
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile method for the preparation of carbon–rhodium hybrid catalysts on graphene and carbon black†
Chin Min
Wong
ab,
D. Barney
Walker
ab,
Alexander H.
Soeriyadi
a,
J. Justin
Gooding
a and
Barbara A.
Messerle
*ab
aDepartment of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia
bDepartment of Chemistry and Biomolecular Sciences, Macquarie University, Sydney, NSW 2109, Australia. E-mail: barbara.messerle@mq.edu.au
First published on 8th December 2015
Abstract
Strategies for combining the selectivity and efficiency of homogeneous organometallic catalysts with the versatility of heterogeneous catalysts are urgently needed. Herein a direct and modular methodology is presented that provides rapid access to well-defined carbon–rhodium hybrid catalysts. A pre-synthesized Rh(I) complex containing a carbene-triazole ligand was found to be stable for direct immobilization onto unactivated graphene, carbon black and glassy carbon electrodes. Characterization of the heterogeneous systems using X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), inductively coupled plasma-optical emission spectroscopy/mass spectrometry (ICP-OES/MS), Raman spectroscopy, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) confirmed the well-defined nature of the hybrid catalysts. The hybrid catalysts show excellent activity, comparable to that of the homogeneous system for the hydrosilylation of diphenylacetylene, with turnover numbers ranging from 5000 to 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. These catalysts are the best reported to date for the hydrosilylation of diphenylacetylene. In common with conventional heterogeneous catalysts, high reusability, due to a lack of Rh metal leaching, was also observed for all carbon–rhodium complexes under investigation.
000. These catalysts are the best reported to date for the hydrosilylation of diphenylacetylene. In common with conventional heterogeneous catalysts, high reusability, due to a lack of Rh metal leaching, was also observed for all carbon–rhodium complexes under investigation.
Introduction
The assembly of highly efficient and selective homogeneous systems onto heterogeneous solid materials is a facile route to the development of optimized and recyclable hybrid systems with well-defined active sites.1–3 While a variety of catalysts covalently tethered to carbon based scaffolds (e.g. activated carbon, graphene/graphene oxide and carbon nanotubes) and other well known supports (e.g. aluminosilicates, organic resins and metal organic frameworks) have been reported,4–12 the fabrication of these hybrid systems is often complicated, requiring multiple reaction steps.The development of hybrid catalysts consisting of well-defined organometallic complexes on solid supports can be established using both non-covalent interactions or covalent bonding. In terms of producing hybrid catalysts that are covalently bonded to the solid support, the synthesis route typically occurs via at least two steps, where initially a precursor ligand is attached to the surface then – in a second and separate step – a transition metal centre is introduced. The first step is normally dictated by the nature of the functional groups on the solid surface. For example, reactive Si–OH moieties on silica surfaces can be effectively used to initiate ligand immobilization.13,14 However, certain supports require additional surface activation in order to achieve the functional groups that are needed for ligand modification. This is a common strategy for functionalization of graphene surfaces, whereby pre-oxidation of graphene to graphene oxide (GO) is often necessary prior to functionalization with an appropriate ligand motif.15–17 At times, reduction back to reduced GO is also performed after ligand immobilization and complexation with the metal precursor.18–20 This complicated multistep assembly to the hybrid system is not ideal for pristine materials, such as pure graphene, where it is desirable that its regular, conjugated surface is retained.21–24 The second step (complexation of metal with the immobilized ligand) in the stepwise assembly of hybrid catalysts could create an additional complication, as incomplete complexation of the immobilized ligand can occur. Furthermore, the common use of an excess of metal precursors increases the chance of nonspecific adsorption of metals onto the surface.
In designing new hybrid catalysts, it would be highly desirable to have a simple, general method for attaching optimized metal complexes directly to a carbon support without the need for surface pre-activation or post-immobilization treatment with excess precious metal precursors. While direct functionalization of graphene is possible using aryl diazonium-based precursors,25–28 the harsh conditions required by this reaction are typically not compatible with finely tuned catalytically-active metal complexes. Herein we describe a modular protocol for the direct hybridization of a pre-formed Rh(I) catalyst with the surface of unactivated carbon materials (Scheme 1). We demonstrate the potential widespread utility of this method by functionalizing two different high surface area carbon allotropes, graphene (G) and carbon black (CB) and show that these are highly efficient catalysts. Furthermore, it is demonstrated that even low surface area glassy carbon (GC) electrodes can be readily transformed into functionalized, recyclable catalytic systems using this efficient methodology.
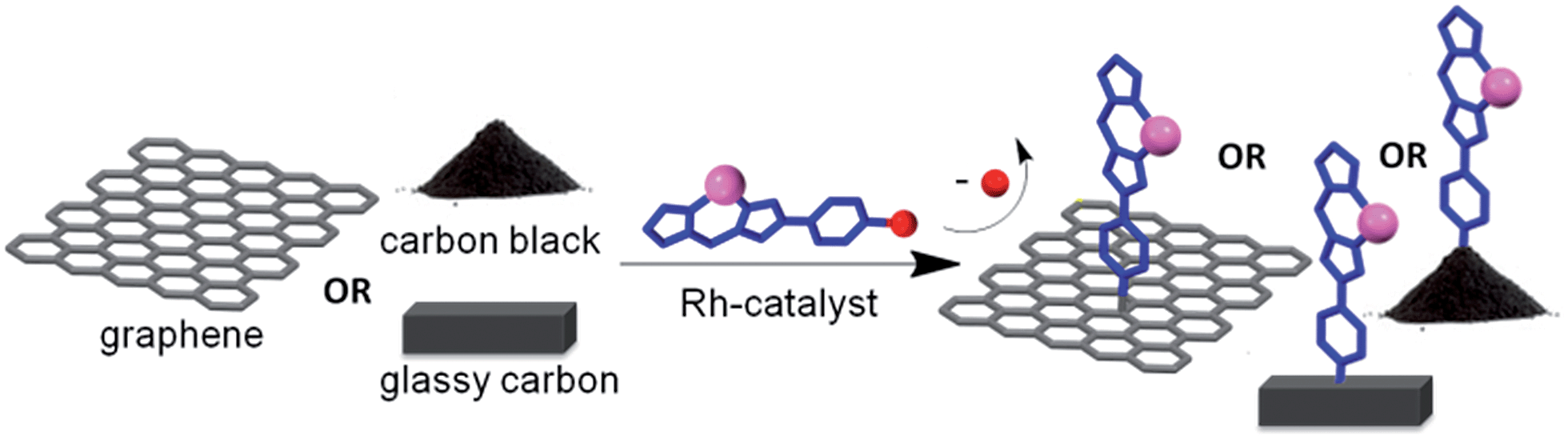 | ||
| Scheme 1 Illustration of the direct attachment of catalyst to G, CB or GC without the need for oxidatively introduced functional groups. | ||
Results and discussion
Here, we introduce our novel homogeneous catalyst, [2Rh(CO)2]BPh4 (Fig. 1) onto the carbon supports by directly immobilizing the pre-synthesized Rh(I) complex containing an aniline-functionalized bidentate carbene-triazole ligand, [1Rh(COD)]BPh4 (COD = cyclooctadiene) via the in situ generation of the diazonium moiety (Scheme 2). The aniline functionalized complex [1Rh(COD)]BPh4, in an acidic solution of nitromethane and sodium nitrite was added to a GC electrode, or stirred with CB or G, for 24 h. The carbon materials were then washed repeatedly, and suspended in a mixture of dichloromethane and pentane under an atmosphere of CO. The GC electrode was washed thoroughly and dried under a gentle flow of nitrogen to give [GC-2Rh(CO)2]. [CB-2Rh(CO)2] or [G-2Rh(CO)2] were isolated as black powders after copious washing and drying under vacuum at 25 °C overnight.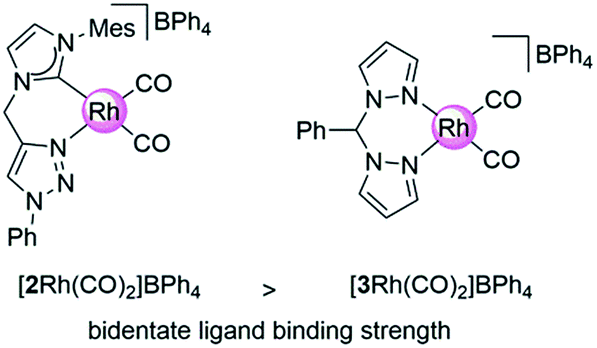 | ||
| Fig. 1 Homogeneous Rh(I) complexes containing carbene-triazole (2) and bis-pyrazole (3) ligands. | ||
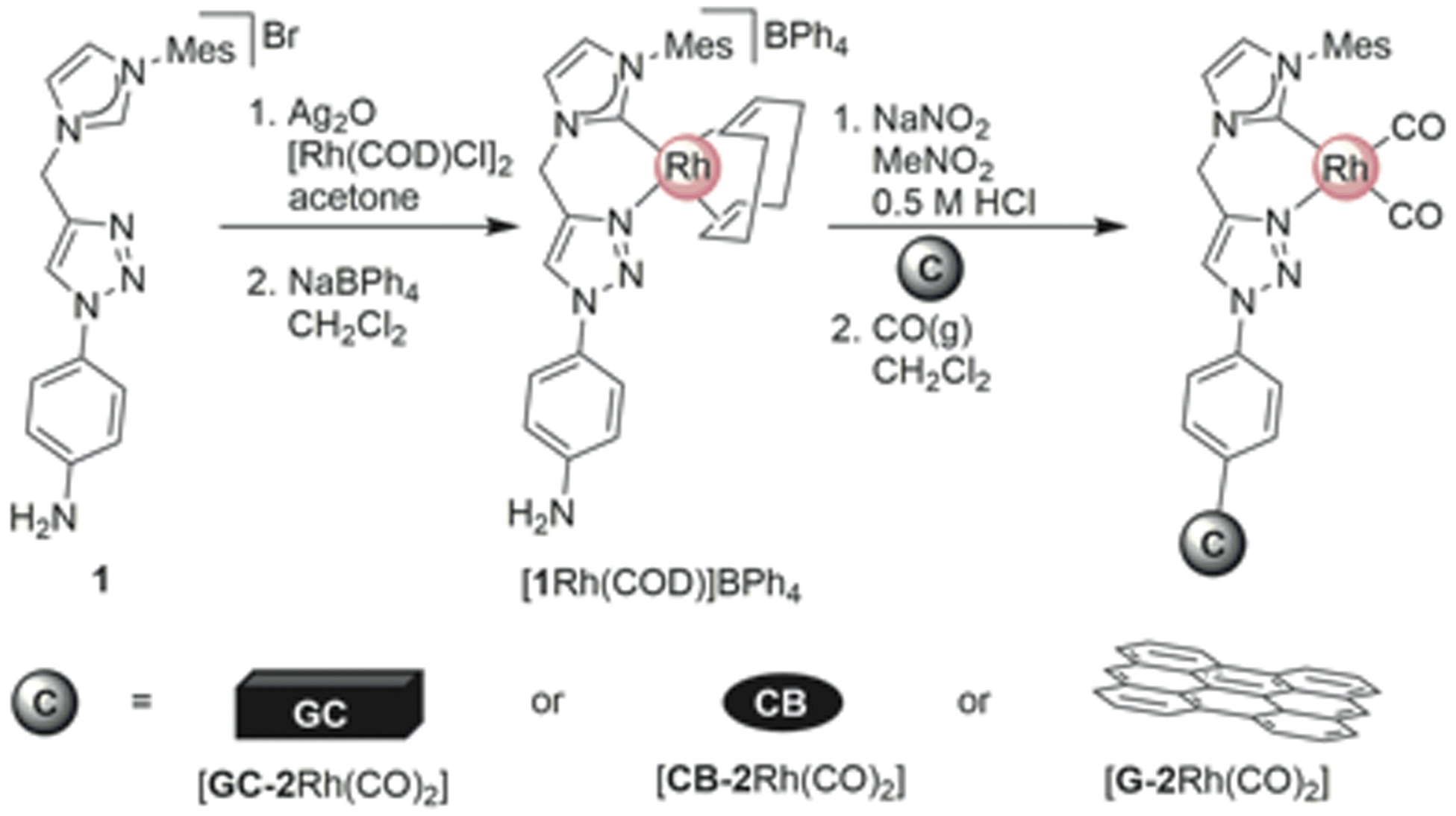 | ||
| Scheme 2 Synthesis of [GC-2Rh(CO)2], [CB-2Rh(CO)2] and [G-2Rh(CO)2]. | ||
On application as a catalyst for hydroamination, our previously employed carbon supported bis-pyrazole system29,30 (Fig. 1) delivered remarkably high turnover numbers29 but underwent some leaching of the Rh metal centre over time. The novel Rh(I) catalyst attached to the carbon surface here contains a carbene-triazole ligand system that has a high binding affinity for the Rh ion in order to create recyclable hybrid catalysts that exhibit minimal metal leaching.
While [GC-2Rh(CO)2] was characterized using X-ray photoelectron spectroscopy (XPS), [CB-2Rh(CO)2] and [G-2Rh(CO)2] were characterized using XPS, thermogravimetric analysis (TGA), inductively coupled plasma-optical emission spectroscopy/mass spectrometry (ICP-OES/MS), Raman spectroscopy, scanning electron microscopy (SEM) and/or transmission electron microscopy (TEM).
In the XP spectra31 of [G-2Rh(CO)2] (Fig. 2), the appearance of the N1s peak at ca. 402 eV32 and Rh3d5/2 peak at ca. 310 eV were observed. N1s and Rh3d peaks at similar binding energies were observed in the XP spectra of [GC-2Rh(CO)2], [CB-2Rh(CO)2] and the unattached complex [2Rh(CO)2]BPh4 (see all XPS figures of hybrid complexes, unmodified carbon surfaces and homogeneous complex in the ESI, section SI-2†), consistent with analogous hybrid Rh(I) complexes on GC and CB reported previously.29,30 The atomic ratio of N:Rh for [GC-2Rh(CO)2] and [G-2Rh(CO)2] was found to be close to 5:1 as expected, which statistically means that only individual complexes are immobilized on the surfaces. In the case of [CB-2Rh(CO)2], the ratio of N:Rh was found to be 3.8:1 suggesting that there may be a small amount of adventitious physisorbed Rh on the CB surface.
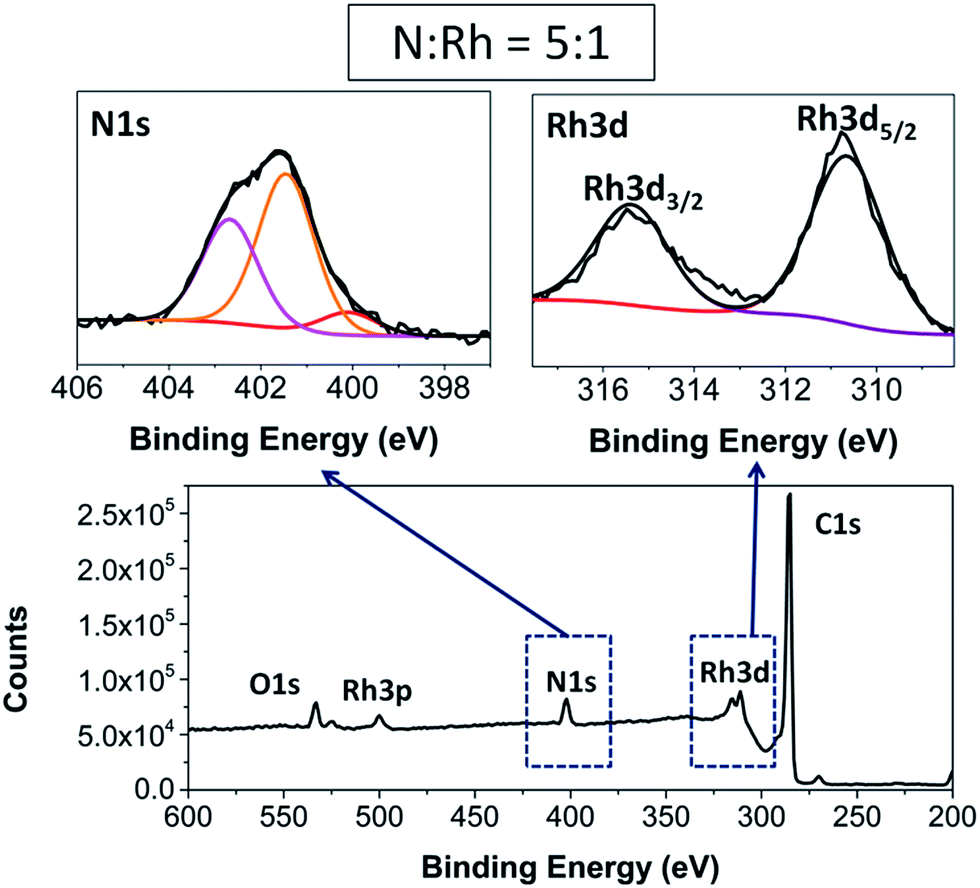 | ||
| Fig. 2 XP survey scan of [G-2Rh(CO)2] and selected narrow scan of N1s and Rh3d. | ||
The thermal stability of [CB-2Rh(CO)2] and [G-2Rh(CO)2] was analyzed against that of unmodified CB and G using TGA under an atmosphere of nitrogen (Fig. 3). The thermogram of [CB-2Rh(CO)2] displayed two weight loss processes at ca. 100–470 and 470–490 °C giving a total weight loss of 13% while the thermogram for [G-2Rh(CO)2] displayed one weight loss process between ca. 350–650 °C with a total weight loss of 7% (see ESI, section SI-3†). Additionally, we observed that the thermogram of [2Rh(CO)2]BPh4 only and a mixture of [2Rh(CO)2]BPh4 and carbon black (see ESI, Fig. S12†) showed a two weight loss trend from 100–380 °C due to the burn-off of the unattached metal complex similar to what was observed from the thermogram of [CB-2Rh(CO)2].
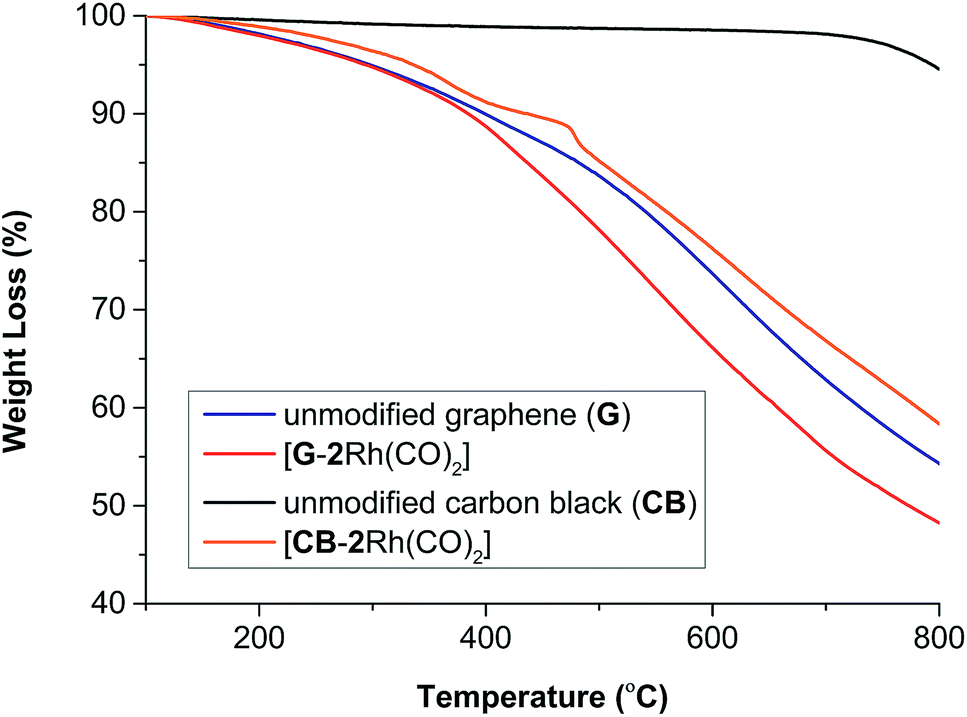 | ||
| Fig. 3 TGA curves for unmodified CB and G, [G-2Rh(CO)2] and [CB-2Rh(CO)2] recorded under an atmosphere of nitrogen. | ||
The quantitative amount of Rh on CB and G obtained using ICP-OES was found to be 2.5 wt% for [CB-2Rh(CO)2] and 0.84 wt% for [G-2Rh(CO)2]. This corresponds well to the estimated amount of Rh in [CB-2Rh(CO)2] (2.6 wt%) and [G-2Rh(CO)2] (1.3 wt%), calculated from the total weight loss obtained from TGA. For [GC-2Rh(CO)2], cyclic voltammetry was used to determine the quantity of Rh complexes immobilized on the GC electrode to be 3.4 × 10−10 mol cm−2 or 1.2 × 10−4 wt% cm−2 (see ESI, section SI-5†).
SEM and TEM images of [G-2Rh(CO)2] showed that the integrity and morphology of the 2D structure is retained after treatment for immobilization of the Rh complex, and is similar to images of unmodified G (see ESI, section SI-8/9†). The Raman spectra of [CB-2Rh(CO)2] and [G-2Rh(CO)2] showed a slight increase in the defects (D) band compared to the graphite (G) band following immobilization of the complex onto the surfaces (see ESI, section SI-7†), which indicates that both carbon–rhodium hybrids contained covalent linkages to the carbon black or graphene surface respectively, consistent with observations for similar covalent functionalization on carbon materials.33,34 An increase in the defects (D) band compared to the graphite (G) band of the carbon samples can be observed when comparing the peak intensity ratio of the D to G band of unmodified graphene, which was 0.79. After immobilization to form [G-2Rh(CO)2], the peak intensity ratio of the D to G band increased to 0.82. Similarly, the peak intensity ratio of the D to G band for [CB-2Rh(CO)2] was 1.0 which was higher when compared to the peak intensity ratio of D to G band of the unmodified carbon black of 0.92.
To summarize the surface characterization, we have demonstrated a straight-forward and effective method for the direct immobilization of Rh(I) complexes containing a mixed bidentate carbene-triazole ligand system onto carbon surfaces. The well-defined complexes were covalently anchored on GC, CB and G, showing that this method is versatile enough to be performed on various carbon surfaces with different surface areas and properties.
Catalysis
Following characterization of the hybrid catalysts, the efficiency of homogeneous complex [2Rh(CO)2]BPh4 and hybrid complexes [GC-2Rh(CO)2], [CB-2Rh(CO)2] and [G-2Rh(CO)2] were evaluated as catalysts for the hydrosilylation of diphenylacetylene (4) and triethylsilane at 50 °C and/or 25 °C using 0.02 mol% of Rh relative to the quantity of 4 (Scheme 3).35 Transition metal-catalyzed hydrosilylation of alkynes allows the formation of alkenylsilanes in a straightforward, atom efficient and convenient method. Alkenylsilanes are versatile organosilicon compounds that can be used for further transformations in many organic synthesis reactions.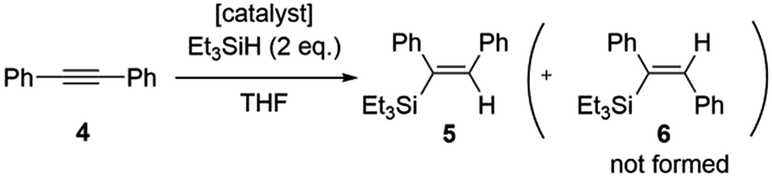 | ||
| Scheme 3 Hydrosilylation reaction of diphenylacetylene 4 and triethylsilane. | ||
At 50 °C, [CB-2Rh(CO)2] and [2Rh(CO)2]BPh4 were both highly efficient catalysts, with the reaction reaching complete conversion of substrate 4 to product 5 in just 0.25 h (Fig. 4). While [G-2Rh(CO)2] was slightly less efficient compared to [CB-2Rh(CO)2] and [2Rh(CO)2]BPh4, the reaction still reached 56% conversion in 0.5 h and complete conversion in 1 h. The hydrosilylation reaction was repeated at 25 °C (to decrease the reaction rate to a more readily monitored rate) with [2Rh(CO)2]BPh4 and [CB-2Rh(CO)2] for better comparison. [CB-2Rh(CO)2] and [2Rh(CO)2]BPh4 each displayed very similar efficiency as catalysts for the reaction (Fig. 4). The reaction using [CB-2Rh(CO)2] as catalyst was slightly slower, reaching 77% conversion to product 5 in 1 h compared to the complete conversion obtained using [2Rh(CO)2]BPh4 in the same time frame. This slightly lower efficiency of [CB-2Rh(CO)2] could be due to the time required for diffusion of substrate to the carbon black support compared to the direct mixing of the two in the homogeneous reaction mixture. On completion, all reactions had turnover numbers (TON) close to 5000 (see ESI, Table S14†). These catalysts are the most efficient reported to date for the hydrosilylation of disubstituted alkyne, both homogeneous36–40 and heterogeneous.41–45
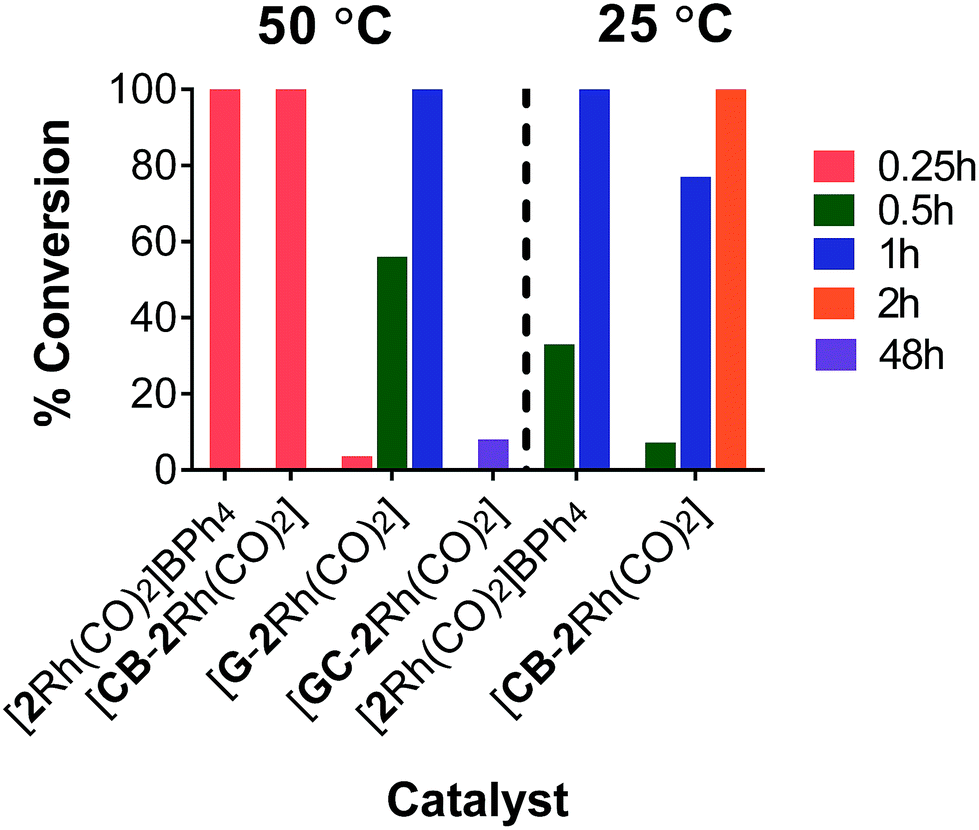 | ||
| Fig. 4 Graph depicting the % conversion at times 0.25, 0.5, 1, 2 and 48 h for the hydrosilylation reaction using [2Rh(CO)2]BPh4, [CB-2Rh(CO)2], [G-2Rh(CO)2] and [GC-2Rh(CO)2] at 50 °C or 25 °C. | ||
On immobilizing the complex on a GC support ([GC-2Rh(CO)2]), it was found that although the reaction was slow and showed only 8% conversion to product 5 in 48 h (Fig. 4), the TON for this reaction reached the very high value of 48000 in that time. This correlates well with the fact that the GC electrode has a plate-like flat structure with a limited surface area (2.4 cm2) and hence holds only a small number of moles of immobilized catalyst relative to the substrate whereas there was a much higher mole ratio of catalyst to substrate immobilized on the high surface area CB and G. The high TON observed here for [GC-2Rh(CO)2] is very similar to the turnover numbers we reported previously for GC supported catalysts.29
In a series of control experiments at 50 °C, when using no catalyst or unmodified CB and G as the catalyst, the hydrosilylation reaction did not proceed even after 24 h (Table 1). In order to confirm that the carbon surfaces are covalently attached to the Rh(I) complexes, the homogeneous complex [2Rh(COD)]BPh4, which lacks the aniline functional group that [1Rh(COD)]BPh4 contains for the purpose of attachment, was mixed together with some unmodified G. The mixture was then treated using the same approach used to synthesize the hybrid catalysts, and the isolated G powder was tested for the hydrosilylation reaction. There was <0.08% conversion observed after 24 h, confirming that without the aniline functionalized group on the ligand, a covalent hybrid catalyst could not be assembled using the established synthesis.
Recyclability of hybrid catalysts
Initially, [GC-2Rh(CO)2] was investigated for the hydrosilylation reaction over the course of three reaction cycles. After each run, the GC electrode was analyzed by XPS. The ratio of N:Rh on the surface of the GC remained constant at ca. 4.5:1 (see ESI, Table S15†), and this confirmed that the Rh complex remained on the GC surface even after multiple runs.
The low level of conversion to product 5 (<11%) obtained over an extended reaction time (48 h) when using [GC-2Rh(CO)2] led to the investigation of the efficiency of hybrid catalysts on G and CB. In the recycling experiments for [CB-2Rh(CO)2] (1.6 mol%) and [G-2Rh(CO)2] (0.3 mol%), the hybrid catalysts were recycled over three 24 h cycles, and the filtrate and washings after each run were analyzed by ICP-MS to determine the Rh content. Both [G-2Rh(CO)2] and [CB-2Rh(CO)2] had excellent recycling ability (Fig. 5(a)), with complete conversion to product 5 after each run. The excellent recyclability was reinforced by ICP-MS analysis of the washings (Fig. 5(b)). When using [G-2Rh(CO)2] as the catalyst, the ICP-MS showed hardly any leaching after each run, with less than 6% loss of Rh from the G surface after three runs. When using [CB-2Rh(CO)2] as catalyst instead, an initial 12% loss of Rh from the CB surface was observed after the first cycle (Fig. 5(b)), consistent with the initial loss of the physisorbed Rh. This physisorbed Rh could not be removed prior to catalysis by normal washing procedures, and most likely requires the reaction conditions of the catalysis reaction to enable its' removal. After the second and third cycle, there was only a total of 3% loss of Rh observed. Again, such results confirm the stability of the immobilized Rh complex on the CB surface. These results also suggest that the use of the mixed bidentate carbene-triazole ligand system allows an enhanced capacity to recycle the Rh(I) complexes compared to our previously described hybrid system.29,30
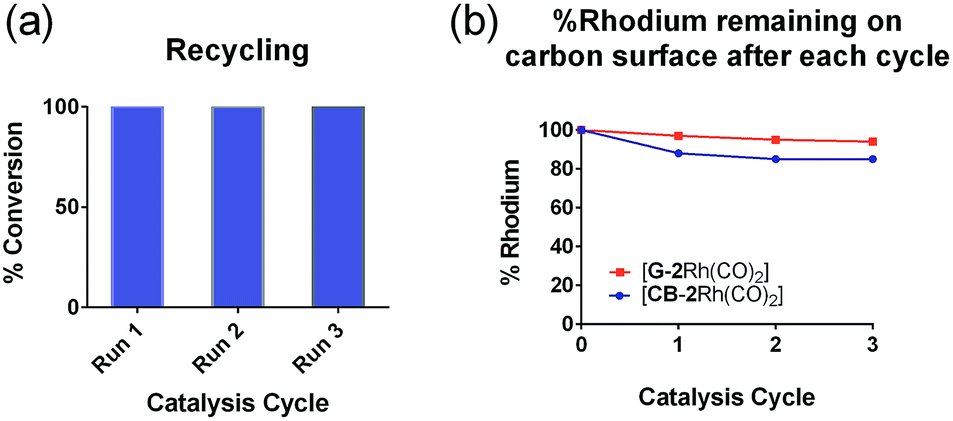 | ||
| Fig. 5 Graphs depicting: (a) the % conversions of the hydrosilylation reaction for three reaction cycles when using [CB-2Rh(CO)2] and [G-2Rh(CO)2)], which are shown on the same graph and (b) the % rhodium remaining on the carbon surfaces after each reaction cycle. | ||
To further investigate the recyclability of the hybrid catalysts on G and CB, [CB-2Rh(CO)2] and [G-2Rh(CO)2] were reused over 10 cycles of the catalysis reaction. Each cycle was tested after 4 h at 25 °C (Fig. 6). [G-2Rh(CO)2] was found to be an extremely recyclable heterogeneous catalyst and complete (>98%) conversions were obtained for all 10 cycles (Fig. 6(b)). When recycling [CB-2Rh(CO)2], complete conversion (>98%) of the reaction was obtained for the first 7 cycles. Over the following 8–10 cycles, there was a slight 10–20% reduction in product formation (Fig. 6(a)), which suggests that [CB-2Rh(CO)2] is slightly less recyclable compared to [G-2Rh(CO)2]. The high recyclability of the hybrid catalysts shown here supports the benefit of immobilizing the highly efficient and well-defined homogeneous catalyst [2Rh(CO)2]BPh4 on the surface. [2Rh(CO)2]BPh4 is also shown to be a highly efficient homogeneous catalyst for the hydrosilylation of diphenylacetylene as it can catalyze up to 5 cycles of extra substrate addition before reaching a saturation point in the reaction mixture (see ESI, Fig. S22†). The development of the hybrid catalysts from the optimized homogeneous catalyst allows us to exploit the reusability of the homogeneous catalyst, providing us with highly recyclable hybrid catalyst that also allows for easy product separation.
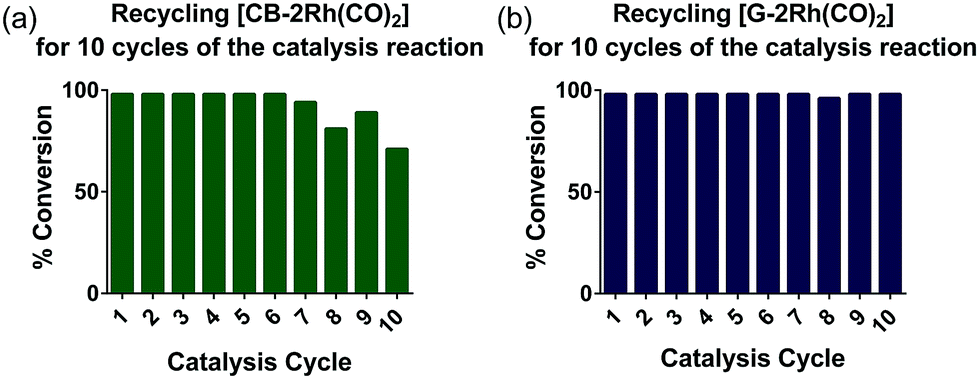 | ||
| Fig. 6 Graphs depicting: (a) the % conversions of the hydrosilylation reaction for 10 reaction cycles when using [CB-2Rh(CO)2] and (b) the % conversions of the hydrosilylation reaction for 10 reaction cycles when using [G-2Rh(CO)2]. | ||
Conclusions
In conclusion, during the investigations of a series of hybrid complexes on G, CB and GC as catalysts for the hydrosilylation of diphenylacetylene, it was found that [CB-2Rh(CO)2] was the most efficient catalyst and [G-2Rh(CO)2] was slightly less efficient than [CB-2Rh(CO)2]. While [GC-2Rh(CO)2] did not reach the same conversion levels over time, the very high TON achieved demonstrated excellent efficiency of this catalyst as well. The results also showed hybrid complex [CB-2Rh(CO)2] was as efficient as the homogeneous complex [2Rh(CO)2]BPh4 in catalyzing this reaction. The catalysts investigated here could be utilized in very small amounts to give high TONs ranging from 5000 to 48000. These catalysts are the most efficient of the homogeneous and heterogeneous catalysts reported to date for promoting the hydrosilylation of disubstituted alkyne. Finally, the recycling experiments also showed that the hybrid catalysts could be recycled for at least 10 times with hardly any loss of Rh from the surface. [G-2Rh(CO)2] was found to be slightly more recyclable compared to [CB-2Rh(CO)2]. All the results demonstrate the stability and reusability of the hydrid Rh(I) complexes containing the carbene-triazole mixed bidentate ligand system.
Experimental
General experimental and materials
Unless otherwise stated, all manipulations were carried out under inert atmosphere using standard Schlenk techniques or in a nitrogen or argon-filled Braun glovebox. All reagents were purchased from Aldrich Chemical Company Inc. or Alfa Aesar Inc. and used as received unless otherwise noted. Graphene46 was contributed by Professor John Stride (UNSW). Glassy carbon electrodes (1 × 1 × 0.1 cm3) were purchased from Goodfellow. Carbon black (XC-72R) was supplied by Cabot Corporation. RhCl3·xH2O was purchased from Precious Metals Online PMO P/L. For the purposes of air sensitive manipulations and in the preparation of air sensitive metal complexes, tetrahydrofuran, dichloromethane, acetonitrile and pentane were dispensed from a PuraSolv solvent purification system. Methanol was distilled from magnesium methoxide under an atmosphere of nitrogen or argon. The bulk compressed gases argon (>99.999%) and carbon monoxide (>99.5%) were obtained from Air Liquide and used as received. Nitrogen gas for Schlenk line operation comes from in-house liquid nitrogen boil-off. 1-Azido-4-nitrobenzene,47 [Rh(COD)Cl]2,48 1-mesityl-3-(prop-2-yn-1-yl)-1H-imidazol-3-ium bromide49 and 1-mesityl-3-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-1H-imidazol-3-ium tetraphenylborate49 were synthesized using literature procedures. 1H and 13C{1H} NMR spectra were recorded on Bruker DMX400, DMX500 and DMX600 spectrometers operating at 400, 500 and 600 MHz (1H) respectively and 100, 125 and 150 MHz (13C) respectively. 1H and 13C NMR chemical shifts were referenced internally to residual solvent resonances. Unless otherwise stated, spectra were recorded at 298 K and chemical shifts (δ), with uncertainties ±0.01 Hz for 1H and ±0.05 Hz for 13C, are quoted in parts per million, ppm. Coupling constants (J) are quoted in Hz and have uncertainties of ±0.05 Hz for 1H–1H and ±0.5 Hz for 13C–103Rh and 13C–11B. Numbering and figures corresponding to NMR characterization data are located in the ESI, Table S1.† Deuterated solvents were purchased from Cambridge Stable Isotopes and used as received. Air sensitive NMR samples were prepared in an inert gas glovebox or by vacuum transfer of deuterated solvents into NMR tubes fitted with a Young's Teflon valve. For air sensitive NMR samples, dichloromethane-d2 and chloroform-d were distilled over calcium hydride. Microanalyses were carried out at the Campbell Micro-analytical Laboratory, University of Otago, New Zealand or at the Research School of Chemistry, The Australian National University, Canberra, Australia. Mass spectra were acquired using a Thermo LTQ Orbitrap XL located in the Bioanalytical Mass Spectrometry Facility (BMSF) in UNSW. M is defined as the molecular weight of the compound of interest or cationic fragment for cationic metal complexes.1-Mesityl-3-((1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)-1H-imidazol-3-ium bromide, nitro functionalized ligand precursor to ligand 1
1-Mesityl-3-(prop-2-yn-1-yl)-1H-imidazol-3-ium bromide (1020 mg, 3.33 mmol) was reacted with 1-azido-4-nitrobenzene (547 mg, 3.33 mmol) in a deoxygenated solution of isopropanol:water (2:1, 50 mL). After stirring for 5 minutes, sodium ascorbate (132 mg, 0.666 mmol, 20 mol%) followed by copper sulfate pentahydrate (41.3 mg, 0.165 mmol, 5 mol%) was added to the orange mixture. The reaction mixture was stirred for 24 hours to give an orange-red precipitate in an orange solution. The precipitate was collected via filtration and washed thoroughly with a saturated solution of Na2EDTA until the filtrate turned from green to colourless. The precipitate was then dried under vacuum to give the ligand as a red solid. The ligand can be recrystallized from dichloromethane:pentane as red crystals (712 mg, 46%). 1H NMR (400 MHz, CDCl3): δ 10.31 (br s, 1H, Im-H2), 9.63 (s, 1H, Tz-H5′), 8.38 (d, 3J = 9.3 Hz, 2H, m-CH of ArNO2), 8.13 (d, 3J = 9.3 Hz, 2H, o-CH of ArNO2), 8.08 (t, 3J = 1.5 Hz, 1H, Im-H5), 7.14 (t, 3J = 1.5 Hz, 1H, Im-H4), 6.98 (s, 2H, m-CH of Mes), 6.22 (s, 2H, CH2), 2.32 (s, 3H, p-CH3 of Mes), 2.04 (s, 6H, o-CH3 of Mes) ppm; 13C{1H} NMR (100 MHz, CDCl3): 147.5 (Cq of PhNO2), 142.0 (Tz-C4′), 141.7 (p-CCH3 of Mes), 140.9 (ipso-C to NH2 of PhNH2), 138.1 (Im-C2), 134.2 (o-CCH3 of Mes), 130.6 (ipso-C of Mes), 130.0 (m-CH of Mes), 125.6 (m-CH of ArNO2), 124.9 (Tz-C5′), 123.4 (Im-C5), 123.3 (Im-C4), 120.9 (o-CH of ArNO2), 44.5 (CH2), 21.2 (p-CH3 of Mes), 17.8 (o-CH3 of Mes) ppm; HRMS (ESI+, MeOH): m/z (%): calculated for [C21H23N6O2]+ = [M]+ = 389.1721; found [M]+ = 389.1718 (100%) amu; elemental analysis: found C, 51.85; H, 4.68; N, 17.21; calculated for C21H23BrN6O2·0.25CH2Cl2: C, 52.03; H, 4.42; N, 17.13%.
3-((1-(4-Aminophenyl)-1H-1,2,3-triazol-4-yl)methyl)-1-mesityl-1H-imidazol-3-ium bromide, (1)
The nitro functionalized ligand precursor above (605 mg, 1.29 mmol) was dissolved in methanol (30 mL). Pd/C (10% w/w, 54.4 mg, 0.0511 mmol, 4 mol%) was then added to the orange solution. After stirring for 5 minutes, hydrazine hydrate (1.3 mL, 26.8 mmol) was added slowly to the reaction mixture. The red-black mixture was heated at reflux under argon overnight. The mixture was filtered through Celite® which was washed thoroughly with excess methanol. The filtrate was dried with anhydrous magnesium sulfate, filtered and the solvent was evaporated to give the crude product as a yellow oil. The crude product was recrystallized from a dichloromethane:pentane mixture to yield ligand 1 as a fluffy pale yellow solid (526 mg, 93%). 1H NMR (400 MHz, CDCl3): δ 10.4 (br s, 1H, Im-H2), 8.93 (s, 1H, Tz-H5′), 8.03 (apparent t, 3J = 1.5 Hz, 1H, Im-H5), 7.50 (d, 3J = 8.8 Hz, 2H, o-CH of ArNH2), 7.10 (apparent t, 3J = 1.5 Hz, 1H, Im-H4), 7.00 (s, 2H, m-CH of Mes), 6.74 (d, 3J = 8.5 Hz, 2H, m-CH of ArNH2), 6.23 (s, 2H, CH2), 3.93 (s, 2H, NH2), 2.34 (s, 3H, p-CH3 of Mes), 2.05 (s, 6H, o-CH3 of Mes) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 147.5 (Cq of ArNH2), 141.7 (p-CCH3 of Mes), 140.7 (Tz-C4′), 137.9 (Im-C2), 134.3 (o-CCH3 of Mes), 130.7 (ipso-C of Mes), 130.04 (m-CH of Mes), 128.2 (ipso-C to NH2 of ArNH2), 123.8 (Tz-C5′), 123.3 (Im-C5), 122.94 (Im-C4), 122.3 (o-CH of ArNH2), 115.4 (m-CH of ArNH2), 44.7 (CH2), 21.2 (p-CH3 of Mes), 17.8 (o-CH3 of Mes) ppm; HRMS (ESI+, MeOH): m/z (%): calculated for [C21H23N6]+ = [M]+ = 359.1979; found [M]+ = 359.1974 (100%) amu; elemental analysis: found C, 57.70; H, 5.55; N, 19.03; calculated for C21H23BrN6: C, 57.41; H, 5.28; N, 19.13%.
[1Rh(COD)]BPh4
Ligand 1 (189 mg, 0.429 mmol) was dissolved partially as a suspension in acetone (25 mL). Ag2O (87.6 mg, 0.378 mmol) was added to the suspension prior to heating the reaction mixture at reflux under argon for one hour. After cooling the reaction mixture to room temperature, [Rh(COD)Cl]2 (106 mg, 0.214 mmol) was added. The reaction mixture was heated again at reflux for another one hour and then stirred at room temperature overnight under argon. The mixture was filtered through Celite® which was washed thoroughly with excess acetone. The solvent was removed in vacuo and the crude product was dissolved in dichloromethane (15 mL). NaBPh4 (147 mg, 0.430 mmol) was added and white solid precipitates immediately. The reaction mixture was stirred vigorously at room temperature for 45 minutes to ensure complete reaction. The mixture was filtered through Celite® which was washed thoroughly with dichloromethane. The solution was concentrated in vacuo to ca. 2 mL which was added slowly dropwise to a vigorously stirred solution of pentane (30 mL) in an ice bath. Yellow precipitate forms immediately. After five minutes of stirring, the solvent was filtered and the precipitate dried under vacuum to give [1Rh(COD)]BPh4 as a bright yellow solid (323 mg, 85%). 1H NMR (400 MHz, CD2Cl2): δ 7.40 (m, 8H, o-CH of BPh4), 7.24 (d, 3JH–H = 8.9 Hz, 2H, o-CH of ArNH2), 7.04 (m, 2H, m-CH of Mes overlapped with Tz-H5′), 7.04 (s, 1H, Tz-H5′ overlapped with m-CH of Mes), 7.00 (t, 3JH–H = 7.5 Hz, 8H, m-CH of BPh4), 6.89 (d, 3JH4–H5 = 1.9 Hz, 1H, Im-H5), 6.85 (t, 3JH–H = 7.1 Hz, 3H, p-CH of BPh4), 6.75 (d, 3JH4–H5 = 1.9 Hz, 1H, Im-H4), 6.72 (d, 3JH–H = 8.9 Hz, 2H, m-CH of ArNH2), 4.92 (m, 2H, CH of COD), 4.76 (s, 2H, CH2), 4.02 (s, 2H, NH2), 3.64 (m, 2H, CH of COD), 2.37 (s, 3H, p-CH3 of Mes), 2.25 (m, 2H, CH2 of COD), 2.10 (s, 6H, o-CH3 of Mes), 1.97 (m, 6H, CH2 of COD) ppm; 13C{1H} NMR (100 MHz, CD2Cl2): δ 176.2 (d, 1JRh–C = 51.2 Hz, Im-C2), 164.4 (q, 1JB–C = 49.3 Hz, o-CH of BPh4), 149.0 (Cq of ArNH2), 140.1 (p-CCH3 of Mes), 140.0 (Cq of Tz), 136.3 (br s, o-CH of BPh4), 135.9 (o-CCH3 of Mes), 135.5 (ipso-C of Mes), 129.5 (m-CH of Mes), 127.1 (ipso-C to NH2 of ArNH2), 126.2 (q, 4JB–H = 2.6 Hz, m-CH of BPh4), 123.1 (Im-C4), 122.6 (o-CH of ArNH2), 122.5 (Im-C5), 122.3 (p-CH of BPh4), 121.2 (Tz-C5′), 115.3 (m-CH of ArNH2), 96.7 (d, 2JRh–C = 7.9 Hz, 2C, CH of COD), 79.1 (d, 2JRh–C = 12.4 Hz, 2C, CH of COD), 45.1 (CH2), 32.6 (2C, CH2 of COD), 29.2 (2C, CH2 of COD), 21.2 (p-CH3 of Mes), 18.6 (o-CH3 of Mes) ppm; HRMS (ESI+, MeOH): m/z (%): calculated for [C29H34N6Rh]+ = [M]+ = 569.1900; found [M]+ = 569.1893 (100%); calculated for [M − C8H11Rh]+ = 359.1979; found [M − C8H11Rh]+ = 359.1977 (78%) amu; elemental analysis: found C, 71.62; H, 6.09; N, 9.44; calculated for C53H54BN6Rh: C, 71.62; H, 6.12; N, 9.46%.[2Rh(COD)]BPh4
Sodium ethoxide (14.7 mg, 0.225 mmol) was added to a stirred suspension of [Rh(COD)Cl]2 (53.5 mg, 0.109 mmol) in ethanol (10 mL). The orange-yellow mixture was stirred at room temperature for 10 minutes. A solution of 1-mesityl-3-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-1H-imidazol-3-ium tetraphenylborate ligand4 (144 mg, 0.217 mmol) in tetrahydrofuran (20 mL) was added slowly dropwise into the stirred suspension. The reaction mixture was stirred at room temperature for three hours. The solvent was removed in vacuo to give the crude product as a bright yellow solid. The crude product was dissolved in dichloromethane and filtered through Celite® which was washed thoroughly with excess dichloromethane. The solvent was concentrated to ca. 2 mL. Pentane (25 mL) was then added slowly and a bright yellow solid precipitates. The solvent was filtered off and the solid was dried under vacuum to yield [2Rh(COD)]BPh4 as a bright yellow solid (126 mg, 64%). 1H NMR (400 MHz, CDCl3): δ 7.55 (m, 8H, o-CH of BPh4), 7.48 (m, 3H, o- and p-CH of Ph), 7.38 (m, 2H, m-CH of Ph), 7.02 (s, 2H, m-CH of Mes overlapped with m-CH of BPh4), 7.00 (t, 3JH–H = 7.2 Hz, 8H, m-CH of BPh4 overlapped with m-CH of Mes), 6.84 (t, 3JH–H = 7.3 Hz, 4H, p-CH of BPh4), 6.76 (d, 3JH4–H5 = 1.8 Hz, 1H, Im-H5), 6.65 (d, 3JH4–H5 = 1.8 Hz, 1H, Im-H4), 6.58 (s, 1H, Tz-H5′), 4.89 (m, 2H, CH of COD), 4.44 (s, 2H, CH2), 3.63 (m, 2H, CH of COD), 2.38 (s, 3H, o-CH3 of Mes), 2.25 (m, 2H, CH2 of COD), 2.09 (s, 6H, p-CH3 of Mes), 1.96 (m, 6H, CH2 of COD) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 175.3 (d, 1JRh–C = 52.7 Hz, Im-C2), 164.4 (q, 1JB–C = 49.6 Hz, ipso-C of BPh4), 140.1 (Cq of Tz), 139.8 (p-CCH3 of Mes), 136.3 (br s, o-CH of BPh4), 135.8 (o-CCH3 of Mes), 135.6 (ipso-C of Ph), 135.2 (ipso-C of Mes), 130.1 (p-CH of Ph), 130.0 (o-CH of Ph), 129.4 (m-CH of Mes), 126.0 (q, 4JB–C = 2.8 Hz, m-CH of BPh4), 122.8 (Im-C5), 122.6 (Im-C4), 122.1 (p-CH of BPh4), 121.8 (Tz-C5′), 120.8 (m-CH of Ph), 96.3 (d, 2JRh–C = 7.8 Hz, 2C, CH of COD), 78.9 (d, 2JRh–C = 12.5 Hz, 2C, CH of COD), 44.6 (CH2), 32.4 (2C, CH2 of COD), 29.0 (2C, CH2 of COD), 21.2 (p-CH3 of Mes), 18.6 (o-CH3 of Mes) ppm; HRMS (ESI+, MeOH): m/z (%): calculated for [C29H33N5Rh]+ = [M]+ = 554.1786, found [M]+ = 554.1776 (100%) amu; elemental analysis: found C, 69.76; H, 6.19 and N, 7.80; calculated for C53H53BN5Rh·0.5CH2Cl2: C, 70.13; H, 5.94 and N, 7.64%.[2Rh(CO)2]BPh4
[2Rh(COD)]BPh4 (117 mg, 0.133 mmol) was dissolved in dichloromethane (15 mL). The yellow solution was degassed via three cycles of freeze–pump–thaw. The reaction mixture was placed under an atmosphere of carbon monoxide gas in a balloon. The solution turned pale yellow in colour after stirring at room temperature for one hour. Pentane (45 mL) was added and yellow solid precipitates in solution with vigorous stirring. The solvent was filtered and the solid was washed with pentane (2 × 2 mL). The precipitate was dried in vacuo to yield [2Rh(CO)2]BPh4 as a pale yellow solid (75.6 mg, 70%). 1H NMR (500 MHz, CD2Cl2): δ 7.59 (m, 3H, o- and p-CH of Ph), 7.53 (m, m-CH of Ph), 7.44 (m, 8H, o-CH of BPh4), 7.06 (s, 2H, m-CH of Mes), 6.98 (t, 3JH–H = 7.5 Hz, 8H, m-CH of BPh4 overlapped with Im-H4), 6.97 (d, 3JH4–H5 = 1.8 Hz, 1H, Im-H4 overlapped with m-CH of BPh4), 6.93 (d, 3JH4–H5 = 1.8 Hz, 1H, Im-H5), 6.82 (t, 3JH–H = 7.2 Hz, 4H, p-CH of BPh4), 6.70 (s, 1H, Tz-H5′), 4.41 (s, 2H, CH2), 2.38 (s, 3H, p-CH3), 2.06 (s, 6H, o-CH3) ppm; 13C{1H} NMR (125 MHz, CD2Cl2): δ 184.7 (d, 1JRh–C = 55.9 Hz, CO), 184.6 (d, 1JRh–C = 70.6 Hz, CO), 172.1 (d, 1JRh–C = 46.9 Hz, Im-C2), 164.5 (q, 1JB–C = 49.3 Hz, ipso-C of BPh4), 141.2 (p-CCH3 of Mes), 140.8 (Cq of Tz), 136.2 (br s, o-CH of BPh4), 136.1 (o-CCH3), 135.7 (ipso-C of Ph), 135.2 (ipso-C of Mes), 131.1 (p-CH of Ph), 130.5 (o-CH of Ph), 129.9 (m-CH of Mes), 126.4 (q, 4JB–C = 2.6 Hz, m-CH of BPh4), 124.1 (Im-C5), 123.4 (Im-C4), 122.5 (p-CH of BPh4), 122.3 (Tz-C5′), 121.4 (m-CH of Ph), 44.6 (CH2), 21.3 (p-CH3 of Mes), 18.4 (o-CH3 of Mes) ppm; HRMS (ESI+, MeOH): m/z (%): calculated for [C23H21N5O2Rh]+ = [M]+ = 502.0750; found [M]+ = 502.0703 (100%) amu; FTIR (solid) νCO = 2083, 2019 (s) cm−1; elemental analysis: found C, 66.42; H, 4.95; N, 8.49; calculated for C47H41BN5O2Rh·0.5CH2Cl2: C, 66.03; H, 4.90; N, 8.52%.Immobilization of [CB-2Rh(CO)2] and [G-2Rh(CO)2]
The complex [1Rh(COD)]BPh4 (24.8 mg, 0.0279 mmol) was dissolved in a degassed solution of nitromethane (10 mL). The mixture was cooled in an ice bath for 15 minutes before concentrated hydrochloric acid (0.5 mL, 0.5 M) was added dropwise into the stirred yellow solution while keeping the mixture between 0–5 °C. Sodium nitrite (1.90 mg, 0.0279 mmol) was then added and the mixture was stirred vigorously for another 5 minutes. After taking the mixture out of the ice bath, carbon black or graphene (50 mg) was added while the mixture was stirred vigorously to ensure that the carbon powders was diffused in solution properly. The mixture was allowed to gradually warm up to room temperature. The mixture was stirred under nitrogen at room temperature overnight. The modified carbon powder was separated from the reaction media via centrifugation (4000 rpm) for 5 minutes. It was consecutively washed and stirred vigorously with a methanol:MilliQ water (50:50, 20 mL) and methanol (5 × 10 mL) followed by centrifugation after each washing. The washed powder was dried under vacuum in a dessicator for 24 hours. The modified carbon powders was then dispersed with vigorous stirring in a dichloromethane:pentane solution (1:1, 20 mL). The mixture was degassed via three cycles of freeze–pump–thaw and then placed under an atmosphere of carbon monoxide in a balloon (1 atm). The mixture was left to react for one hour before the modified powder was separated from the reaction solvent via centrifugation. The modified powder was washed and stirred vigorously with pentane (3 × 10 mL) and methanol (3 × 10 mL) followed by centrifugation after each washing. The washed powder was then dried under vacuum in the dessicator for 24 hours to give either [CB-2Rh(CO)2] (37.2 mg, 72%) and [G-2Rh(CO)2] (32.0 mg, 68%).
Immobilization of [GC-2Rh(CO)2]
The glassy carbon electrode was polished sequentially in alumina slurries (distilled water suspension) of 1.0, 0.3 and 0.05 μm on micropolishing cloth (Buehler, IL, USA) and rinsed with distilled water between each step. It was then washed thoroughly with ethanol and soaked for 30 min in dichloromethane. The complex [1Rh(COD)]BPh4 (24.8 mg, 0.0279 mmol) was dissolved in a degassed solution of nitromethane (10 mL). The mixture was cooled in an ice bath for 5 minutes before concentrated hydrochloric acid (0.5 mL, 0.5 M) was added dropwise into the stirred yellow solution while keeping the mixture between 0–5 °C. Sodium nitrite (1.90 mg, 0.0279 mmol) was then added followed by the glassy carbon (GC) electrode. The mixture was left to sit in the ice bath for a further hour before allowing the mixture to gradually warm up to room temperature and remained at room temperature overnight. The modified GC electrodes was separated from the reaction media and washed with MilliQ water (5 mL). It was then consecutively washed with a methanol:MilliQ water (50:50, 20 mL) and methanol (5 × 10 mL). The washed modified GC electrode was dried under a gentle flow of nitrogen. The modified GC electrode was then dispersed in a dichloromethane:pentane solution (1:1, 20 mL). The mixture was degassed via three cycles of freeze–pump–thaw and then carbon monoxide was slowly bubbled into the mixture for 15 minutes. The modified GC electrode was washed with pentane (3 × 10 mL) and then methanol (3 × 10 mL). The washed powder was then dried under a gentle flow of nitrogen to give with [GC-2Rh(CO)2].
Procedure for hydrosilylation reaction using the homogeneous catalyst, glassy carbon (GC), carbon black (CB) and graphene (G) hybrid catalysts
The reactions were performed in glass v-vials fitted with a magnetic stirring bar and a Teflon screw-caps. For the glassy carbon hybrid catalyst, the electrode (1 × 1 × 0.1 cm), diphenylacetylene (0.1 M), triethylsilane (0.2 M) and tetrahydrofuran (2 mL) were weighed into the glass vial in the glovebox. For the homogenous catalyst (0.3 mg), carbon black (0.8 mg) or graphene (0.5 mg) hybrid catalysts, the catalyst (0.02 mol% Rh relative to amount of diphenylacetylene) and diphenylacetylene (3.3 M) were weighed in air into the vial. The vials was then brought into the glovebox and triethylsilane (6.6 M) and tetrahydrofuran (0.06–0.6 mL) was added. The reactions vials were stirred and heated in an oil bath at 50 °C for 0.25–24 hours. At set time intervals until completion of the reaction, the reaction vial was removed from the oil bath and immediately cooled in an ice bath to room temperature.For the reaction vial containing the glassy carbon hybrid catalyst, the reaction mixture was removed from the vial and solvent evaporated. The product was dissolved in CDCl3 for NMR analysis. The reaction vial containing the hybrid catalysts on carbon black and graphene was centrifuged and the supernatant was collected into a round bottom flask. The solvent was evaporated and CDCl3 was added to dissolve the products for NMR analysis. In all cases, the product conversions were calculated by integration of the chosen product peak in the 1H NMR spectrum against the integration of the chosen starting material (diphenylacetylene) peak.
During the recycling experiments, the glassy carbon electrode catalyst was removed from the reaction mixture, washed with methanol thoroughly (6 mL) and the electrode was dried under a gentle flow of nitrogen for 1 minute and reused subsequently. The carbon–rhodium catalysts on carbon black and graphene was washed with methanol (3 × 2 mL) and centrifuged to remove the solvent used in between each cycle. The carbon–rhodium catalyst was then dried under vacuum and reused in the next cycle. All supernatant and washings were collected and analyzed by ICP-MS.
Acknowledgements
Financial support from the University of New South Wales and the Australian Research Council are gratefully acknowledged. This research was supported under the Australian Research Council's Discovery Projects funding scheme (project numbers DP130101838 and DP150103065). We are grateful to Prof. John Stride for providing the graphene and Cabot Corporation for the donation of the carbon black (Vulcan XC-72R). We thank Dr Karen Privat of the Mark Wainwright Analytical Centre (UNSW) for collecting the SEM images and Mr Fei Han for collecting the TEM images. CMW thanks the Australian Government for the award of an International Postgraduate Research Scholarship (IPRS).Notes and references
- Heterogenized Homogeneous Catalysts for Fine Chemicals Production, ed. P. Barbaro and F. Liguori, Springer, Dordrecht, The Netherlands, 2010 Search PubMed.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- C. W. Jones, Top. Catal., 2010, 53, 942–952 CrossRef CAS.
- A. Schaetz, M. Zeltner and W. J. Stark, ACS Catal., 2012, 2, 1267–1284 CrossRef CAS.
- C. Freire, C. Pereira and S. Rebelo, Catalysis, 2012, 24, 116–203 CAS.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- D. Gajan and C. Coperet, New J. Chem., 2011, 35, 2403–2408 RSC.
- U. Diaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083–4097 RSC.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- Carbon Materials For Catalysis, ed. P. Serp and J. L. Figueiredo, 2009 Search PubMed.
- E. Auer, A. Freund, J. Pietsch and T. Tacke, Appl. Catal., A, 1998, 173, 259–271 CrossRef CAS.
- F. Rascon, R. Wischert and C. Coperet, Chem. Sci., 2011, 2, 1449–1456 RSC.
- M. C. Kung, M. V. Riofski, M. N. Missaghi and H. H. Kung, Chem. Commun., 2014, 50, 3262–3276 RSC.
- S. Kumar, P. Kumar and S. L. Jain, J. Mater. Chem. A, 2014, 2, 18861–18866 CAS.
- Q. Zhao, Y. Zhu, Z. Sun, Y. Li, G. Zhang, F. Zhang and X. Fan, J. Mater. Chem. A, 2015, 3, 2609–2616 CAS.
- H. Su, Z. Li, Q. Huo, J. Guan and Q. Kan, RSC Adv., 2014, 4, 9990–9996 RSC.
- Y.-T. Xi, P.-J. Wei, R.-C. Wang and J.-G. Liu, Chem. Commun., 2015, 51, 7455–7458 RSC.
- M. Blanco, P. Alvarez, C. Blanco, M. V. Jimenez, J. Fernandez-Tornos, J. J. Perez-Torrente, L. A. Oro and R. Menendez, Carbon, 2015, 83, 21–31 CrossRef CAS.
- N. Shang, S. Gao, C. Feng, H. Zhang, C. Wang and Z. Wang, RSC Adv., 2013, 3, 21863–21868 RSC.
- J. K. Wassei and R. B. Kaner, Acc. Chem. Res., 2013, 46, 2244–2253 CrossRef CAS PubMed.
- D. K. James and J. M. Tour, Acc. Chem. Res., 2013, 46, 2307–2318 CrossRef CAS PubMed.
- B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54–75 CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- G. L. C. Paulus, Q. H. Wang and M. S. Strano, Acc. Chem. Res., 2013, 46, 160–170 CrossRef CAS PubMed.
- P. Huang, L. Jing, H. Zhu and X. Gao, Acc. Chem. Res., 2013, 46, 43–52 CrossRef CAS PubMed.
- C. Peng, Y. Xiong, Z. Liu, F. Zhang, E. Ou, J. Qian, Y. Xiong and W. Xu, Appl. Surf. Sci., 2013, 280, 914–919 CrossRef CAS.
- E. Bekyarova, M. E. Itkis, P. Ramesh, C. Berger, M. Sprinkle, W. A. de Heer and R. C. Haddon, J. Am. Chem. Soc., 2009, 131, 1336–1337 CrossRef CAS PubMed.
- A. A. Tregubov, K. Q. Vuong, E. Luais, J. J. Gooding and B. A. Messerle, J. Am. Chem. Soc., 2013, 135, 16429–16437 CrossRef CAS PubMed.
- A. A. Tregubov, D. B. Walker, K. Q. Vuong, J. J. Gooding and B. A. Messerle, Dalton Trans., 2015, 44, 7917–7926 RSC.
- The nature of the counter-ions for the positively charged immobilized complexes could not be established unambiguously by XPS, which was consistent with previous reports: D. R. Laws, J. Sheats, A. L. Rheingold and W. E. Geiger, Langmuir, 2010, 26, 15010–15021 CrossRef CAS PubMed.
- We also observed in all hybrid cases, a very minor peak within the N1s peak at about 399–400 eV indicative of the azo bond (N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) formation due to some incomplete diazonium reaction and this is consistent with previous reports: A. L. Gui, E. Luais, J. R. Peterson and J. J. Gooding, ACS Appl. Mater. Interfaces, 2013, 5, 4827–4835 CAS.
N) formation due to some incomplete diazonium reaction and this is consistent with previous reports: A. L. Gui, E. Luais, J. R. Peterson and J. J. Gooding, ACS Appl. Mater. Interfaces, 2013, 5, 4827–4835 CAS. - J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinruck, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- In all cases, the E isomer (5) of the alkene product was formed in >99:1 ratio to the Z isomer (6).
- J. Berding, J. A. van Paridon, V. H. S. van Rixel and E. Bouwman, Eur. J. Inorg. Chem., 2011, 2450–2458 CrossRef CAS.
- M. Blug, X.-F. Le Goff, N. Mezailles and P. Le Floch, Organometallics, 2009, 28, 2360–2362 CrossRef CAS.
- L. Yong, K. Kirleis and H. Butenschoen, Adv. Synth. Catal., 2006, 348, 833–836 CrossRef CAS.
- M. R. Chaulagain, G. M. Mahandru and J. Montgomery, Tetrahedron, 2006, 62, 7560–7566 CrossRef CAS.
- N. Imlinger, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2005, 690, 4433–4440 CrossRef CAS.
- W. Guo, R. Pleixats, A. Shafir and T. Parella, Adv. Synth. Catal., 2015, 357, 89–99 CrossRef CAS.
- M. Planellas, W. Guo, F. Alonso, M. Yus, A. Shafir, R. Pleixats and T. Parella, Adv. Synth. Catal., 2014, 356, 179–188 CrossRef CAS.
- R. Cano, M. Yus and D. J. Ramon, ACS Catal., 2012, 2, 1070–1078 CrossRef CAS.
- F. Alonso, R. Buitrago, Y. Moglie, J. Ruiz-Martinez, A. Sepulveda-Escribano and M. Yus, J. Organomet. Chem., 2010, 696, 368–372 CrossRef.
- M. Chauhan, B. J. Hauck, L. P. Keller and P. Boudjouk, J. Organomet. Chem., 2002, 645, 1–13 CrossRef CAS.
- M. Choucair, P. Thordarson and J. A. Stride, Nat. Nanotechnol., 2009, 4, 30–33 CrossRef CAS PubMed.
- S. W. Kwok, J. R. Fotsing, R. J. Fraser, V. O. Rodionov and V. V. Fokin, Org. Lett., 2010, 12, 4217–4219 CrossRef CAS PubMed.
- G. Giordano and R. H. Crabtree, Inorg. Synth., 1990, 28, 88–90 CrossRef CAS.
- K. Q. Vuong, M. G. Timerbulatova, M. B. Peterson, M. Bhadbhade and B. A. Messerle, Dalton Trans., 2013, 42, 14298–14308 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional schemes, figures and tables, characterization of the complexes and hybrid complexes on carbon surfaces, XPS, TGA, ICP-MS/OES, IR, Raman, SEM, TEM and electrochemical data and calculations are included. Full catalysis and recycling tables are also included. See DOI: 10.1039/c5sc03787e |
| This journal is © The Royal Society of Chemistry 2016 |