
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anticancer metallohelices: nanomolar potency and high selectivity†
Rebecca A.
Kaner
ab,
Simon J.
Allison
c,
Alan D.
Faulkner
a,
Roger M.
Phillips
*c,
David I.
Roper
e,
Samantha L.
Shepherd
c,
Daniel H.
Simpson
ae,
Nicholas R.
Waterfield
d and
Peter
Scott
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: peter.scott@warwick.ac.uk
bInstitute of Advanced Study, University of Warwick, CV4 7HS, UK
cSchool of Applied Sciences, University of Huddersfield, Huddersfield, HD1 3DH, UK
dWarwick Medical School, University of Warwick, Coventry, CV4 7AL, UK
eSchool of Life Sciences, University of Warwick, Coventry, CV4 7AL, UK
First published on 26th October 2015
Abstract
A range of new helicate-like architectures have been prepared via highly diastereoselective self-assembly using readily accessible starting materials. Six pairs of enantiomers [Fe2L3]Cl4·nH2O (L = various bidentate ditopic ligands NN–NN) show very good water solubility and stability. Their activity against a range of cancer cell lines in vitro is structure-dependent and gives IC50 values as low as 40 nM. In an isogenic pair of HCT116 colorectal cancer cells, preferential activity was observed against cell lines that lack functional p53. Selectivity is also excellent, and against healthy human retinal pigment epithelial (ARPE19) and lung fibroblast (WI38) cells IC50 values are nearly three orders of magnitude higher. Cisplatin is unselective in the same tests. The compounds also appear to have low general toxicity in a number of models: there is little if any antimicrobial activity against methicillin-resistant Staphylococcus aureus and Escherichia coli; Acanthamoeba polyphaga is unaffected at 25 μg mL−1 (12.5 μM); Manduca sexta larvae showed clear evidence of systemic distribution of the drug, and rather than any observation of adverse effects they exhibited a significant mean weight gain vs. controls. Investigation of the mode of action revealed no significant interaction of the molecules with DNA, and stimulation of substantial cell death by apoptosis.
Introduction
The main purpose of current anticancer therapies is to eradicate tumour cells without damaging overall patient health. However, side effects limit the dosage of chemotherapeutic drugs which may be safely applied, and as a result, cancer cells often remain. This leads to poor outcomes in the clinic and the evolution of drug-resistant tumours.1 Hence, while the potency of a drug is a very important consideration, drug selectivity towards cancer cells is key to ensuring both safety and effectiveness.2 While we might hope that more effective cancer chemotherapies would come from drugs designed to address specific biomolecular targets,2 this is far from uniformly the case.3 Such drugs may be too targeted since tumours can circumvent the blockade of a specific pathway by switching to another – so-called tumour plasticity.4 Compounds with polypharmacology (action against multiple targets) are thus currently of considerable interest to the pharmaceutical industry. This coincides with the resurgence of phenotypic drug discovery,5–7 where the targets of a drug are established after the observation of the useful biological effect. This strategy has led to a disproportionately high number of first-in-class drugs with novel mechanisms of action (1999–2008)8,9 The accompanying challenge for synthetic chemistry is to discover, perhaps without reference to some specific biomolecular target, new classes of drug candidates which are both potent and selective.Lehn recognized the potential of helicates in medicinal chemistry,10 and this was borne out in early studies, particularly in the area of cancer.11–15 We have argued,16 however, that in order for helicates to be capable of translation to the clinic a number of criteria need to be addressed: optical purity and stability, solubility and chemical stability in water, availability on a practical scale, and synthetic diversity. Our recent work has attempted to address these matters17 using a new strategy whereby the absolute configurations of individual metal centres are controlled18 and linked together to form the prototype helicate-like architectures of Fig. 1. Of these flexicates,19 [Fe2L13]4+ contains a diamine linker while [Fe2L2a3]4+ is based on a dialdehyde.19 Promising results were reported in a number of disease areas,16,19–21 including good activity against a range of cancer cell lines.20 Here we report the discovery of a new series of highly potent (40 nM) anticancer compounds of the dialdehyde class related to [Fe2L2a3]4+ that preferentially kill cancer cells that lack functional p53, are nearly three orders of magnitude less toxic to healthy human cell lines tested and have low toxicity to microbes, amoeba and caterpillar larvae.
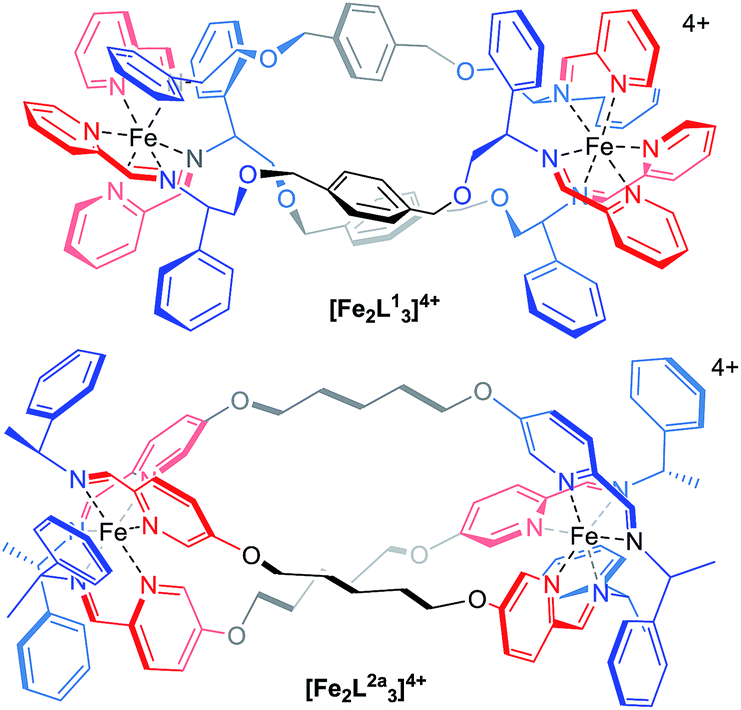 | ||
| Fig. 1 Structure of flexicates [Fe2L13]4+ and [Fe2LA3]4+.19 | ||
While DNA does not appear to be the target, the compounds are triggering significant apoptotic cell death as part of their mode of action.
Results
Synthesis of ligands and ZnII systems
The dialdehyde units of Fig. 2(a) and (b) include various linker rigidities and orientations designed to probe structural viability and biological activity. They were synthesized via simple etherifications of 5-hydoxypicolinaldehyde.22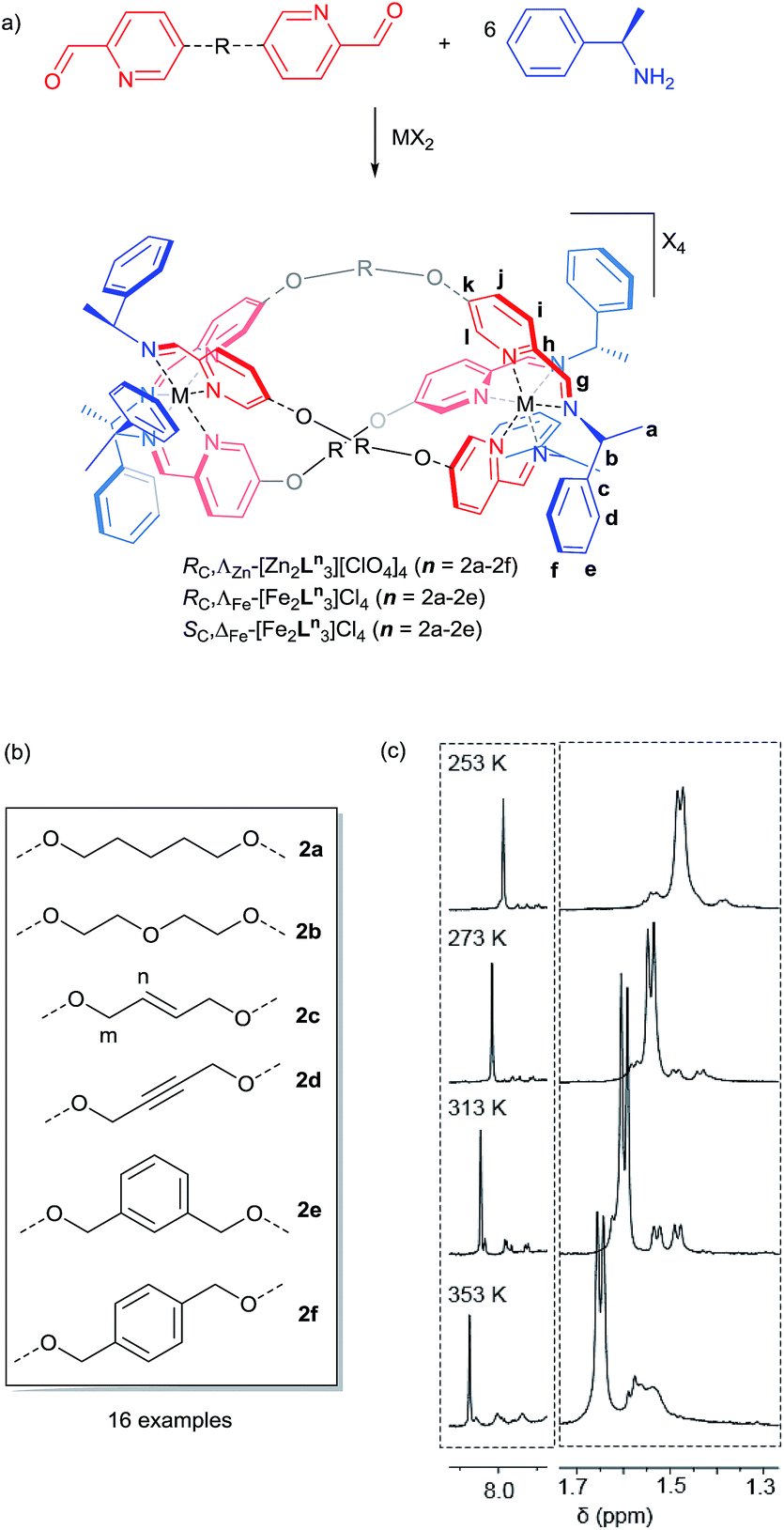 | ||
| Fig. 2 (a) Synthesis of new flexicates; (b) linking groups used in this study; (c) sections of 1H NMR spectra of ΛZn-[Zn2L2e3][ClO4]4·4H2O at 253–353 K in d3-acetonitrile showing the higher equilibrium population of a minor asymmetric conformer at higher temperatures. | ||
Treatment with Zn(ClO4)2·6H2O and (R)-1-phenylethan-1-amine, in appropriate proportions, led to the rapid self-assembly of the bimetallic flexicates in acetonitrile solution at ambient temperature. For the majority of these new ZnII complexes NMR spectra indicated that within the limits of the experiment single diastereomers were formed (vide infra).
The sole exception was the 1,3-phenylene system ΛZn-[Zn2L2e3][ClO4]4·4H2O, which gave more complex 1H NMR spectra [Fig. 2(c)]. At 253 K the phenethylamine methyl group doublet region 1.4–1.7 ppm contains one more intense doublet and two broader signals in the ratio ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. The proportion of the minor species increases with temperature and the resonances sharpen somewhat, such that by 313 K two of the smaller doublets corresponding to the minor species are relatively sharp and resolved while a third overlaps with the main resonance. By 353 K the smaller peaks had again broadened considerably and the ratio of the two sets of resonances was ca. 10:9. The imine region (8.5–7.6 ppm) behaved in a corresponding manner (253 K, three peaks in ratio 10:1:1:1; 353 K, ratio 10:3:3:3). These observations are consistent with the presence of two species – one of high-symmetry and one low – in thermodynamic equilibrium (ratio ca. 1:0.3 at low temperature, increasing to almost 1:1 at high temperature) but with the involvement of other related conformers particularly at higher temperatures. The processes leading to the observed NMR behaviour may correspond to exchange between these conformers, or indeed between isostructural low symmetry species. While the spectra are not sufficiently well resolved to determine kinetic parameters, we sought to investigate this molecular system by computational means.
:1:1. The proportion of the minor species increases with temperature and the resonances sharpen somewhat, such that by 313 K two of the smaller doublets corresponding to the minor species are relatively sharp and resolved while a third overlaps with the main resonance. By 353 K the smaller peaks had again broadened considerably and the ratio of the two sets of resonances was ca. 10:9. The imine region (8.5–7.6 ppm) behaved in a corresponding manner (253 K, three peaks in ratio 10:1:1:1; 353 K, ratio 10:3:3:3). These observations are consistent with the presence of two species – one of high-symmetry and one low – in thermodynamic equilibrium (ratio ca. 1:0.3 at low temperature, increasing to almost 1:1 at high temperature) but with the involvement of other related conformers particularly at higher temperatures. The processes leading to the observed NMR behaviour may correspond to exchange between these conformers, or indeed between isostructural low symmetry species. While the spectra are not sufficiently well resolved to determine kinetic parameters, we sought to investigate this molecular system by computational means.
Computational studies
Following extensive searching, six conformers of ΛZn-[Zn2L2e3]4+ were located and minimised [Fig. 3]. These fell into two classes: those where the three m-xylenyl groups were oriented away from the central cavity i.e. exo, and those where one such group was oriented endo. No conformers were observed in which two or three m-xylenyl groups were oriented into the cavity – this caused too much torsional and steric strain. Structure endo1 was found to be the lowest in energy, the next lowest being endo2 (ca. +5 kcal) which differs only in the fold of one of the linkers. For these structures the Zn–Zn distances are ca. 11.7 and 11.8 Å respectively. The structure exo1 (+7 kcal) has a large central cavity but a similar Zn–Zn distance (11.8 Å). The structure exo2 (+8 kcal) has a considerably shorter Zn–Zn distance at ca. 9.5 Å with accompanying concertinaed fold. Furthermore, higher energy conformers exo3 and exo4 differed principally in how the m-xylenyl groups folded towards each metal centre. Both were found to have a short Zn–Zn distance of 9.4 and 9.5 Å respectively. | ||
| Fig. 3 DFT calculated structures, relative energies (compared to endo1) and Zn–Zn distances of the six conformational isomers of ΛZn-[Zn2L2e3][ClO4]4. | ||
While prediction of an equilibrium population from the above calculations is complicated by statistical and entropic contributions from the total number of possible structures and the differences in structural flexibility, the detection of two distinct structural classes is clearly consistent with observations in solution.
We propose that the species detected by NMR displaying high symmetry (D3) comprises exo conformations while the asymmetric (C1) species is endo. Examination of the structures indicates that the barrier to conversion within the exo or endo manifolds would be low since it would involve relatively simple concertina-type processes, but conversion between exo and endo conformations requires the rotation of the m-xylenyl linker through a strained, high energy transition state.
Synthesis of water soluble compounds
Pairs of water-soluble FeII flexicate enantiomers [Fe2Ln3]Cl4 (n = 2b–2e) were synthesised in high yield by heating the appropriate dialdehyde linker with either (R)- or (S)-1-phenylethan-1-amine and FeCl2 in methanol. 1H-NMR spectra were similar though slightly broader than the analogous ZnII perchlorate complexes and are consistent, along with 13C-NMR [Fig. 4 and ESI†] and circular dichroism spectra (ESI†) with the presence of single, stable, non-racemising diastereomers in solution, although unsurprisingly [Fe2L2e3]Cl4 exists as a similar mixture of conformers to the Zn analogue above. The complexes gave excellent electrospray mass spectrometry data e.g. ΛFe-[Fe2L2c3]Cl4 gave a strong peak at m/z 420.17 Da for the tetracationic ion. The formula weights of the panel of complexes, including levels of hydration, were determined by correlation of NMR, IR, thermogravimetric and elemental analyses (see ESI†). The p-xylenyl system [Fe2L2f3]Cl4 displayed poor solubility in water and methanol and could not be fully characterised.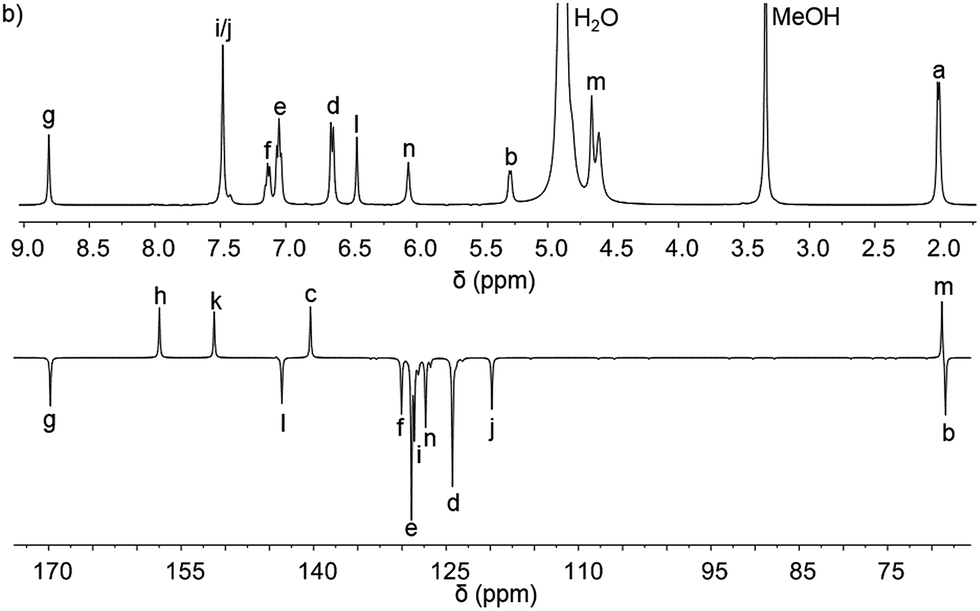 | ||
| Fig. 4 1H and 13C{1H}. NMR spectra of ΛFe-[Fe2L2c3]Cl4·9H2O at 298 K in d4-methanol; see Fig. 2 for key. | ||
Stability in aqueous media
Absorbance spectra indicated that little decomposition of the flexicates occurred in water at pH 7 over weeks, but half-lives for decomposition could readily be recorded in hydrochloric acid (0.2 M) via the 540 nm MLCT absorbance band of the complex. Even under such conditions, first order kinetic plots gave t1/2 values in the region 10–20 h. This very favourable aqueous stability of flexicates probably arises from the presence of extensive (hydrophobic) π-stacking.23Biological activity & selectivity
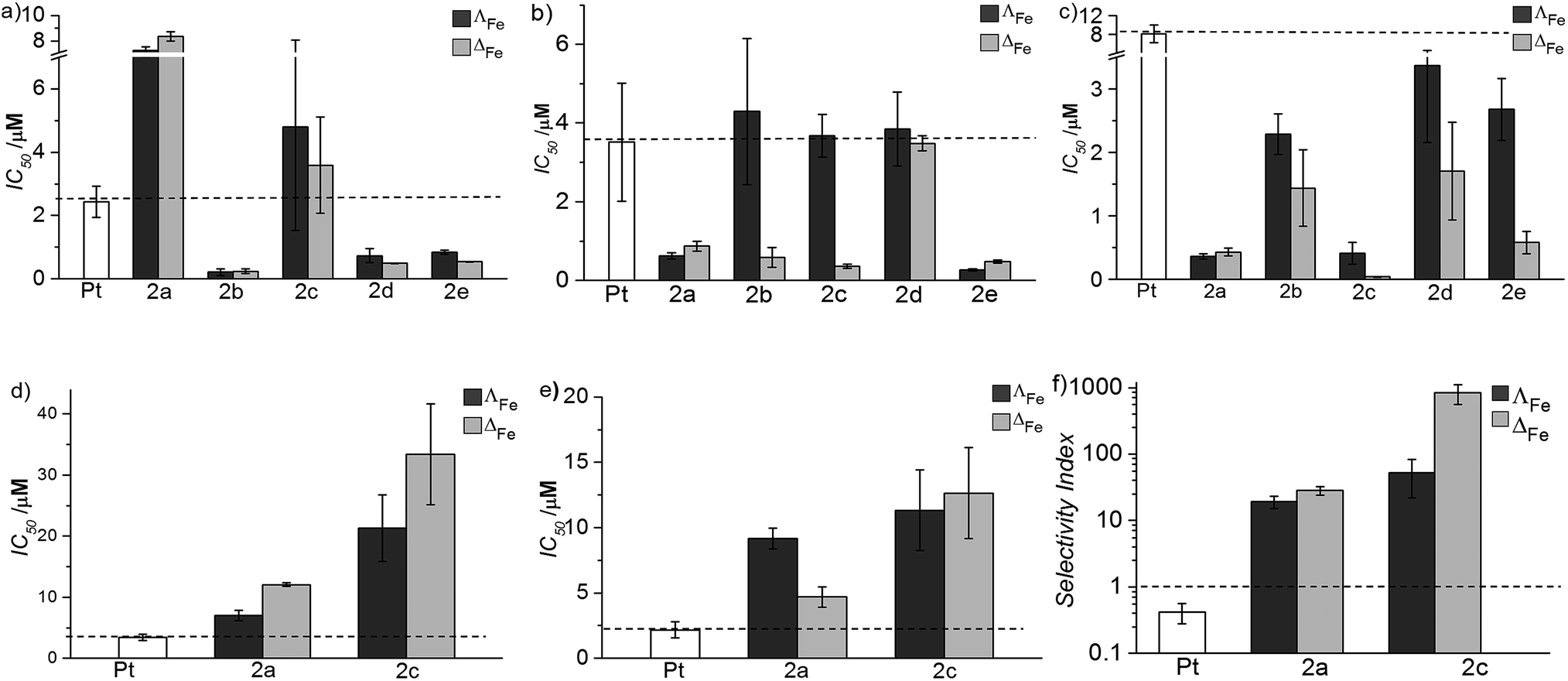 | ||
| Fig. 5 IC50 values of cisplatin (Pt, white) and flexicates [Fe2Ln3]Cl4 (n = 2a–2e) (ΛFe – dark grey, ΔFe – light grey) against (a) MDA-MB-468, (b) HCT116 p53+/+, (c) HCT116 p53−/−, (d) ARPE19 and (e) W138 cells over 96 h; dotted line highlights the relative sensitivity of test compounds compared to cisplatin, (f) in vitro selectivity index (IC50 ARPE19/IC50 HCT116 p53−/−) – broken line at selectivity index = 1 represents no difference in IC50 between tumour and normal cells – note log scale on y axis. | ||
The new flexicates had very modest antimicrobial activity (Table 1) or did not significantly inhibit microbial growth at concentrations well over 3 orders of magnitude higher than the IC50 values observed in cancer cells.
| Compound | MRSA MIC | E. coli MIC | ||
|---|---|---|---|---|
| (μg ml−1) | (μM) | (μg ml−1) | (μM) | |
| Kanamycin | 1 | 2 | 2 | 4 |
| ΛFe-[Fe2L2a3]Cl4 | 64 | 35 | >128 | 70 |
| ΛFe-[Fe2L2b3]Cl4 | 128 | 70 | >128 | 70 |
| ΛFe-[Fe2L2c3]Cl4 | 128 | 70 | >128 | 70 |
| ΛFe-[Fe2L2d3]Cl4 | >128 | 70 | 64 | 35 |
| ΛFe-[Fe2L2e3]Cl4 | 64 | 35 | 128 | 70 |
Oral and systemic toxicity of these flexicates was assessed in neonate larvae of Manduca sexta.31 Caterpillars which had ingested a solution of 25 μg mL−1 [12.5 μM], [Fe2L2a3]Cl4 in artificial diet for 7 d [Fe2L2a3]Cl4 showed comparable weight gain to controls suggesting no oral toxicity and no adverse effect on feeding behaviour (Fig. 6). Interestingly the larvae exhibited an increased mean weight gain of approximately 30% (P < 1). Also we noted that larvae that ingested the flexicate solutions turned a bright purple colour over the course of the assay, suggesting that these compounds were persisting in the insect and not being rapidly metabolized or excreted.
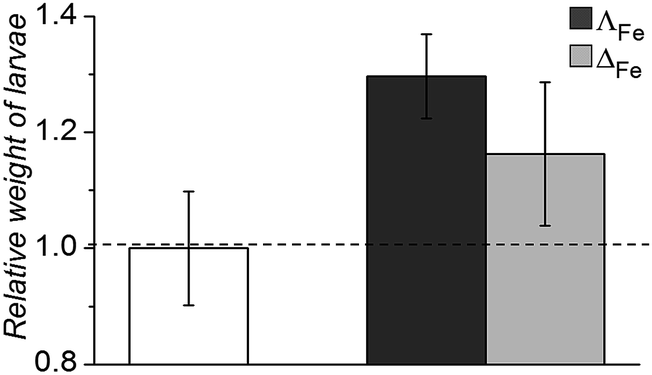 | ||
| Fig. 6 Relative weight of Manduca sexta larvae after treatment (7 d) with [Fe2L2a3]Cl4 (ΛFe – dark grey, ΔFe – light grey) compared to an untreated control. | ||
Systemic toxicity was further tested by injection of 50 μg (0.25 μM) of the compounds directly into the hemocoel of cohorts of 5th instar larvae (n = 3). The cohorts were then allowed to continue feeding. Despite becoming purple, all larvae proceeded to develop into the pupal diapause stage as per the buffer control injections.
Mode of action
The mode or modes of action of such a new and different system will require intensive investigation and is likely to involve multiple targets and pathways. Here, we describe two preliminary studies towards this end.Isolated ct-DNA (0.5 mg mL−1) was mixed with each flexicate (7.5 μM) in buffered conditions (10 mM tris, 1 mM EDTA at pH 7.0), to give 10 bases: 1 flexicate complex, and the absorbance at 260 nm between 25 °C and 90 °C was recorded (0.4 °C min−1). Tm for each experiment was calculated from the first derivative of a Boltzmann sigmoidal fit of the plot of absorbance versus temperature.
T m of untreated ct-DNA (0.25 mg mL−1 in 10 mM tris, 1 mM EDTA at pH 7.0) was measured to be 68.3 ± 0.5 °C. Most of the new flexicates had no significant effect on the denaturation of ct-DNA (Fig. 7); the small (ΔT ca. 1 °C) reduction for L2e enantiomers can be ascribed to an electrostatic effect.34 We are therefore satisfied that DNA is unlikely to be the target of this panel of compounds.
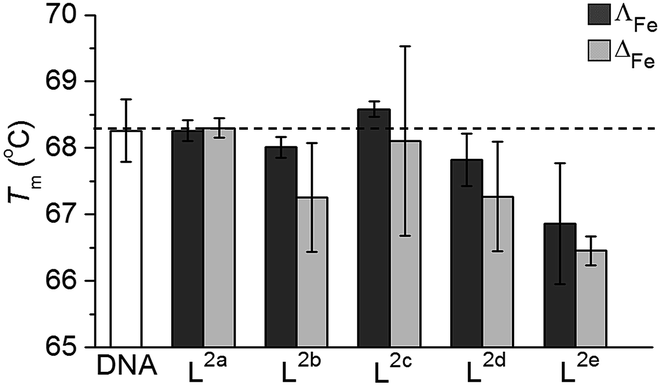 | ||
| Fig. 7 Effect on Tm of linear ct-DNA (DNA, white) from interactions [Fe2Ln3]Cl4 (n = 2a–2e) (ΔFe – light grey, ΛFe – dark grey) in 1 mM Trizma base (10 base pairs of DNA to 1 flexicate complex). | ||
HCT116 p53+/+ cancer cells (24 h post-seeding) were incubated in fresh media containing flexicate or no flexicate (control) and were then analysed after 48 h for levels of apoptosis and necrosis. As cells start to undergo apoptosis, one of the first cellular changes is the externalisation of the membrane protein phosphatidylserine (PS). This can be detected and quantified by fluorescently labelled annexin V37,38 which can selectively bind externally exposed PS but is membrane-impermeable. This enables cells in the early stages of apoptosis to be distinguished from necrotic cells and cells in the late stages of apoptosis both of which have lost membrane integrity and will therefore also stain with the membrane-impermeable DNA stain propidium idodide.39
Flexicates ΛFe-[Fe2L2a3]Cl4 and ΔFe-[Fe2L2a3]Cl4 were tested and both induced significant levels of apoptosis that were ∼2.6 fold (ΛFe-[Fe2L2a3]Cl4) and ∼4.4 fold (ΔFe-[Fe2L2a3]Cl4) above background control levels in the HCT116 cancer cells at 48h (Fig. 8). A significant proportion of late apoptotic/necrotic cells were also detectable by 48 h, with levels ∼2.3–2.5 fold above background control levels (Fig. 8). These preliminary investigations indicate induction of apoptosis by these new flexicates as part of their mode of action.
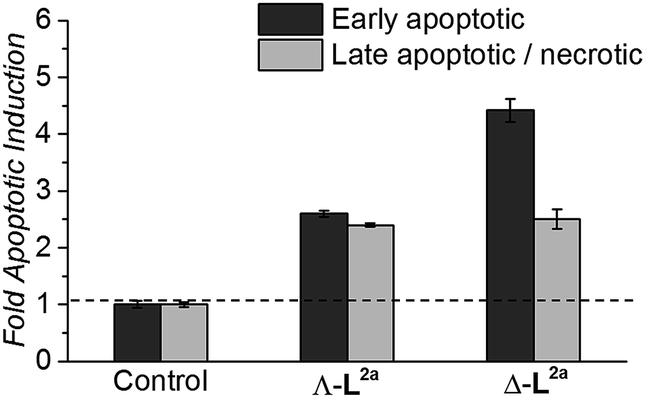 | ||
| Fig. 8 Induction of apoptosis in HCT116 p53+/+ cancer cells by 48 h treatment with the indicated flexicates. Fold change shown relative to basal apoptotic levels in untreated HCT116 control cells. | ||
Experimental
Synthesis
(E)-5,5′-(But-2-ene-1,4-diylbis(oxy))dipicolinaldehyde (0.13 g, 0.44 mmol) and (R)-1-phenylethan-1-amine (0.11 g, 0.88 mmol) were dissolved in acetonitrile (10 mL) with ZnII perchlorate hexahydrate (0.11 g, 0.29 mmol) and the solution was stirred at ambient temperature for 20 h. Ethyl acetate was added drop-wise to cause precipitation of a white crystalline solid, ΛZn-[Zn2L2c3][ClO4]4·10H2O. Yield 0.214 g, 57%. 1H NMR (400 MHz, 298 K, CD3CN) δH 8.06 (6H, s, CHN), 7.49 (6H, dd, 3JHH = 8.5 Hz, 4JHH = 3.5 Hz), 7.36 (6H, d, 3JHH = 8.5 Hz), 7.14 (6H, d, 3JHH = 3.5 Hz), 7.09 (6H, t, 3JHH = 8.0 Hz), 6.95 (12H, t, 3JHH = 7.5 Hz), 6.64 (12H, d, 3JHH = 7.0 Hz, Ar), 6.12 (6H, m), 5.38 (6H, q, 3JHH = 6.5 Hz, CH), 4.64 (12H, s, CH2), 1.61 (18H, d, 3JHH = 6.5 Hz). 13C{1H} NMR (101 MHz, 298 K, CD3CN) δC 161.8 (CHN), 159.6, 142.0, 139.9, 139.4, 132.3 (Ar), 129.7 (CH), 129.4, 128.5, 126.4, 122.7 (Ar), 69.7 (CH2), 64.7 (CH), 23.6 (CH3). MS (ESI) m/z 411 [Zn2L3]4+. IR ν cm−1 2976 w, 1570 m, 1316 m, 1225 m, 1082 s, 762 w, 703 m, 653 m. Elemental analysis found (calculated for C96H96Cl4N12O22Zn2·10H2O) % C 51.08 (51.88), H 4.82 (5.26), N 7.38 (7.56).(E)-5,5′-(But-2-ene-1,4-diylbis(oxy))dipicolinaldehyde (0.1 g, 0.32 mmol) and (R)-1-phenylethan-1-amine (0.08 g, 0.63 mmol) were dissolved in methanol with FeII chloride (0.03 g, 0.21 mmol). The solution was stirred at reflux (75 °C) for 48 h and all volatiles were removed under reduced pressure to yield a dark purple solid, ΛFe-[Fe2L2c3]Cl4·9H2O. Yield 0.388 g, 97%. 1H NMR (400 MHz, 298 K, MeOD) δH 8.80 (6H, s, CHN), 7.47 (6H, s), 7.13 (6H, t, 3JHH = 7.0 Hz), 7.04 (12H, t, 3JHH = 7.0 Hz), 6.64 (12H, d, 3JHH = 7.0 Hz), 6.44 (6H, s), 6.05 (6H, s, Ar), 5.26 (6H, q, 3JHH = 6.0 Hz, CH), 4.65 (12H, s, CH2), 4.59 (6H, br s, CH), 1.99 (18H, d, 3JHH = 6.0 Hz, CH3). 13C{1H} NMR (101 MHz, 298 K, MeOD) δC 171.0 (CHN), 158.8, 152.5, 144.8, 141.7, 131.2, 130.1, 129.8, 128.5, 125.5 (Ar), 121.0 (CH), 70.0 (CH2), 69.6 (CH), 26.1 (CH3). MS (ESI) m/z 406 [Fe2L3]4+, 505 [L + H].IR ν cm−1 3352 br s, 2970 br s, 1589 s, 1557 w, 1488 w, 1381 m, 1299 s, 1067 m, 1028 m, 760 m, 699 s, 562 w. Elemental analysis found (calculated for C96H96Cl4Fe2N12O6·9H2O) % C 59.92 (59.76), H 5.84 (5.96), N 8.63 (8.71).
Molecular modelling
Models of a number of possible conformers of ΛZn-[Zn2L2e3][ClO4]4 were constructed and optimised. Starting points for geometry optimisations were taken from crystallographic data. Monometallic structures were first optimised using the B3LYP-D3(BJ)40 functional and the 6–31g* basis set, with convergence criteria of 0.0001 a.u. as implemented in the Firefly quantum chemistry package,41 which is partially based on the GAMESS(US) source code.42 Bimetallic systems were optimised using ligand field molecular mechanics (LFMM)43 as implemented in the DommiMOE program,44 before being annealed at 500 K for 1 ns prior to re-optimisation. Single point energy calculations of all structures were performed using the B3LYP-D3(BJ)40 functional and the deff2-TZVP basis set with energy convergence criteria of 0.0001 a.u. as implemented in the Firefly quantum chemistry package.41 The calculations were conducted by employing the RIJCOSX approximation with SCF convergence criteria set to ‘tight’, both of which are defined internally as part of the ORCA DFT quantum chemistry package.45 Where relevant, acetonitrile solvate correction was performed using the conductor-like screening model (COSMO)46 as implemented in ORCA.45Biological activity
MIC values were established using a macro broth dilution method in cation-adjusted Müller–Hinton (MH) broth. 96-well plates (200 μL of 128 μg mL−1, 64 μM) complex in MH broth, diluted 2n μg mL−1, inoculated with each bacterial strain (103 cfu mL−1) were sealed and growth was monitored over 20 h at 37 °C with an iEMS 96-well plate reader (see ESI†).IC50 values were determined by incubating cells in 96-well plates (2.0 × 103 cells per well) for 24 h at 37 °C, 5% CO2 prior to drug exposure. Compounds were added (100 μM to 5 nM in cell medium) for a further 96 h. 3-(4,5-Dimethylthiazol-1-yl)-2,5-diphenyl tetrazolium bromide solution (0.5 mg mL−1, 20 μL per well) was added for a final 4 h. Upon completion all solutions were aspirated, dimethyl sulfoxide (150 μl) was added and absorbance (540 nm) was recorded with a Thermo Scientific Multiskan EX microplate photometer.
Oral toxicity was established by feeding cohorts of Manduca sexta31 one-day-old neonate larvae with each flexicate (25 μg mL−1 in artificial wheat germ diet) for 7 d at 28 °C and weighing to assess growth rate. Systemic toxicity assays47 were conducted by injecting an ethanol (70% v/v) swabbed region of first day fifth instar M. sexta larvae with each flexicate (0.5 mg mL−1 [0.25 μM] in PBS), before allowing them to continue feeding for 7 d at 28 °C, using physical stimulus to assess their status.
Mode of action
Denaturation of ct-DNA was measured by mixing ct-DNA (0.5 mg mL−1, 7.5 × 10−5 per base, as determined by absorbance at 200 nm) with each complex (7.5 μM) in buffered conditions (10 mM tris, 1 mM EDTA at pH 7.0) to give 10 base: 1 complex. The absorbance at 260 nm as a function of temperature (every 1 °C, 25–90 °C) was measured in a 1 cm masked quartz cuvette at a rate of 0.4 °C min−1 and run in triplicate. Tm was calculated from the first derivative of a Boltzmann sigmoidal fit of the plot of absorbance at 260 nm against temperature for each complex.Induction of apoptosis was determined by incubating HCT116 p53+/+ cells (5 × 105 cells/flask, 10 mL complete RPMI-1640 medium) for 24 h at 37 °C in 5% CO2, before treating with each flexicate (20 μM in fresh complete media for 48 h) or fresh media containing no drug (control). The supernatant containing any non-adhered, floating cells was then collected and pooled with cells harvested by trypsinisation. This pooled single cell suspension was washed twice with PBS and incubated with propidium iodide and Annexin-V-FLUOS (Roche) to stain apoptotic cells in accordance with the manufacturer's instructions. The proportion of early stage apoptotic cells and late stage apoptotic/necrotic cells were then quantified by flow cytometry as previously described.37,38
Conclusions
Our approach to metallohelix assembly has allowed us to generate a panel of biologically-compatible enantiomers incorporating various bridging groups. This was possible because in this so-called flexicate platform the stereochemistry of the metal complex units is predetermined very efficiently and largely independently of the bridges, and by a mechanism that also provides water-compatibility.23,48 In contrast, in a conventional “helication” approach the bridging units are structure-determining, so a mechanism of stereoselection would need to be designed for each example. A further advantage of the flexicate platform is beginning to emerge in that we may be able to develop asymmetric molecules from symmetric ligands via the kinds of conformational abnormalities caused by bridges that partially oppose the predetermined stereochemistry e.g.L2e. We have already shown that asymmetric (as opposed to merely chiral) optically pure assemblies are available using directional ligands.49 Further, this modular self-assembling system will allow us to probe the effects of peripheral functionality and lipophilicity.The activity of these new assemblies against cancer cells is strongly dependent on structure, with a range of potencies from 30 μM to as low as 40 nM. The most active compound ΔFe-[Fe2L2c3]Cl4 shows a selectivity index (versus healthy cell lines) approaching 103, demonstrating superiority over the clinically used anticancer drug cisplatin in vitro (SI < 1). This selectivity is substantiated in tests with various models; bacteria and amoeba exposed to high concentrations were essentially unaffected, and in Manduca sexta larvae, where the systemic stability of the drug is evidenced, there is arguably a pro-biotic effect i.e. the insects appear to thrive.
In respect of mechanism or mode of action, the lack of binding to DNA indicates that this is unlikely to be the general target in this panel. In fact only one early flexicate19 ([Fe2L13]4+, Fig. 1) in our growing library shows significant interactions with nucleic acids, and while there are fascinating selectivities with various motifs19,32,33 there is no DNA damage akin to that induced by e.g. platinum drugs and alkylators.34,49 Instead, relevant examples of protein interaction and enzyme inhibition have been characterised.21,32 To achieve drug safety and cancer selectivity, mechanistic classes which do not involve induction of DNA damage are attractive, and this may well be the source of the excellent selectivities we describe in this manuscript. Mode of action studies indicate that these compounds can induce substantial cell death by apoptosis independent of any DNA damage. Extensive studies are now required to understand how this complex process, normally subverted in cancers, is induced by these compounds. The above observations of remarkable selectivity alongside very high potency and large enantiomeric differences are however all consistent with a subtle mechanism involving the targeting of oncogenic drivers.
Acknowledgements
We thank The University of Warwick and Institute of Advanced Study for support of this work. ADF and DHS were supported by EPSRC doctoral studentships.Notes and references
- A. Persidis, Nat. Biotechnol., 1999, 17, 94–95 CrossRef CAS PubMed.
- Y. Cao, R. A. DePinho, M. Ernst and K. Vousden, Nat. Rev. Cancer, 2011, 11, 749–754 CrossRef CAS PubMed.
- H. M. W. Verheul and H. M. Pinedo, Nat. Rev. Cancer, 2007, 7, 475–485 CrossRef CAS PubMed.
- C. E. Meacham and S. J. Morrison, Nature, 2013, 501, 328–337 CrossRef CAS PubMed.
- U. S. Eggert, Nat. Chem. Biol., 2013, 9, 206–209 CrossRef CAS PubMed.
- J. A. Lee, M. T. Uhlik, C. M. Moxham, D. Tomandl and D. J. Sall, J. Med. Chem., 2012, 55, 4527–4538 CrossRef CAS PubMed.
- F. Sams-Dodd, Drug Discovery Today, 2013, 18, 211–217 CrossRef PubMed.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed.
- J. G. Moffat, J. Rudolph and D. Bailey, Nat. Rev. Drug Discovery, 2014, 13, 588–602 CrossRef CAS PubMed.
- J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS.
- J. M. C. A. Kerckhoffs, J. C. Peberdy, I. Meistermann, L. J. Childs, C. J. Isaac, C. R. Pearmund, V. Reudegger, S. Khalid, N. W. Alcock, M. J. Hannon and A. Rodger, Dalton Trans., 2007, 734–742 RSC.
- J. Malina, M. J. Hannon and V. Brabec, Nucleic Acids Res., 2008, 36, 3630–3638 CrossRef CAS PubMed.
- A. D. Richards, A. Rodger, M. J. Hannon and A. Bolhuis, Int. J. Antimicrob. Agents, 2009, 33, 469–472 CrossRef CAS PubMed.
- L. Cardo, V. Sadovnikova, S. Phongtongpasuk, N. J. Hodges and M. J. Hannon, Chem. Commun., 2011, 47, 6575–6577 RSC.
- H. Yu, M. Li, G. Liu, J. Geng, J. Wang, J. Ren, C. Zhao and X. Qu, Chem. Sci., 2012, 3, 3145–3153 RSC.
- S. E. Howson and P. Scott, Dalton Trans., 2011, 40, 10268–10277 RSC.
- R. A. Kaner and P. Scott, Future Med. Chem., 2015, 7, 1–4 CrossRef CAS PubMed.
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. van Gorkum and P. Scott, Chem. Commun., 2009, 1727–1729 RSC.
- S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2012, 4, 31–36 CrossRef CAS PubMed.
- V. Brabec, S. E. Howson, R. A. Kaner, R. M. Lord, J. Malina, R. M. Phillips, Q. M. A. Abdallah, P. C. McGowan, A. Rodger and P. Scott, Chem. Sci., 2013, 4, 4407–4416 RSC.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS PubMed.
- M. Seredyuk, A. B. Gaspar, V. Ksenofontov, Y. Galyametdinov, J. Kusz and P. Gütlich, J. Am. Chem. Soc., 2008, 130, 1431–1439 CrossRef CAS PubMed.
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. J. Deeth, A. D. Faulkner, D. H. Simpson and P. Scott, Dalton Trans., 2011, 40, 10416–10433 RSC.
- S. Pathak, M. J. Siciliano, R. Cailleau, C. L. Wiseman and T. C. Hsu, J. Natl. Cancer Inst., 1979, 62, 263–271 CAS.
- F. Bunz, P. M. Hwang, C. Torrance, T. Waldman, Y. Zhang, L. Dillehay, J. Williams, C. Lengauer, K. W. Kinzler and B. Vogelstein, J. Clin. Invest., 1999, 104, 263–269 CrossRef CAS PubMed.
- T. Soussi and C. Beroud, Nat. Rev. Cancer, 2001, 1, 233–239 CrossRef CAS PubMed.
- K. C. Dunn, A. E. Aotaki-Keen, F. R. Putkey and L. M. Hjelmeland, Exp. Eye Res., 1996, 62, 155–170 CrossRef CAS PubMed.
- A. D. Kennedy, M. Otto, K. R. Braughton, A. R. Whitney, L. Chen, B. Mathema, J. R. Mediavilla, K. A. Byrne, L. D. Parkins, F. C. Tenover, B. N. Kreiswirth, J. M. Musser and F. R. DeLeo, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1327–1332 CrossRef CAS PubMed.
- A. D. Kennedy, S. F. Porcella, C. Martens, A. R. Whitney, K. R. Braughton, L. Chen, C. T. Craig, F. C. Tenover, B. N. Kreiswirth, J. M. Musser and F. R. DeLeo, J. Clin. Microbiol., 2010, 48, 4504–4511 CrossRef CAS PubMed.
- J. M. Andrews, J. Antimicrob. Chemother., 2001, 48(1), 5–16 CrossRef CAS PubMed.
- S. E. Reynolds, S. F. Nottingham and A. E. Stephens, J. Insect Physiol., 1985, 31, 119–127 CrossRef.
- J. Malina, P. Scott and V. Brabec, Nucleic Acids Res., 2015 DOI:10.1093/nar/gkv438.
- J. Malina, P. Scott and V. Brabec, Dalton Trans., 2015, 44, 14656–14665 RSC.
- V. Brabec, S. E. Howson, R. A. Kaner, R. M. Lord, J. Malina, R. M. Phillips, Q. M. A. Abdallah, P. C. McGowan, A. Rodger and P. Scott, Chem. Sci., 2013, 4, 4407–4416 RSC.
- S. W. G. Tait, G. Ichim and D. R. Green, J. Cell Sci., 2014, 127, 2135–2144 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- S. J. Allison and J. Milner, Cell Cycle, 2007, 6, 2669–2677 CrossRef CAS PubMed.
- S. J. Allison, J. R. P. Knight, C. Granchi, R. Rani, F. Minutolo, J. Milner and R. M. Phillips, Oncogenesis, 2014, 3, e102 CrossRef CAS PubMed.
- Apoptosis, Cytotoxicity and Cell Proliferation, 4th edn, ed. Hans-Jürgen Rode, Roche Diagnostics GmbH, Mannheim, Germany, 2008 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. A. Granovsky, Firefly version 7.1.G, http://www.classic.chem.msu.su/gran/firefly/index.html Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- R. J. Deeth, A. Anastasi, C. Diedrich and K. Randell, Coord. Chem. Rev., 2009, 253, 795–816 CrossRef CAS.
- R. J. Deeth, N. Fey and B. Williams–Hubbard, J. Comput. Chem., 2005, 26, 123–130 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- P. J. Daborn, N. Waterfield, C. P. Silva, C. P. Y. Au, S. Sharma and R. H. ffrench-Constant, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10742–10747 CrossRef CAS PubMed.
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. van Gorkum and P. Scott, Chem. Commun., 2009, 1727–1729 RSC.
- A. D. Faulkner, R. A. Kaner, Q. M. A. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797–803 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, syntheses, biophysical analyses, antimicrobial, anticancer, toxicity, and mechanistic studies. See DOI: 10.1039/c5sc03677a |
| This journal is © The Royal Society of Chemistry 2016 |