas the complexes (Helix HQT i + 3)Co2+ and (Helix HQT i + 3)Ni2+ remained intact (Fig. S17–20†). From this experiment, we can propose that the selectivity for Cu2+ is thermodynamically driven with regard to Zn2+ and Mn2+, and kinetically driven with regard to Co2+ and Ni2+.
Extraction of Cu2+ from a mixture solution by helix HQT i + 3
One immediate application for this selective recognition of Cu2+ is its extraction from a mixed metal ions solution. Selective extraction of metal ions, and specifically of Cu2+, is important for processes such as metal ion(s) overload that is toxic to living cells,16 Cu2+ contamination in cell cultures, and chemical reactions that involve several metal reagents or catalysts, which require the removal of Cu2+ at any stage of the reaction. Reports about chelators that show high selectivity for Cu2+ (ref. 17) focus on its detection in cells with the aim of studying its mechanism of action, rather than focusing on its extraction from various media. Recently, several peptoid chelators were described, demonstrating the removal of Cr6+ from aqueous media5g and of Cd2+ from biological media,5l but not of Cu2+. These peptoids were identified using combinatorial peptoid libraries rather than via rational design, which is in the heart of this study.The selective extraction of Cu2+ from a mixture solution containing 1 equivalent of Cu2+ and 10 equivalents of Co2+, Zn2+, Mn2+ and Ni2+ was estimated by inductively coupled plasma (ICP) measurements.18 Following the reaction of Helix HQT i + 3 with the mixture solution in aqueous methanol, the solvent was removed, the solid residue was re-dissolved in water and the obtained precipitate was separated from the solution by centrifugation. ICP analysis of the precipitated metallopeptoid revealed the exclusive presence of Cu with negligible amounts of the other metals (Fig. 4A). ICP analysis of the filtrate showed high concentrations of Co, Zn, Mn and Ni together with an insignificant amount of Cu (Fig. 4B). The ICP experiments demonstrate the biomimicry of Helix HQT i + 3 being able not only to select a specific metal ion from a mixture containing high concentrations of neighboring metal ions, but also to remove it from this mixture solution, leaving the other metal ions intact, even when it is present in much smaller concentrations than the other metal ions.
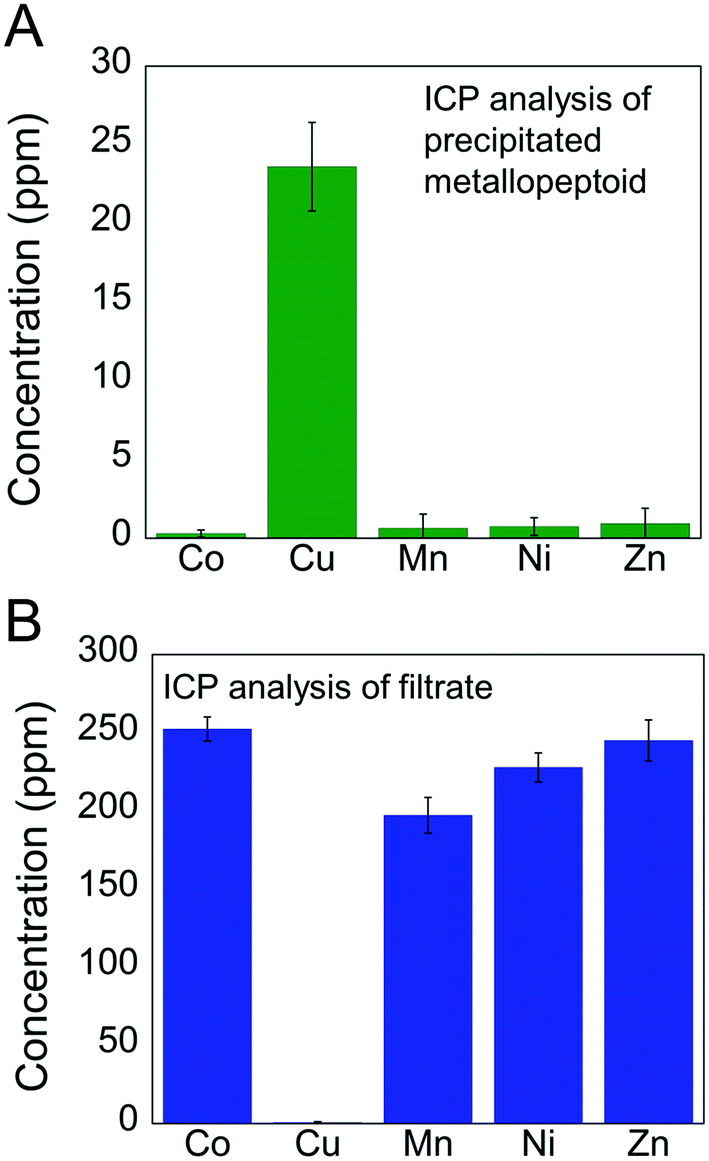 | ||
| Fig. 4 Selective extraction of Cu2+: (A) ICP analysis of the precipitate and (B) of the filtrate obtained from the reaction of Helix HQT i + 3 with a mixture of Cu2+ (1 equivalent, 0.5 mM) and the metal ions Co2+, Mn2+, Ni2+ and Zn2+ (10 equivalent, 5 mM each). Standard errors are represented by error bars, with experiments number = 3. | ||
The role of peptoid helicity and ligands pre-organization in the selective recognition of Cu2+
Furthermore, we wished to explore whether the high affinity and selectivity of Helix HQT i + 3 towards Cu2+ arises from its secondary structure and/or from the pre-organization of HQ and Terpy at positions i and i + 3. For this purpose, four control peptoids were synthesized and purified: the unstructured hexamer Nonhelix HQT i + 3, containing HQ and Terpy at positions i and i + 3 in addition to the non-structure directing groups benzyl and methoxyethyl in the other positions along the backbone; the dimer DI HQT, having only the two ligands HQ and Terpy; and the two helical hexamers Helix HQT i + 2 and Helix HQT i + 4 in which HQ and Terpy are not pre-organized to face the same side of helix (Fig. 5A). The association constants of the control peptoids with Cu2+ were calculated under the same conditions as described above and the values obtained for the formation of (Nonhelix HQT i + 3)Cu, (DI HQT)Cu, (Helix HQT i + 2)Cu and (Helix HQT i + 4)Cu were K = 1.16 ± 0.53 × 1012, 1.36 ± 0.66 × 1012, 1.50 ± 0.37 × 1012 and 4.12 ± 0.84 × 1011 M−1, respectively (Fig. 5B). All these values were about one order of magnitude smaller compared with the value calculated for (Helix HQT i + 3)Cu, suggesting that both the helicity and the ligands' pre-organization play a role in the high selectivity of Helix HQT i + 3 towards the binding of Cu2+.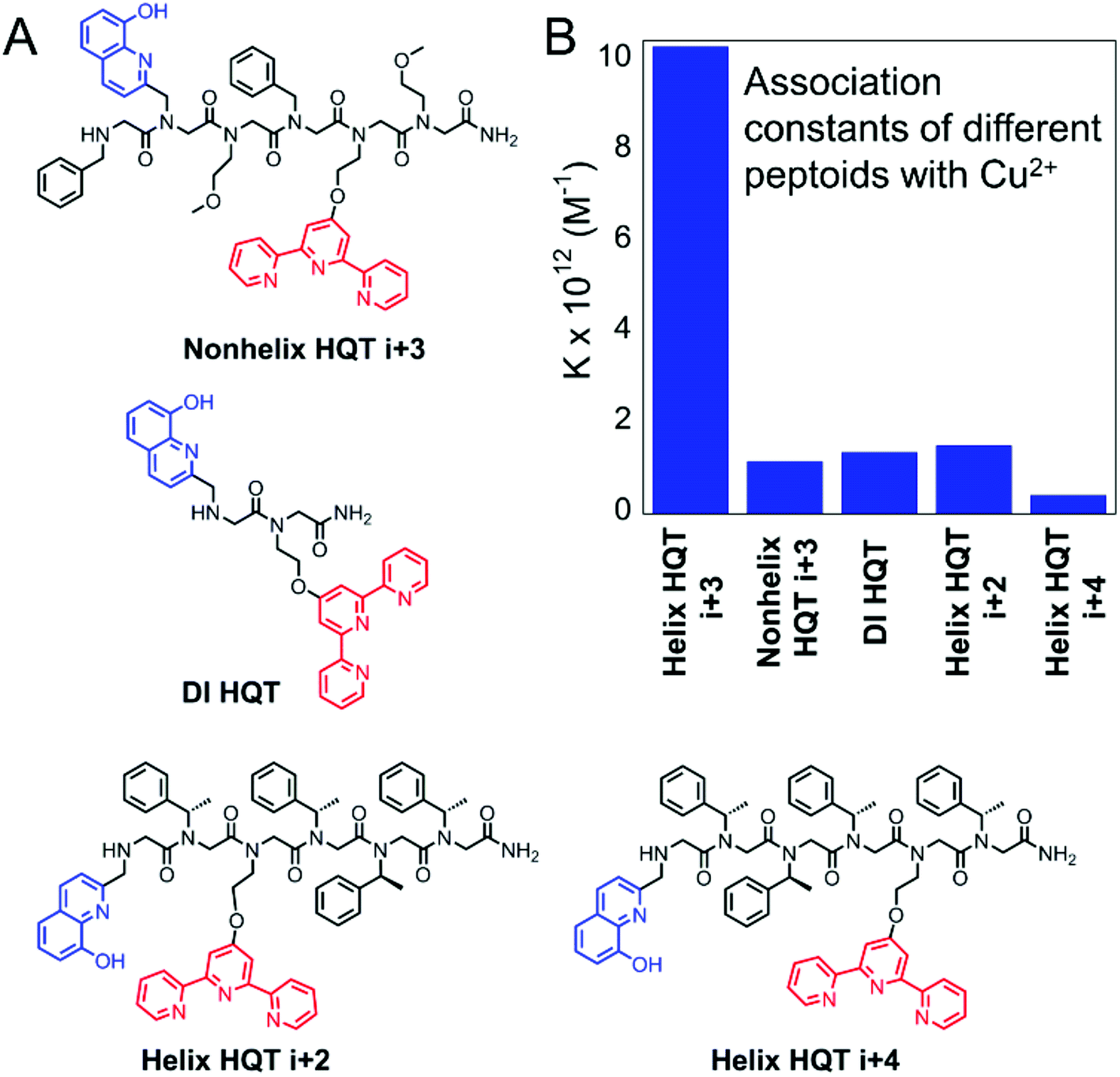 | ||
| Fig. 5 Sequences of the control peptoids (A) and their association constants for the formation of their corresponding Cu complexes (B). | ||
EPR measurements, conducted in the solid state at rt., indicated the presence of Cu2+ in all four complexes. The values of the hyperfine splitting for (Nonhelix HQT i + 3)Cu, (DI HQT)Cu, (Helix HQT i + 2)Cu and (Helix HQT i + 4)Cu were 162 G, 166 G, 150 G and 158 G, respectively (Table 1). All these values are lower than the value for (Helix HQT i + 3)Cu (175 G), implying that some (or all) of the control peptoid complexes exhibit tetragonal geometries.1 To probe this point, we conducted an EPR measurement for H26, which has two HQ ligands and was shown to bind Cu2+ in a pseudotetrahedral geometry.5b The measurement, which was done in the same conditions as with the other (peptoid)Cu complexes, provided a value of 150 G (Table 1), which is identical to that of (Helix HQT i + 2)Cu and close to the hyperfine splitting measured for (Helix HQT i + 4)Cu. It was previously suggested that the trans-planar configuration of Cu(hydroxyquinolate)2 is not possible in the intramolecular (H26)Cu complex because of the steric interactions arising from its helical structure that prohibit a square planar geometry.5b We therefore suggest that in the case of the sterically hindered helices (Helix HQT i + 2) and (Helix HQT i + 4), in which HQ and Terpy are not pre-organized, their Cu2+ complexes adopt tetragonal coordination geometries (i.e. by binding to only four out of the five N- and O-coordination atoms) because their hyperfine splitting values resemble the value of the pseudotetrahedral (H26)Cu complex, and therefore they are less selective towards Cu2+.
| Complex/EPR data | A ∥[G] | g ⊥ | g ∥ |
|---|---|---|---|
| a All measurements were performed in the solid state at rt., with TEMPO as a reference (g = 2.0058). | |||
| (Helix HQT i + 3)Cu | 175 | 2.070 | 2.23 |
| (Nonhelix HQT i + 3)Cu | 162 | 2.070 | 2.23 |
| (DI HQT)Cu | 166 | 2.070 | 2.21 |
| (Helix HQT i + 2)Cu | 150 | 2.070 | 2.25 |
| (Helix HQT i + 4)Cu | 158 | 2.070 | 2.25 |
| (H26)Cu | 150 | 2.071 | 2.25 |
To evaluate the selectivity of the four peptoids towards Cu2+ from a mixture solution containing the metal ions Cu2+, Co2+, Zn2+, Fe3+, Mn2+ and Ni2+, 1 equivalent of that mixture in MeOH:H2O 4:1 was added to each one of these four peptoids and the UV-Vis spectra were obtained. The obtained four UV-Vis spectra were different from the spectra of their copper complexes, suggesting that there is no selective recognition of Cu2+ by these four peptoids. These results were further proved by ESI MS analysis of these solutions, which demonstrated the formation of other metal complexes in addition to (Helix HQT i + 3)Cu; for example, the ESI MS spectrum of Nonhelix HQT i + 3 showed the mass of a Co2+ complex in addition to the Cu2+ complex, and the spectrum of DI HQT showed the mass of a Zn2+ complex in addition to the Cu2+ complex. Moreover, calculating the association constants of these four control peptoids with each metal ion from the mixture15 showed that the values for the formation of the complexes with Cu2+, Co2+ and Zn2+ are all in the same order of magnitude, supporting the low selectivity for Cu2+ (Table S4†). Overall, our observations support the biomimetic character of our system, demonstrating that control over both the helical structure and the pre-organization of HQ and Terpy is crucial for recognition.
Selective recognition of two different metal ions by helix HQT i + 3
According to the abovementioned results, only intramolecular binding of all the tested metal ions (excluding Fe3+) by Helix HQT i + 3 can be achieved in aqueous methanol. One possible explanation could be that both methanol and water are able to coordinate as an additional ligand to the metal ions that do not stabilize the square pyramid geometry (e.g. cobalt ions), forming other stable penta- or hexa-coordinated complexes (e.g. octahedral geometry). Thus, the selective intermolecular binding of two different metal ions in distinct binding sites is impossible in these conditions. To enable such intermolecular binding, we sought to conduct the binding experiments in a different solvent that is less coordinative than water and/or methanol such as acetonitrile.19 Indeed, adding 1 equivalent mixture solution containing the metal ions Cu2+, Co2+, Zn2+, Fe3+, Mn2+ and Ni2+ in acetonitrile to Helix HQT i + 3 produced a UV-Vis spectrum that was different from the spectrum of (Helix HQT i + 3)Cu (Fig. 6A), suggesting that the binding in acetonitrile is not selective towards Cu2+. This was further supported by ICP experiments showing low selectivity for Cu2+ in acetonitrile (Fig. 6B and C). In addition, to facilitate intermolecular binding, we thought to explore it using two different approaches: (i) the step approach, in which 1 equivalent of a metal ion other than Cu2+, e.g. Zn2+ or Fe3+, will be added to 2 equivalents of the peptoid, aiming for selective binding to two Terpy ligands, followed by the addition of Cu2+ to be bound to the two HQ ligands (kinetic control); and (ii) the mixture approach, in which the peptoid will be treated with a mixture solution containing 0.5 equivalent of Cu2+ and 0.5 equivalent of the other metal ion under some heating and/or longer reaction time (thermodynamic control), targeting simultaneous binding of the two ions: Cu2+ to two HQ ligands and the other ion to two Terpy ligands.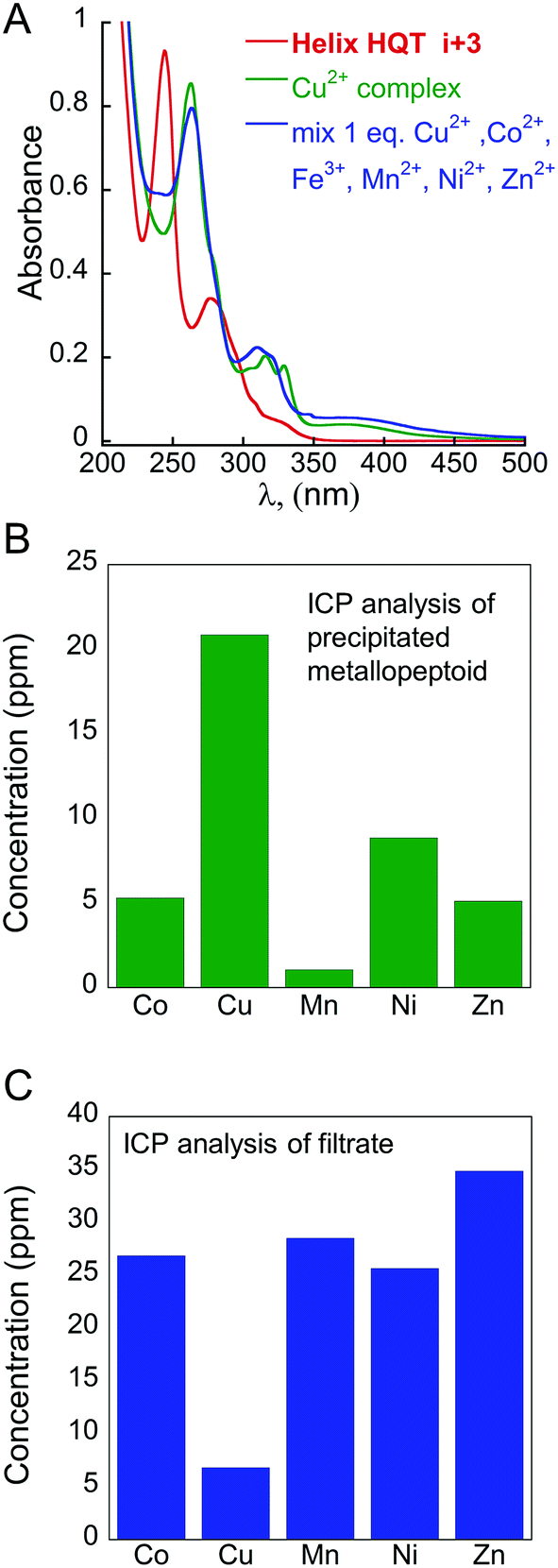 | ||
| Fig. 6 Non-selective binding of Cu2+ in acetonitrile: (A) UV-Vis spectra of Helix HQT i + 3 (red line), its Cu complex (green line) and the complex formed from a mixture solution of 1:1 Cu2+:other metal ions (blue line). (B) ICP analysis of the precipitate and (C) filtrate obtained from the reaction of Helix HQT i + 3 with a mixture of Cu2+ (1 equivalent, 1 mM) and the metal ions Co2+, Mn2+, Ni2+ and Zn2+ (1 equivalent, 1 mM each). | ||
Starting with the step approach, 1 equivalent of metal-free peptoid Helix HQT i + 3 in acetonitrile was treated with 0.5 equivalent of Co2+, Zn2+, Fe3+, Ni2+ or Mn2+ in a UV cuvette, and the changes in the absorption bands near λ = 245 and 278 nm of HQ and Terpy ligands, respectively, were recorded. Upon addition of Co2+, Zn2+ or Fe3+, the band near 278 nm disappeared and new absorption bands near λ = 307 nm, λ = 312 and 323 nm, and λ = 316 and 559 nm, respectively, were obtained (Fig. 7A–C, black and red lines). It can be noted that no change in the absorption band near λ = 245 nm was recorded, reflecting the exclusive binding of each of these three metal ions to Terpy and the formation of (Terpy)2M complexes (see ESI†).20 In contrast, upon addition of either Ni2+ or Mn2+, the absorbance bands of both Terpy and HQ disappeared simultaneously and new bands near λ = 270, 312 and 325 nm, and λ = 263, 312 and 323 nm, respectively, were obtained, reflecting the intramolecular binding of these ions and the formation of complexes of the type (Helix HQT i + 3)M. Subsequent addition of 0.5 equivalents of Cu2+ to each of the (Terpy)2M complexes (M = Co2+, Zn2+ or Fe3+) resulted in the disappearance of the band at 245 nm and the appearance of a new absorption band at λ = 266 nm, indicating the formation of (HQ)2Cu complex (Fig. 7A and B, green lines and 7C, blue line).
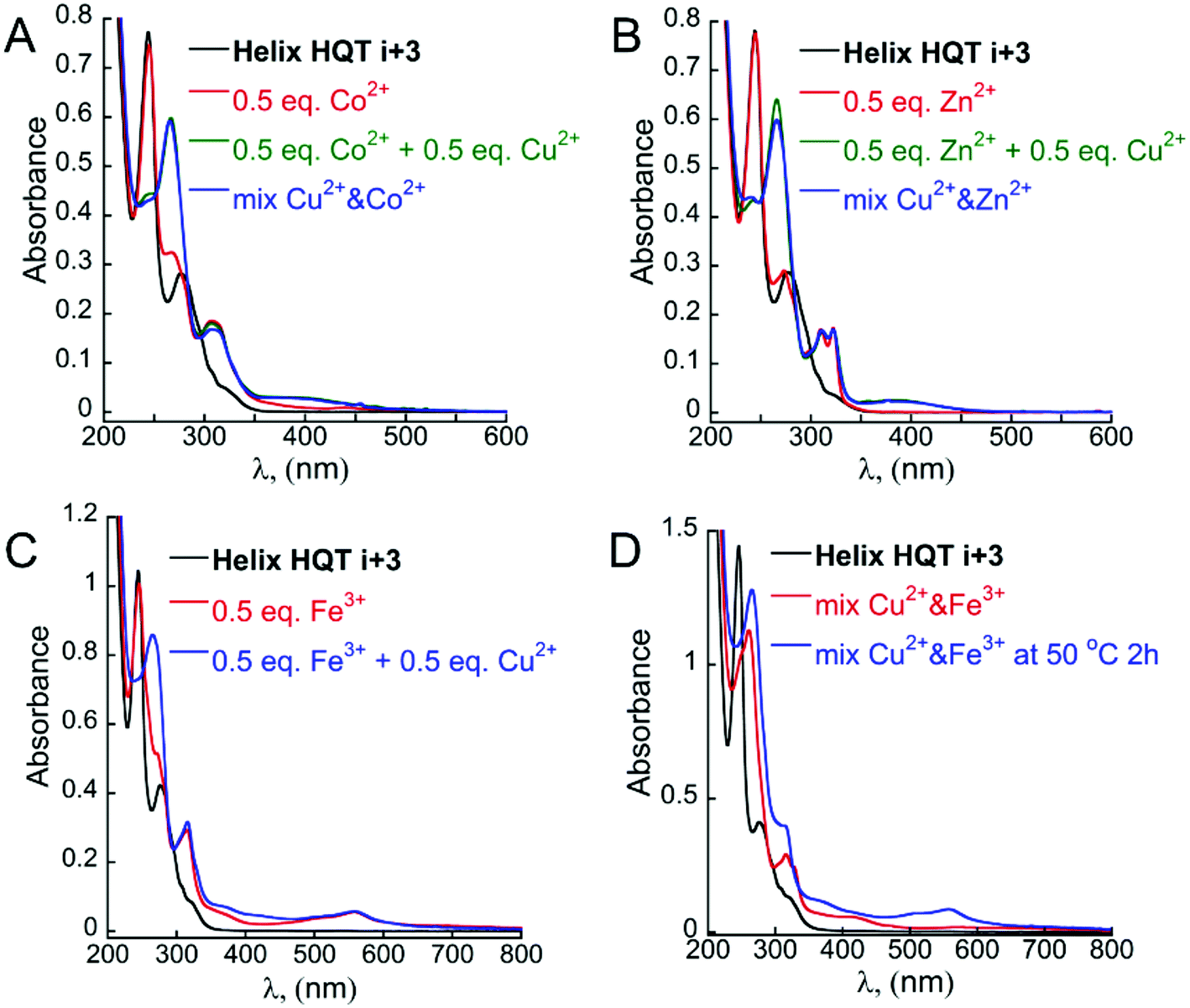 | ||
| Fig. 7 UV-Vis spectra of Helix HQT i + 3 (black line), its 2:1 peptoid:M Co (A), Zn (B) or Fe (C) complexes (red line) and its 2:2 peptoid:M complexes formed via the step approach (A and B green lines and C, blue line) or the mixture approach (A and B blue lines; D, red and blue lines). | ||
Overall, this approach enabled the formation of the heteronuclear bimetallic duplexes (Helix HQT i + 3)2MCu (M = Co2+, Zn2+ or Fe3+) as demonstrated by the UV-Vis spectra and supported by MS analysis (ESI†). The CD spectra of (Helix HQT i + 3)2MCu (M = Co2+, Zn2+ or Fe3+) in acetonitrile was similar to that of (Helix HQT i + 3)Cu that was measured in aqueous methanol, showing a double minima characteristic of a peptoid helix and exciton couplet CD peaks (Fig. S8†). EPR analysis of (Helix HQT i + 3)2ZnCu in the solid state at rt. clearly indicated the presence of Cu in this complex. The Hamiltonian parameters obtained from the simulated spectra were g∥ = 2.24, g⊥ = 2.065 and A∥ = 155 G. It can be noted that this hyperfine splitting resembles the one obtained for the tetragonal complex (H26)Cu (150 G, Table 1), supporting our observations from the UV-Vis spectra that copper is bound to the two HQ ligands of the hetero bimetallic complex (Helix HQT i + 3)2ZnCu.
Interestingly, titrating the acetonitrile solution of (Helix HQT i + 3)2ZnCu with additional aliquots of Cu2+ solution resulted in a gradual shift in the absorbance bands until the full disappearance of the bands at λ = 266, 312 and 323 nm, corresponding to the complex (Helix HQT i + 3)ZnCu, and the appearance of bands near λ = 262, 316 and 329 nm (Fig. 8), which indicated the formation of the complex (Helix HQT i + 3)Cu. These results demonstrate the biomimetic ability of Helix HQT i + 3 to modify its binding mode by adjusting it to changes in its environment (i.e. excess of one metal ion over another one). Similar titrations of (Helix HQT i + 3)2CoCu and (Helix HQT i + 3)2FeCu did not lead to any significant changes in their UV-Vis spectra, suggesting that these complexes are more stable as duplexes, probably due to the highly stable octahedral geometry that both ions form with 2 equivalents of Terpy.
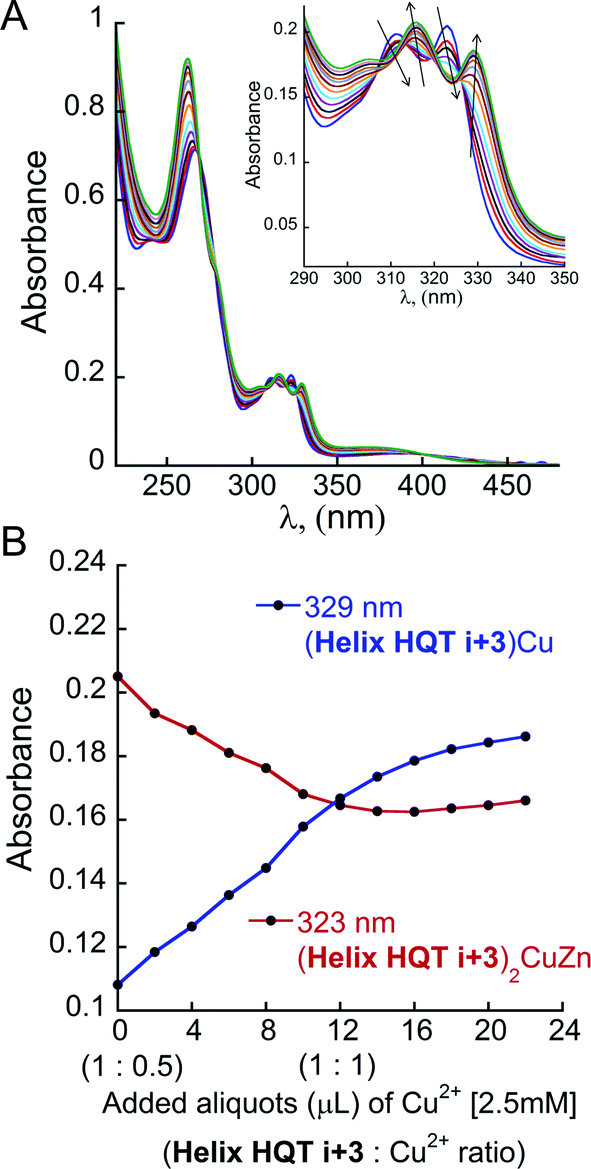 | ||
| Fig. 8 (A) UV-Vis titration of (Helix HQT i + 3)2ZnCu (blue line, 8 μM) with Cu2+ (2 μL aliquots of 2.5 mM) in 3 mL acetonitrile. Inset: the UV-Vis spectrum in the region between 290 and 350 nm. (B) Representation of the decrease in the absorbance of (Helix HQT i + 3)2ZnCu complex at λ = 323 nm (red line) and the simultaneous increase in the absorbance of (Helix HQT i + 3)Cu complex at λ = 329 nm (blue line) during the titration. | ||
Continuing with the mixture approach, 1 equivalent of metal-free peptoid Helix HQT i + 3 in acetonitrile was treated with a mixture containing 0.5 equivalents (from each ion) of (i) Co2+ and Cu2+, (ii) Zn2+ and Cu2+ or (iii) Fe3+ and Cu2+ in a UV cuvette. We then followed the changes in the absorption bands near λ = 245 and 278 nm of HQ and Terpy ligands, respectively. In the case of Co2+ and Cu2+, we were pleased to observe that both of these bands disappeared simultaneously, while two new bands near λ = 266 and 307 nm appeared, indicating the generation of the complexes (HQ)2Cu and (Terpy)2Co and the overall formation of the metallopeptoid duplex (Helix HQT i + 3)2CoCu (Fig. 7A, blue line). It can be noted that this spectrum was identical to the spectrum obtained when Co2+ and Cu2+ were added separately to Helix HQT i + 3 following the step approach. The existence of (Helix HQT i + 3)2CoCu was further confirmed by ESI-MS (ESI†). The same observations were obtained with the mixture solution of Zn2+ and Cu2+ (Fig. 7B, blue line), indicating that also in this case a metallopeptoid duplex binding the two different metal ions in distinct sites was formed, and the existence of (Helix HQT i + 3)2ZnCu was further confirmed by ESI-MS (ESI†). Using the mixture solution of Fe3+ and Cu2+, however, resulted in the appearance of absorbance bands near λ = 262, 316 and 328 nm, whereas the absorbance band near λ = 559 was missing (Fig. 7D, red line). In fact, this UV-Vis spectrum was identical to the one obtained upon the addition of only Cu2+ ion, indicating that no peptoid duplex was formed. These results suggest that (Helix HQT i + 3)Cu is both the kinetic and the thermodynamic product in these conditions. We therefore sought to thermodynamically control this transformation by stirring the reaction mixture (peptoid and metal ions) for extended periods of time, but unfortunately, no change in the UV-Vis spectrum was recorded even after 52 h. Only upon stirring the reaction mixture at 50 °C for 2 hours was a change in the solution color from colorless to light pink (indicative of a Terpy–Fe complex) observed, and the UV spectrum showed the disappearance of the bands near λ = 259 and 328 nm and the appearance of the bands near λ = 266 and 559 nm (Fig. 7D, blue line). These findings suggested that the peptoid duplex (Helix HQT i + 3)2FeCu can be formed under thermodynamic conditions, as was further supported by MS.
The role of peptoid helicity and ligands' pre-organization in the selective recognition of two different metal ions
Finally, we set up to determine whether the pre-organization of HQ and Terpy within the peptoid Helix HQT i + 3 also affects its unique binding with Co2+ or Zn2+, which enables the formation of heteronuclear metallopeptoid duplexes at rt. For this purpose, we have tested the ability of the four control peptoids (Fig. 5A) to bind two metal ions, Cu2+ and either Co2+ or Zn2+, using UV-Vis spectroscopy. Starting with the step approach, we have noticed that upon addition of 0.5 equivalent of either Co2+ or Zn2+ to each of the control peptoids, both Terpy and HQ absorption bands were changed, indicating that the metal ions bind simultaneously to both HQ and Terpy towards the formation of intramolecular complexes (see ESI†). These results demonstrated that unlike Helix HQT i + 3, the control peptoids are not able to selectively bind Co2+ or Zn2+ exclusively in the Terpy site; therefore, the step approach cannot be applied for generating (peptoid)2CuM complexes. Therefore, we conducted UV-Vis experiments using mixtures of either Cu2+ and Zn2+ or Co2+ and Cu2+. Interestingly, in both cases, the addition of a solution containing 0.5 equivalent of each of Cu2+ and Zn2+ or Cu2+ and Co2+ to 1 equivalent of each peptoid resulted in a simultaneous but only partial decrease of the absorbance bands corresponding to both Terpy and HQ. Only upon treating these solutions with an additional portion of 0.5 equivalent of each of Cu2+ and Zn2+ or Cu2+ and Co2+, a full decrease in the absorbance bands corresponding to both Terpy and HQ was obtained (see ESI†). These results suggest that a mixture of products was generated with each of the control peptoids, most probably two different intramolecular (peptoid)M complexes. Thus, both the helicity and the pre-organization of Terpy and HQ are important factors also in the creation of the heteronuclear peptoid duplexes (peptoid)2CuM.Conclusions
In this study, we have demonstrated the rational design of a biomimetic oligomer peptoid, Helix HQT i + 3, which can perform the recognition processes as follows: (1) selective binding of Cu2+ ion and its extraction from a solution containing the ions Co2+, Zn2+, Fe3+, Mn2+ and Ni2+ in high concentrations, resulting in the intramolecular complex (Helix HQT i + 3)Cu; (2) selective binding of two different metal ions, each at a specific metal-binding site, from a mixture containing both ions, to generate the intermolecular bimetallic duplexes (Helix HQT i + 3)CuM (M = Co2+, Zn2+, and Fe3+); and (3) selective transition from an intermolecular to an intramolecular binding as the metal-binding peptoid adjusts its coordination properties to changes in its environment (i.e. excess of one metal ion over another one). The high selectivity in all these cases reflects the unique ability of this peptoid to mimic the recognition properties of metal-binding biopolymers. These properties, together with our current efforts to increase the water solubility of this peptoid and to design similar peptoids for selective binding of Zn2+, might enable various applications such as chelate therapy and selective catalysis.21Experimental
Materials
Rink amide resin was purchased from Novabiochem; trifluoroacetic acid (TFA), zinc acetate dehydrate and nickel acetate tetrahydrate were purchased from Alfa Aesar; 8-hydroxy-2-quinolinecarbonitrile, (S)-(−)-1-phenylethylamine (Nspe), 4′-chloro-2,2′:6′,2′′-terpyridine and manganese acetate tetrahydrate were purchased from Acros; bromoacetic acid, cobalt acetate tetrahydrate and copper acetate monohydrate were purchased from MERCK; N,N′-diisopropylcarbodiimide (DIC), piperidine, 2-methoxyethylamine, benzylamine, acetonitrile (ACN) and water and HPLC grade solvents were purchased from Sigma-Aldrich; iron perchlorate hydrate was purchased from Strem Chemicals; dimethylformamide (DMF) and methanol (MeOH) solvents were purchased from Bio-Lab Ltd. These reagents and solvents were used without additional purification. 2,2′:6′,2′′-Terpyridine amine (Nterpy) and 8-hydroxy-2-quinolinemethylamine (Nhq) were synthesized according to previously published procedures.13Synthesis and purification of the peptoid oligomers
Peptoid oligomers were synthesized manually at room temperature on Rink amide resin using a variation of a previously reported peptoid sub-monomer protocol.13 Peptoid synthesis was carried out with alternating bromoacylation and amine displacement steps until peptoid oligomers of the desired sequence were obtained. In our case, after incorporation of 8-hydroxy-2-quinolinemethylamine, 0.17 mL of a 1.2 M solution of bromoacetic acid, 0.04 mL of neat N,N′-diisopropylcarbodiimide (DIC) and 0.29 mL of DMF were added to the resin and mixed at room temperature for 20 minutes.13 When the desired sequence was achieved, the peptoid products were cleaved from the resin by treatment with 95% trifluoroacetic acid (TFA) in water (50 mL g−1 resin) for 30 minutes. After filtration, the cleavage mixture was concentrated by rotary evaporation under reduced pressure. Cleaved samples were then re-suspended in 50% acetonitrile in water and lyophilized to powders. Peptoids were purified by preparative High Performance Liquid Chromatography (HPLC) using a C18 column. Products were detected by UV absorbance at 230 nm during a linear gradient conducted from 5% to 95% solvent B (0.1% TFA in HPLC grade acetonitrile) over solvent A (0.1% TFA in HPLC grade water) in 50 minutes with a flow rate of 5 mL min−1. Purified peptoid oligomers were analyzed by reversed-phase HPLC (C18 column) with a linear gradient of 5–95% ACN in water (0.1% TFA) over 10 min at a flow rate of 700 μL min−1 and 214 nm of UV absorbance. Mass spectrometry of peptoid oligomers was performed on Waters LCT Premier mass spectrometer under electrospray ionization (ESI), direct probe ACN:H2O (70:30), flow rate 0.3 mL min−1 or on Advion expression CMS mass spectrometer under electrospray ionization (ESI), direct probe ACN:H2O (95:5), flow rate 0.2 mL min−1.
UV-Vis spectroscopy
Titration experiments of the peptoid Helix HQT i + 3 with the metal ions (Co2+, Cu2+, Ni2+, Mn2+, Zn2+, and Fe3+) were followed by UV-Vis in two different solvents. In a typical experiment, 10–15 μL of a peptoid solution (5 mM in MeOH or ACN) was diluted in 3 mL 4:1 MeOH:H2O or acetonitrile solution (to get 17–26 μM concentration) and then sequentially titrated with 2 μL aliquots of a metal ion (2.5 or 5 mM in H2O), in multiple steps, until the binding was completed. In the selectivity experiments, solutions containing mixtures of metal ions (1 equivalent of Cu2+ and 1–20 equivalents of Co2+, Zn2+, Mn2+, Ni2+ and Fe3+, which is of 10 μL of 5 mM and 2–40 μL of 25 mM, respectively) in 3 mL of 4:1 MeOH:H2O or acetonitrile were first measured as a blank. Then, peptoid Helix HQT i + 3 was added (10 μL, 5 mM) and the spectrum was measured again. UV measurements were performed using an Agilent Cary 60 UV-Vis spectrophotometer, a double beam, Czerny–Turner monochromator.
Synthesis of metal complexes for MS analysis
Samples for MS analysis were prepared shortly before measurements. In a typical experiment, a solution of peptoid oligomers (100–200 μL of 0.05 mM) in MeOH or ACN was treated with metal solution (5 mM in H2O or ACN) and the mixture was stirred for 30 minutes prior to MS analysis. Mass spectrometry analysis of the metal complexes was performed on a Waters LCT Premier mass spectrometer under electrospray ionization (ESI), direct probe ACN:H2O (70:30), flow rate 0.3 mL min−1.
Circular dichroism spectroscopy
Approximately, 500 μL solutions (5 mM in methanol or ACN) of lyophilized peptoids powders were prepared immediately before CD measurements. CD scans were performed at 25 °C at a concentration of 100 μM in a solution of methanol/water 4:1 or ACN. The spectra were obtained by averaging 4 scans per sample in a fused quartz cell (path length = 0.1 cm). Scans were performed over the 320 to 190 nm region using 50 nm min−1 scan rate. CD measurements were performed using a circular dichroism spectrometer Model Jasco 810 Spectropolarimeter and Applied Photophysics Chirascan.
Synthesis of Cu(II) complexes for EPR analysis
Copper complexes for EPR were prepared in methanol (0.4 mL) by addition of 1.2 equivalents of copper acetate to Helix HQT i + 3 (6.4 mg, 0.0053 mmol), Helix HQT i + 4 (6.1 mg, 0.005 mmol), Helix HQT i + 2 (4.2 mg, 0.0035 mmol), Nonhelix HQT i + 3 (5.8 mg, 0.0053 mmol) and DI HQT (4 mg, 0.007 mmol) and stirring the solution for 1 hour. A green solid was precipitated after the addition of NH4PF6 (0.08 mL of 1 M aqueous solution). The precipitates were isolated by centrifugation, washed twice with water and lyophilized overnight. (Helix i + 3)Cu was obtained in 76% yield (5.1 mg), (Helix HQT i + 4)Cu 79% yield (5.0 mg), (Helix HQT i + 2)Cu 81% yield (3.6 mg), (Nonhelix HQT i + 3)Cu 80% (4.9 mg), DI HQT-Cu 70% yield (3.1 mg). (Helix HQT i + 3)2ZnCu complex was synthesized by addition of 0.5 equivalents of Zn acetate in ACN (5 mM) to the solution of Helix HQT i + 3 in ACN (5 mg, 21 mM). The solution was stirred for 10 min and 0.5 equivalents of Cu in ACN (5 mM) was added and the mixture was stirred for 30 min. Then, 500 μL of water was added, the solvent was lyophilized overnight, the complex was washed twice with water and dried by lyophilization. Mass of the complex = 4.02 mg, yield: 72%. EPR spectra were obtained on a Bruker EMX-10/12 X-band (ν = 9.4 GHz) digital EPR spectrometer. All spectra of peptoid copper complexes were obtained at room temperature from solid state with (2,2,6,6-tetramethyl-1-piperidinyl)oxidanyl (TEMPO, g = 2.0059) in an inner tube for determination of the g-factor. Spectra processing and simulation were performed with a Bruker WIN-EPR and SimFonia Software.Inductively coupled plasma (ICP) experiments
To a solution of 1 equivalents of Cu2+ (0.5 mM in MeOH:H2O 4:1) and 10 equivalents of Co2+, Mn2+, Ni2+, Fe3+ and Zn2+ (5 mM each in MeOH:H2O 4:1), 6 mg of peptoid Helix HQT i + 3 were added and the mixture was allowed to shake for 30 min. The solvent was then evaporated and the water solution was lyophilized overnight. To the lyophilized powder, 1 mL of H2O was added; the mixture was shaken for 5 min and centrifuged 15 min to separate the solution and the precipitate. After the centrifugation, the water was removed and this process was repeated another 9 times. To the precipitated metallopeptoid, 0.2 mL of 69% nitric acid HNO3 was added, the mixture was mixed thoroughly for 30 minutes, followed by the addition of water to get 10 mL final volume. This experiment was done 3 times. When ACN was used as a solvent, 1 equivalent each of Cu2+, Co2+, Mn2+, Ni2+, Fe3+ and Zn2+ was added (1 mM each in ACN:H2O 17:3) to 5.6 mg of peptoid Helix HQT i + 3. All solutions were filtered by 0.2 μm filters prior to ICP analysis by a Thermo Scientific iCAP 6000 ICP-OES analyzer. Wavelengths used for the detection of the different metal ions were as follows: 324.7 nm for Cu, 228.6 nm for Co, 257.6 nm for Mn, 231.6 nm for Ni, 213.8 nm for Zn.
Binding constants calculations
The association constants for metal binding were measured using UV-Vis spectroscopy by titration of 2 μL aliquots of a metal ion solution (2 mM in H2O) into a 3 mL solution of the peptoid (typically 6–8 μM) in MeOH:H2O 1:5. The binding was followed by obtaining the UV-Vis spectrum from 200 to 500 nm as a function of the total added metal ions.
The metal binding to the reported peptoids can be described by the equilibrium as follows:
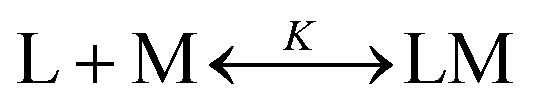
Defining [L] = [L0] − [LM]
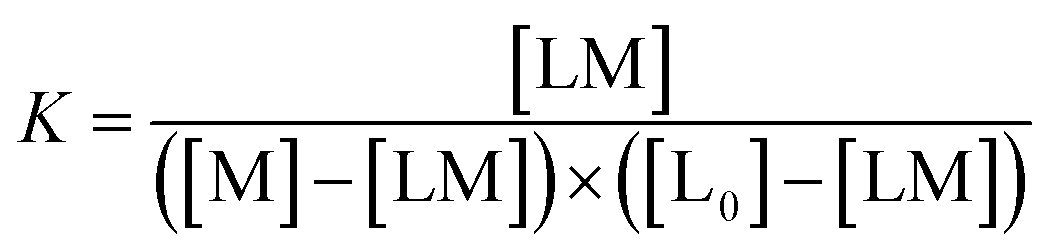
The real solution to [LM] is
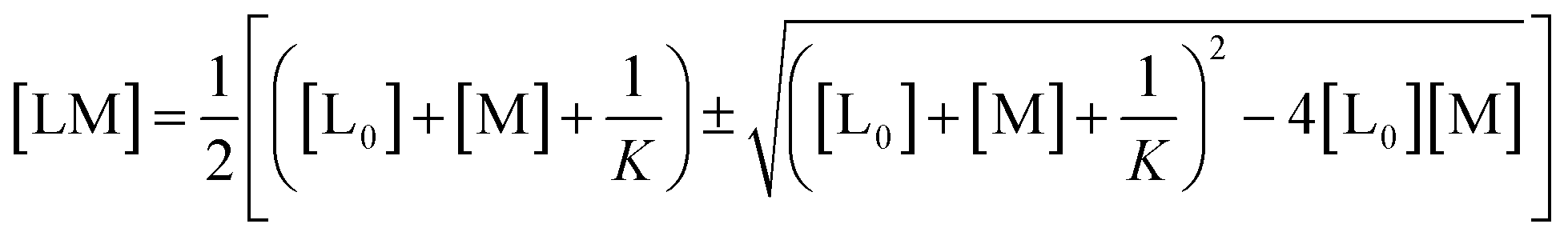 | (1) |
Now,
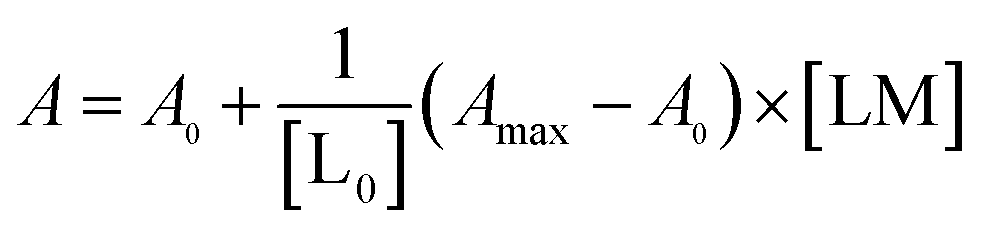 | (2) |
Substitution of [LM] in eqn (2) with the expression in eqn (1), gives
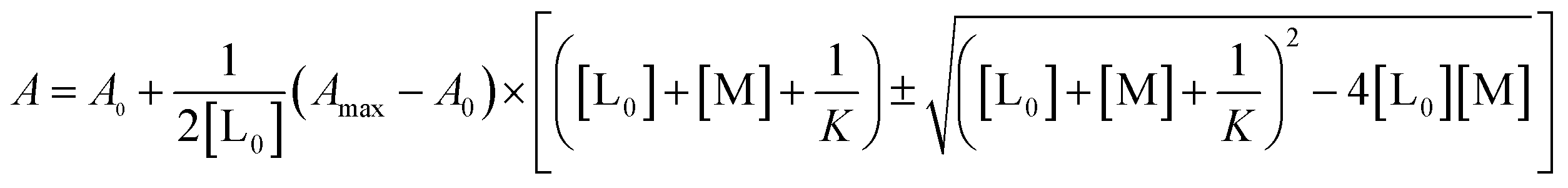 | (3) |
The results were fitted by a nonlinear regression (curve fit)5b using GraphPad Prism® software.
Acknowledgements
The research leading to these results has received funding from the European Union's-Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 333034-MC--MF STRC AND FCN. The authors thank Mrs Larisa Panz for her enormous assistance with the various MS measurements and their analysis, Dr Boris Tumanskii for his assistance with the EPR measurements and to Mrs Malka Kochva for her assistance with the ICP measurements. Maria Baskin thanks the Schulich foundation for her PhD fellowship.References
- M. Zastrow and V. L. Pecoraro, Coord. Chem. Rev., 2013, 257, 2565–2588 CrossRef CAS PubMed.
- (a) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS; (b) Foldamers: structure, properties, and applications, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) D. Seebach and J. Cardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS PubMed; (d) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed; (e) S. A. Fowler and H. E. Blackwell, Org. Biomol. Chem., 2009, 7, 1508–1524 RSC; (f) C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- (a) G. Maayan, Eur. J. Org. Chem., 2009, 5699–5710 CrossRef CAS; (b) Metallofoldamers. Supramolecular Architectures from Helicates to biomimetics, ed. G. Maayan and M. Albrecht, John Wiley& Sons, Ltd, 2013 Search PubMed.
- (a) F. Zhang, S. Bai, G. P. A. Yap, V. Tarwade and J. M. Fox, J. Am. Chem. Soc., 2005, 127, 10590 CrossRef CAS PubMed; (b) Z. Dong Jr, R. J. Karpowicz, S. Bai, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2006, 128, 14242 CrossRef PubMed; (c) Y. Zhao and Z. Zhong, J. Am. Chem. Soc., 2006, 128, 9988 CrossRef CAS PubMed; (d) R. Métivier, I. Leray and B. Valeur, Chem.–Eur. J., 2004, 10, 4480 CrossRef PubMed; (e) Y. Zhao and Z. Zhong, Org. Lett., 2006, 8, 4715 CrossRef CAS PubMed; (f) G. Lelais, D. Seebach, B. Jaun, I. Raveendra, R. I. Mathad, O. Flögel, F. Rossi, M. Campo and A. Wortmann, Helv. Chim. Acta, 2006, 89, 361 CrossRef CAS; (g) C. E. Schafmeister, L. G. Belasco and P. H. Brown, Chem.–Eur. J., 2008, 14, 6406 CrossRef CAS PubMed; (h) Z. Ma, Y. A. Skorik and C. Achim, Inorg. Chem., 2011, 50, 6083 CrossRef CAS PubMed; (i) S. Tashiro, K. Matsuoka, A. Minoda and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 13123 CrossRef CAS PubMed; (j) J. P. Miller, M. S. Melicher and A. Schepartz, J. Am. Chem. Soc., 2014, 136, 14726 CrossRef CAS PubMed.
- (a) B. C. Lee, T. K. Chu, K. A. Dill and R. N. Zuckermann, J. Am. Chem. Soc., 2008, 130, 8847–8855 CrossRef CAS PubMed; (b) G. Maayan, M. D. Ward and K. Kirshenbaum, Chem. Commun., 2009, 56–58 RSC; (c) N. Maulucci, I. Izzo, G. Bifulco, A. Aliberti, C. De Cola, D. Comegna, C. Gaeta, A. Napolitano, C. Pizza, C. Tedesco, D. Flot and F. De Riccardis, Chem. Commun., 2008, 3927–3929 RSC; (d) C. De Cola, S. Licen, D. Comegna, E. Cafaro, G. Bifulco, I. Izzo, P. Tecilla and F. De Riccardis, Org. Biomol. Chem., 2009, 7, 2851 RSC; (e) G. Della Sala, B. Nardone, F. De Riccardis and I. Izzo, Org. Biomol. Chem., 2013, 11, 726–731 RSC; (f) I. Izzo, G. Ianniello, C. De Cola, B. Nardone, L. Erra, G. Vaughan, C. Tedesco and F. De Riccardis, Org. Lett., 2013, 15, 598–601 CrossRef CAS PubMed; (g) A. S. Knight, E. Y. Zhou, J. G. Pelton and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17488–17493 CrossRef CAS PubMed; (h) C. De Cola, G. Fiorillo, A. Meli, S. Aime, E. Gianolio, I. Izzo and F. De Riccardis, Org. Biomol. Chem., 2014, 12, 424 RSC; (i) C. Tedesco, L. Erra, I. Izzo and F. De Riccardis, CrystEngComm, 2014, 16, 3667–3687 RSC; (j) T. Zabrodski, M. Baskin, J. K. Prathap and G. Maayan, Synlett, 2014, 25, A–F Search PubMed; (k) M. Baskin and G. Maayan, Biopolymers, 2015, 104, 577–584 CrossRef CAS PubMed; (l) A. S. Knight, E. Y. Zhou and M. B. Francis, Chem. Sci., 2015, 6, 4042–4048 RSC.
- (a) A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27, 5665–5691 CrossRef CAS PubMed; (b) R. N. Zuckermann, Biopolymers, 2011, 96, 545–555 CrossRef CAS PubMed; (c) K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS PubMed; (d) C. W. Wu, T. J. Sanborn, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 6778–6784 CrossRef CAS PubMed; (e) S. B. Y. Shin, B. Yoo, L. Todaro and K. Kirshenbaum, J. Am. Chem. Soc., 2007, 129, 3218–3225 CrossRef CAS PubMed; (f) C. W. Wu, K. Kirshenbaum, T. J. Sanborn, J. A. Patch, K. Huang, K. A. Dill, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2003, 125, 13525–13530 CrossRef CAS PubMed; (g) B. C. Gorske, B. L. Bastian, G. D. Geske and H. E. Blackwell, J. Am. Chem. Soc., 2007, 129, 8928–8929 CrossRef CAS PubMed; (h) N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, J. Am. Chem. Soc., 2008, 130, 16622–16632 CrossRef CAS PubMed; (i) R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Org. Chem., 2010, 75, 6068–6078 CrossRef PubMed; (j) B. Paul, G. L. Butterfoss, M. G. Boswell, M. L. Huang, R. Bonneau, C. Wolf and K. Kirshenbaum, Org. Lett., 2012, 14, 926–929 CrossRef CAS PubMed; (k) B. Paul, G. L. Butterfoss, M. G. Boswell, P. D. Renfrew, F. G. Yeung, N. H. Shah, C. Wolf, R. Bonneau and K. Kirshenbaum, J. Am. Chem. Soc., 2011, 133, 10910–11091 CrossRef CAS PubMed; (l) O. Roy, C. Caumes, Y. Esvan, C. Didierjean, S. Faure and C. Taillefumier, Org. Lett., 2013, 15, 2246–2249 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. W. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- (a) A. E. Barron and R. N. Zuckermann, Curr. Opin. Chem. Biol., 1999, 3, 681–687 CrossRef CAS PubMed; (b) S. L. Seurynck, J. A. Patch and A. E. Barron, Chem. Biol., 2005, 12, 77–88 CrossRef CAS PubMed; (c) J. A. Patch and A. E. Barron, Curr. Opin. Chem. Biol., 2002, 6, 872–877 CrossRef CAS PubMed; (d) J. Seo, B. C. Lee and R. N. Zuckermann, in Comprehensive Biomaterials, ed. P. Ducheyne, K. E. Healy, D. W. Hutmacher, D. W. Grainger and C. J. Kirkpatrick, Elsevier, 2011, vol. 2, pp. 53–76 Search PubMed.
- (a) T. J. Sanborn, C. W. Wu, R. N. Zuckermann and A. E. Barron, Biopolymers, 2002, 63, 12–20 CrossRef CAS PubMed; (b) J. Sun, G. M. Stone, N. P. Balsara and R. N. Zuckermann, Macromolecules, 2012, 45, 5151–5156 CrossRef CAS; (c) G. Maayan and L.-K. Liu, Pept. Sci., 2011, 96, 679–687 CrossRef CAS PubMed.
- P. J. Kaniraj and G. Maayan, Chem. Commun., 2015, 51, 11096 RSC.
- This work, see ESI Table S4 and Fig. S58.†.
- (a) S. F. Sousa, A. B. Lopes, P. A. Fernandes and M. J. Ramos, Dalton Trans., 2009, 7946–7956 RSC; (b) C. Andreini, I. Bertini and G. Cavallaro, PLoS One, 2011, 6, e26325 CAS; (c) A. M. Goforth, M. A. Tershansy, M. D. Smith, L. Peterson Jr, J. G. Kelley, W. J. I. DeBenedetti and H.-C. zur Loye, J. Am. Chem. Soc., 2011, 133, 603–612 CrossRef CAS PubMed; (d) N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC; (e) L. Mishra, K. Bindu and S. Bhattacharya, Spectrochim. Acta, Part A, 2005, 61, 807–813 CrossRef PubMed.
- G. Maayan, B. Yoo and K. Kirshenbaum, Tetrahedron Lett., 2008, 49, 335–338 CrossRef CAS PubMed.
- (a) J. Peisach and W. E. Blumberg, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef CAS PubMed; (b) N. Wei, N. N. Murthy and K. D. Karlin, Inorg. Chem., 1994, 44, 6093–6100 CrossRef; (c) B. Lucches, K. S. Humphreys, D.-H. Lee, C. D. Incarvito, R. D. Sommer, A. L. Rheingold and K. D. Karlin, Inorg. Chem., 2004, 43, 5987–5998 CrossRef PubMed.
- The association constant of the intramolecular complex with Fe3+ was not calculated due to our observation that this complex was not formed but rather an intermolecular binding occurs. Addition of 1 equiv. of Fe3+ solution to 1 equiv. of the peptoid solution yielded the 2:2 peptoid:Fe complex (Helix HQT i + 3)2Fe2, such that one Fe3+ ion binds two Terpy ligands and one Fe3+ ion binds two HQ ligands (as supported by MS, Fig. S71 and S72†).
- (a) O. Andersen, Chem. Rev., 1999, 99, 2683–2710 CrossRef CAS PubMed; (b) K. Kalia and S. J. S. Flora, J. Occup. Health, 2005, 47, 1–21 CrossRef CAS PubMed; (c) R. D. Hancock, Chem. Soc. Rev., 2013, 42, 1500–1524 RSC; (d) P. V. Bernhardt, Dalton Trans., 2007, 3214–3220 RSC; (e) K. M. Lincoln, P. Gonzalez, T. E. Richardson, D. A. Julovich, R. Saunders, J. W. Simpkins and K. N. Green, Chem. Commun., 2013, 49, 2712–2714 RSC; (f) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- (a) H.-H. Liu and Y. Chen, Eur. J. Org. Chem., 2009, 5261–5265 CrossRef CAS; (b) H.-H. Wang, L. Xue, Z.-J. Fang, G.-P. Li and H. Jiang, New J. Chem., 2010, 34, 1239–1242 RSC; (c) W.-J. Shi, J.-Y. Liu and D. K. P. Ng, Chem.–Asian J., 2012, 7, 196–200 CrossRef CAS PubMed; (d) S. Goswami, D. Sen and N. K. Das, Org. Lett., 2010, 12, 856–859 CrossRef CAS PubMed; (e) L. Jiao, J. Li, S. Zhang, C. Wei, E. Hao and M. G. H. Vicente, New J. Chem., 2009, 33, 1888–1893 RSC; (f) S.-P. Wu, R.-Y. Huang and K.-J. Du, Dalton Trans., 2009, 4735–4740 RSC; (g) R. Sheng, P. Wang, Y. Gao, Y. Wu, W. Liu, J. Ma, H. Li and S. Wu, Org. Lett., 2008, 10, 5015–5018 CrossRef CAS PubMed; (h) S. Mukherjee, P. Mal and H. Stoeckli-Evans, Polyhedron, 2014, 73, 87–97 CrossRef CAS; (i) S. Khatua, J. Kang and D. G. Churchill, New J. Chem., 2010, 34, 1163–1169 RSC; (j) V. S. Jisha, A. J. Thomas and D. Ramaiah, J. Org. Chem., 2009, 74, 6667–6673 CrossRef CAS PubMed; (k) S. Sarkar, M. Chatti and V. Mahalingam, Chem.–Eur. J., 2014, 20, 3311–3316 CrossRef CAS PubMed; (l) S. Khatua, S. H. Choi, J. Lee, J. O. Huh, Y. Do and D. G. Churchill, Inorg. Chem., 2009, 48, 1799–1801 CrossRef CAS PubMed; (m) M. Kumar, N. Kumar, V. Bhalla, P. R. Sharma and T. Kaur, Org. Lett., 2012, 14, 406–409 CrossRef CAS PubMed; (n) M. Yu, M. Shi, Z. Chen, F. Li, X. Li, Y. Gao, J. Xu, H. Yang, Z. Zhou, T. Yi and C. Huang, Chem.–Eur. J., 2008, 14, 6892–6900 CrossRef CAS PubMed; (o) R. K. Pathak, V. K. Hinge, P. Mondala and C. P. Rao, Dalton Trans., 2012, 41, 10652–10660 RSC; (p) W. Wang, Q. Wen, Y. Zhang, X. Fei, Y. Li, O. Yang and X. Xu, Dalton Trans., 2013, 42, 1827–1833 RSC; (q) Z.-L. Gong and Y.-W. Zhong, Organometallics, 2013, 32, 7495–7502 CrossRef CAS; (r) D. Maity, A. K. Manna, D. Karthigeyan, T. K. Kundu, S. K. Pati and T. Govindaraju, Chem.–Eur. J., 2011, 17, 11152–11161 CrossRef CAS PubMed.
- ICP measurements require thorough filtration of the samples prior to analysis (see the Experimental section for full description of the samples preparation). We have noticed that the iron salt does not go through the filter, thus, it was not included in the ICP measurements.
- R. Diaz-Torres and S. Alvarez, Dalton Trans., 2011, 40, 10742–10750 RSC.
- We note here that titrations of 1 equiv. (Helix HQT i + 3)2Co or (Helix HQT i + 3)2Zn solution with 0.5 equiv. of either Co2+ or Zn2+ solution, respectively led to binding of these additional ions at the HQ site affording the final complexes (Helix HQT i + 3)2Co2 and (Helix HQT i + 3)2Zn2, respectively (as supported by UV, Fig. S28 and S29†).
- (a) M. L. Zastrow and V. L. Pecoraro, Biochemistry, 2014, 53, 957–978 CrossRef CAS PubMed; (b) M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118–123 CrossRef CAS PubMed; (c) M. L. Zastrow and V. L. Pecoraro, J. Am. Chem. Soc., 2013, 135, 5895–5903 CrossRef CAS PubMed; (d) W. J. Song and F. A. Tezcan, Science, 2014, 346, 1525–1528 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04358a |
| This journal is © The Royal Society of Chemistry 2016 |