
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirecting group assisted meta-hydroxylation by C–H activation†
Arun
Maji
,
Bangaru
Bhaskararao
,
Santanu
Singha
,
Raghavan B.
Sunoj
* and
Debabrata
Maiti
*
Department of Chemistry, IIT Bombay, Powai, Mumbai-400076, India. E-mail: dmaiti@chem.iitb.ac.in; sunoj@chem.iitb.ac.in
First published on 21st January 2016
Abstract
meta-Hydroxylated cores are ubiquitous in natural products. Herein, we disclose the first template assisted meta-hydroxylation reaction. Experimental and in silico studies helped us to gain valuable mechanistic insights, including the role of the hexafluoroisopropanol (HFIP) solvent, during C–H hydroxylation. The reactive intermediates, prior to the C–H activation, have been detected by spectroscopic techniques. Additionally, the C–O bond formation has been extended to meta-acetoxylation. The preparation of a phase II quinone reductase activity inducer and a resveratrol precursor illustrated the synthetic significance of the present strategy.
Introduction
The ability to transform a carbon–hydrogen bond to a carbon–heteroatom bond is highly important owing to its involvement in late-stage functionalization of complex natural products, pharmaceuticals and agrochemicals.1 An effective strategy in this domain is yet to achieve its full potential due to the inherent problem of the selectivity and reactivity of the carbon–hydrogen bond.2 Although the concept of directing-group (DG) assistance resolves the issue of the selective functionalization of the proximal C–H bond,3 the distal meta-C–H bond is difficult to activate.2b,4 Hence the intrinsic electronic bias of the substituted core has been utilized to promote meta-C–H functionalization.5 In order to reach the remote meta-C–H bond the idea of a traceless directing group, transient mediators and the concept of H-bonding have been utilized.6 However, regardless of the functionalization, template assisted direct meta-C–H bond activation requires the involvement of a large strained metallacycle intermediate. Therefore success relies on the perfect design of the directing group (DG) linkage, the coordinating site and the choice of metals. In 2012, pioneering work by Yu and co-workers demonstrated the feasibility of the hypothesis using a U-shaped template with a tethered –CN directing group.7 The concept was explored in various arylamine, benzylamine, cinnamic acid, phenol, benzyl alcohol and phenyl acetic acid scaffolds by the same group to elucidate the directing group assisted C–C and C–O bond formations via olefination, arylation, alkylation and acetoxylation reactions.7,8 Furthermore, the concept of optimal coordination by a –CN directing group has been extended by the Tan group, our group and recently by the Li group towards benzylsilyl, benzylsulphonyl, phenyl acetic acid and phenylethyl amine derivatives.9 Despite significant progress in template assisted meta selective C–C bond formation, the generation of a C–X (heteroatom) bond at the meta position is still in its infancy.Following our initial report on the meta-olefination of the phenylacetic acid core (Scheme 1),9b an arylmethane sulphonyl moiety was noted to mitigate the trans-esterification issues noted in earlier work.9c Moreover meta-C–O bond formation with phenylacetic acid was found to be problematic. Therefore, we planned a meta-hydroxylation reaction with an arylmethane sulphonyl core as the model substrate. Despite the successful acetoxylation of arylamine,8c benzylamine and indoline scaffolds8d by Yu and coworkers, a single-step meta-hydroxylation protocol is yet to be reported.
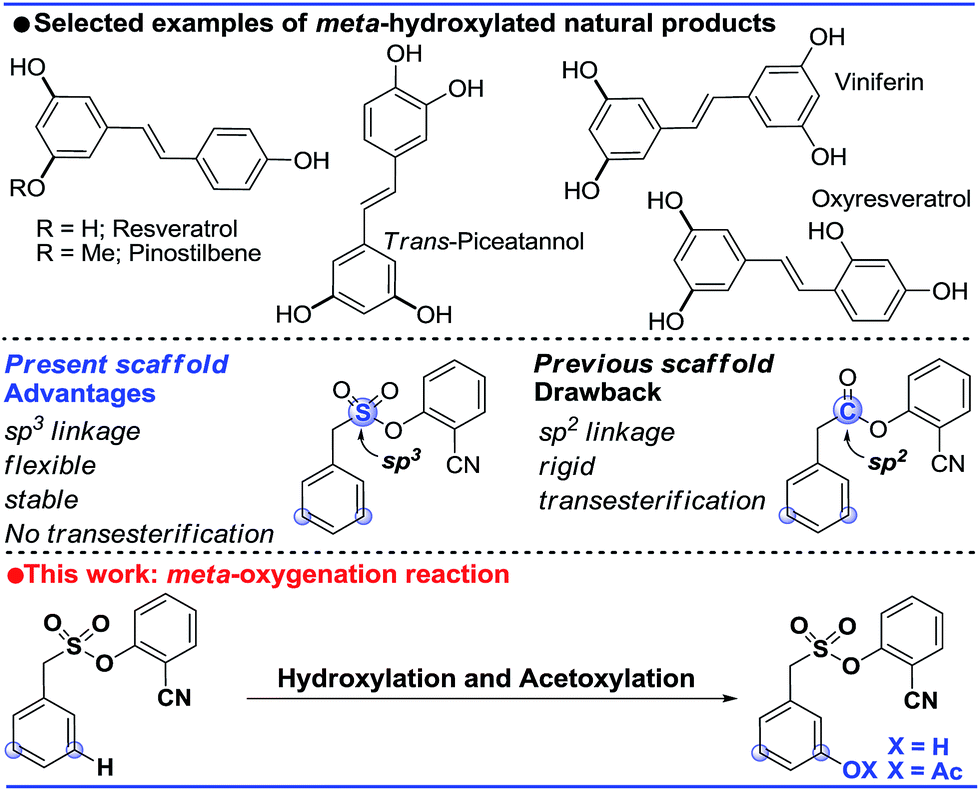 | ||
| Scheme 1 Overview of the present work. | ||
Results and discussion
Our initial attempts at direct hydroxylation failed owing to the intolerance of the cyano directing group towards strong oxidizing conditions (Table 1). Hence we envisioned installing an oxygenating group under mild oxidizing conditions followed by its in situ hydrolysis.10 With PhI(TFA)2 (4 equiv.) as the hydroxylating agent,11 and N-formyl-glycine (For-Gly-OH, 25 mol%) as the ligand for Pd(OAc)2 (10 mol%) in HFIP solvent, the desired meta-hydroxylated compound was obtained in a 78% (isolated, 74%) yield and with excellent selectivity (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
|
|
|||
|---|---|---|---|
| Catalysta | –OH source | Yield | |
| a Catalyst loading 10 mol%. b 0.5 mL of (CF3CO)2O added. c 70 °C was maintained for all the reactions; all the reactions were performed on a 0.2 mmol scale. | |||
| 1 | PdCl2/Pd(OAc)2 | TBHP (4 eq.) | 0 |
| 2 | PdCl2 | H2O2 (4 eq.) | 0 |
| 3 | PdCl2 | NHPI (2 eq.) | 0 |
| 4 | Cu(OAc)2 | (PhCO)2O (2 eq.), HFIP (1 mL) | 0 |
| 5 | PdCl2/Pd(OAc)2 | TEMPO (2 eq.) | 0 |
| 6 | Cu(OAc)2 | TBAI(2 eq.), Ag2CO3(2 eq.) | 0 |
| 7 | PdCl2 | K2S2O8 (2 eq.), CF3COOH (0.5 mL) | 0 |
| 8 | Pd(OAc)2 | Na2S2O8 (2 eq.); dioxane (1 mL) | 0 |
| 9b | Pd(OAc)2 | PhI(TFA)2(4 eq.), (CF3CO)2O | 11 |
| 10 | Pd(OAc)2 | PhI(TFA)2 (4 eq.), HFIP (1 mL) | 78 |

Using the optimized conditions, the scope of the reaction was explored (Table 2). Electron rich alkyl substituted arenes were hydroxylated with good yield and excellent selectivity (1b, 1c and 1g). Steric crowding in the close vicinity of the meta-C–H bond was found to affect the formation of the palladated intermediate. Moving from –Me (1b) to –iPr (1c) as the para substituent, the yield declined from 77% to 63%. In disubstituted systems, replacing chloro (1h) with bromo (1i) enhanced the selectivity towards the less hindered meta-C–H bond (1h, 65% and 1i, 63%). Groups such as –OPh, –OCF3, and –OMe were also tolerated.
| a These yields are based on the recovered starting material. b All the reactions were performed on a 0.2 mmol scale with Pd(OAc)2 (0.1 equiv.), For-Gly-OH (0.25 equiv.), PhI(TFA)2 (4 equiv.), and HFIP (1 mL). All the selectivity ratios (meta:others) were obtained from the crude reaction mixtures using 1,3,5-trimethoxybenzene as a reference. (m:m′). |
|---|
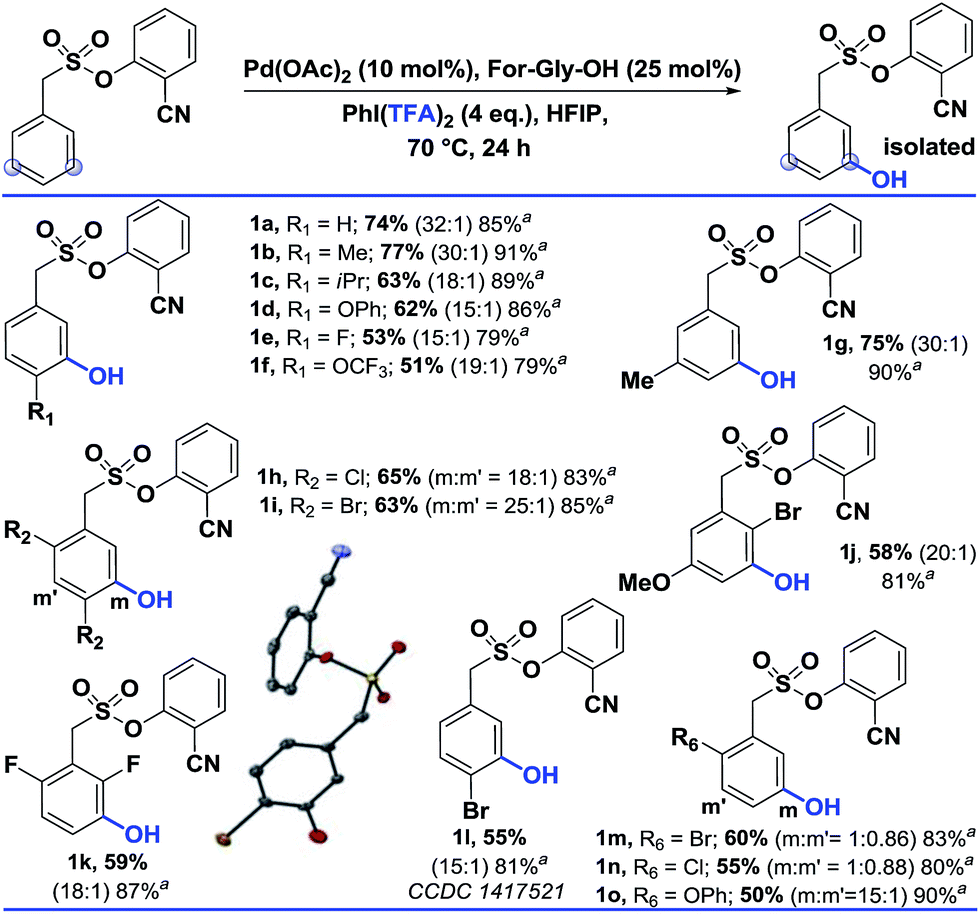
|
Interestingly, changing the acetoxylating agent from PhI(TFA)2 to PhI(OAc)2 led to the formation of the meta-acetoxylated compound as the sole product. Due to the importance of the meta-acetoxylated compounds, the protocol was optimized and the scope was explored. For meta-acetoxylation, N-tert-butyloxycarbonyl-alanine (Boc-Ala-OH) was found to be the best ligand for palladium. A similar reactivity trend to that of hydroxylation was observed for acetoxylation (Table 3).
| a These yields are based on the recovered starting material. b All the reactions were performed on a 0.2 mmol scale with Pd(OAc)2 (0.1 equiv.), Boc-Ala-OH (0.25 equiv.), PhI(OAc)2 (4 equiv.), and HFIP (1 mL) all the selectivity ratios (meta:others) were obtained from the crude reaction mixtures using 1,3,5-trimethoxybenzene as a reference. |
|---|
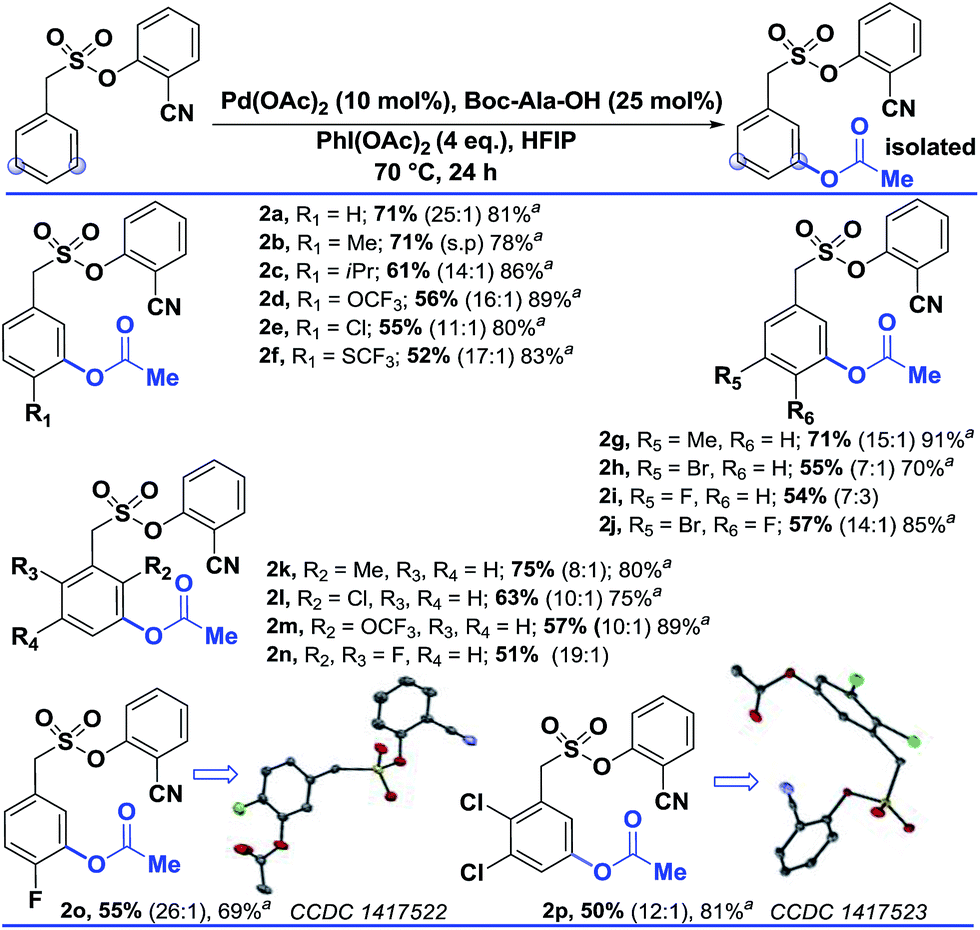
|
Notably, in the standard one-pot approach no dihydroxylation or diacetoxylation products were detected. Although hydroxyl groups are known to direct the metal to the proximal C–H bond, no such difunctionalization was observed under the present reaction conditions.
Arguably, the difference in ligand environment, competitive coordination with the –CN and hindered deprotonation of the phenol under the mildly acidic conditions of the reaction mixture reduces the scope for undesired functionalization of the product.14
Intermediate detection
The interesting switch from hydroxylation to acetoxylation upon changing the –R group on PhI(OOCR)2 from –CF3 to –CH3 can be justified by the reduced electrophilicity of the ester carbonyl in the acetate as compared to that in the trifluoroacetate, which disfavors hydrolysis and thus the product remains acetoxylated. This observation is additionally supported by a computational study (vide infra).Moreover the quantitative conversion of an independently synthesized meta-trifluoroacetoxylated substrate to the meta-hydroxy compound under the standard reaction conditions (Scheme 3) supported our initial hypothesis for the hydroxylation strategy.12 Furthermore, the intermediacy of the trifluoroacetoxylated compound as a hydroxyl precursor has been intercepted through NMR study (Scheme 2).
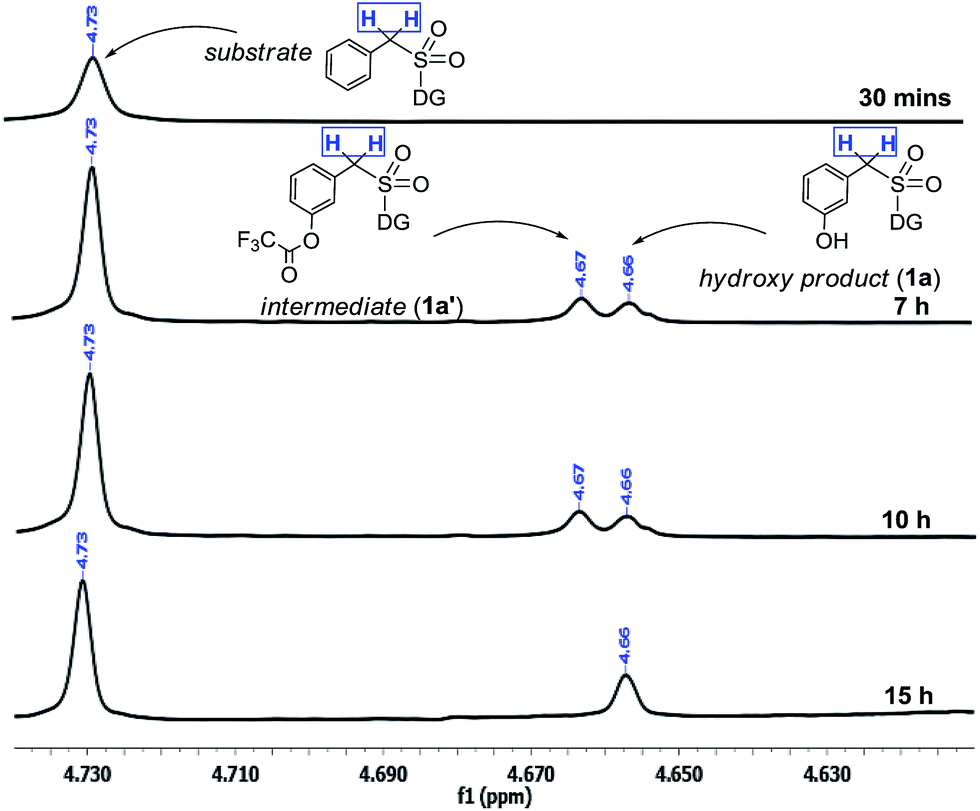 | ||
| Scheme 2 Reaction profile of meta-hydroxylation. aReactions were performed with 1.5 equiv. of PhI(TFA)2. | ||
 | ||
| Scheme 3 Generation of hydroxyl compounds via hydrolysis. | ||
Mechanistic study
During the optimization of the reaction conditions, HFIP and the N-protected aminocarboxylic acid ligand were found to be indispensible. To gain better insights into the mechanistic complexity, the reaction was monitored by NMR. Interestingly, in the presence of stoichiometric Pd(OAc)2 and the ligand, the addition of the substrate intensified the acetic acid (–CH3) peak, which indicates the release of acetic acid from the metal complex (Scheme 4).12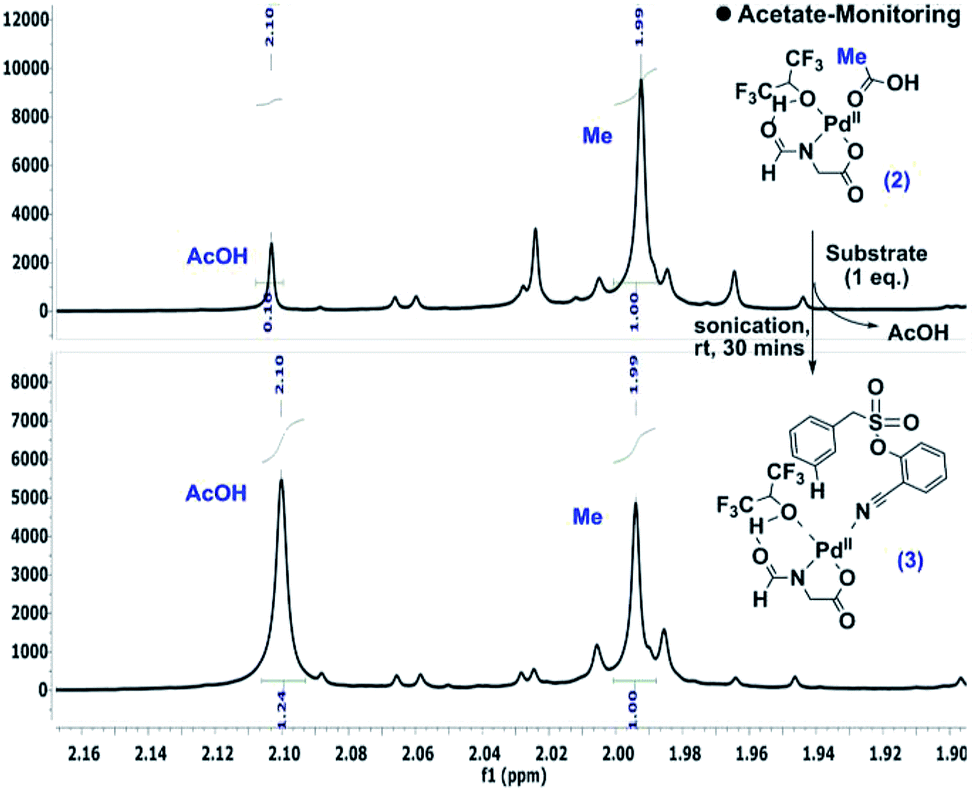 | ||
| Scheme 4 Detection of acetate release.10 | ||
Upon standing, the splitting pattern of the target aromatic ring was found to be disturbed (Scheme 5). Control experiments revealed that the palladium is essential for such splitting variation in the aromatic region. Such an observation can be visualized as the weak interaction between the substrate and palladium, likely the CN-anchored one, which eventually leads to the C–H activation.
 | ||
| Scheme 5 Evidence in support of Pd–arene interaction. | ||
The interaction between the palladium and the substrate could also be observed from the variation in the C–N bond stretching frequency. Upon addition of stoichiometric Pd(OAc)2 to the substrate, the –CN peak position shifted from 2237 cm−1 to 2247 cm−1, which further shifted to 2250 cm−1 upon addition of a stoichiometric amount of ligand. Moreover, solvated (MeOH and MeCN) For-Gly-OH ligated palladium species could be identified through mass spectroscopy.
All of these observations pointed towards a facile interaction of the substrate, ligand and HFIP with the palladium centre under ambient temperature. Despite these interactions, none of the desired product was formed at room temperature. This could be indicative of the fact that the C–H activation and the ensuing steps are more energy demanding.
The kinetic study showed a first-order rate dependency on the palladium and the substrate, whereas a zero-order dependency was observed for PhI(TFA)2 (Fig. 1).12 The isotope labelling experiment showed a high value of kinetic isotope effect (KIE = 3.02) (Scheme 6). Such a high value of KIE unambiguously establishes C–H activation as the rate limiting step.
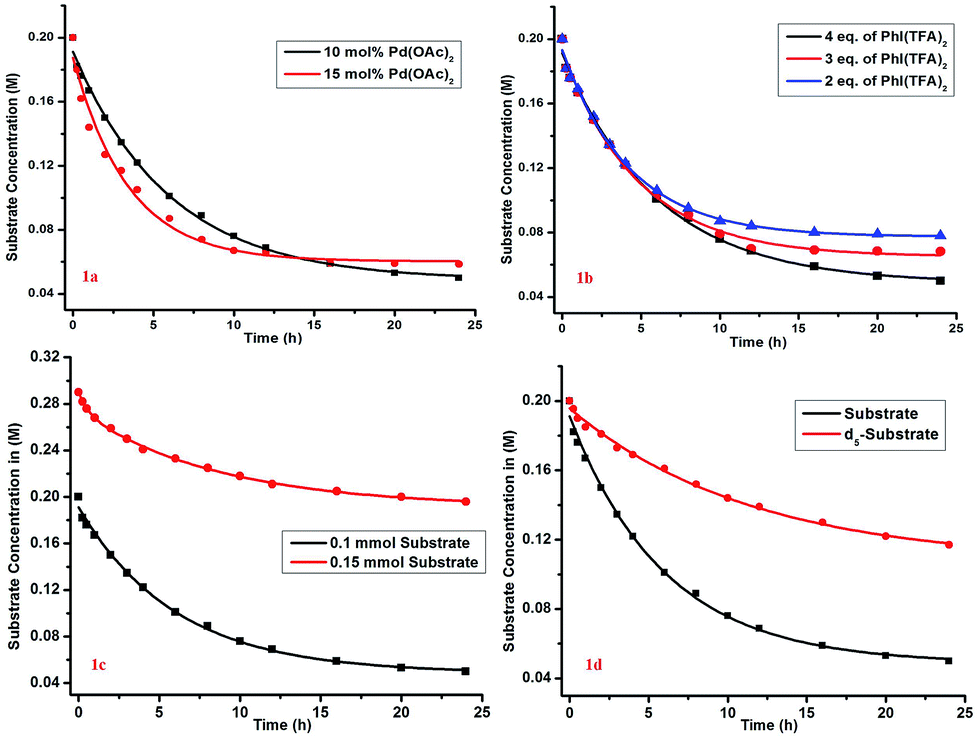 | ||
| Fig. 1 Order determination: (a) overlay of reaction profile with 10 mol% and 15 mol% Pd; (b) reaction profiles with different PhI(TFA)2 loadings. The reactions were performed with 0.1 mmol substrate, 10 mol% Pd, 25 mol% ligand and 0.5 mL of HFIP with variation in PhI(TFA)2 only as 2 eq., 3 eq. and 4 eq. (c) order determination w.r.t to substrate (order = 1). Reactions are done on a 0.1 and 0.15 mmol scale (d) determination of the kinetic isotope effect (kH/kD = 3.02). Reactions are done with the model substrate and d5-substrate on a 0.1 mmol scale. In (c) and (d) all the reactions are performed with 10 mol% catalyst, 25 mol% ligand, 4 equiv. of PhI(OTFA)2 and 0.5 mL (0.1 mmol scale) of HFIP at 70 °C. | ||
 | ||
| Scheme 6 Determination of the kinetic isotope effect. | ||
DFT calculation
Guided by these experimental observations, we have also conducted density functional theory computations to arrive at a plausible mechanistic pathway (Scheme 7). It is worth mentioning that the meta selective olefination pathway has been computationally investigated jointly by the Yu, Wu and Houk groups to illustrate the role of the N-protected amino acid in remote C–H activation.15 However, the meta selective C–O bond formation reaction is yet to be addressed using in silico studies. Contextually, DFT studies were carried out using the SMD/M06/6-31G** level of theory to locate key intermediates and transition states involved in the catalytic cycle.16 Two ligands, namely For-Gly-OH and Boc-Ala-OH, were used in our computational study.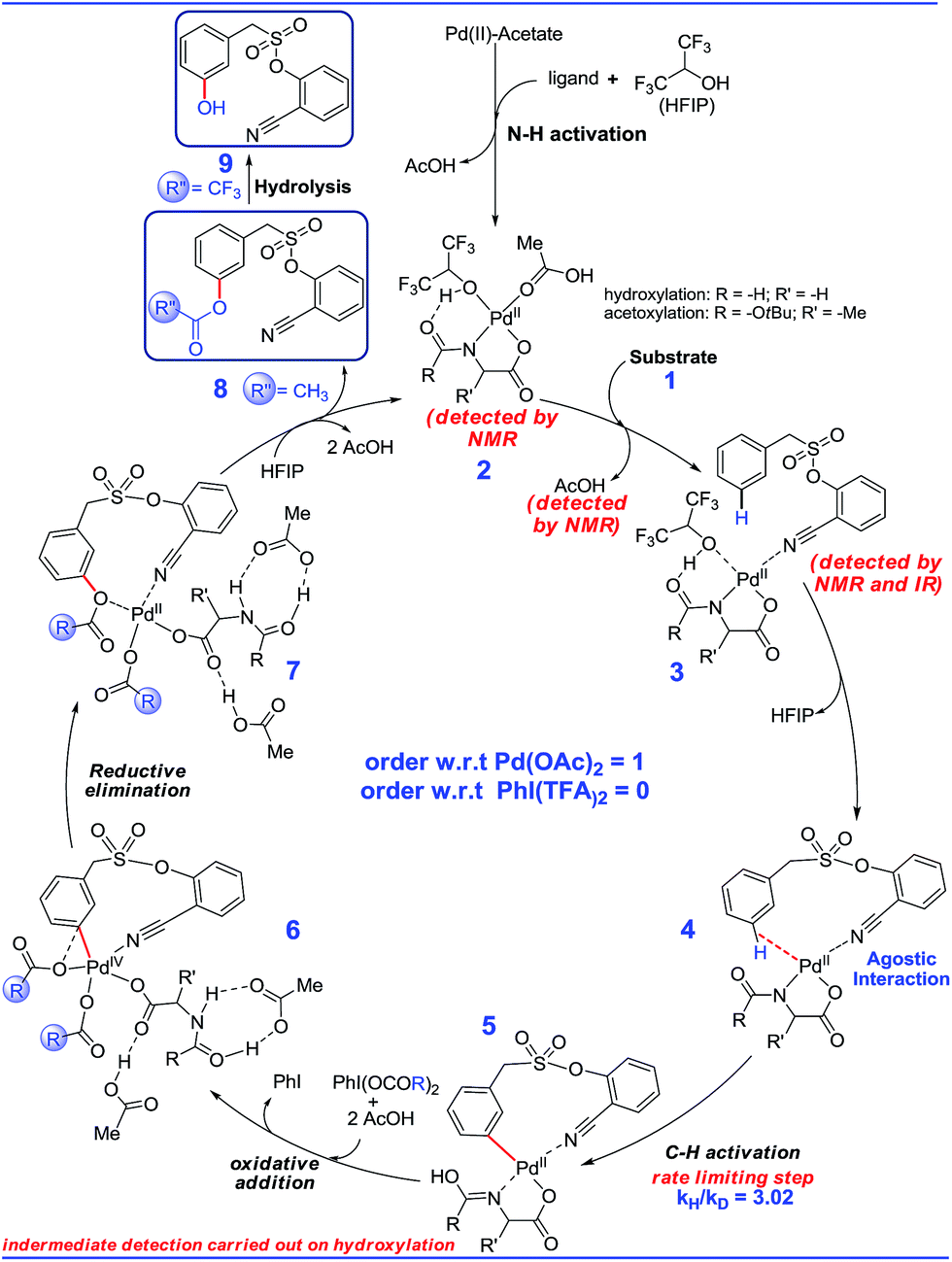 | ||
| Scheme 7 A plausible mechanism. | ||
First, different likely active species were examined by varying the ligand combinations around the Pd(II). Deprotonation of the carboxyl and amino groups of the ligand by the Pd-bound acetates led to the formation of an amino acid chelated Pd(II) intermediate (b; Fig. 2),17 and labile acetic acid ligands on the Pd center. Experimentally, HFIP was found to be vital for the title reaction. Hence the solvent HFIP was probed to study its potential influence on the energetics of the reaction. Notably, the HFIP binding, as in intermediate 2, was 1.5 kcal mol−1 lower than the corresponding acetic acid binding.18 This prediction is in accordance with our NMR identification of intermediate 2. The active species generated through the binding of the amino acid ligand to Pd(II) participated in the catalytic cycle as described below. The major mechanistic steps consist of (a) substrate uptake through ligand exchange, (b) ligand-assisted C–H activation, (c) oxidative addition to PhI(OCOR)2, and (d) reductive elimination leading to Caryl–O bond formation (Scheme 7).
 | ||
| Fig. 2 Gibbs free energy profile (kcal mol−1) of the meta hydroxylation obtained at the SMD/M06/6-31G**, LANL2DZ(Pd) level of theory. | ||
Substrate binding through the cyano group has been considered to direct palladium to the meta-C–H bond as in intermediate 3. Although intermediate 3 could have either neutral acetic acid or HFIP coordination, HFIP was found to be moderately preferred.19 Among the various possibilities for ligand assisted C–H activation, the abstraction of the Caryl–H proton by the amino acid ligand (Fig. 3) was found to be much more preferred (by 4.8 kcal mol−1) over the generally proposed acetate-assisted C–H activation.20
 | ||
| Fig. 3 Transition state geometries of two critical steps involved in the meta-hydroxylation. Selected hydrogen atoms shown only (distances are in Å). | ||
The palladated aryl thus formed underwent oxidative insertion to yield penta-coordinate Pd(IV) intermediate 6. In the following vital step, reductive elimination (RE) led to the formation of the Caryl–OCOR bond (R = CH3 or CF3). In the most preferred RE transition state, mono-dentate binding of the amino acid ligand (Fig. 3) was found to be preferred over the chelated form.21 At this stage of the reaction, the uptake of an HFIP molecule could assist the release of the product from the catalyst–product complex (7) to regenerate the active species (2).22 A more interesting aspect at this juncture is the fate of the resulting acetoxylated or trifluoro-acetoxylated product. Experimental observation suggests that the trifluroacetylated product is hydrolysed to yield a meta-hydroxylated compound whereas the acetylated product continues to remain as it is under identical reaction conditions. The origin of this kind of product distribution is traced to the lower energy transition state for acid catalyzed hydrolysis in the case of the trifluoroacetate.23
Application
Although the DG promoted meta-C–H functionalization, its removal will be crucial for the synthetic utility of such a strategy. In this context, both the acetoxylated and hydroxylated compounds formed hydroxyl-stilbenes via Julia olefination (10, Scheme 8). An unsymmetric trisubstituted phenol was also generated using a similar approach (11, Scheme 8). | ||
| Scheme 8 Applicatory removal of the DG. | ||
Further sequential acetoxylation and hydroxylation resulted in the formation of a potential resveratrol precursor (12). A phase II “Quinone Reductase” (QR) activity inducer (13) has also been synthesized using the present protocol (Scheme 9).24
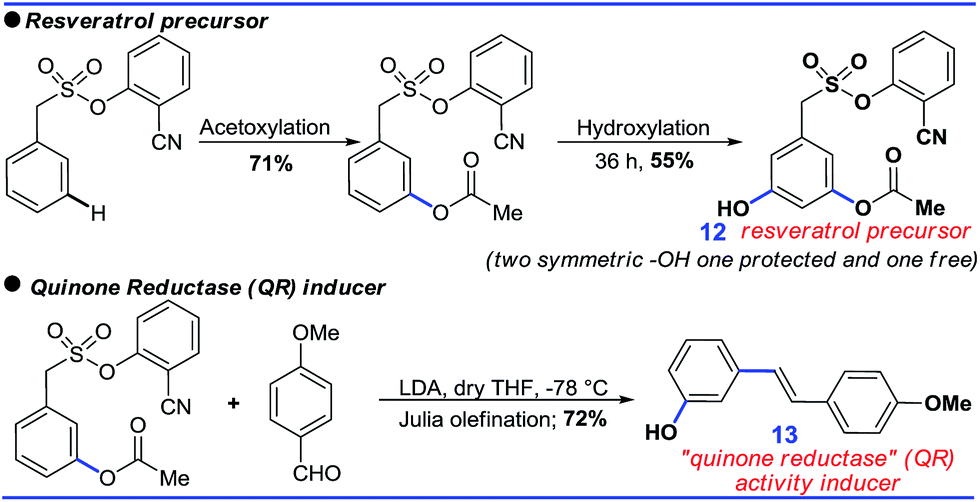 | ||
| Scheme 9 Application of the protocol. | ||
Conclusions
In summary, we have developed the first template assisted palladium catalyzed direct meta-hydroxylation strategy, which was further extended to a meta-acetoxylation reaction. Interestingly, the variation of R (R = H, F) on the acetoxylating agent PhI(OOCR3)2 led to the formation of different target molecules under similar reaction conditions. Mechanistic studies revealed the interesting role of the HFIP in the catalytic cycle. The approach has also been utilized for the synthesis of unsymmetrically substituted phenols and a QR activity inducer to demonstrate the synthetic utility of the protocol.Acknowledgements
This activity is supported by SERB, India (EMR/2015/000164). Generous computing time from IIT Bombay supercomputing is acknowledged.Notes and references
- (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (b) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed; (c) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654–2672 CrossRef CAS PubMed; (d) D. Y. K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452–9474 CrossRef CAS PubMed; (e) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed; (f) J. Chao, H. Li, K. W. Cheng, M. S. Yu, R. C. Chang and M. Wang, J. Nutr. Biochem., 2010, 21, 482–489 CrossRef CAS PubMed.
- (a) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (b) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (c) M. H. Holthausen, T. Mahdi, C. Schlepphorst, L. J. Hounjet, J. J. Weigand and D. W. Stephan, Chem. Commun., 2014, 50, 10038–10040 RSC; (d) G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4409–4412 CrossRef PubMed; (e) G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed; (f) M. Tobisu and N. Chatani, Science, 2014, 343, 850–851 CrossRef CAS PubMed; (g) L. T. Pilarski, N. Selander, D. Böse and K. J. Szabó, Org. Lett., 2009, 11, 5518–5521 CrossRef CAS PubMed; (h) H. Grennberg and J. E. Bäckvall, Chem.–Eur. J., 1998, 4, 1083–1089 CrossRef CAS.
- (a) D. Zhao, C. Nimphius, M. Lindale and F. Glorius, Org. Lett., 2013, 15, 4504–4507 CrossRef CAS PubMed; (b) G. D. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722–17725 CrossRef PubMed; (c) H. Wang, N. Schröder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386–5389 CrossRef CAS PubMed; (d) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2013, 15, 3014–3017 CrossRef CAS PubMed; (e) P. Fang, M. Li and H. Ge, J. Am. Chem. Soc., 2010, 132, 11898–11899 CrossRef CAS PubMed; (f) C. P. Ting and T. J. Maimone, Angew. Chem., Int. Ed., 2014, 53, 3115–3119 CrossRef CAS PubMed; (g) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (h) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (i) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (j) M. S. Sigman and E. W. Werner, Acc. Chem. Res., 2012, 45, 874–884 CrossRef CAS PubMed; (k) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef CAS PubMed; (l) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (m) Z. Chen, B. Wang, Z. Wang, G. Zhu and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 2027–2031 CrossRef CAS PubMed; (n) W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910–1913 CrossRef CAS PubMed.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- (a) J. Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (b) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (c) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed; (d) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed; (e) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463–466 CrossRef CAS PubMed; (f) F. Juliá-Hernández, M. Simonetti and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 11458–11460 CrossRef PubMed; (g) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (h) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed; (i) J. Schranck, A. Tlili and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 9426–9428 CrossRef CAS PubMed; (j) A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef PubMed.
- (a) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429–9432 CrossRef CAS PubMed; (b) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS PubMed; (c) J. Luo, S. Preciado and I. Larrosa, Chem. Commun., 2015, 51, 3127–3130 RSC; (d) J. Luo, S. Preciado, S. O. Araromi and I. Larrosa, Chem.–Asian J., 2015, 6, 5595–5600 Search PubMed; (e) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (f) X.-C. Wang, W. Gong, L. Z. Fang, R. Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed; (g) Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed; (h) P. X. Shen, X. C. Wang, P. Wang, R. Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS PubMed.
- D. Leow, G. Li, T. S. Mei and J. Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- (a) H. X. Dai, G. Li, X. G. Zhang, A. F. Stepan and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed; (b) L. Wan, N. Dastbaravardeh, G. Li and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056–18059 CrossRef CAS PubMed; (c) R. Y. Tang, G. Li and J. Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed; (d) G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R. Y. Tang, M. Movassaghi and J. Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 CrossRef CAS PubMed; (e) Y. Deng and J. Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888–891 CrossRef CAS PubMed.
- (a) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed; (b) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed; (c) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed; (d) S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595–5600 RSC.
- (a) N. Itoh, T. Sakamoto, E. Miyazawa and Y. Kikugawa, J. Org. Chem., 2002, 67, 7424–7428 CrossRef PubMed; (b) R. M. Moriarty, J. Org. Chem., 2005, 70, 2893–2903 CrossRef CAS PubMed; (c) V. S. Thirunavukkarasu, J. Hubrich and L. Ackermann, Org. Lett., 2012, 14, 4210–4213 CrossRef CAS PubMed; (d) D. J. Gallardo and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350–9353 CrossRef PubMed; (e) H. Y. Zhang, H. M. Yi, G. W. Wang, B. Yang and S. D. Yang, Org. Lett., 2013, 15, 6186–6189 CrossRef CAS PubMed; (f) F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718–720 CrossRef CAS PubMed; (g) Y. M. Xing, L. Zhang and D. C. Fang, Organometallics, 2015, 34, 770–777 CrossRef CAS; (h) C. Cheng, S. Liu and G. Zhu, J. Org. Chem., 2015, 80, 7604–7612 CrossRef CAS PubMed; (i) Q. R. Liu, C. X. Pan, X. P. Ma, D. L. Mo and G. F. Su, J. Org. Chem., 2015, 80, 6496–6501 CrossRef CAS PubMed; (j) C. Yang, X. G. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2015, 80, 5320–5328 CrossRef CAS PubMed; (k) T. Zhou, F. X. Luo, M. Y. Yang and Z. J. Shi, J. Am. Chem. Soc., 2015, 137, 14586–14589 CrossRef CAS PubMed; (l) S. Z. Sun, M. Shang, H. L. Wang, H. X. Lin, H. X. Dai and J. Q. Yu, J. Org. Chem., 2015, 80, 8843–8848 CrossRef CAS PubMed; (m) Y. H. Zhang and J. Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654–14655 CrossRef CAS PubMed.
- (a) P. Y. Choy and F. Y. Kwong, Org. Lett., 2013, 15, 270–273 CrossRef CAS PubMed; (b) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070–13074 CrossRef CAS PubMed.
- See ESI† for more details.
- Details of CCDC 1417521 (1l), 1417522 (2o), and 1417523 (2q) are available via www.ccdc.cam.ac.uk/data_ request/cif.
- U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669–12673 CrossRef CAS PubMed.
- (a) G. J. Cheng, Y. F. Yang, P. Liu, P. Chen, T. Y. Sun, G. Li, X. G. Zhang, K. N. Houk, J. Q. Yu and Y. D. Wu, J. Am. Chem. Soc., 2014, 136, 894–897 CrossRef CAS PubMed; (b) Y. F. Yang, G. J. Cheng, P. Liu, D. Leow, T. Y. Sun, P. Chen, X. G. Zhang, J. Q. Yu, Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed.
- M. J. Frisch et. al. (a) Full details of the computational methods are provided in the ESI.†; (b) Gaussian09 quantum chemical suite has been used for the computations.
- (a) G.-J. Cheng, Y.-F. Yang, P. Liu, D. Leow, T. Y. Sun, P. Chen, X. Zhang, J. Q. Yu, Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 894 CrossRef CAS PubMed; (b) More details in ESI Fig. S4 and S9.†.
- The replacement of both acetic acid ligands by two HFIP molecules is energetically higher than substitution of just one HFIP (See Fig. S5 and S10 in the ESI†).
- Link to the results and relative energies of intermediates in the ESI. [Fig. S6 and S11†].
- (a) We have also compared the ortho as well as para C–H activation to learn that the meta is energetically most preferred. See Tables S20, 21, Fig. S7 and S12 in the ESI.†; (b) D. G. Musaev, A. Kaledin, B.-F. Shi and J. Q. Yu, J. Am. Chem. Soc., 2011, 134, 1690 CrossRef PubMed.
- (a) More details of the RE possibilities are provided in Fig. S8 and S13 in the ESI.† For examples of RE for a penta coordinated Pd(IV) see ; (b) D. M. Crumpton and K. I. Goldberg, J. Am. Chem. Soc., 2000, 122, 962 CrossRef CAS; (c) A. R. Dick, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 12790 CrossRef CAS PubMed.
- More details are provided in the ESI (See Fig. S7, S8 and Table S20†).
- (a) More details are provided in Fig. S15 in the ESI.†; (b) R. Gómez-Bombarelli, E. Calle and J. Casado, J. Org. Chem., 2013, 78, 6880 CrossRef PubMed.
- W. Zhang and M. L. Go, Eur. J. Med. Chem., 2007, 42, 841–850 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1417521–1417523. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04060d |
| This journal is © The Royal Society of Chemistry 2016 |