
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing pattern and dynamics of disulfide bridges using synthesis and NMR of an ion channel blocker peptide toxin with multiple diselenide bonds†
Krisztina
Fehér‡
ab,
István
Timári‡
a,
Kinga
Rákosi‡
c,
János
Szolomájer
c,
Tünde Z.
Illyés
d,
Adam
Bartok
e,
Zoltan
Varga
ef,
Gyorgy
Panyi
eg,
Gábor K.
Tóth
*c and
Katalin E.
Kövér
*a
aDepartment of Inorganic and Analytical Chemistry, University of Debrecen, Egyetem tér 1, H-4032, Debrecen, Hungary. E-mail: kover@science.unideb.hu
bDepartment of Organic and Macromolecular Chemistry, Ghent University, Kringslaan 281 S4, 9000, Ghent, Belgium
cDepartment of Medical Chemistry, University of Szeged, Dóm tér 8, H-6720, Szeged, Hungary. E-mail: toth.gabor@med.u-szeged.hu
dDepartment of Organic Chemistry, University of Debrecen, Egyetem tér 1, H-4032, Debrecen, Hungary
eDepartment of Biophysics and Cell Biology, University of Debrecen, Egyetem tér 1, H-4012, Debrecen, Hungary
fMTA-DE-NAP B Ion Channel Structure-Function Research Group, Egyetem tér 1, H-4032, Debrecen, Hungary
gMTA-DE Cell Biology and Signaling Research Group, University of Debrecen, Egyetem tér 1, H-4032, Debrecen, Hungary
First published on 21st December 2015
Abstract
Anuroctoxin (AnTx), a 35-amino-acid scorpion toxin containing four disulfide bridges, is a high affinity blocker of the voltage-gated potassium channel Kv1.3, but also blocks Kv1.2. To improve potential therapeutic use of the toxin, we have designed a double substituted analog, [N17A/F32T]-AnTx, which showed comparable Kv1.3 affinity to the wild-type peptide, but also a 2500-fold increase in the selectivity for Kv1.3 over Kv1.2. In the present study we have achieved the chemical synthesis of a Sec-analog in which all cysteine (Cys) residues have been replaced by selenocysteine (Sec) forming four diselenide bonds. To the best of our knowledge this is the first time to replace, by chemical synthesis, all disulfide bonds with isosteric diselenides in a peptide/protein. Gratifyingly, the key pharmacological properties of the Sec-[N17A/F32T]-AnTx are retained since the peptide is functionally active. We also propose here a combined experimental and theoretical approach including NOE- and 77Se-based NMR supplemented by MD simulations for conformational and dynamic characterization of the Sec-[N17A/F32T]-AnTx. Using this combined approach allowed us to attain unequivocal assignment of all four diselenide bonds and supplemental MD simulations allowed characterization of the conformational dynamics around each disulfide/diselenide bridge.
Introduction
The disulfide pattern of polypeptides containing multiple disulfide bridges is of considerable interest because the presence of disulfide bonds is essential for maintaining the tertiary structure responsible for the observed biological activity. Therefore, a key issue in the structural characterization of these peptides is an unambiguous determination of the pattern of disulfide pairing. Since small, flexible disulfide-rich peptides are typically difficult to crystallize, therefore nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) are the most commonly used techniques for elucidating their structures.1MS determination of a disulfide bonding pattern is a challenging task primarily relying on manual interpretation.2,3 It requires either enzymatic digestion of the protein where disulfide scrambling may take place, or chemical modification if the target protein is cysteine rich, or else contains an unexpected folding pattern with unknown disulfide bonds.
NMR provides powerful tools for determining the three-dimensional structures of these small proteins and peptides, but, due to the unfavorable NMR properties of sulfur isotopes, disulfide bonds often present a ‘blind spot’ in NMR structural investigations.1 Experimental observations supported by DFT quantum-chemical calculations proved that the redox state of cysteine residues can be safely disclosed on the basis of Cys 13Cβ chemical shifts.4 These data, however, do not provide information about the disulfide bridge connectivity. Heretofore the most commonly used approach for determining S–S connectivity relied on the detection of interresidue dipolar (NOE) interactions between the β-methylene protons of the covalently linked cysteine residues.5 Detection of these weak inter-disulfide NOE contacts is often hampered by spin-diffusion effects due to the strong intraresidue cross-relaxation between the geminal beta protons of Cys and/or in some cases by an enhanced conformational flexibility around the disulfide bonds, leading to an ambiguous assignment of the disulfide network. However, such uncertainties could be resolved by stereospecific deuterium labeling of the cysteine beta protons at the cost of laborious isotope labeling. Besides, in the case of closely packed disulfide bonds, the NOE contacts can be also ambiguous and lead to incorrect pairing of Cys residues. Alternatively, as has been recently suggested, replacement of the NMR-inactive 32S nucleus by 77Se possessing more favorable magnetic properties6,7 – by mutation of cysteine into selenocysteine (Sec) residues8–13 – may allow the detection of diselenide pairing directly through the three-bond 3J(1H, 77Se) couplings across the diselenide bond.5 Importantly, it was also confirmed that the isosteric replacement of the disulfide bond with diselenide has no significant effect, in general, on the three-dimensional structure and the associated biological activity of peptides.14 In addition, using 77Se NMR both the conformational and dynamic properties of diselenide (disulfide) bonds can be explored.
The Sec residues can be introduced into peptides or proteins either by chemical synthesis11,12,14–16 or by recombinant expression.7,9,10,17 Using chemical synthesis, substitution of only one or two disulfide bridges has been reported in most cases whereas multiple disulfide bonds can be replaced with diselenides by applying recombinant expression. Recently, an increasing interest also has emerged for the synthesis of selenium-containing polymers due to their potential applicability as biomaterials.18,19
Biological background
Peptide toxins form an important class of disulfide bond containing peptides and proteins. One particular group of peptide toxins isolated from scorpions and other venomous animals targets Kv1.3, the voltage-gated ion channel of human T lymphocytes.20 The Kv1.3 channel plays a crucial role in antigen-induced activation and proliferation of human T cells in general, and in those of effector memory T cells in particular.21 These latter cells are responsible for tissue damage in certain autoimmune diseases such as multiple sclerosis, type I diabetes, and rheumatoid arthritis.22 High affinity and selectivity Kv1.3 inhibitors persistently inhibit the proliferation of effector memory T cells thereby ameliorating the symptoms of the disease in animal models. This indicates the great therapeutic value of the Kv1.3 inhibitors.22–24Anuroctoxin (AnTx), a 35-amino-acid scorpion toxin having four disulfide bridges, is a high affinity blocker of Kv1.3 channel, but also blocks Kv1.2.25 Improving selectivity of the toxin for Kv1.3 over Kv1.2 while keeping the high affinity for Kv1.3 opens the door to the toxin's therapeutic use. In line with these, we have constructed a double substituted analog of AnTx, [N17A/F32T]-AnTx, which is characterized by comparable Kv1.3 affinity to the wild-type peptide but shows a 2500-fold increase in the selectivity for Kv1.3 over Kv1.2 and other ion channels tested (Kv1.1 and KCa3.1).26 Thus, [N17A/F32T]-AnTx may serve as an excellent template for a novel therapeutically applicable peptide, and, interestingly, understanding the structural features of this peptide may lead to a better understanding of the nature of the selectivity of scorpion toxins between Kv1.3 and Kv1.2.
In the present work we report the chemical synthesis of a Sec-analog of [N17A/F32T]-AnTx during which – for the first time to our knowledge – the replacement of all four disulfide bonds with isosteric diselenide bonds has been achieved. Using a combined NOE- and 77Se NMR-based approach unequivocal assignment of the diselenide (disulfide)-bond connectivities was also accomplished. Moreover, the significant broadening of some of the 77Se resonances revealed the presence of slow conformational exchange processes which have been subsequently interpreted by molecular dynamics (MD) simulations.
Results and discussion
Synthesis of the Sec-analog of [N17A/F32T]-AnTx mutant
For the preparation of the anuroctoxin derived peptides, both the Boc and Fmoc chemistry were used previously with success.26 Since the stability of several Boc compatible Se protecting groups is significantly lower than the corresponding sulfur protecting groups (e.g., BnSe− is a much better leaving group than the corresponding sulfur derivative12,27), for the solid-phase synthesis of selenocysteine analog of [N17A/F32T]-AnTx mutant, the Fmoc/tBu strategy was applied. Correspondingly, the related Nα-Fmoc derivative of the 4-methoxy-benzyl protected selenocysteine8 was synthesized and used as replacement for all eight cysteines in the original sequence. For the linear Sec(Mob) protected [N17A/F32T]-AnTx analog, a standard synthetic protocol was applied with DCC/HOBt (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as coupling agent in a 3-fold molar excess. The removal of the Mob group from the selenocysteine residues can be carried out, according to previous studies, under various conditions, e.g., acidic treatment, I2, or applying a sub-stoichiometric amount of 2,2′-dithiobis(5-nitropyridine). With regard to the complexity of the amino acid sequence of the peptide, we decided to achieve the cleavage from the resin together with the protecting groups commonly used in Fmoc/tBu synthesis and removal of the Mob group from the selenocysteine side-chains in a two-step protocol. Acidolytic cleavage of the Sec(Mob) peptide from the resin was performed under various conditions (TFA/H2O/TIS cocktail in 93/5/2, v/v, at low temperature for 2 h; TFA/H2O/TIS cocktail in 93/5/2, v/v at room temperature for 2.5 h) and in both cases the crude product obtained consisted mainly of the compound with all eight Mob groups intact besides compounds which had partially removed one, two, or even three Mob groups. In order to avoid incomplete removal of the side-chain protecting groups, and considering the fact that it follows Mob group de-protection, we decided to carry on with this complex reaction mixture. For Mob de-protection/cyclization we tried procedures well established in cysteine chemistry,27,28 like the DMSO/TFA (v/v) procedure (10% DMSO/TFA and 4% DMSO/TFA) and the I2-mediated method (added quantitatively or in excess), but in all cases we obtained a very complex mixture of compounds from which the desired diselenide-bridged peptide could not be isolated. Cleavage of the p-methoxybenzyl group and intramolecular oxidation to the diselenide peptide was successfully achieved with the more gentle 2,2′-dithiobis(5-nitropyridine) (DTNP) in TFA. Incubation of the protected Sec-containing peptide in TFA with DTNP resulted in effective protecting group removal and its substitution by the 2-(5-nitropyridyl) (Npys) group derived from DTNP fragmentation, independent of the number of DTNP equivalents and peptide concentration used. After test reactions, a final 4 mM peptide concentration in TFA was used with 1 equiv. DTNP incubation for 1 h. The reaction was quenched via precipitation into cold diethyl ether and, after isolation of the precipitate, the crude material was treated with one equivalent of L-cysteine in pH 8 NH4OAc buffer solution in order to induce collapse into the desired multiple diselenide-peptide. HPLC analysis of the resulting solution showed an almost instantaneous transformation of the hemi-reduced intermediate into the multiple diselenide compound (Scheme 1).
:1) as coupling agent in a 3-fold molar excess. The removal of the Mob group from the selenocysteine residues can be carried out, according to previous studies, under various conditions, e.g., acidic treatment, I2, or applying a sub-stoichiometric amount of 2,2′-dithiobis(5-nitropyridine). With regard to the complexity of the amino acid sequence of the peptide, we decided to achieve the cleavage from the resin together with the protecting groups commonly used in Fmoc/tBu synthesis and removal of the Mob group from the selenocysteine side-chains in a two-step protocol. Acidolytic cleavage of the Sec(Mob) peptide from the resin was performed under various conditions (TFA/H2O/TIS cocktail in 93/5/2, v/v, at low temperature for 2 h; TFA/H2O/TIS cocktail in 93/5/2, v/v at room temperature for 2.5 h) and in both cases the crude product obtained consisted mainly of the compound with all eight Mob groups intact besides compounds which had partially removed one, two, or even three Mob groups. In order to avoid incomplete removal of the side-chain protecting groups, and considering the fact that it follows Mob group de-protection, we decided to carry on with this complex reaction mixture. For Mob de-protection/cyclization we tried procedures well established in cysteine chemistry,27,28 like the DMSO/TFA (v/v) procedure (10% DMSO/TFA and 4% DMSO/TFA) and the I2-mediated method (added quantitatively or in excess), but in all cases we obtained a very complex mixture of compounds from which the desired diselenide-bridged peptide could not be isolated. Cleavage of the p-methoxybenzyl group and intramolecular oxidation to the diselenide peptide was successfully achieved with the more gentle 2,2′-dithiobis(5-nitropyridine) (DTNP) in TFA. Incubation of the protected Sec-containing peptide in TFA with DTNP resulted in effective protecting group removal and its substitution by the 2-(5-nitropyridyl) (Npys) group derived from DTNP fragmentation, independent of the number of DTNP equivalents and peptide concentration used. After test reactions, a final 4 mM peptide concentration in TFA was used with 1 equiv. DTNP incubation for 1 h. The reaction was quenched via precipitation into cold diethyl ether and, after isolation of the precipitate, the crude material was treated with one equivalent of L-cysteine in pH 8 NH4OAc buffer solution in order to induce collapse into the desired multiple diselenide-peptide. HPLC analysis of the resulting solution showed an almost instantaneous transformation of the hemi-reduced intermediate into the multiple diselenide compound (Scheme 1).
 | ||
| Scheme 1 Amino acid sequence of Sec-[N17A/F32T]-AnTx with four diselenide bonds. | ||
The HPLC trace of the folded, purified tetra diselenide toxin analog is shown on Fig. S1.† The spectra of mass spectrometric analyses of the multiple selenocysteine peptide recorded with quadrupole ESI and Q-TOF spectrometers are depicted in Fig. S2–S5.† The characteristic isotopic selenium abundance (Fig. S5†) is more complicated than the sulfur containing ones, but a careful study led to the justification of the correct molecular ion (Fig. S4†). Subsequent NMR studies and biological measurements of the isolated main product proved that all four diselenide bridges are completely analogous with the disulfide connectivities from the native [N17A/F32T]-AnTx peptide.
The Sec-analog of [N17A/F32T]-AnTx retains its biological activity
Prior to obtaining detailed structural information one key question remained to be answered, namely, whether the Sec-analog of [N17A/F32T]-AnTx still blocks the Kv1.3 channel with high affinity. To this end, the Sec-analog was tested on Kv1.3 expressed in human peripheral blood lymphocytes. Fig. 1 shows that the peptide in 3 nM concentration blocks approx. 75% of the peak current (Fig. 1A), which is comparable to the inhibition of Kv1.3 with the double mutated peptide [N17A/F32T]-AnTx at identical concentration (89% inhibition in the latter case,26Fig. 1C). Fig. 1B shows that the block of Kv1.3 develops quickly and it is rapidly reversible by perfusing the bath with toxin-free solution. The dose–response relationship obtained for Kv1.3 inhibition by Sec-[N17A/F32T]-AnTx (Fig. 1D) indicates an IC50 of 1.54 nM, which is comparable to that of [N17A/F32T]-AnTx (IC50 = 0.63 nM, dashed line). The Hill coefficient was close to 1 for both peptides (H = 0.93 for Sec-[N17A/F32T]-AnTx and H = 0.88 for [N17A/F32T]-AnTx) which argues that the peptide:channel stoichiometry of inhibition is 1:1, as predicted by currently accepted models of toxin binding to the channels.29
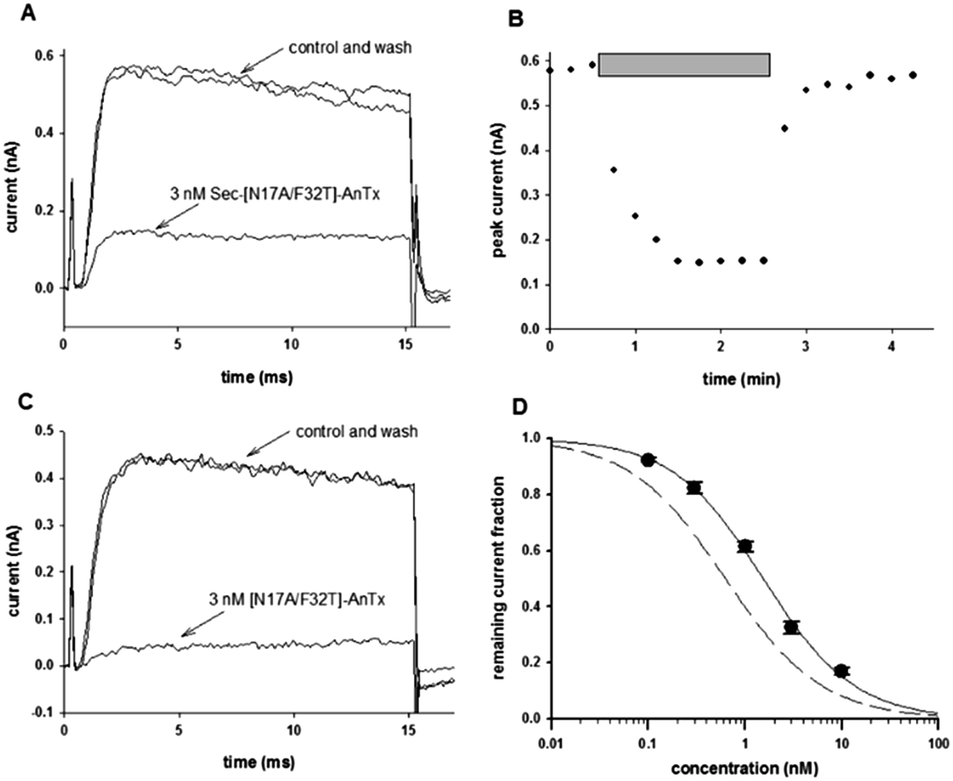 | ||
| Fig. 1 Effect of AnTx variants on Kv1.3 channel. Whole-cell Kv1.3 current traces were measured in activated human lymphocytes in whole cell configuration. Cells were held at −100 mV then depolarized to +50 mV for 15 ms every 15 s. The bath was perfused continuously. (A), The traces show the K+ current before the application of the toxin (control), after the equilibration of the block in the presence of 3 nM Sec-[N17A/F32T]-AnTx (as indicated) and after recovery from block during the perfusion of the bath with toxin-free solution (wash). (B), The blocking effect was fully reversible. Peak K+ currents were determined and plotted as a function of time. Grey bars mark the presence of 3 nM Sec-[N17A/F32T]-AnTx in the bath solution. Other experimental details are as in A. (C), The traces show the K+ current before the application of the toxin (control), after the equilibration of the block in the presence of 3 nM [N17A/F32T]-AnTx (as indicated) and after recovery from block during the perfusion of the bath with toxin-free solution (wash). (D), Concentration-dependence of K+ current block by Sec-[N17A/F32T]-AnTx. The remaining fraction of the Kv1.3 current was calculated as I/I0, where I0 and I are the peak K+ currents measured in the control solution and upon reaching equilibrium block during bath perfusion with the test solution containing the toxin at indicated concentrations, respectively. The voltage protocol and other experimental conditions were the same as in (A). The superimposed solid line is the Hill equations fitted to the data points (see Experimental). The best fit yielded IC50 = 1.54 nM, and H = 0.93. Error bars indicate SEM (n = 3–5). The dashed line shows the dose–response relationship of [N17A/F32T]-AnTx which is characterized by IC50 = 0.63 nM and H = 0.88 (data from ref. 26). | ||
In summary, the key pharmacological property of [N17A/F32T]-AnTx, i.e., the high affinity block of the Kv1.3 current, is retained in Sec-[N17A/F32T]-AnTx so the peptide is functionally active.
NMR investigation of the Sec-analog of [N17A/F32T]-AnTx
Almost complete assignments of 1H, 13C and 15N resonances of Sec-mutant have been achieved using standard solution state NMR techniques. The 1H resonances were assigned based on two-dimensional (2D) homonuclear correlation spectra including 1H-1H COSY, TOCSY and NOESY. Whereas the well-resolved cross peaks of 1H-13C and 1H-15N HSQC spectra allowed the assignment of 13C and 15N resonances (see Table S1 in the ESI†), it should be mentioned that some of the Hβ–Cβ cross peaks, such as of Sec4, Sec10 and Sec24 residues, are significantly broadened and have, therefore, lower intensities in the 1H-13C HSQC correlation map (Fig. 2); this is indicative of slow conformational dynamics occurring in the vicinity of those residues. Note that similar exchange mediated line-broadening was observed in the 1H-15N HSQC spectrum for T5, Q8, H9, U10, T11 and K30 residues (data not shown). It was also found that the Sec-derivative possesses almost identical 1H, 13C, and 15N chemical shifts as the S-containing peptide (selected regions of 1H-13C HSQC spectra of the two peptide analogs are shown in Fig. S6†), suggesting high similarity of their three-dimensional structures. | ||
| Fig. 2 The Hβ/Cβ region of 1H-13C HSQC spectrum of Sec-[N17A/F32T]-AnTx. Broadened (lower intensity) Hβ–Cβ cross peaks of Sec4, Sec10 and Sec24 residues are indicated by blue asterisks. Hβ–Cβ cross peaks of the remaining Sec residues are labelled by red asterisks. | ||
The oxidized state of all eight Sec residues incorporated into the peptide could be unequivocally proved on the basis of the characteristic Hβ/Cβ and Se chemical shifts of the Sec residues (Table S1†). The MS data also supported the formation of four intramolecular diselenide bonds.
To establish the location of bridged residues, 1H-1H 2D NOESY spectra were recorded with 150 ms and 300 ms mixing. However, even using longer mixing time (300 ms), NOE cross peaks between the β-methylene protons of the covalently linked Sec residues could be hardly detected. This may be due partly to the strong intraresidue cross-relaxation between the geminal β-protons of Sec, partly to the line broadening caused by slow conformational dynamics around the Se–Se bridge, and also to the severe signal overlap in the region of interest. Specifically, inspection of the NOESY spectrum shown in Fig. 3 reveals only one Se–Se bond unequivocally, which can be assigned to the Sec4–Sec24 bridge. Unambiguous assignment of the other inter-Sec(Hβ) cross peaks is hampered by the strong signal overlap.
 | ||
| Fig. 3 Partial contour plot of 1H-1H NOESY spectrum (500 MHz, τm = 300 ms) of Sec-[N17A/F32T]-AnTx. NOE cross peaks between the β-methylene protons of covalently linked Sec residues are indicated by arrows. | ||
To take advantage of our ‘Se NMR-spies’ incorporated into the peptide sequence, multiple bond 1H-77Se correlation spectra were recorded using standard HMQC or HMBC experiments. Sites around disulfide/diselenide bridges in peptides/proteins, however, are often regions of conformational exchange. The exchange process, in general, can be detrimental to the quality of NMR spectra, resulting in broad and very weak, or even unobservable signals, and which may be detrimental to structure elucidation and, in particular, to the NMR-based characterization and assignment of disulfide/diselenide bonds. To our disappointment, even after several attempts using a different experimental setup (see ESI† for details and selected examples), only a few and exclusively intraresidue 1H-77Se correlations could be observed, probably due to conformational exchange line broadening.
To improve the sensitivity of 1H-77Se multiple bond correlation measurements, a modified, 77Se-decoupled variant of the CPMG-HSQMBC experiment30–32 has been developed in our group. This experiment considerably reduces the adverse line broadening effect of conformational exchange and also efficiently suppresses the undesired co-evolution of proton–proton couplings (such as the large geminal coupling between the Hβ protons of Sec) during the long scalar coupling evolution period matched to the intra- and interresidue multiple bond 1H-77Se couplings. To minimize heating of the probe electronics and the sample during the long duration of CPMG pulse train, low-power composite 180° pulses were applied on both the proton and Se nuclei. Note that, similarly, CPMG pulse trains have been successfully employed in HSQC experiments to reduce the undesired effect of exchange processes and, as a consequence, for better observation of exchange broadened signals in nucleic acids and proteins.33–35
The details of the experiment proposed here (scheme in Fig. S7† and Bruker code of the pulse sequence are given in ESI†) and comparison of its performance to the classical HMQC/HMBC methods are demonstrated in the ESI (Fig. S8–S10†). Recording several 1H-77Se CPMG-HSQMBC spectra with the sequence of Fig. S7† at different temperatures and using evolution times matched to different long-range couplings, we found that the largest number of connectivities could be detected at 318 K with the coupling evolution time set to 30–35 ms. The resulting spectrum shown in Fig. 4 allowed unambiguous assignment of all eight Se-resonances (as given on the projection at left), though some of the correlations are weak due to exchange broadening as noticed. Nevertheless, the cross peaks enlarged and shown in insets at lower intensity threshold could be assigned to provide direct evidence of two additional Se–Se pairings, between Sec14–Sec31 and Sec19–Sec34 residues, respectively.
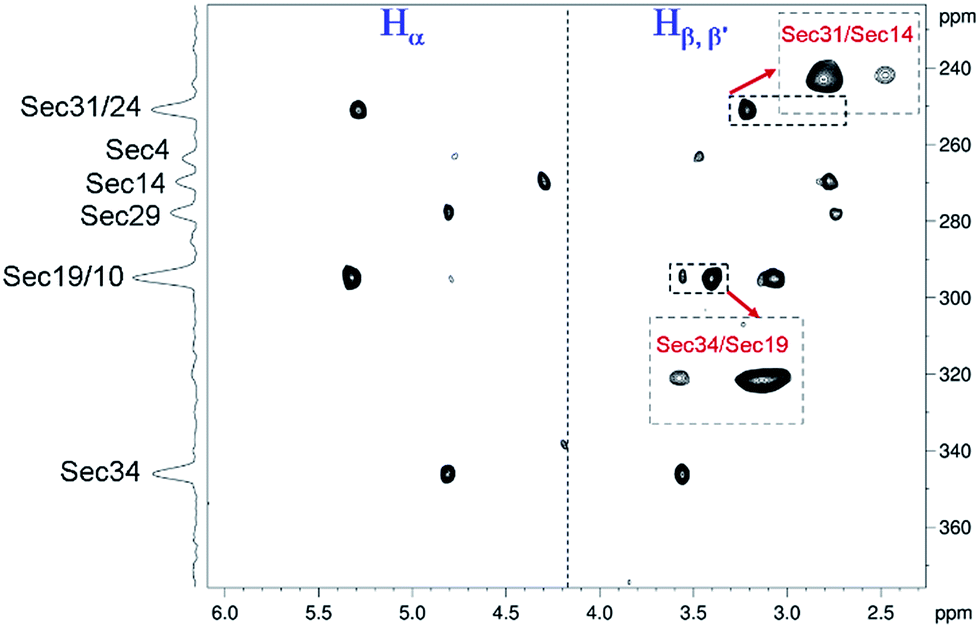 | ||
| Fig. 4 1H-77Se multiple bond correlation spectrum of Sec-[N17A/F32T]-AnTx collected with a refocused 77Se-decoupled CPMG-HSQMBC experiment at 318 K and natural abundance of 77Se isotope. Heteronuclear coupling evolution time was set to 33 ms. Assignment of 77Se resonances are given on the projection at left side. Interresidue 1H-77Se cross peaks detected are shown in insets at lower intensity threshold and provide direct evidence of Se–Se bridges between Sec14–Sec31 and Sec19–Sec34 residues, respectively. | ||
Since the chemical shifts (Cβ, Se) of Sec-s already confirmed that all eight Sec residues are involved in diselenide bonds, thus the remaining, unassigned Se–Se connectivity could be unambiguously assigned to the Sec10 and Sec29 residues.
Molecular dynamics (MD) simulation
MD calculations were found to be useful tools to analyze disulfide bridge connectivities in small disulfide-rich proteins.36,37 In order to find an explanation for the observed NMR behaviour of peptide sites around diselenide bridges, we carried out a 100 ns molecular dynamics (MD) simulation on the [N17A/F32T]-AnTx peptide. Since replacement of disulfide bridges with diselenide bonds has been demonstrated to be bioisoteric due to high similarity between the conformational properties of sulfur and selenium,11 the simulations were carried out on the disulfide bridge containing peptide. The χ3 angle has taken up conformations around the canonical ±90° and has mostly persisted in the conformation presented in the starting structure shown in Fig. 5a. The dynamic behavior of the different S–S bridges in the trajectory could be classified into three groups according to the conformation along the side chain χ2 dihedral angles of the cysteine residues (Fig. 5b and c): the conformation around the 14–31 bridge was static apart from small fluctuations, the 19–34 bridge was most dynamic with frequent transitions between different states, and the bridges 10–29 and 4–24 showed intermediate dynamic behavior with less frequent or a single transition between conformations. Thus, the strong intra/inter-residue HSQMBC correlations for 77Se resonances in bridge 14–31 are due to the lack of any exchange process for the nuclei involved, while the nuclei in bridge 19–34 may experience fast exchange line narrowing. The weaker correlations for the connectivities in bridges 10–29 and 4–24 could be attributed to the presence of intermediate conformational exchange broadening.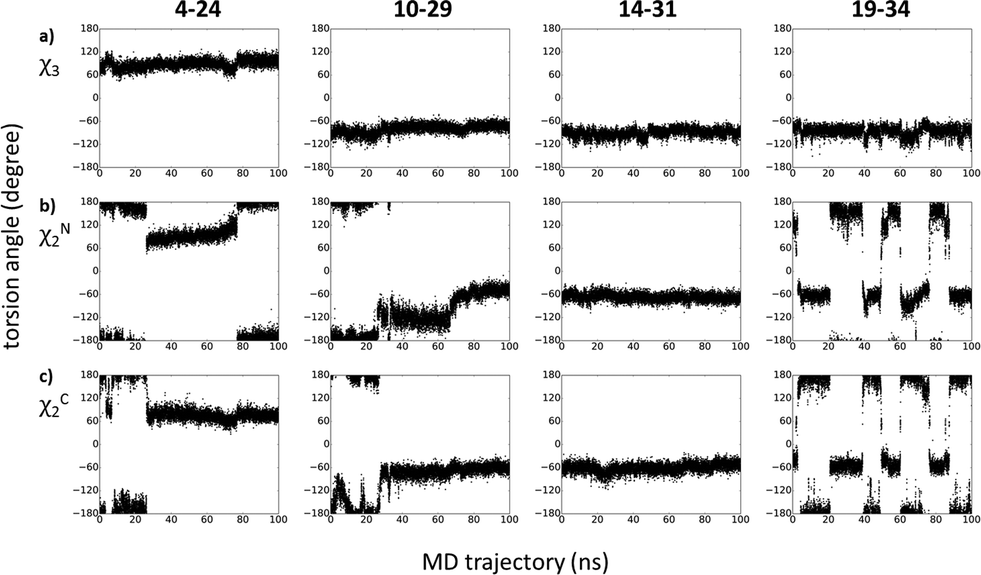 | ||
| Fig. 5 Development of torsion angles along the trajectory for disulfide bridges 4–24, 10-29, 14–31 and 19–34. The first row (a) shows the χ3 angle is the torsion angle around the disulfide bond (as defined by atoms CBN-SGN-SGC-CBC). The χ2 sidechain torsion angle is shown for (b) the N terminal (as defined by atoms CAN-CBN-SGN-SGC) and (c) the C terminal residue (as defined by atoms SGN-SGC-CBC-CAC) of the cysteine pair in the bridge. | ||
Conclusions
In summary, a Sec-analog of the biologically active [N17A/F32T]-AnTx peptide toxin containing four diselenide bonds has been produced with chemical synthesis. To the best of our knowledge, this is the first time all disulfide bonds in a protein were replaced by chemical synthesis with isosteric diselenides. We found that the Sec-derivative displayed similar voltage-gated potassium channel (Kv1.3) blocking potency and almost identical 1H, 13C and 15N chemical shifts as the S-containing peptide, suggesting high similarity of their three-dimensional structures including their Se–Se/S–S bridge pattern, respectively.Using a combined NMR approach relying on characteristic chemical shifts, 1H-1H NOEs and 1H-77Se multiple bond connectivities, the diselenide network of the Sec-analog was unequivocally disclosed. To improve the sensitivity of 1H-77Se correlation NMR experiment for peptides with multiple diselenide bonds and in the presence of exchange broadening, a modified CPMG-HSQMBC pulse sequence was designed and applied. The CPMG-cycles employed efficiently reduce the loss of spin coherence due to both chemical exchange and undesired evolution of proton–proton couplings.
Finally, NMR experiments complemented with MD simulations allowed us to explore and characterize the conformational dynamics of diselenide/disulfide bonds.
Experimental
Electrophysiology
Kv1.3 currents were elicited by 15 ms long depolarization impulses to +50 mV from a holding potential of −100 mV every 15 s. To acquire and analyze the measured data a pClamp10 software package was used. Current traces were lowpass-filtered by the analog four-pole Bessel filters of the amplifiers. The effect of the toxin in a given concentration was determined as remaining current fraction (RF = I/I0, where I0 is the peak current in the absence of the toxin and I is the peak current at equilibrium block at a given toxin concentration). Points on the dose–response curves represent the mean of 5 independent measurements where the error bars represent the S.E.M. Data points were fitted with a two-parameter Hill equation, RF = IC50H/(IC50H + [Tx]H), where IC50 is the dissociation constant, H is the Hill coefficient and [Tx] is the toxin concentration.
NMR spectroscopy
3.9 mg peptide toxin was dissolved in 280 μL buffer (90:10 H2O/D2O, 10 mM Na3PO4, pH 5.7, 50 mM NaCl, 1 mM NaN3) for all NMR runs whereas 7.9 mg was disssolved in D2O buffer for 77Se NMR measurements and transferred into a Shigemi NMR tube. All NMR spectra were recorded on an Avance II 500 (Bruker, Rheinstetten, Germany) spectrometer at 303 K, if not indicated otherwise. 1H chemical shifts are referenced to sodium 2,2-dimethyl-2-silapentane-5-sulfonate (0 ppm), while heteronuclear shifts are referenced indirectly based on the gyromagnetic ratios for 13C, 15N, and 77Se, respectively. All heteronuclear correlation spectra (1H-13C and 1H-15N HSQC, selenium-decoupled 1H-77Se CPMG-HSQMBC, HMBC and HMQC) were acquired at natural abundance. The 1H-77Se multiple bond correlation spectra were recorded at 283, 303, 313, and 318 K, respectively using scalar coupling evolution time matched to 10, 12, 15, 17, 20, and 30 Hz, typically in overnight experiments. 2D 1H-1H NOESY spectra using a Watergate sequence for water suppression were recorded with 150 and 300 ms mixing, respectively. TOCSY mixing was set to 80 ms. The 1H, 13C, 15N, and 77Se 90° hard pulses were of 11.5, 13.0, 29.0, and 15.0 μs duration, respectively.
Computational methods
All MD simulations were performed using AMBER version 12.38 The AMBER ff99SB force field39 for the peptides and the TIP3P model40 for water were used. The cut-off used for non-bonded interactions was 8 Å. The particle-mesh Ewald41 procedure was used to describe long-range electrostatic interactions with a maximal grid spacing of 1 Å. Periodic boundary conditions were applied with a truncated octahedron geometry. The SHAKE42 algorithm was used to keep the bond lengths of hydrogen atoms rigid allowing a timestep of 2 fs to be used.Since there is no selenium atom type implemented in the chosen force field, we used sulfur in place of selenium. Comparison of the properties of sulfur and selenium shows that the structure and dynamics of the selenium-containing peptide should be very similar to the sulfur-containing one. This assumption was confirmed by comparison of the 1H, 13C, and 15N chemical shifts, which were strongly correlated.
000 steps was performed to remove bad contacts switching from steepest descent to a conjugate gradient algorithm after 100 steps. After minimization at a constant pressure MD was carried out to equilibrate the system for 0.5 ns, during which the density stabilized at around 0.9965 g cm−3, and the temperature settled at ca. 300 K. Subsequently, a classical constant total energy MD producing micro canonical NVE ensemble was carried out for 100 ns.
All simulations were run in a single Nvidia GTX680 GPU using the GPU43,44 implementation of the pmemd provided in the AMBER12 simulation package. Using the described protocols a 100 ns-long calculation of the peptide took about 25 h to complete.
000 coordinate snapshots were saved for analysis in all cases. The trajectories were analyzed with ptraj45 and visualized in VMD.46 The stability of the trajectories were analyzed using the mdout_analyser.py.
Acknowledgements
Financial support from TÁMOP-4.2.2/A-11/1/KONV-2012-0025 (K. E. K. and G. P.) and TÁMOP-4.2.2/A-11/1/KONV-2012-0035 (G. K. T.) as well as OTKA K 75904 and OTKA NK 101337 (G. P.), OTKA K 105459 (K. E. K.), KTIA_NAP_13-2-2015–0009 (Z. V.) and KTIA_13_NAP-A-III/8 (G. K. T.) are gratefully acknowledged. K. F. is a Marie-Curie Carrier Integration Grant fellow (PCIG10-GA-2011-303917), Z. V. is a Bolyai Fellowship and I. T. is a Richter Gedeon Talentum Alapítvány Ph.D. scholarship awardee.Notes and references
- K. E. Kövér and G. Batta, in Amino Acids, Peptides and Proteins: Volume 38, The Royal Society of Chemistry, 2014, vol. 38, pp. 37–59 Search PubMed.
- P. L. Tsai, S. F. Chen and S. Y. Huang, Rev. Anal. Chem., 2013, 32, 257–268 CrossRef CAS.
- K. A. Makepeace, J. J. Serpa, E. V. Petrotchenko and C. H. Borchers, Methods, 2015, 89, 74–78 CrossRef CAS PubMed.
- D. Sharma and K. Rajarathnam, J. Biomol. NMR, 2000, 18, 165–171 CrossRef CAS PubMed.
- M. Mobli and G. F. King, Toxicon, 2010, 56, 849–854 CrossRef CAS PubMed.
- H. Duddeck, Annu. Rep. NMR Spectrosc., 2004, 52, 105–166 CrossRef CAS.
- S. A. Schaefer, M. Dong, R. P. Rubenstein, W. A. Wilkie, B. J. Bahnson, C. Thorpe and S. Rozovsky, J. Mol. Biol., 2013, 425, 222–231 CrossRef CAS PubMed.
- T. Koide, H. Itoh, A. Otaka, H. Yasui, M. Kuroda, N. Esaki, K. Soda and N. Fujii, Chem. Pharm. Bull., 1993, 41, 502–506 CrossRef CAS PubMed.
- S. Muller, H. Senn, B. Gsell, W. Vetter, C. Baron and A. Bock, Biochemistry, 1994, 33, 3404–3412 CrossRef CAS PubMed.
- S. Pegoraro, S. Fiori, S. Rudolph-Bohner, T. X. Watanabe and L. Moroder, J. Mol. Biol., 1998, 284, 779–792 CrossRef CAS PubMed.
- L. Moroder, J. Pept. Sci., 2005, 11, 187–214 CrossRef CAS PubMed.
- M. Muttenthaler and P. F. Alewood, J. Pept. Sci., 2008, 14, 1223–1239 CrossRef CAS PubMed.
- A. D. de Araujo, B. Callaghan, S. T. Nevin, N. L. Daly, D. J. Craik, M. Moretta, G. Hopping, M. J. Christie, D. J. Adams and P. F. Alewood, Angew. Chem., Int. Ed., 2011, 50, 6527–6529 CrossRef PubMed.
- C. J. Armishaw, N. L. Daly, S. T. Nevin, D. J. Adams, D. J. Craik and P. F. Alewood, J. Biol. Chem., 2006, 281, 14136–14143 CrossRef CAS PubMed.
- D. Besse, F. Siedler, T. Diercks, H. Kessler and L. Moroder, Angew. Chem., Int. Ed., 1997, 36, 883–885 CrossRef CAS.
- A. D. de Araujo, M. Mobli, J. Castro, A. M. Harrington, I. Vetter, Z. Dekan, M. Muttenthaler, J. J. Wan, R. J. Lewis, G. F. King, S. M. Brierley and P. F. Alewood, Nat. Commun., 2014, 5, 3165 Search PubMed.
- M. P. Strub, F. Hoh, J. F. Sanchez, J. M. Strub, A. Bock, A. Aumelas and C. Dumas, Structure, 2003, 11, 1359–1367 CrossRef CAS PubMed.
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed.
- G. Panyi, L. D. Possani, R. C. Rodriguez de la Vega, R. Gaspar and Z. Varga, Curr. Pharm. Des., 2006, 12, 2199–2220 CrossRef CAS PubMed.
- H. Wulff, P. A. Calabresi, R. Allie, S. Yun, M. Pennington, C. Beeton and K. G. Chandy, J. Clin. Invest., 2003, 111, 1703–1713 CrossRef CAS PubMed.
- C. Beeton, H. Wulff, N. E. Standifer, P. Azam, K. M. Mullen, M. W. Pennington, A. Kolski-Andreaco, E. Wei, A. Grino, D. R. Counts, P. H. Wang, C. J. Leehealey, S. Andrews, A. Sankaranarayanan, D. Homerick, W. W. Roeck, J. Tehranzadeh, K. L. Stanhope, P. Zimin, P. J. Havel, S. Griffey, H. G. Knaus, G. T. Nepom, G. A. Gutman, P. A. Calabresi and K. G. Chandy, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17414–17419 CrossRef CAS PubMed.
- M. D. Cahalan and K. G. Chandy, Immunol. Rev., 2009, 231, 59–87 CrossRef CAS PubMed.
- V. Chi, M. W. Pennington, R. S. Norton, E. J. Tarcha, L. M. Londono, B. Sims-Fahey, S. K. Upadhyay, J. T. Lakey, S. Iadonato, H. Wulff, C. Beeton and K. G. Chandy, Toxicon, 2012, 59, 529–546 CrossRef CAS PubMed.
- M. Bagdany, C. V. F. Batista, N. A. Valdez-Cruz, A. Somodi, R. C. R. de la Vega, A. F. Licea, Z. Varga, R. Gaspar, L. D. Possani and G. Panyi, Mol. Pharmacol., 2005, 67, 1034–1044 CrossRef CAS PubMed.
- A. Bartok, K. Fehér, A. Bodor, K. Rákosi, G. K. Tóth, K. E. Kövér, G. Panyi and Z. Varga, Sci. Rep., 2015, 5, 18397 CrossRef CAS PubMed.
- A. L. Schroll, R. J. Hondal and S. Flemer, J. Pept. Sci., 2012, 18, 155–162 CrossRef CAS PubMed.
- K. M. Harris, S. Flemer and R. J. Hondal, J. Pept. Sci., 2007, 13, 81–93 CrossRef CAS PubMed.
- S. A. Goldstein and C. Miller, Biophys. J., 1993, 65, 1613–1619 CrossRef CAS PubMed.
- R. T. Williamson, B. L. Marquez, W. H. Gerwick and K. E. Kövér, Magn. Reson. Chem., 2000, 38, 265–273 CrossRef CAS.
- K. E. Kövér, G. Batta and K. Fehér, J. Magn. Reson., 2006, 181, 89–97 CrossRef PubMed.
- S. Boros and K. E. Kövér, Magn. Reson. Chem., 2011, 49, 106–110 CrossRef CAS PubMed.
- L. Mueller, P. Legault and A. Pardi, J. Am. Chem. Soc., 1995, 117, 11043–11048 CrossRef CAS.
- F. A. A. Mulder, C. A. E. M. Spronk, M. Slijper, R. Kaptein and R. Boelens, J. Biomol. NMR, 1996, 8, 223–228 CrossRef CAS PubMed.
- P. Barraud, F. Dardel and C. Tisne, Cron. Chim., 2008, 11, 474–479 CrossRef CAS.
- C. Combelles, J. Gracy, A. Heitz, D. J. Craik and L. Chiche, Proteins: Struct., Funct., Bioinf., 2008, 73, 87–103 CrossRef CAS PubMed.
- M. M. Castellanos and C. M. Colina, J. Phys. Chem. B, 2013, 117, 11895–11905 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. I. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolosvary, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, Amber 12 Reference Manual, University of California, San Francisco, 2012 Search PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- S. Le Grand, A. W. Gotz and R. C. Walker, Comput. Phys. Commun., 2013, 184, 374–380 CrossRef CAS.
- R. Salomon-Ferrer, A. W. Gotz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthesis and analytical characterization of the Sec-analog of [N17A/F32T]-AnTx mutant including quadrupole ESI and ESI-TOF MS spectra, overlay of 1H-13C HSQC spectra of the double mutant [N17A/F32T]-AnTx and its Sec-analog, scheme and Bruker code of the 77Se-decoupled 1H-77Se CPMG-HSQMBC pulse sequence and comparison of its performance to the classical HMQC method, 1H, 13C, 15N and 77Se resonance assignment of Sec-[N17A/F32T]-AnTx peptide. See DOI: 10.1039/c5sc03995a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |