
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quasi-type II CuInS2/CdS core/shell quantum dots†
Kaifeng
Wu‡
a,
Guijie
Liang‡
b,
Degui
Kong
c,
Jinquan
Chen
a,
Zheyuan
Chen
a,
Xinhe
Shan
a,
James R.
McBride
d and
Tianquan
Lian
*a
aDepartment of Chemistry, Emory University, 1515 Dickey Drive, NE, Atlanta, Georgia 30322, USA. E-mail: tlian@emory.edu
bHubei Key Laboratory of Low Dimensional Optoelectronic Materials and Devices, Hubei University of Arts and Science, Xiangyang 441053, Hubei Province, P. R. China
cCollege of Electronic Engineering, Heilongjiang University, Harbin 150080, P. R. China
dDepartment of Chemistry, The Vanderbilt Institute of Nanoscale Science and Engineering, Vanderbilt University, Nashville TN 37235, USA
First published on 12th November 2015
Abstract
Ternary chalcopyrite CuInS2 quantum dots (QDs) have been extensively studied in recent years as an alternative to conventional QDs for solar energy conversion applications. However, compared with the well-established photophysics in prototypical CdSe QDs, much less is known about the excited properties of CuInS2 QDs. In this work, using ultrafast spectroscopy, we showed that both conduction band (CB) edge electrons and copper vacancy (VCu) localized holes were susceptible to surface trappings in CuInS2 QDs. These trap states could be effectively passivated by forming quasi-type II CuInS2/CdS core/shell QDs, leading to a single-exciton (with electrons delocalized among CuInS2/CdS CB and holes localized in VCu) half lifetime of as long as 450 ns. Because of reduced electron–hole overlap in quasi-type II QDs, Auger recombination of multiple excitons was also suppressed and the bi-exciton lifetime was prolonged to 42 ps in CuInS2/CdS QDs from 10 ps in CuInS2 QDs. These demonstrated advantages, including passivated trap states, long single and multiple exciton lifetimes, suggest that quasi-type II CuInS2/CdS QDs are promising materials for photovoltaic and photocatalytic applications.
Introduction
Ternary chalcopyrite CuInS2 has a direct bulk bandgap of 1.5 eV,1 which matches well with the solar spectrum for photovoltaic or photocatalytic applications.2–6 The absorption and emission of colloidal CuInS2 quantum dots (QDs) can cover most of the visible and near IR range by tuning their sizes.7–9 Moreover, recently developed non-injection and high chemical yield colloidal syntheses of mono-dispersed CuInS2 QDs are promising for scaled-up production in industry.4,9 For these reasons, CuInS2 QDs have been extensively studied in recent years as an alternative to conventional II–VI CdX (X = S, Se, Te) QDs. CuInS2 QDs sensitized solar cells (QDSSCs) with power conversion efficiencies higher than 5% have been reported,3,6,10 exceeding those of CdSe QDSSCs.11 The radiative lifetime in CuInS2 QDs is found to be surprisingly long (∼300 ns),4,12–17 which was generally attributed to slow recombination between the conduction band (CB) edge electrons and holes trapped in defect states likely associated with Cu vacancies (VCu).4,17,18 Additionally, there have been reports about other defects,9,19 such as sulfur vacancy (VS), indium vacancy (VIn), interstitial copper (Cui), copper site substituted by indium (InCu), and indium site substituted by copper (CuIn). These off-stoichiometry defects, typical for CuInS2 because of its ternary chemical composition,19 make its excited state dynamics more complicated and much less well understood than prototypical CdSe QDs.20–23In addition to off-stoichiometry defects in the bulk, surface states are also found to play a significant role in carrier trapping for CuInS2 QDs,4,16,24 which reduces charge separation efficiencies in related photovoltaic and photocatalytic devices. Coating QDs with another material to form core/shell hetero QDs has been an effective approach to mitigating surface trapping states.25,26 Furthermore, core/shell QDs can lead to new properties that cannot be achieved in single component QDs.25,27–33 Depending on the relative alignment of the CB and valence band (VB) positions of the core and shell materials, core/shell QDs can be type I (in which the CB and VB of the core are nested between those of the shell25,29), type II (when they are staggered with respect to each other34,35), or quasi-type II (when either their CB or VB band edge positions are similar26,36,37). For CuInS2 QDs, it has been demonstrated that surface trapping can be suppressed and therefore photoluminescence (PL) quantum yields (QYs) can be strongly enhanced by coating with ZnS or CdS shells.4,16,24,38 It is well known that CuInS2/ZnS QDs have type I band alignment where both the lowest energy electron and hole wavefunctions are confined in the core so that the effect of surface states is reduced. However, for photovoltaic and photocatalytic applications, (quasi-)type II QDs are more suitable because their (partially) separated electron and hole wavefunctions can significantly prolong single and multiple exciton lifetimes.35,39,40 The CB and VB offsets between CuInS2 and CdS are ∼0.05 eV and 0.95 eV (Scheme 1),41 respectively. In principle, these offsets allow the formation of quasi-type II core/shell QDs where holes are well confined in CuInS2 and electrons are delocalized among CuInS2 and CdS (Scheme 1).
 | ||
| Scheme 1 (Left) schematic structure and (right) quasi-type II band alignment in CuInS2/CdS core/shell QDs. Bulk band edges of CuInS2 and CdS (gray dashed lines), electron and hole energy levels in the core and shell (black solid lines), hole trapping levels associated with Cu vacancy (VCu, green dashed line), lowest energy transitions in the CdS (B1) and CuInS2 (B2). In the lowest excited state, electron and hole wavefunctions are delocalized among core and shell and localized in the core (VCu), respectively. | ||
In this work, we present the first direct evidence for quasi-type II band alignment in CuInS2/CdS core/shell QDs and demonstrate their advantages for light harvesting and charge separation through comparison with core-only CuInS2 QDs. We show that both CB edge electrons and VCu localized holes were susceptible to surface trapping in CuInS2 QDs and these traps can be efficiently suppressed in CuInS2/CdS QDs to achieve a long-lived single exciton state (with electrons delocalized among CuInS2/CdS CB and holes localized in VCu) with a half lifetime as long as 450 ns. Bi-exciton lifetime was prolonged from 10 ps in CuInS2 QDs to 42 ps in CuInS2/CdS QDs due to reduced electron–hole interactions in such quasi-type II QDs.
Sample preparation and characterizations
CuInS2 (CIS) core and CuInS2/CdS (CIS/CdS) core/shell QDs were synthesized using a literature-reported method.4 Details can be found in the ESI.†Fig. 1a and b are representative Transmission Electron Microscopy (TEM) images of CIS core and CIS/CdS core/shell QDs, respectively. The CIS core exhibited typical tetrahedral shapes and the average size or edge length (standard deviations) was 3.34 (±0.95) nm. The size distribution histogram of CIS core is shown in Fig. S1a.† After CdS shell growth, the average size of core/shell QDs increased to 4.25 (±1.22) nm (Fig. S1b†). According to previous studies, CIS cores were possibly partially etched during shell growth4 and therefore we were not able to determine the core and shell sizes in our core/shell QDs. In the ESI,† we provided additional characterizations of the core/shell QDs using high-angle annular dark-field (HAADF) scanning TEM (Fig. S2†) and energy-dispersive X-ray spectroscopy (EDX, Fig. S3†). The EDX images showed spatially overlapped Cu, In, S, and Cd elements, consistent with uniform shell formation. Unfortunately, the differentiation between core and shell was still hindered by limited spatial resolution (∼0.5 nm) of EDX measurements, overlapping cadmium and indium X-ray peaks, and by the rate of electron beam-induced damage.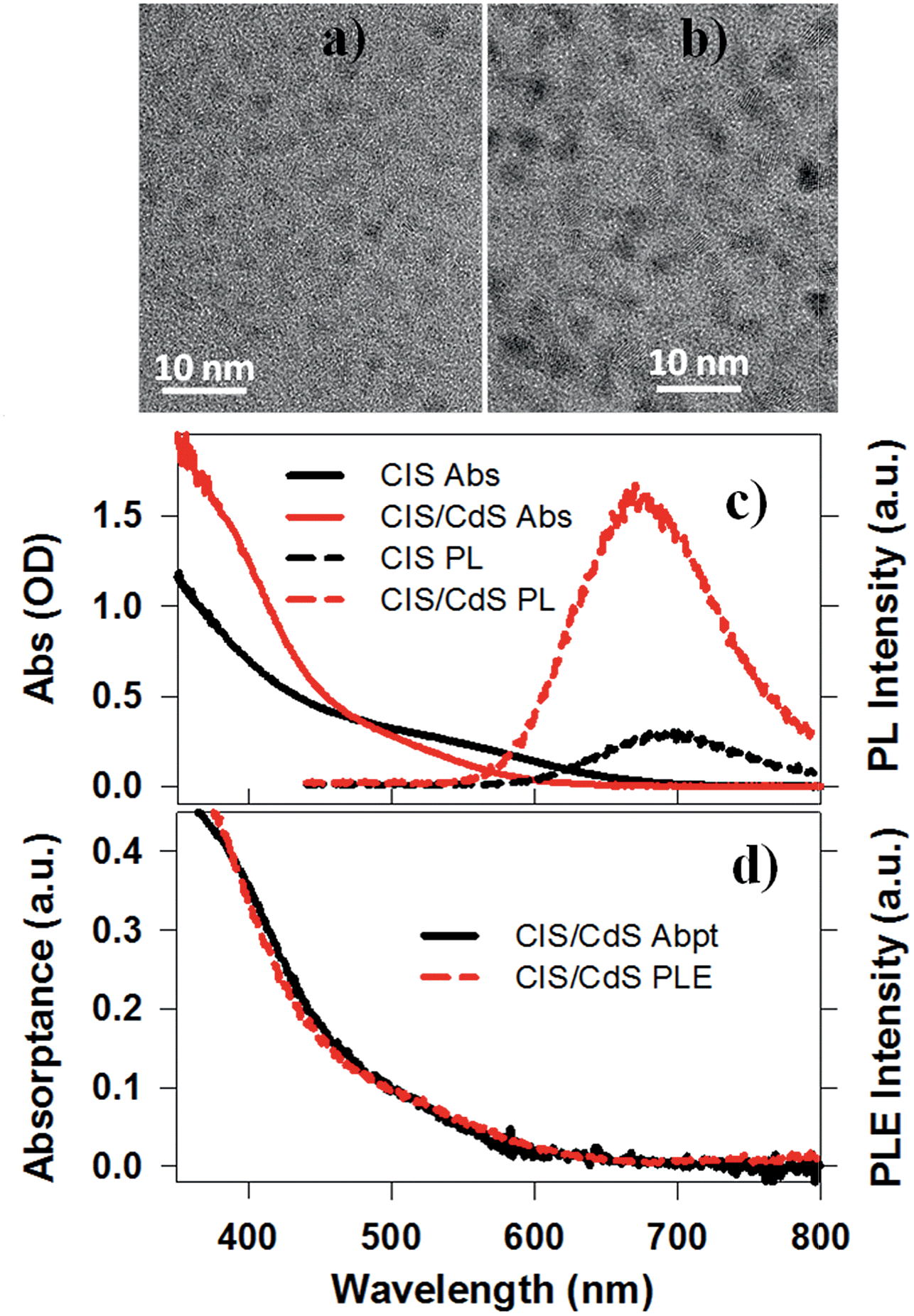 | ||
| Fig. 1 TEM images and absorption, PL and PLE spectra. Representative TEM images of (a) CuInS2 core, and (b) CuInS2/CdS core/shell QDs. (c) Absorption (solid lines) and photoluminescence (PL, dashed lines) spectra of CuInS2 core (black lines) and CuInS2/CdS core/shell QDs (red lines) dispersed in heptane. (d) Comparison between normalized absorptance (black solid line) and photoluminescence excitation (PLE, red dashed line) spectra of CuInS2/CdS core/shell QDs. They were normalized at the low energy side (500–600 nm). | ||
The UV-vis absorption spectra of CIS core and CIS/CdS core/shell QDs dispersed in heptane are shown in Fig. 1c. Unlike typical CdSe QDs, discrete exciton peaks were not observed for CIS. In ternary CIS QDs, there exist heterogeneous distributions of nanocrystal shapes, tetragonal lattice distortions, and compositional off-stoichiometry,9 all of which can contribute to inhomogeneous broadening of the exciton bands. As will be discussed below (Fig. 3), the lowest energy exciton peaks at ∼580 nm and ∼520 nm for CIS core and CIS/CdS core/shell QDs, respectively, can be clearly observed in transient absorption spectra. The blue shift of the exciton peak in CIS/CdS core/shell QDs could result from core etching during shell coating or alloying and cation exchange between core and shell, consistent with previous observations.4,42,43 If the core size and composition had remained unchanged during the shell growth, a red-shift should have been observed due to a reduction in the confinement energy.25,26 In addition to the blue-shift, the CIS/CdS QDs showed enhanced absorption in at <450 nm, which can be attributed to the absorption of the CdS shell.
Photoluminescence (PL) spectra of CIS and CIS/CdS QDs measured with 400 nm excitation are also shown in Fig. 1c. The CIS QDs showed a PL peak at ∼690 nm, which was red-shifted from the absorption peak (580 nm) by 0.34 eV. This Stokes-shift was almost an order of magnitude larger than in CdSe QDs and could be attributed to radiative recombination between CB electrons and VCu localized holes above the VB edge.4,17,18 The PL peak of CIS/CdS QDs (∼670 nm) was also blue-shifted from that of CIS QDs, consistent with absorption spectrum changes. As shown in Fig. 1d, the PL excitation (PLE) spectrum of CIS/CdS QDs (monitored the emission at 670 ± 1 nm), matched well with the absorptance spectrum (representing the percentage of absorbed photons), indicating that the PL quantum efficiency is independent on excitation wavelength. This result suggests that there was a negligible contribution of isolated CdS QDs and there was negligible exciton trapping at the CdS shell in CdSe/CdS QDs.44,45
Ultrafast spectroscopic studies
To identify the band alignment in CIS/CdS QDs, we used a pump wavelength at 490 nm, which selectively excited the CIS core but avoided excitation of CdS shell.36,50 The resultant TA spectra (Fig. 2a) showed a bleach of the lowest energy exciton band of CIS core at ∼520 nm (labelled as B2) and an additional bleach of the exciton band in CdS shell at ∼400 nm (labelled as B1). It has been well established experimentally that exciton bleach in CdS QDs is dominated by state filling of CB electron levels.20,52 Similar spectral assignment was found to be applicable to CIS QDs4 and we have confirmed this assignment by showing that the bleach features could be completely removed in the presence of electron acceptors (see Fig. S10, ESI† for details). As shown in Fig. 2b, the kinetics of B1 and B2 match well with each other, showing a half-life of ∼600 ns. The fitting parameters are listed in Table S1.† The simultaneous formation of long-lived B1 and B2 bleaches with the same decay kinetics after selective excitation of B2 can only be explained by a quasi-type II band alignment indicated in Scheme 1: the lowest electron level is degenerate among the CIS core and CdS shell. The energy difference between the B1 and B2 transitions (∼0.62 eV) reflects the large VB edge offset in these materials.
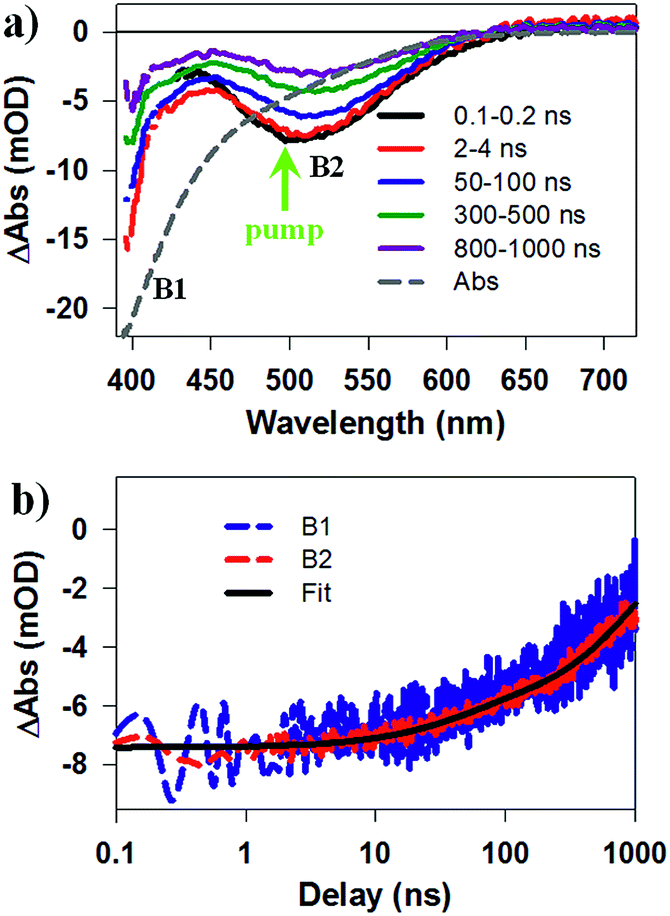 | ||
| Fig. 2 Transient Absorption (TA) spectra and kinetics of CuInS2/CdS core/shell QDs measured at 490 nm excitation. (a) TA spectra at indicated time delays (from 0.1 to 1000 ns). Static absorption spectrum (gray dashed line) is inverted and scaled for comparison. B1, B2 and excitation wavelength (green arrow) are labeled. (b) Comparison of TA kinetics of B1 (blue dashed line) and B2 (red dashed line) features and their multi-exponential fit (black solid line). | ||
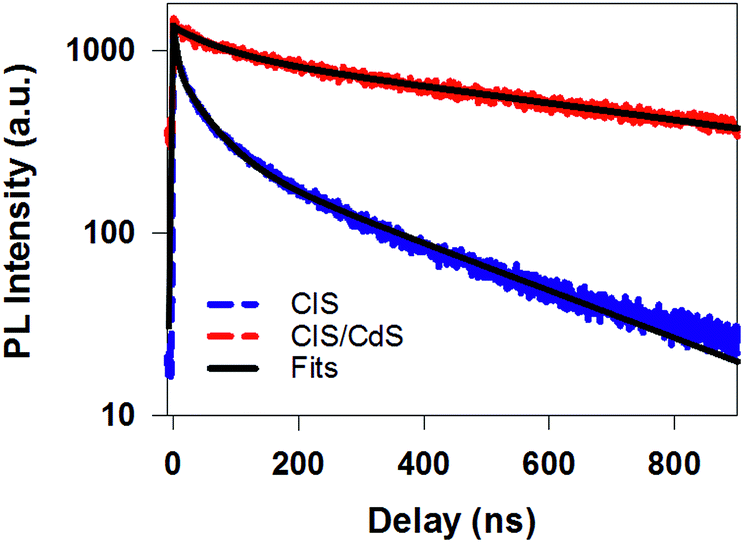 | ||
| Fig. 3 Time-resolved PL decay kinetics for CuInS2 core (blue dashed line) and CuInS2/CdS core/shell QDs (red dashed line) measured with 400 nm excitation. Their multi-exponential fits are shown in black solid lines and fitting parameters are listed in Table S1.† | ||
While PL decay can be caused by both electron or hole dynamics, TA can be used to investigate electron and/or hole dynamics separately.53 TA spectra of CIS QDs (Fig. 4a) measured with 400 nm excitation were dominated by an exciton bleach feature at ∼580 nm (XB) that can be attributed to state filling of the lowest energy CB electron level. TA spectra of CIS/CdS QDs (Fig. 4b) measured with 400 nm excitation showed B1 and B2 bleach features at ∼400 nm and 520 nm, respectively, consistent with those measured at 490 nm excitation (Fig. 2a). For both CIS and CIS/CdS QDs, there is an additional photoinduced absorption (PA) feature to the red of bleach features. The kinetics of PA is the same as the exciton bleach features in both samples (Fig. S4†). Similar broad and featureless PA signals have also been observed in many QDs, including CdSe,54–56 CdS,36 InP,50,57 and Cd3P2,58 and can be attributed to either holes or electrons or both. In Fig. 4c, we compared the kinetics of XB bleach recovery in CIS QDs with PL decay kinetics by normalizing them at long delay time (>100 ns), when the decay kinetics are the same and are dominated by recombination of CB electron with the trapped hole. The comparison showed that the PL decay was faster than exciton bleach in the time range of ∼1–100 ns. Since exciton bleach is determined by the CB electron, the fast components in its recovery kinetics suggest the presence of electron trapping processes. Furthermore, we attributed the much faster PL decay to hole trapping process in CIS QDs. There might exist hole trapping processes on faster time scale but not resolved due to the limited instrument response function (IRF ∼ 500 ps) of PL decay measurements. Our combined TA and PL decay measurements on CIS QDs revealed for the first time that both the CB edge electrons and the localized holes in Vcu are susceptible to surface trappings.
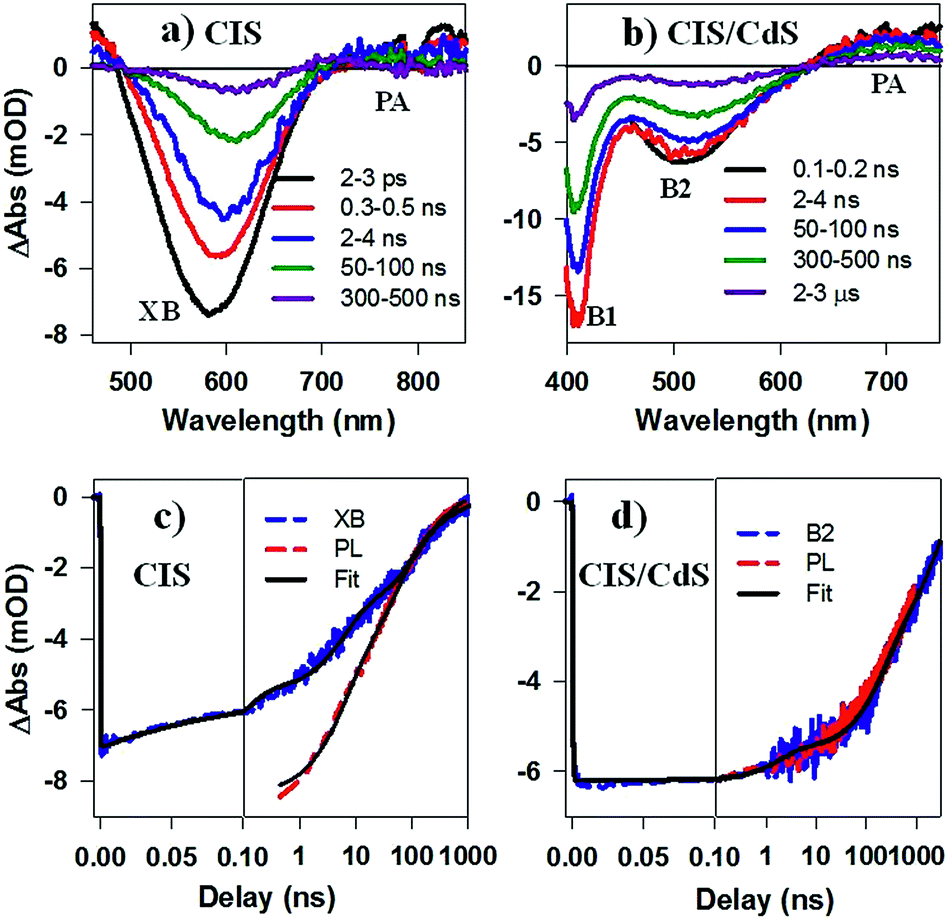 | ||
| Fig. 4 TA spectra of (a) CuInS2 core, and (b) CuInS2/CdS core/shell QDs at indicated delays after 400 nm excitation. Comparison of the kinetics of exciton bleach recovery (XB or B2, blue dashed line) and PL decay (red dashed lines) in (c) CuInS2 core, and (d) CuInS2/CdS core/shell QDs. Their multi-exponential fits are shown in black solid lines. | ||
The B2 kinetics in CIS/CdS QDs measured with 400 nm excitation is shown in Fig. 4d. B1 and B2 showed the same kinetics and were the same as those measured at 490 nm excitation (Fig. S5†). Compared with XB kinetics in CIS QDs (half-life 9.8 ± 0.6 ns), B2 in CIS/CdS QDs is much longer lived (half-life 450 ± 20 ns), suggesting both effective passivation of electron traps due to shell growth and reduction of electron–hole recombination rates due to quasi-type II band alignment. Moreover, B2 bleach and PL decay kinetics in CIS/CdS QDs matched well with each other, suggesting that nearly all the hole trapping processes have been removed and the only decay channel for holes was through recombination with electrons, radiatively or nonradiatively. Thus, our results provided direct evidence for the passivation of both electron and hole traps in CIS/CdS QDs through the growth of CdS shell.
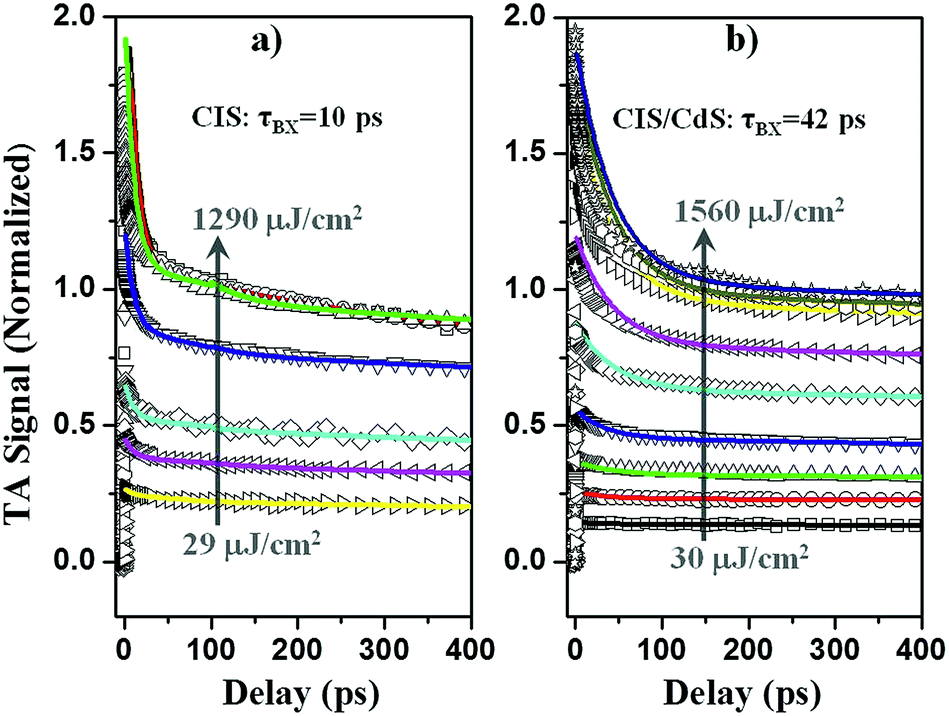 | ||
| Fig. 5 Normalized exciton bleach TA kinetics (open symbols) at different excitation intensities for (a) CuInS2 core, and (b) CuInS2/CdS core/shell QDs. The solid lines are fits to a multicarrier annihilation model described in the main text. | ||
To determine multiple exciton AR rates, we first quantify the number of initially excited excitons in the QDs at different excitation powers by analyzing the power dependence of the XB signal amplitude. It is assumed that the probability of a QD absorbing n photons, f(n), follows the Poisson distribution: f(n) = wne−w/n!, where w, the average number of excitons per QD, is proportional to the excitation density.20,36,62,63P(n, t) denotes the probability of finding QDs with n excitons at time t. At early delay time t0 ∼ 1 ps, prior to AR, P(n, t0) = f(n). At long delay time, tL = 200 ps, when AR is completed and only single exciton states remain, P(1, tL) = 1 − f(0), and P(n > 1, tL) = 0; the transient XB signal amplitude, ΔA(tL), is proportional to the number of excited QDs: ΔA(tL) = α[1 − f(0)], where α is a scaling factor. It is convenient to define a normalized transient signal at tL:36,58,63,64
| ΔS(tL) = ΔA(tL)/α = 1 − e−w | (1) |
The scaling factor α can be determined by realizing that ΔS(tL) approaches 1 at high excitation intensities (w ≫ 1), when all QDs are excited. Similarly, normalized transient signals at all delay times, ΔS(t) = ΔA(t)/α, can also be obtained and are shown in Fig. 5. The initial normalized transient bleach signal at t0 is given by:36,58,63,64
| ΔS(t0) = P(1, t0) + 2[1 − P(0, t0) − P(1, t0)] = 2 − (2 + w)e−w | (2) |
The normalized transient signal at any delay time is given by:36,58,63,64
| ΔS(t) = P(1, t) + 2[1 − P(0, t) − P(1, t)] | (3) |
The first and second terms represent the probability of single and multiple exciton (n > 1) states, respectively. Assuming that n-exciton state can only decay by sequential Auger recombination (to n − 1 exciton state with time constant τn), the time-dependent distribution of multi-exciton states in QDs is described by a set of coupled rate equations:36,59,63,65,66
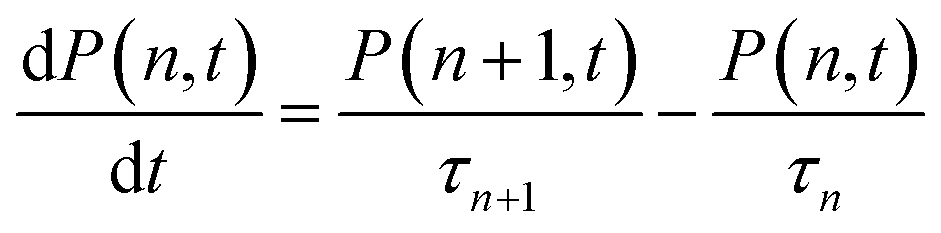 | (4) |
It has been demonstrated that the Auger recombination time of n-exciton states in QDs can be well described by a statistical scaling law: (τn−1 = n2(n − 1)τ2−1/4), where τ2 is the bi-exciton AR lifetime constant.36,59,63,67 With the initial exciton population distribution obtained from fitting Fig. S8† and the bi-exciton lifetime as the only fitting parameter, the transient kinetics in Fig. 5 can be globally fitted by eqn (3) and (4), with emphasis on the lowest four powers. At higher powers, transient kinetics deviate from the model for the following reasons: first, although stirred, some of the QDs are likely photo-charged and show additional charging induced exciton decay channels;68–70 second, when n is large, Auger lifetime of n-exciton states in QDs might not obey the simple statistical scaling law.71 From the fit, we obtain bi-exciton lifetimes of 10 ± 2 ps and 42 ± 5 ps for CIS and CIS/CdS QDs, respectively. Another simple but not as rigorous way to extract bi-exciton lifetimes is to take the difference between kinetics at two lowest powers and attribute it to bi-exciton AR process.59 The bi-exciton lifetimes obtained from this simpler procedure are similar to the fitting results described above (Fig. S9†).
Therefore, we demonstrate that bi-exciton lifetime in CIS/CdS QDs is ∼4 times longer than in CIS QDs, which can be attributed to reduced wavefunction overlap between VCu localized holes in the CIS core and delocalized electrons among the CIS core and CdS shell. Suppressed AR in QDs is significant for their many applications. For instance, when using QDs to deliver multiple electrons or holes to an adsorbed catalyst molecule for solar fuel generation, interfacial charge transfer needs to compete with AR.36,63,64,72,73 Also, QDs are often unintentionally charged in devices such as QDSSCs, for which reason electron injection yield can be compromised by AR.74 In addition, AR is generally considered to be responsible for single QD PL blinking75,76 and gain decay in QD lasing processes.77 Therefore, suppressed AR in CIS/CdS QDs can improve their performances in these applications.
Conclusions
In conclusion, we have presented a comprehensive ultrafast spectroscopic study of quasi-type II CIS/CdS core/shell QDs and demonstrated their improved light harvesting properties over CIS core only QDs. The CIS/CdS core/shell structures have a quasi-type II band alignment with a lowest energy CB electron level extending throughout the core and shell and a large VB offset. This band alignment is confirmed by the observation that the selective excitation of lowest CIS exciton band simultaneously generates long-lived CIS and CdS exciton bleaches. Through both ultrafast TA and time-resolved PL decay measurements, we show that electron and holes trapping states in CIS core are effectively passivated by the CdS shell and the single exciton state lifetime was prolonged to 450 ns as a result of reduced electron–hole overlap in quasi-type II CIS/CdS QDs. For the same reason, Auger recombination of multiple excitons was suppressed and the bi-exciton lifetime was extended to 42 ps in CIS/CdS QDs from 10 ps in CIS QDs. These findings suggest that quasi-type II CIS/CdS QDs are a promising material for photovoltaic and photocatalytic applications.Acknowledgements
T.L. acknowledge the financial support from the National Science Foundation (CHE-1309817) J.R.M. acknowledges funding by the National Science Foundation (CHE-1213758). STEM and EDS images were acquired using an FEI Tecnai Osiris electron microscope supported by the National Science Foundation (EPS-1004083).Notes and references
- T. Omata, K. Nose and S. Otsuka-Yao-Matsuo, J. Appl. Phys., 2009, 105, 073106 CrossRef.
- H. McDaniel, N. Fuke, J. M. Pietryga and V. I. Klimov, J. Phys. Chem. Lett., 2013, 4, 355–361 CrossRef CAS PubMed.
- H. McDaniel, N. Fuke, N. S. Makarov, J. M. Pietryga and V. I. Klimov, Nat. Commun., 2013, 4 Search PubMed.
- L. Li, A. Pandey, D. J. Werder, B. P. Khanal, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2011, 133, 1176–1179 CrossRef CAS PubMed.
- M. G. Panthani, C. J. Stolle, D. K. Reid, D. J. Rhee, T. B. Harvey, V. A. Akhavan, Y. Yu and B. A. Korgel, J. Phys. Chem. Lett., 2013, 4, 2030–2034 CrossRef CAS PubMed.
- Z. Pan, I. Mora-Seró, Q. Shen, H. Zhang, Y. Li, K. Zhao, J. Wang, X. Zhong and J. Bisquert, J. Am. Chem. Soc., 2014, 136, 9203–9210 CrossRef CAS PubMed.
- H. Zhong, Y. Zhou, M. Ye, Y. He, J. Ye, C. He, C. Yang and Y. Li, Chem. Mater., 2008, 20, 6434–6443 CrossRef CAS.
- R. Xie, M. Rutherford and X. Peng, J. Am. Chem. Soc., 2009, 131, 5691–5697 CrossRef CAS PubMed.
- H. Z. Zhong, S. S. Lo, T. Mirkovic, Y. C. Li, Y. Q. Ding, Y. F. Li and G. D. Scholes, ACS Nano, 2010, 4, 5253–5262 CrossRef CAS PubMed.
- J. Luo, H. Wei, Q. Huang, X. Hu, H. Zhao, R. Yu, D. Li, Y. Luo and Q. Meng, Chem. Commun., 2013, 49, 3881–3883 RSC.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CAS.
- Y.-K. Kim, S.-H. Ahn, K. Chung, Y.-S. Cho and C.-J. Choi, J. Mater. Chem., 2012, 22, 1516–1520 RSC.
- V. K. Komarala, C. Xie, Y. Wang, J. Xu and M. Xiao, J. Appl. Phys., 2012, 111, 124314 CrossRef.
- A. Shi, X. Wang, X. Meng, X. Liu, H. Li and J. Zhao, J. Lumin., 2012, 132, 1819–1823 CrossRef CAS.
- J. Seo, S. Raut, M. Abdel-Fattah, Q. Rice, B. Tabibi, R. Rich, R. Fudala, I. Gryczynski, Z. Gryczynski, W.-J. Kim, S. Jung and R. Hyun, J. Appl. Phys., 2013, 114, 094310 CrossRef.
- J. Sun, D. Zhu, J. Zhao, M. Ikezawa, X. Wang and Y. Masumoto, Appl. Phys. Lett., 2014, 104, 023118 CrossRef.
- K. E. Knowles, H. D. Nelson, T. B. Kilburn and D. R. Gamelin, J. Am. Chem. Soc., 2015, 137, 13138–13147 CrossRef CAS PubMed.
- W. D. Rice, H. McDaniel, V. I. Klimov and S. A. Crooker, J. Phys. Chem. Lett., 2014, 5, 4105–4109 CrossRef CAS PubMed.
- W. Liu, Y. Zhang, J. Zhao, Y. Feng, D. Wang, T. Zhang, W. Gao, H. Chu, J. Yin, Y. Wang, J. Zhao and W. W. Yu, J. Lumin., 2015, 162, 191–196 CrossRef CAS.
- V. I. Klimov, J. Phys. Chem. B, 2000, 104, 6112–6123 CrossRef CAS.
- V. I. Klimov, Annu. Rev. Phys. Chem., 2007, 58, 635–673 CrossRef CAS PubMed.
- D. J. Norris and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16338–16346 CrossRef CAS.
- D. J. Norris, A. L. Efros, M. Rosen and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16347–16354 CrossRef CAS.
- R. Bose, G. H. Ahmed, E. Alarousu, M. R. Parida, A. L. Abdelhady, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. C, 2015, 119, 3439–3446 CAS.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468–471 CrossRef CAS.
- J. J. Li, Y. A. Wang, W. Z. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. G. Peng, J. Am. Chem. Soc., 2003, 125, 12567–12575 CrossRef CAS PubMed.
- D. J. Milliron, S. M. Hughes, Y. Cui, L. Manna, J. Li, L.-W. Wang and A. Paul Alivisatos, Nature, 2004, 430, 190–195 CrossRef CAS PubMed.
- P. Reiss, M. Protière and L. Li, Small, 2009, 5, 154–168 CrossRef CAS PubMed.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019–7029 CrossRef CAS.
- G. D. Scholes, Adv. Funct. Mater., 2008, 18, 1157–1172 CrossRef CAS.
- F. Shieh, A. E. Saunders and B. A. Korgel, J. Phys. Chem. B, 2005, 109, 8538–8542 CrossRef CAS PubMed.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2009, 43, 190–200 CrossRef PubMed.
- S. Kim, B. Fisher, H.-J. Eisler and M. Bawendi, J. Am. Chem. Soc., 2003, 125, 11466–11467 CrossRef CAS PubMed.
- S. A. Ivanov, A. Piryatinski, J. Nanda, S. Tretiak, K. R. Zavadil, W. O. Wallace, D. Werder and V. I. Klimov, J. Am. Chem. Soc., 2007, 129, 11708–11719 CrossRef CAS PubMed.
- H. Zhu, N. Song, W. Rodríguez-Córdoba and T. Lian, J. Am. Chem. Soc., 2012, 134, 4250–4257 CrossRef CAS PubMed.
- O. Chen, J. Zhao, V. P. Chauhan, J. Cui, C. Wong, D. K. Harris, H. Wei, H.-S. Han, D. Fukumura, R. K. Jain and M. G. Bawendi, Nat. Mater., 2013, 12, 445–451 CrossRef CAS PubMed.
- K. E. Knowles, T. B. Kilburn, D. G. Alzate, S. McDowall and D. R. Gamelin, Chem. Commun., 2015, 51, 9129–9132 RSC.
- D. Oron, M. Kazes and U. Banin, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035330 CrossRef.
- H. Zhu, N. Song and T. Lian, J. Am. Chem. Soc., 2011, 133, 8762–8771 CrossRef CAS PubMed.
- M. Sun, D. Zhu, W. Ji, P. Jing, X. Wang, W. Xiang and J. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 12681–12688 CAS.
- D.-E. Nam, W.-S. Song and H. Yang, J. Mater. Chem., 2011, 21, 18220–18226 RSC.
- W.-S. Song and H. Yang, Chem. Mater., 2012, 24, 1961–1967 CrossRef CAS.
- K. Wu, L. J. Hill, J. Chen, J. R. McBride, N. G. Pavlopolous, N. E. Richey, J. Pyun and T. Lian, ACS Nano, 2015, 9, 4591–4599 CrossRef CAS PubMed.
- K. Wu, Q. Li, Y. Jia, J. R. McBride, Z.-x. Xie and T. Lian, ACS Nano, 2015, 9, 961–968 CrossRef CAS PubMed.
- P. Peng, D. J. Milliron, S. M. Hughes, J. C. Johnson, A. P. Alivisatos and R. J. Saykally, Nano Lett., 2005, 5, 1809–1813 CrossRef CAS PubMed.
- C. J. Dooley, S. D. Dimitrov and T. Fiebig, J. Phys. Chem. C, 2008, 112, 12074–12076 CAS.
- M. G. Lupo, F. Della Sala, L. Carbone, M. Zavelani-Rossi, A. Fiore, L. Lüer, D. Polli, R. Cingolani, L. Manna and G. Lanzani, Nano Lett., 2008, 8, 4582–4587 CrossRef CAS PubMed.
- N. N. Hewa-Kasakarage, P. Z. El-Khoury, A. N. Tarnovsky, M. Kirsanova, I. Nemitz, A. Nemchinov and M. Zamkov, ACS Nano, 2010, 4, 1837–1844 CrossRef CAS PubMed.
- K. Wu, N. Song, Z. Liu, H. Zhu, W. Rodríguez-Córdoba and T. Lian, J. Phys. Chem. A, 2013, 117, 7561–7570 CrossRef CAS PubMed.
- K. Wu, W. E. Rodríguez-Córdoba, Z. Liu, H. Zhu and T. Lian, ACS Nano, 2013, 7, 7173–7185 CrossRef CAS PubMed.
- H. Zhu, Y. Yang, K. Hyeon-Deuk, M. Califano, N. Song, Y. Wang, W. Zhang, O. V. Prezhdo and T. Lian, Nano Lett., 2014, 14, 1263–1269 CrossRef CAS PubMed.
- K. Wu, H. Zhu, Z. Liu, W. Rodríguez-Córdoba and T. Lian, J. Am. Chem. Soc., 2012, 134, 10337–10340 CrossRef CAS PubMed.
- J. E. Huang, Z. Q. Huang, S. Y. Jin and T. Q. Lian, J. Phys. Chem. C, 2008, 112, 19734–19738 CAS.
- E. A. McArthur, A. J. Morris-Cohen, K. E. Knowles and E. A. Weiss, J. Phys. Chem. B, 2010, 114, 14514–14520 CrossRef CAS PubMed.
- P. Tyagi and P. Kambhampati, J. Chem. Phys., 2011, 134, 094706–094710 CrossRef PubMed.
- J. L. Blackburn, R. J. Ellingson, O. I. Micic and A. J. Nozik, J. Phys. Chem. B, 2003, 107, 102–109 CrossRef CAS.
- K. Wu, Z. Liu, H. Zhu and T. Lian, J. Phys. Chem. A, 2013, 117, 6362–6372 CrossRef CAS PubMed.
- V. I. Klimov, A. A. Mikhailovsky, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Science, 2000, 287, 1011–1013 CrossRef CAS PubMed.
- Y. Chen, J. Vela, H. Htoon, J. L. Casson, D. J. Werder, D. A. Bussian, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 5026–5027 CrossRef CAS PubMed.
- F. Garcia-Santamaria, Y. F. Chen, J. Vela, R. D. Schaller, J. A. Hollingsworth and V. I. Klimov, Nano Lett., 2009, 9, 3482–3488 CrossRef CAS PubMed.
- J. Huang, Z. Huang, Y. Yang, H. Zhu and T. Lian, J. Am. Chem. Soc., 2010, 132, 4858–4864 CrossRef CAS PubMed.
- H. Zhu and T. Lian, J. Am. Chem. Soc., 2012, 134, 11289–11297 CrossRef CAS PubMed.
- J. Huang, Z. Q. Huang, Y. Yang, H. M. Zhu and T. Q. Lian, J. Am. Chem. Soc., 2010, 132, 4858–4864 CrossRef CAS PubMed.
- M. Hilczer and M. Tachiya, J. Phys. Chem. C, 2009, 113, 18451–18454 CAS.
- A. V. Barzykin and M. Tachiya, J. Phys.: Condens. Matter, 2007, 19, 065105 CrossRef.
- V. I. Klimov, J. A. McGuire, R. D. Schaller and V. I. Rupasov, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195324 CrossRef.
- J. A. McGuire, M. Sykora, J. Joo, J. M. Pietryga and V. I. Klimov, Nano Lett., 2010, 10, 2049–2057 CrossRef CAS PubMed.
- J. A. McGuire, M. Sykora, I. Robel, L. A. Padilha, J. Joo, J. M. Pietryga and V. I. Klimov, ACS Nano, 2010, 4, 6087–6097 CrossRef CAS PubMed.
- M. C. Beard, J. Phys. Chem. Lett., 2011, 2, 1282–1288 CrossRef CAS PubMed.
- V. I. Klimov, J. A. McGuire, R. D. Schaller and V. I. Rupasov, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77 Search PubMed.
- Y. Yang, W. Rodríguez-Córdoba and T. Lian, Nano Lett., 2012, 12, 4235–4241 CrossRef CAS PubMed.
- V. V. Matylitsky, L. Dworak, V. V. Breus, T. Basché and J. Wachtveitl, J. Am. Chem. Soc., 2009, 131, 2424–2425 CrossRef CAS PubMed.
- H. Zhu, N. Song and T. Lian, J. Am. Chem. Soc., 2013, 135, 11461–11464 CrossRef CAS PubMed.
- P. Frantsuzov, M. Kuno, B. Janko and R. A. Marcus, Nat. Phys., 2008, 4, 519–522 CrossRef.
- X. Wang, X. Ren, K. Kahen, M. A. Hahn, M. Rajeswaran, S. Maccagnano-Zacher, J. Silcox, G. E. Cragg, A. L. Efros and T. D. Krauss, Nature, 2009, 459, 686–689 CrossRef CAS PubMed.
- V. I. Klimov, A. A. Mikhailovsky, S. Xu, A. Malko, J. A. Hollingsworth, C. A. Leatherdale, H. J. Eisler and M. G. Bawendi, Science, 2000, 290, 314–317 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Sample synthesis and characterizations, TA set-ups, additional transient absorption spectra and kinetics, fitting procedures and fitting parameters. See DOI: 10.1039/c5sc03715h |
| ‡ K. Wu and G. Liang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |