
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dichotomous mechanistic behavior in Narasaka–Heck cyclizations: electron rich Pd-catalysts generate iminyl radicals†
Nicholas J.
Race
,
Adele
Faulkner
,
Megan H.
Shaw
and
John F.
Bower
 *
*
School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: john.bower@bris.ac.uk; Fax: +44 (0)117 925 1295
First published on 1st December 2015
Abstract
Pd-catalyzed cyclizations of oxime esters with pendant alkenes are subject to an unusual ligand controlled mechanistic divergence. Pd-systems modified with electron deficient phosphines (e.g. P(3,5-(CF3)2C6H3)3) promote efficient aza-Heck cyclization, wherein C–N bond formation occurs via alkene imino-palladation. Conversely, electron rich ligands, such as P(t-Bu)3, cause deviation to a SET pathway and, in these cases, C–N bond formation occurs via cyclization of an iminyl radical. A series of mechanistic experiments differentiate the two pathways and the scope of the hybrid organometallic radical cyclization is outlined. This study represents a rare example in Pd-catalysis where selection between dichotomous mechanistic manifolds is facilitated solely by choice of phosphine ligand.
Introduction
Palladium-catalyzed processes are fundamental to organic synthesis, and it is estimated that one in five C–C bond formations employed in commercial syntheses of new drugs are reliant on this technology.1 Significant and continuing efforts are focused on the development of diverse phosphine ligands to enhance key mechanistic steps, such as oxidative addition.2 Consequently, the overall efficiency of a given process is strongly dependent on the exact choice of P-based ligand. However, cases where this choice causes deviation from common two electron redox processes to one electron, radical-based pathways are rare.3 This is despite the well documented, but underappreciated propensity of Pd(0)-systems to undergo single electron transfer (SET) oxidative addition in certain contexts,4 and the emergence of a series of hybrid organometallic-radical methodologies that invoke the intermediacy of Pd(I)-complexes (Scheme 1A).5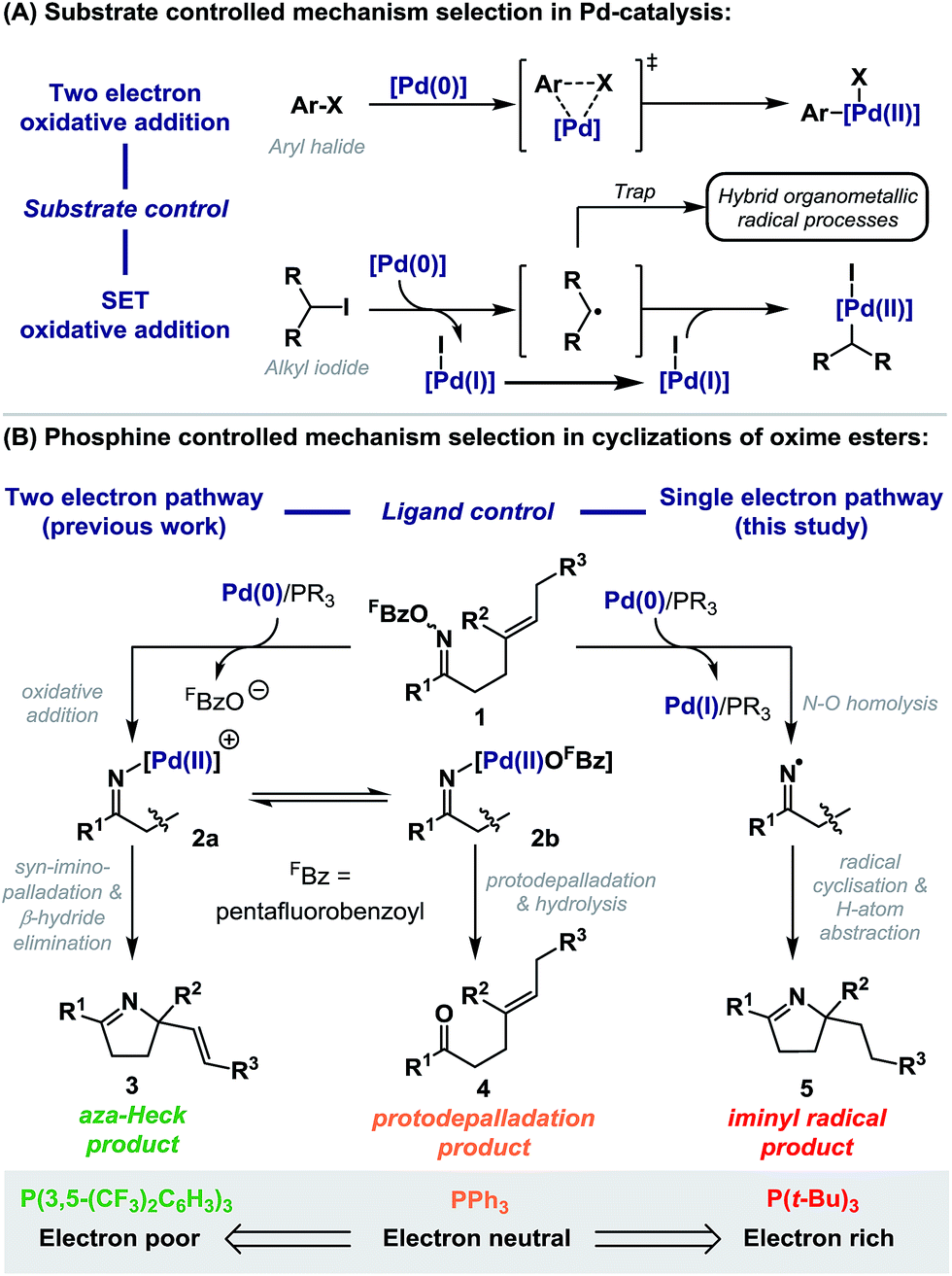 | ||
| Scheme 1 | ||
We have reported a range of aza-Heck methodologies that involve oxidative addition of Pd(0)-catalysts into the N–O bond of O-pentafluorobenzoyl oxime esters 1 (Scheme 1B, two electron pathway).6–8 Addition of the resulting imino-Pd intermediate 2a across a pendant alkene leads to aza-Heck (3)6,7 or cascade products.8 For these processes, electron deficient ligand systems, especially P(3,5-(CF3)2C6H3)3, are most effective, as they enhance migratory insertion and suppress protodepalladation of 2a/b; this latter pathway leads to the corresponding ketone 4 and predominates with electron neutral ligand systems, such as PPh3.7 In this report we disclose that electron rich phosphines (e.g. P(t-Bu)3) do not lead to Heck type products, but instead promote exclusive access to radical manifolds (Scheme 1B, single electron pathway). This has important implications for processes reliant upon the oxidative addition of Pd(0)-catalysts into oxime ester N–O bonds. Indeed, in addition to aza-Heck reactions,6,7 this catalysis platform has enabled diverse methodologies, including alkene 1,2-carboaminations,8 aryl C–H aminations,9 alkene aziridinations,10 alkene 1,2-iodoaminations,11 benzyne 1,2-aminofunctionalizations,12 and C–C bond activations.13 Furthermore, this study provides convenient and unique access to iminyl radical chemistry,14 and in broader terms, represents a rare example in Pd-catalysis where selection between dichotomous mechanistic manifolds is facilitated solely by choice of phosphine ligand.3
Results and discussion
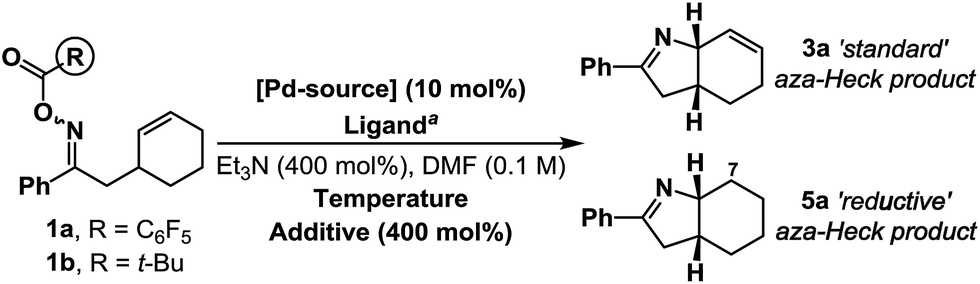
Under optimized aza-Heck conditions, which use P(3,5-(CF3)2C6H3)3 as ligand, cyclization of O-pentafluorobenzoyl oxime ester 1a to alkene 3a occurs in 93% yield (Table 1, entry 3).7a The O-pentafluorobenzoyl group is important, as, following oxidative addition, the pentafluorobenzoate leaving group undergoes facile protodecarboxylation to C6F5H, which drives access to cationic intermediate 2a, as required for cyclization.8 When PPh3 was used as ligand, a 30% yield of 3a was achieved (entry 1) and the mass balance consisted predominantly of the corresponding ketone, which likely arises via protodepalladation of intermediate 2b. For O-pivaloyl oxime ester 1b, cyclization to 3a was not observed using either P(3,5-(CF3)2C6H3)3 or PPh3, and, in both cases, the only identifiable product was the corresponding ketone. Here, the issue is likely that the pivalate leaving group does not dissociate readily after oxidative addition to provide access to key cationic intermediate 2a. Thus, effective aza-Heck cyclization requires both an O-pentafluorobenzoyl oxime ester and an electron deficient phosphine ligand, whereas protodepalladation predominates using electron neutral phosphines and/or weakly dissociating leaving groups. Exposure of either 1a or 1b to the electron rich Pd-system (dt-bpf)PdCl2 did not lead to aza-Heck product 3a or significant quantities of ketone. Instead, adduct 5a was isolated in 72% yield in both cases (entries 4 and 5). 5a is a formal ‘reductive’ aza-Heck product, however, as outlined later, this likely arises via a Pd(0)-triggered radical-based cyclization. The situation appears to be general for a range of electron rich phosphines, including P(t-Bu)3, and PCy3, and other classes of strong donor ligand, such as N-heterocyclic carbenes (entries 6–10). The use of common hydride sources, such as HCO2H, in conjunction with dt-bpf as ligand was detrimental to the yield of 5a. However, common hydrogen atom donors, such as 1,4-cyclohexadiene (1,4-CHD) and γ-terpinene enhanced cyclization efficiency, with the latter providing 5a in 88% yield (entries 12 and 13). These observations provided early evidence for a radical based pathway.15|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Pd-source/ligand | Temp/°C | Additive | 3a (%) | 5a (%) |
a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 [Pd]:ligand for monodentate systems, 1:1 [Pd]:ligand for bidentate systems.
b dt-bpf = 1,1′-bis(di-tert-butylphosphino)ferrocene.
c 5 mol% Pd/L used.
d Isolated yield. :2 [Pd]:ligand for monodentate systems, 1:1 [Pd]:ligand for bidentate systems.
b dt-bpf = 1,1′-bis(di-tert-butylphosphino)ferrocene.
c 5 mol% Pd/L used.
d Isolated yield.
|
||||||
| 1 | C6F5 | Pd2(dba)3/PPh3 | 100 | None | 30 | <5 |
| 2 | C6F5 | Pd2(dba)3/P(3,5-(CF3)2C6H3)3 | 120 | None | 90 | <5 |
| 3 | C6F5 | Pd2(dba)3/P(3,5-(CF3)2C6H3)3c | 60 | None | 93 | <5 |
| 4 | C6F5 | (dt-bpf)PdCl2b | 120 | None | <5 | 72 |
| 5 | t-Bu | (dt-bpf)PdCl2b | 120 | None | <5 | 72 |
| 6 | t-Bu | Pd2(dba)3/S-Phos | 120 | None | <5 | 10 |
| 7 | t-Bu | Pd2(dba)3/P(1-Ad)2n-Bu | 120 | None | <5 | 29 |
| 8 | t-Bu | Pd2(dba)3/P(Cy)3 | 120 | None | <5 | 30 |
| 9 | t-Bu | Pd2(dba)3/P(t-Bu)3 | 120 | None | <5 | 50 |
| 10 | t-Bu | PEPPSI-IPr | 120 | None | <5 | 27 |
| 11 | t-Bu | (dt-bpf)PdCl2b,c | 70 | None | <5 | 67 |
| 12 | t-Bu | (dt-bpf)PdCl2b,c | 70 | 1,4-CHD | <5 | 80 |
| 13 | t-Bu | (dt-bpf)PdCl2b,c | 70 | γ-Terpinene | <5 | 88 |
Cyclization of 1b under optimized ‘reductive’ aza-Heck conditions, but in the presence of TEMPO (150 mol%) provided trapping adduct 6 in 80% yield and 10:1 d.r.; the structure of the major diastereomer was confirmed by single crystal X-ray diffraction (Scheme 2A).16 An analogous trapping experiment on 1a, using optimized ‘standard’ aza-Heck conditions, did not generate 6, and ‘standard’ aza-Heck product 3a was formed in 41% yield. The formation of 6 is consistent with cyclization to generate an alkyl radical at C7 during the conversion of 1a/b to 5a, however, in the absence of exogenous hydrogen atom donors, the source of reductant is unclear (cf.Table 1, entry 11 vs. 13). Cyclization of 1b, under the conditions outlined in Table 1, entry 11, using DMF-d7/Et3N or DMF-d7/Et3N-d15 did not result in appreciable levels of deuterium incorporation in adduct 5a (Scheme 2B). However, in both cases, the yield of 5a was lower than when solely protio-reagents were used. Overall, these observations implicate the feasibility of hydrogen atom abstraction from either DMF, Et3N, or other components of the reaction system (e.g.1a/b or 5a).17
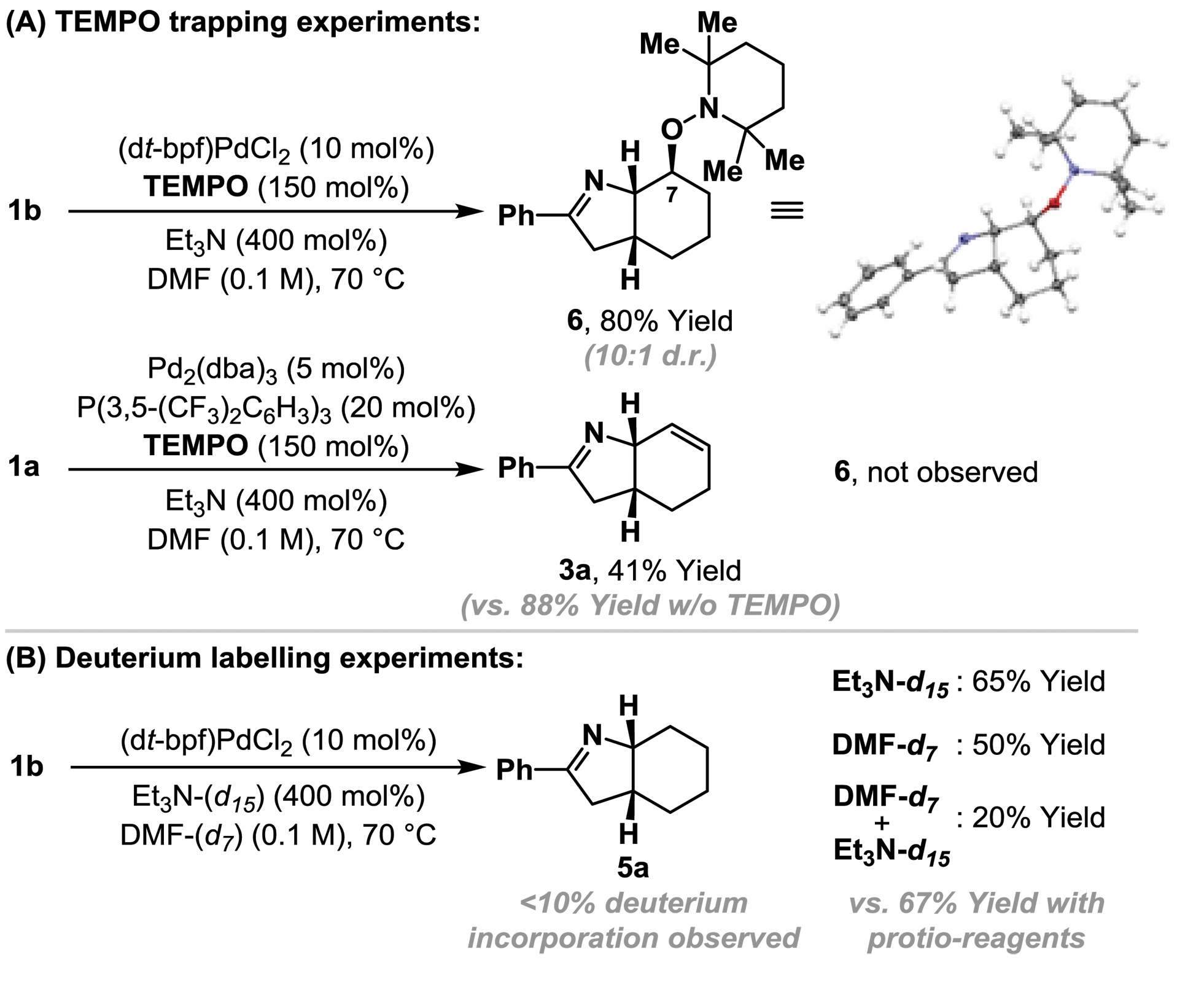 | ||
| Scheme 2 Preliminary mechanistic studies. | ||
TEMPO can trigger radical based pathways when employed as a probe in Pd-catalyzed processes.18 Consequently, the studies outlined in Scheme 2A are not definitive proof for the intermediacy of an alkyl radical during the cyclization of 1a/b to 5a. To provide further evidence, experiments based on Newcomb's radical probe were devised.19 Exposure of O-pivaloyl oxime ester 7a to ‘reductive’ aza-Heck conditions resulted in the formation of dihydropyrrole 10 in 25% yield and as the only observable product (Scheme 3A); the regioselectivity of cyclopropane cleavage was determined by 2D NMR analysis (see the ESI†). The sole formation of 10 is consistent with initial cyclization to alkyl radical 8, which undergoes selective β-scission (via bond b) to generate stabilized benzylic radical 9. Hydrogen atom abstraction from γ-terpinene then yields 10. Cyclization of O-pentafluorobenzoyl oxime ester 7b, under optimized ‘standard’ aza-Heck conditions, resulted in a 51% yield and 1:1 ratio of unstable dihydropyrroles 13a/b (Scheme 3B); the latter was formed as a 5.6:1 mixture of alkene isomers. This result is consistent with an imino-palladation pathway, wherein cyclization generates alkyl-Pd(II) intermediate 11. This is not expected to have significant radical (or carbocationic) character, such that non-selective β-carbon elimination (to 12a/b) ensues en route to 13a/b.20 Products of β-hydride elimination from alkyl-Pd(II) intermediate 11 were not observed. The studies in Scheme 3 provide strong evidence for a radical-based pathway to 5a and an imino-palladation pathway to 3a.
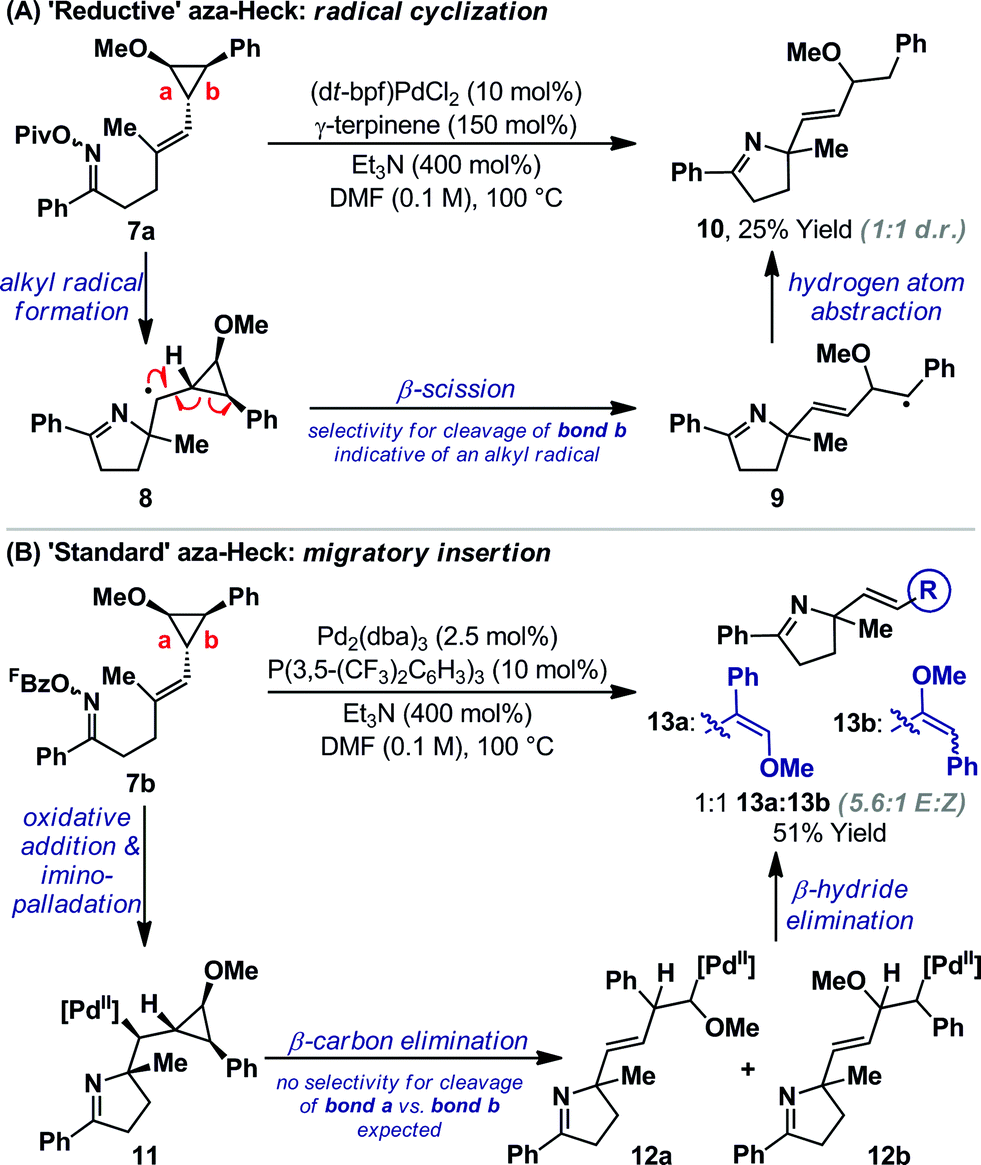 | ||
| Scheme 3 Cyclopropane mechanistic experiments. | ||
The most likely pathway to alkyl radical 8 is via cyclization of an iminyl radical. The generation of these from oxime esters is documented widely,14 however palladium-catalyzed conditions have not been reported. The oxidative addition of PCy3 ligated Pd(0)-systems into oxime ester N–O bonds is known, and both Hartwig and Stahl have characterized associated imino-Pd(II) complexes by X-ray diffraction.9,21 However, little is known about the exact nature of the process and further insights were warranted given that, in the present study, PCy3 leads to radical cyclization product 5a (Table 1, entry 8). To examine this aspect we have employed estrone derived oxime esters 14a/b (Scheme 4); Zard and co-workers have shown that iminyl radicals derived from substrates of this type lead to inversion of the C13 stereocenter.22 Exposure of a DMF solution of O-pivaloyl oxime ester 14a to (dt-bpf)PdCl2 (100 mol%) and Et3N (400 mol%), in the absence of γ-terpinene, resulted in complete consumption of starting material after 15 minutes at 90 °C. After hydrolytic work-up, a 1:5 mixture of estrone derivatives 16a and 16b was isolated in 50% yield. The formation of 16b is consistent with SET from Pd(0) to generate iminyl radical 15a, which undergoes reversible β-scission (via the corresponding nitrile) to afford thermodynamically favored diastereomer 15b. Incomplete inversion of the methyl-substituted stereocenter may be due to quenching of iminyl radical 15a by either hydrogen atom abstraction (from elsewhere in the system) or recombination with Pd(I). An analogous experiment on O-pentafluorobenzoyl oxime ester 14b, using stoichiometric Pd(0)/P(3,5-(CF3)2C6H3)3, generated ketone 16a exclusively in 72% yield. Overall, these results suggest that N–O oxidative addition involving electron rich Pd(0)-systems has significant SET character, whereas electron poor systems insert via a two electron redox pathway. We note that, in certain cases, oxidative addition of Pd(0)-catalysts into alkyl-iodide bonds has been shown to proceed via a SET pathway;4 these observations established that the nature of the substrate can change the mechanistic pathway from that commonly observed for aryl halides (see Scheme 1A). However, the results described in the present study are unique examples where analogous mechanistic deviations are achieved simply by altering the ancillary ligand on Pd.3,23
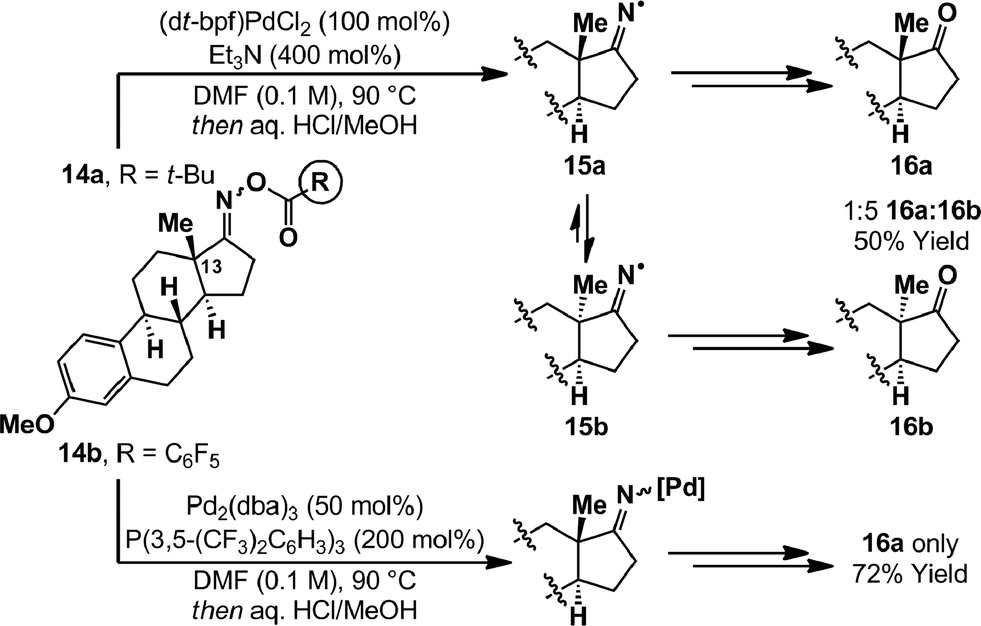 | ||
| Scheme 4 Estrone mechanistic experiments. | ||
Based on the above results, a plausible mechanism for the conversion of 1a/b to 5a is outlined in Scheme 5. Single electron transfer from Pd(0) to oxime ester 1a/b results in N–O cleavage to generate Pd(I) and iminyl radical 17. Studies by the groups of Hartwig and Stahl9,21 suggest that, in principle, recombination could generate imino-Pd(II) intermediate 18, however, this work was conducted on systems without an alkene acceptor. Consequently, in the present case, recombination is ‘interrupted’ by competing and fast 5-exo cyclization to generate alkyl radical 19, which is quenched by hydrogen atom abstraction from γ-terpinene to afford product 5a and bis-allylic radical 20. Hydrogen atom transfer from 20 to Pd(I) generates p-cymene and a Pd(II)-hydride, which undergoes base-induced reductive elimination to Pd(0) to close the catalytic cycle. At the present stage, iminyl radical generation via formation and subsequent N–Pd homolysis of imino-Pd(II) intermediate 18 cannot be discounted. The proposed hybrid organometallic radical mechanism is unusual and adds to a growing body of processes that use late transition metal catalysts to accomplish classical radical processes.5
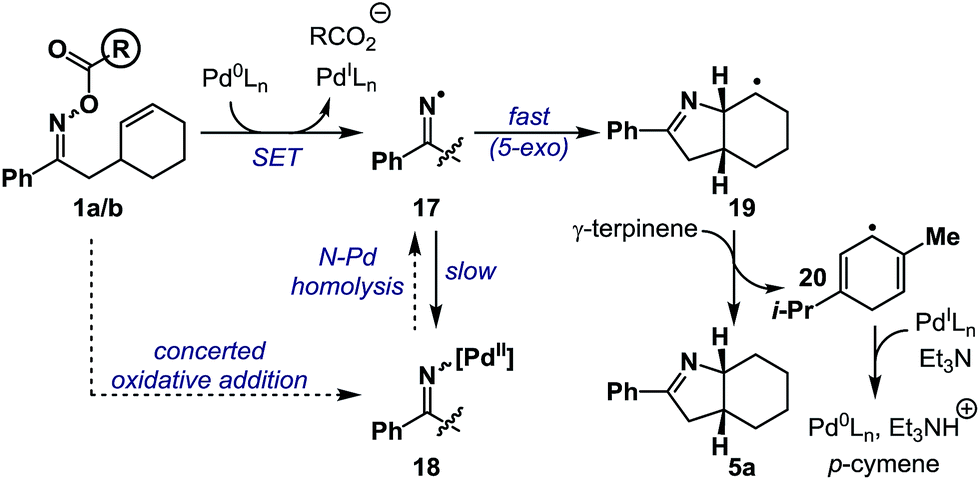 | ||
| Scheme 5 A working mechanistic hypothesis. | ||
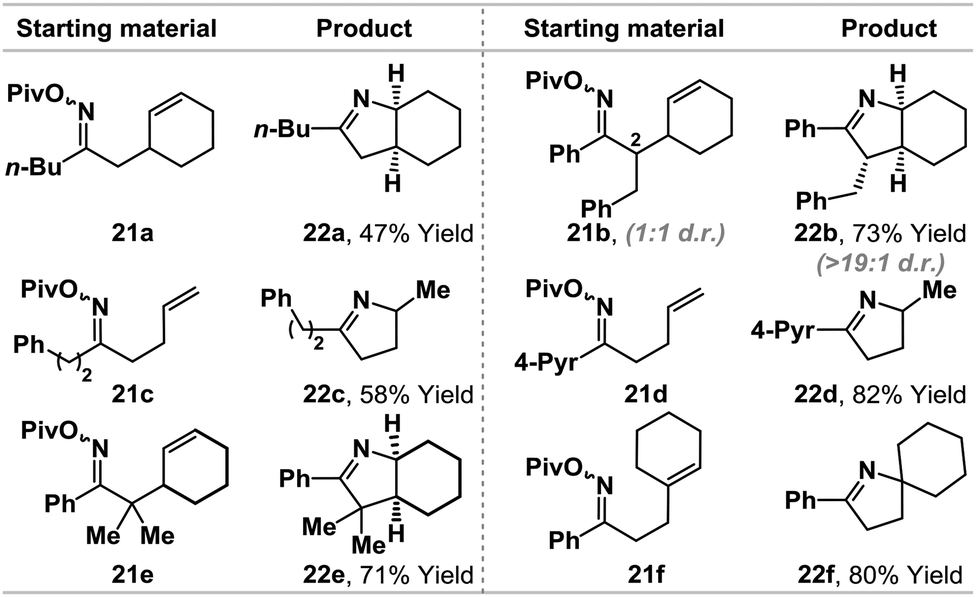
The ability to promote iminyl radical cyclizations using a Pd(0)-catalyst, in combination with γ-terpinene, represents a novel and potentially useful approach to alkene hydroamination. Related iminyl-radical based protocols14 often require specialized (and costly) O-activating groups (e.g. O-Ph)14c,f or toxic (e.g. Bu3SnH/AIBN)14d and/or operationally challenging conditions (e.g. UV/visible light irradiation) that are difficult to scale-up.14e,f In light of the efficiency of the conversion of 1b to 5a, we have conducted a preliminary examination of the scope using a range of pivaloyl oxime esters 21a–f (Table 2). Aryl- and alkyl-substituted oximes esters are tolerated and cyclization occurred in moderate to excellent yields using a range of alkene acceptors. For 21b, cyclization of a 1:1 mixture of diastereomers at C2 provided product 22b in high diastereopurity, likely as a result of post-cyclization epimerization to the thermodynamically favored diastereomer.7a The results outlined in Table 2 show that the present protocol provides a useful entry to iminyl radical chemistry.14
| a Cyclizations were run under the following conditions: (dt-bpf)PdCl2 (5 mol%), γ-terpinene (400 mol%), Et3N (400 mol%), DMF (0.1 M), 70–90 °C, 16–24 h. Full details are given in the ESI. |
|---|
|
Conclusions
In summary, we demonstrate that Pd-catalyzed cyclizations of oxime esters can be partitioned between dichotomous mechanistic manifolds solely through choice of phosphine ligand. Electron rich phosphines promote SET-type oxidative addition, which is ‘interrupted’ at the stage of an iminyl radical to provide hybrid organometallic radical C–N bond forming cyclizations. For electron poor phosphines, N–O oxidative addition proceeds via a ‘conventional’ two electron pathway to generate directly imino-palladium intermediates, which engage pendant alkenes in a Heck-like manner. These mechanistic insights will guide ongoing efforts in our laboratory aimed at providing a general aza-Heck protocol. A wide range of processes are dependent upon aza-Pd(II) intermediates generated by N–O oxidative addition,6–13 and, as such, the studies outlined here are likely to be of importance beyond the immediate area of aza-Heck cyclizations.Note added after first publication
This article replaces the version published on 1st December 2015, which contained errors in Scheme 2.Acknowledgements
We thank the EPSRC (EP/J007455/1) for funding. N.J.R. and M.H.S. thank the Bristol Chemical Synthesis Doctoral Training Centre, funded by the EPSRC (EP/G036764/1), and Syngenta for Ph.D. studentships. N.J.R. thanks the SCI for a postgraduate scholarship. We thank the University of Bristol, School of Chemistry X-ray crystallography service for analysis of 6. J.F.B. is indebted to the Royal Society for a University Research Fellowship.Notes and references
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- Selected reviews: (a) R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283 CrossRef CAS; (b) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS PubMed; (c) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- We are, in fact, unaware of any such examples in Pd-catalysis. For a pertinent example in Cu-catalysis, where the presence or absence of a phosphine ligand dictates the mechanistic pathway, see: B. Zhao, X. Peng, S. Cui and Y. Shi, J. Am. Chem. Soc., 2010, 132, 11009 CrossRef CAS PubMed.
- Selected studies that consider two electron vs. SET pathways for oxidative addition of Pd(0) into C–I bonds: aryl iodides: (a) C. Armatore and F. Pflüger, Organometallics, 1990, 9, 2276 CrossRef. Alkyl iodides: (b) A. V. Kramer and J. A. Osborn, J. Am. Chem. Soc., 1974, 96, 7832 CrossRef CAS; (c) H. Stadtmüller, A. Vaupel, C. E. Tucker, T. Stüdemann and P. Knochel, Chem.–Eur. J., 1996, 2, 1204 CrossRef. Oxidative addition of Pd(0) to alkyl iodides can proceed via either SN2 or SET pathways. For a review, see: (d) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS PubMed.
- Selected recent examples of Pd-catalyzed hybrid organometallic radical processes that employ alkyl iodides: (a) K. S. Bloome, R. L. McMahen and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 20146 CrossRef CAS PubMed; (b) H. Liu, Z. Qiao and X. Jiang, Org. Biomol. Chem., 2012, 10, 7274 RSC; (c) B. M. Monks and S. P. Cook, Angew. Chem., Int. Ed., 2013, 52, 14214 CrossRef CAS PubMed; (d) E. R. Fruchey, B. M. Monks, A. M. Patterson and S. P. Cook, Org. Lett., 2013, 15, 4362 CrossRef CAS PubMed; (e) C. M. McMahon and E. J. Alexanian, Angew. Chem., Int. Ed., 2014, 53, 5974 CrossRef CAS PubMed; (f) A. R. O. Venning, P. T. Bohan and E. J. Alexanian, J. Am. Chem. Soc., 2015, 137, 3731 CrossRef CAS PubMed. For a comprehensive review, see: (g) U. Jahn, Top. Curr. Chem., 2012, 320, 323 CrossRef CAS PubMed.
- Seminal studies: (a) H. Tsutsui and K. Narasaka, Chem. Lett., 1999, 28, 45 CrossRef; (b) H. Tsutsui, M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2002, 75, 1451 CrossRef CAS. Reviews: (c) M. Kitamura and K. Narasaka, Chem. Rec., 2002, 2, 268 CrossRef CAS PubMed; (d) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505 CrossRef CAS.
- Aza-Heck reactions: (a) A. Faulkner and J. F. Bower, Angew. Chem., Int. Ed., 2012, 51, 1675 CrossRef CAS PubMed; (b) N. J. Race and J. F. Bower, Org. Lett., 2013, 15, 4616 CrossRef CAS PubMed; (c) A. Faulkner, J. S. Scott and J. F. Bower, Chem. Commun., 2013, 49, 1521 RSC. For a copper-catalyzed variant, see: (d) A. Faulkner, N. J. Race and J. F. Bower, Chem. Sci., 2014, 5, 2416 RSC.
- Cascade processes: A. Faulkner, J. S. Scott and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 7224 CrossRef CAS PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed.
- K. Okamoto, T. Oda, S. Kohigashi and K. Ohe, Angew. Chem., Int. Ed., 2011, 50, 11470 CrossRef CAS PubMed.
- C. Chen, L. Hou, M. Cheng, J. Su and X. Tong, Angew. Chem., Int. Ed., 2015, 54, 3092 CrossRef CAS PubMed.
- T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572 CrossRef CAS PubMed.
- (a) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 2000, 122, 12049 CrossRef CAS; (b) T. Nishimura, Y. Nishiguchi, Y. Maeda and S. Uemura, J. Org. Chem., 2004, 69, 5342 CrossRef CAS PubMed.
- Selected reviews: (a) J. C. Walton, Acc. Chem. Res., 2014, 47, 1406 CrossRef CAS PubMed; (b) S. Z. Zard, Synlett, 1996, 1148 CrossRef CAS. Selected examples that achieve alkene hydroamination: (c) F. Portela-Cubillo, J. S. Scott and J. C. Walton, Chem. Commun., 2007, 4041 RSC; (d) A.-C. Callier-Dublanchet, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1995, 36, 8791 CrossRef CAS; (e) F. Portela-Cubillo, J. Lymer, E. M. Scanlan, J. S. Scott and J. C. Walton, Tetrahedron, 2008, 64, 11908 CrossRef CAS; (f) J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CAS. Methods that employ visible or UV light irradiation are challenging to scale-up for manufacture.
- For examples of the use of 1,4-CHD and γ-terpinene as hydrogen atom sources, see: A. Gansäuer, A. Barchuk and D. Fielenbach, Synthesis, 2004, 2567 CrossRef.
- An analogous experiment on oxime ester 1a led to compound 6 in 54% yield. O-Pivaloyl oxime esters were selected for mechanistic studies under ‘reductive’ aza-Heck conditions, as they are more atom economical, cheaper and more stable than O-pentafluorobenzoyl variants; these aspects impact upon the wider applicability of the protocol, as outlined in Table 2.
- The propensity for this latter option presumably increases when deuterio-reagents are used. In this scenario, the lower yields observed in Scheme 2B can be attributed to competitive consumption of 1b/5a. DMF is a well-established hydrogen atom source. For example, see: I. Kamiya, H. Tsunoyama, T. Tsukuda and H. Sakurai, Chem. Lett., 2007, 36, 646 CrossRef CAS . Other polar solvents, such as DMAc and NMP, can also be used for the cyclization of 1a/b to 5a; less polar solvents, such as PhMe, lead to low conversions.
- For example, see: A. C. Albéniz, P. Espinet, R. López-Fernández and A. Sen, J. Am. Chem. Soc., 2002, 124, 11278 CrossRef.
- (a) M. Newcomb and D. L. Chestney, J. Am. Chem. Soc., 1994, 116, 9753 CrossRef CAS; (b) M.-H. le Tadic-Biadatti and M. Newcomb, J. Chem. Soc., Perkin Trans. 2, 1996, 1467 RSC.
- R. C. Larock and S. Varaprath, J. Org. Chem., 1984, 49, 3432 CrossRef CAS.
- W. P. Hong, A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 13664 CrossRef CAS PubMed.
- J. Boivin, A.-M. Schiano and S. Z. Zard, Tetrahedron Lett., 1992, 33, 7849 CrossRef CAS.
- Selected recent processes where the choice of phosphine ligand dictates reaction regioselectivity: (a) M.-H. Yang, D. L. Orsi and R. A. Altman, Angew. Chem., Int. Ed., 2015, 54, 2361 CrossRef CAS PubMed; (b) J. Liu, Q. Liu, R. Franke, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2015, 137, 8556 CrossRef CAS PubMed; (c) K. Ohmatsu, M. Ito and T. Ooi, Chem. Commun., 2014, 50, 4554 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all compounds are provided. CCDC 1429194. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04037j |
| This journal is © The Royal Society of Chemistry 2016 |