
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium(III)-bis(imidazolinyl)phenyl catalysts for enantioselective C–H functionalization with ethyl diazoacetate†
N. Mace
Weldy
a,
A. G.
Schafer
a,
C. P.
Owens
a,
C. J.
Herting
a,
A.
Varela-Alvarez
b,
S.
Chen
b,
Z.
Niemeyer
c,
D. G.
Musaev
*b,
M. S.
Sigman
*c,
H. M. L.
Davies
*a and
S. B.
Blakey
*a
aDepartment of Chemistry, Emory University, 1515 Dickey Drive, Atlanta, Georgia 30322, USA. E-mail: sblakey@emory.edu
bEmerson Center for Scientific Computation, Emory University, 1515 Dickey Drive, Atlanta, Georgia 30322, USA
cDepartment of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, Utah 84112, USA
First published on 5th February 2016
Abstract
The intermolecular enantioselective C–H functionalization with acceptor-only metallocarbenes is reported using a new family of Ir(III)-bis(imidazolinyl)phenyl catalysts, developed based on the interplay of experimental and computational insights. The reaction is tolerant of a variety of diazoacetate precursors and is found to be heavily influenced by the steric and electronic properties of the substrate. Phthalan and dihydrofuran derivatives are functionalized in good yields and excellent enantioselectivities.
Introduction
C–H functionalization methods continue to provide elegant solutions to synthetic challenges of natural products and pharmaceuticals.1 In recent years, insertion of metallocarbenes has become particularly powerful.2 The discovery of the donor–acceptor class of carbenes and the development of new dirhodium tetracarboxylate catalysts revolutionized intermolecular C–H insertion reactions.3 The donor group tempers the electrophilicity of the metallocarbene and allows for exquisite regio- and enantioselectivity in these complex reactions (Fig. 1). | ||
| Fig. 1 Comparison of metallocarbene classes. | ||
In contrast, there are no reports of enantioselective intermolecular C–H functionalization reactions utilizing acceptor-only carbene precursors even though these reagents are readily available. Although many catalysts have been developed to promote the mechanistically related enantioselective alkene cyclopropanation using ethyl diazoacetate,4 C–H functionalization with this reagent remains limited to a racemic manifold.5
The challenge in developing catalysts for acceptor-only carbene insertion reactions is two-fold. First, these metallocarbenes are highly electrophilic and reactive. The barrier for the Rh2(OAc)4 catalyzed C–H insertion reaction of methyl diazoacetate into the relatively strong C–H bonds of cyclopentane has been calculated to be only 3.5 kcal mol−1.6 This low barrier to carbene transfer presents an inherent chemoselectivity challenge. Second, the acceptor-only carbene itself is not prochiral, and for enantioselective C–H insertion reactions, the catalyst must provide an environment that differentiates among orientations of the incoming substrate.
Herein, we report the development of an iridium catalyst that promotes enantioselective intermolecular C–H insertion reactions using the prototypical acceptor-only carbene precursor ethyl diazoacetate. Our catalyst design sought to take advantage of the ability of third row transition metals to participate in increased back-bonding to suppress the metallocarbene electrophilicity while also increasing barriers to subsequent group transfer process required for enantioselective catalysis.
Results and discussion
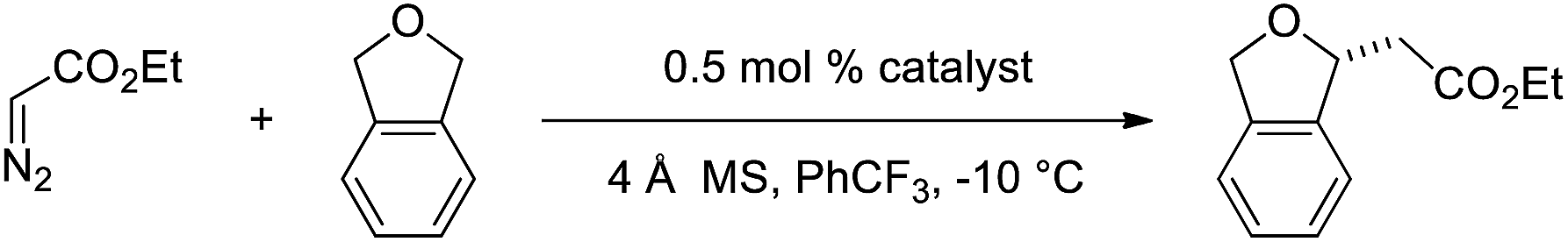
The broad utility of group 9 metal complexes as catalysts for group transfer chemistry led us to adopt iridium as the metal of choice for this study.7 We identified the bis(oxazolinyl)phenyl (phebox) ligand system extensively studied by Nishiyama and coworkers as a readily accessible, modular platform for catalyst development.8 In our initial experiments, we observed Ir(III)-phebox 1 is indeed capable of catalyzing the C–H insertion reaction of ethyl diazoacetate into tetrahydrofuran with good yield (80%) and promising enantioselectivity (eqn (1), 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 er).
:25 er).
Additionally, we demonstrated that it is not necessary to utilize tetrahydrofuran as solvent, and when it is used as a substrate in PhCF3, the enantioselectivity increases (eqn (2), 81:19 er). Diethyl maleate is observed as the major byproduct in this reaction.
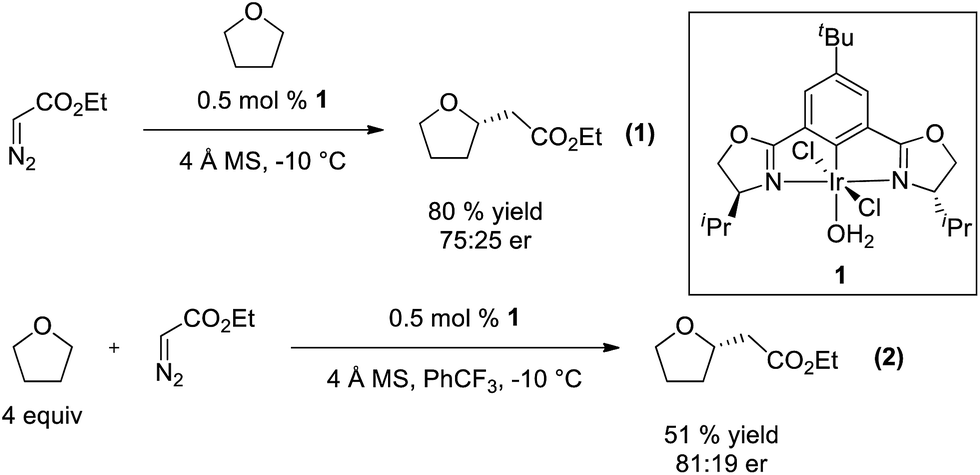
To gain insight and direct catalyst evolution, the structure and free energy of the pro-S and pro-R transition states for Ir(III)-phebox 1 catalyzed ethyl diazoacetate insertion into tetrahydrofuran were calculated using the CPCM-M06L/{LANL08(f)Ir + [6-31G(d,p)]} level of theory (Fig. 2, top). A small ΔΔG‡ value of 0.5 kcal mol−1 was calculated between the pro-S and pro-R transition states TS1 and TS2. Calculations also indicated that the favored transition state TS1 involves a short O1–H2 distance of 2.308 Å due to an electrostatic interaction between the ligand and substrate. When tetrahydrofuran was replaced with phthalan as the substrate in the calculations, the ΔΔG‡ between TS3 and TS4 was predicted to increase to 3.7 kcal mol−1.
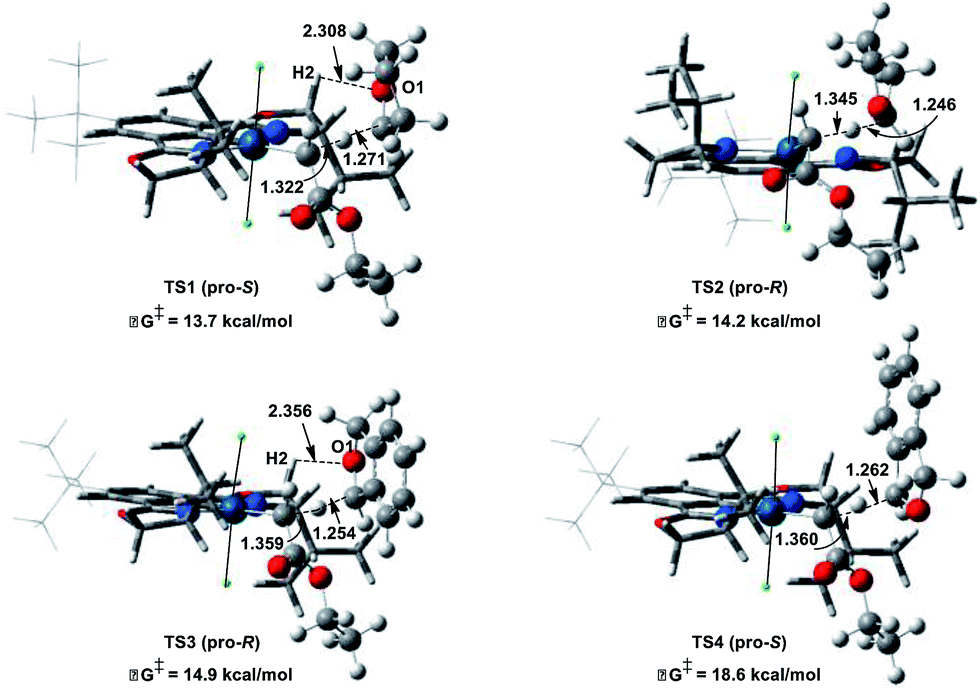 | ||
| Fig. 2 Calculated pro-R and pro-S transition states of ethyl diazoacetate insertion into tetrahydrofuran (top) and phthalan (bottom) with Ir(III)-phebox 1. | ||
Based on this model, we exposed phthalan to reaction conditions with ethyl diazoacetate, and an increased yield of 81% and er of 88:12 were observed (Scheme 1). Additionally, subsequent crystallization of the 4-MeO-Ph ester derivative 2 established that the computational model correctly predicted the major enantiomer formed in this reaction.
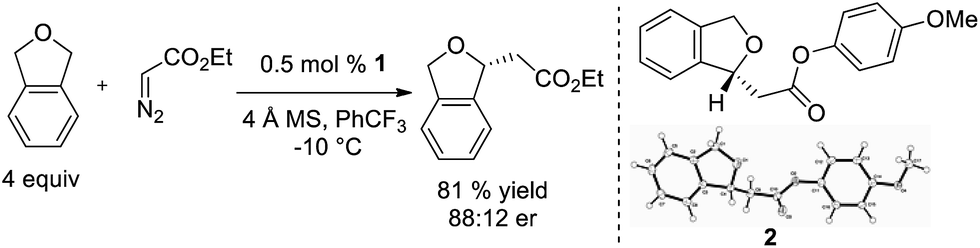 | ||
| Scheme 1 Ir(III)-phebox 1 catalyzed enantioselective insertion of ethyl diazoacetate into phthalan. | ||
Encouraged by the qualitative accuracy of the computational predictions, we utilized this model for further catalyst optimization. Due to the calculated electrostatic interaction between H2 of the ligand and O1 of the substrate in favored transition states TS1 and TS3, we hypothesized that modulating the charge density on H2 could provide further stabilization of the favored transition state and improve enantioselectivity.
In order to influence the charge density on H2, we shifted our attention to the bis(imidazolinyl)phenyl (phebim) ligand system (4) in an effort to modulate the electronics of the imidazoline through an aryl ring at the R2 position (Scheme 2).9 The ligand can be prepared in two steps from commercially available amino alcohols and 5-tert-butylisophthalic acid (3).9b Metalation under standard conditions with IrCl3·3H2O afforded Ir(III)-phebim complexes 5–15.8d,f
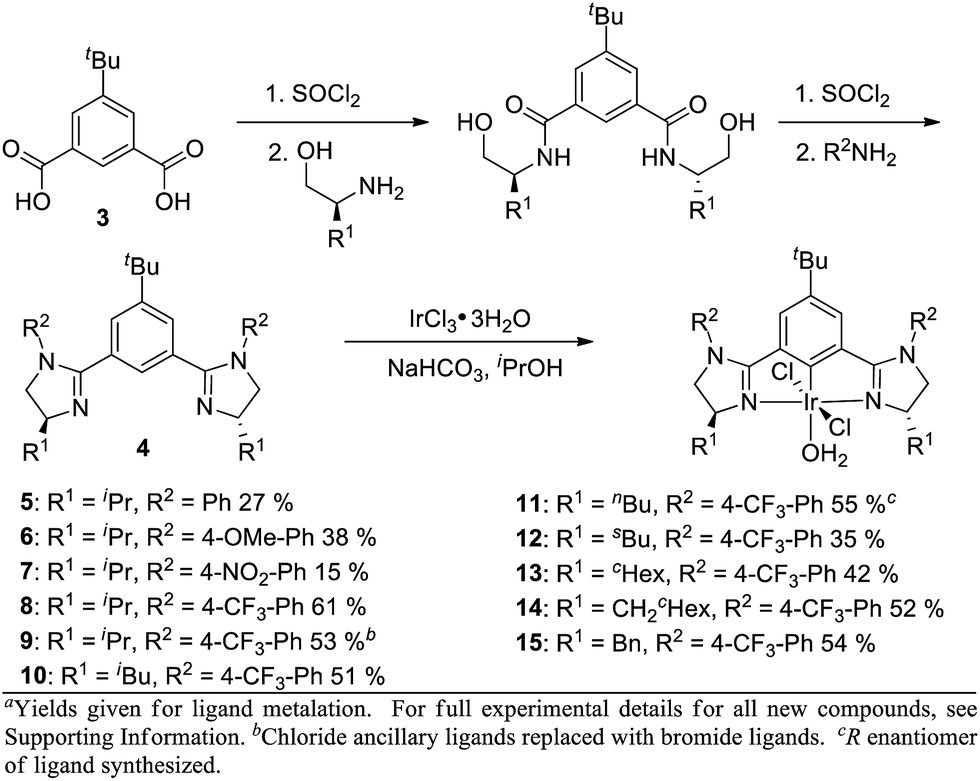 | ||
| Scheme 2 Ir(III)-bis(imidazolinyl)phenyl (phebim) complex synthesisa. | ||
R2-Ph-substituted Ir(III)-phebim 5 was found to catalyze ethyl diazoacetate insertion into phthalan in similar yield and er to phebox 1 (Table 1, entry 1; Scheme 1). Electronic variation at the 4-position of the phenyl ring at R2 had a small but measurable impact on enantioselectivity, with both 4-NO2-Ph-substituted catalyst 7 and 4-CF3-Ph-substituted catalyst 8 giving the best er of 90:10 (entries 2–4). This observation is consistent with the electrostatic control hypothesis suggested by the computational model. Due to the lower yield of the metalation step for 7 (15%), 4-CF3-Ph was adopted as the optimal substituent at R2 (61%). Complex 9 with Br ancilliary ligands improved the yield by 10%, but decreased enantioselectivity (entry 5). The greatest impact on enantioselectivity was achieved by modulating ligand substituent R1, with iBu-substituted catalyst 10 and CH2-cHex-substituted catalyst 14 each improving the er to 95:5 (entries 6 and 10). Though equivalent in enantioselectivity, the use of catalyst 10 gave an enhanced yield relative to 14.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | erc |
| a General conditions: 0.29 M solution of ethyl diazoacetate in PhCF3 was added over 5 h to a mixture of catalyst, phthalan (4 equiv.), and 4 Å MS in PhCF3 at −10 °C. b Isolated yields. c Determined by chiral HPLC. d Catalyst 11 is a member of the opposite enantio-series. | |||
| 1 | 5 | 85 | 88:12 |
| 2 | 6 | 26 | 89:11 |
| 3 | 7 | 70 | 90:10 |
| 4 | 8 | 73 | 90:10 |
| 5 | 9 | 83 | 88:12 |
| 6 | 10 | 95 | 95:5 |
| 7 | 11 | 54 | 13:87d |
| 8 | 12 | 81 | 92:8 |
| 9 | 13 | 83 | 90:10 |
| 10 | 14 | 80 | 95:5 |
| 11 | 15 | 36 | 86:14 |
To further explore if catalysts 10 and 14 were optimal in terms of enantioselectivity for the insertion of ethyl diazoacetate into phthalan, we utilized linear regression mathematical modeling techniques. This was accomplished by collecting steric parameters describing the R1 substituent, including Sterimol, Charton/Taft, and Austel values.10 While the use of Sterimol and Charton values has been explored for analyzing enantioselective reactions,11 the Austel value has not been used previously. Simply stated, this value is a measure of branching in alkyl chains.12 By applying multivariate regression using this parameter set and the observed enantioselectivity, a predictive model was found as depicted in Fig. 3.
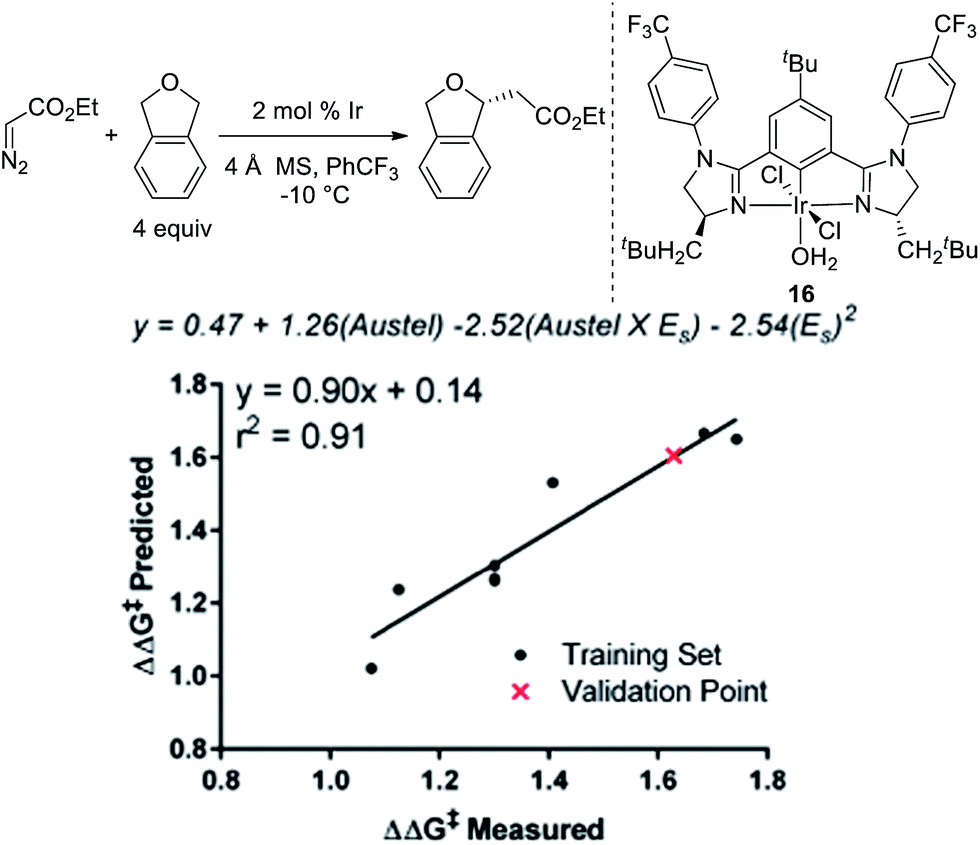 | ||
| Fig. 3 Linear regression modeling of Ir(III)-phebim R1-substitution. | ||
Of particular interest is the conflicting steric terms required to effectively model the reaction. As R1 increases in size, the equation suggests that a maximum can be reached balancing the level of branching as determined by the Austel value and the relative spherical size as determined by the Charton/Taft value (ES). Based on this model, a third comparable catalyst with CH2-tBu-substitution (16) was predicted to give ethyl diazoacetate insertion into phthalan with a modest reduction in er (94:6). This result was confirmed experimentally with catalyst 16 resulting in a yield of 92% and an er of 94:6. The modeling and subsequent experiment suggests that the unusual CH2cHex substituent on the imidazoline ring of the catalyst is indeed optimal for the reaction.
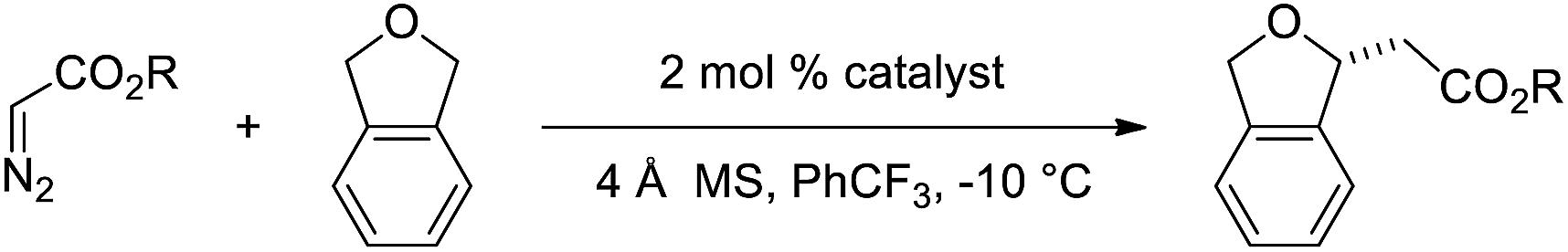
Seeking further improvements in selectivity, and based on the strong influence of the nature of the ester in donor–acceptor metallocarbene C–H insertion, a range of ester substituents were examined for acceptor-only metallocarbene C–H insertion into phthalan with the most selective Ir(III)-phebim catalysts 10 and 14.2b,13 In our system, using methyl diazoacetate resulted in a modest drop in enantioselectivity to 94:6 er with catalyst 14 (Table 2, entry 2). The electronically unique reagent, 2,2,2-trichloroethyl diazoacetate, was found to give the same er as ethyl diazoacetate with 14 (95:5 er, entry 4). Similarly, 2-trimethylsilyl diazoacetate gave C–H insertion in good yield and an er of 94:6 (entry 6). Phenyl diazoacetate gave an enantioselectivity of 93:7 (entry 8), and 4-MeO and 4-CF3 substituted phenyl diazoacetates were tolerated as well, with no significant improvements in yield or er (entries 9–12). In all cases, CH2-cHex-substituted Ir(III)-phebim 14 provided slightly higher enantioselectivity than iBu-substituted Ir(III)-phebim 10, though often coupled with a slightly diminished yield. The consistently higher enantioselectivity led us to establish 14 as the optimal catalyst to further investigate the substrate scope of this methodology.
|
|
||||
|---|---|---|---|---|
| Entry | R | Catalyst | Yieldb (%) | erc |
| a General conditions: 0.29 M solution of ethyl diazoacetate in PhCF3 was added over 5 h to a mixture of catalyst, phthalan (4 equiv.), and 4 Å MS in PhCF3 at −10 °C. b Isolated yields. c Determined by chiral HPLC. d 0.5 mol% catalyst loading. | ||||
| 1d | Me | 10 | 72 | 92:8 |
| 2d | Me | 14 | 69 | 94:6 |
| 3 | CH2CCl3 | 10 | 84 | 94:6 |
| 4 | CH2CCl3 | 14 | 71 | 95:5 |
| 5 | CH2CH2Si(CH3)3 | 10 | 62 | 93:7 |
| 6 | CH2CH2Si(CH3)3 | 14 | 67 | 94:6 |
| 7 | Ph | 10 | 65 | 91:9 |
| 8 | Ph | 14 | 68 | 93:7 |
| 9 | 4-OMe-Ph | 10 | 57 | 91:9 |
| 10 | 4-OMe-Ph | 14 | 70 | 92:8 |
| 11 | 4-CF3-Ph | 10 | 78 | 90:10 |
| 12 | 4-CF3-Ph | 14 | 69 | 92:8 |
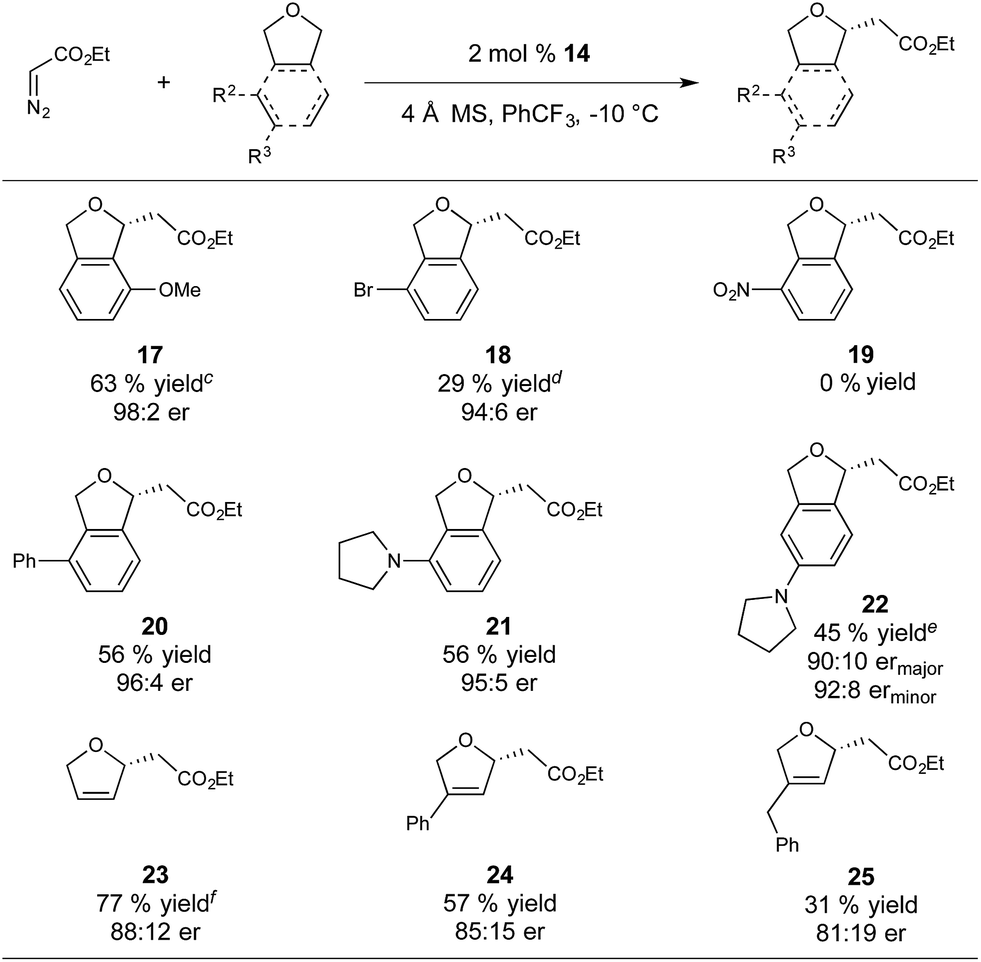
With respect to substrate scope, we subjected a variety of phthalan derivatives to standard C–H insertion conditions with catalyst 14 and ethyl diazoacetate and observed strong electronic and steric influences on reaction efficiency and regioselectivity (Table 3).
|
a General conditions: 0.29 M solution of ethyl diazoacetate in PhCF3 was added over 5 h to a mixture of catalyst, substrate (4 equiv.), and 4 Å MS in PhCF3 at −10 °C.
b Isolated yields are based on EDA.
c The 2,6-regioisomer is also observed in an additional 19% yield and 94:6 er.
d The 2,6-regioisomer is also observed in an additional 10% yield.
e Product was isolated as a 4:1 mixture of the 2,5 (major) and 2,4 (minor) regioisomers.
f 0.5 mol% catalyst loading.
|
|---|
|
In introducing a methoxy substituent at the 3-position of phthalan, the catalyst showed selectivity for insertion at the electronically activated position to give 2,3-substituted phthalan 17 (63% yield), with an excellent er of 98:2 (Table 3). The competing 2,6-substituted regioisomer (sterically favored) was formed in only 19% yield and 94:6 er. The weakly electron withdrawing 3-Br substituent deactivated the substrate towards C–H insertion and delivered 2,6-substituted regioisomer 18 as the major product (39% yield, 3:1 regioselectivity) with excellent enantioselectivity (94:6). In this case steric factors override the weak electronic bias for insertion adjacent to the bromine substituent. Strongly electron withdrawing NO2-substitution at the 3-position resulted in no product formation. Using 3-Ph-substituted phthalan as a substrate afforded good enantioselectivity of 96:4 and yielded the sterically favored 2,6-isomer 20 as the sole product in 56% yield. 3-Pyrrolidinyl-phthalan also showed complete selectivity for the sterically favored 2,6-isomer 21, with the significant steric influence of pyrrolidinyl-substitution at the 3-position overriding the electron-donating effect. Moving the pyrrolidinyl substituent to the 4-position (sterically neutral with respect to potential insertion sites) gave a majority of the electronically favored 2,5-isomer 22, with the 2,4-isomer observed as a minor component of the reaction mixture (45% yield, 4:1 regioselectivity, 90:10 ermajor, 92:8 erminor). Importantly, the Ir(III)-phebim catalyst 14 is active in the presence of these basic amines with no signs of catalyst inhibition or reaction of the metallocarbene with the amine. This observation is particularly significant for potential applications in pharmaceutically relevant molecules.
Insertion into 2,5-dihydrofuran gave 23 in 77% yield and an er of 88:12, with no competing cyclopropanation observed. Consistent with our observations for phthalan-derived substrates, regioselectivity of the insertion is strongly influenced by steric factors. Using 3-Ph-substituted 2,5-dihydrofuran produced 24 in 57% yield and 85:15 er. A greater reduction of yield to 31% was observed when using 3-Bn-substituted 2,5-dihydrofuran to give 25 in 81:19 er.
In addition to enantioselective insertion, kinetic resolution of rac-2-Me-phthalan resulted in formation of the anti-2,7-substituted product 26 primarily, which is consistent with what one would expect based on the computed transition states (Scheme 3 and Fig. 2). Minor amounts of the syn-isomer 27 and 3° C–H insertion product 28 were also formed. The major product 26 was formed in an er of 88:12.
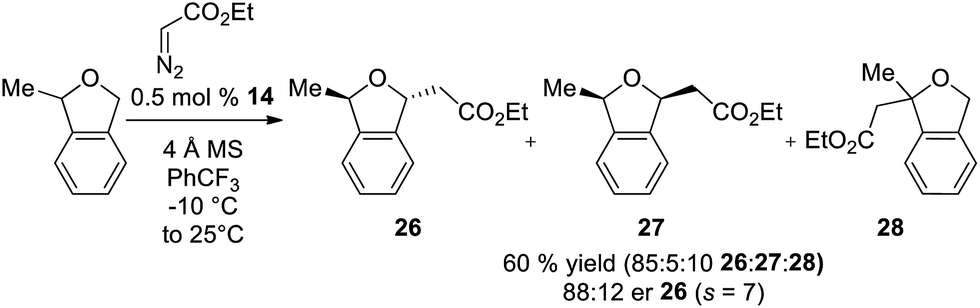 | ||
| Scheme 3 Ir(III)-phebim 14 catalyzed kinetic resolution of rac-2-Me-phthalan with ethyl diazoacetate. | ||
Conclusions
In summary, using a combination of computational study and catalyst design based on fundamental organometallic principles, we have developed an iridium complex capable of mediating highly selective intermolecular functionalization of ethereal C–H bonds with the prototypical acceptor-only carbene precursor ethyl diazoacetate. A variety of phthalan and dihydrofuran derivatives were competent substrates, exhibiting a strong steric and electronic influence on reaction yield and regioselectivity. These results provide a foundation for further expansion in this area, such as C–H insertion with novel, unexplored classes of acceptor-only carbene precursors.Acknowledgements
This research was supported by the National Science Foundation under the Center for Chemical Innovation in Selective C–H Functionalization (CHE1205646). We thank Dr John Bacsa and the Emory X-Ray Center for X-ray structural analysis.Notes and references
- For reviews, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (c) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (d) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (e) D. Y. K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452 CrossRef CAS PubMed; (f) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (g) J. Egger and E. M. Carreira, Nat. Prod. Rep., 2014, 31, 449 RSC.
- For reviews, see: (a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (b) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (c) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (d) H.-Y. Thu, G. S.-M. Tong, J.-S. Huang, S. L.-F. Chan, Q.-H. Deng and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9747 CrossRef CAS PubMed.
- For reviews of donor/acceptor metallocarbene insertion, see: (a) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC; (b) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC.
- (a) H. Nishiyama, Y. Itoh, H. Matsumoto, S.-B. Park and K. Itoh, J. Am. Chem. Soc., 1994, 116, 2223 CrossRef CAS; (b) P. Müller and S. Tohill, Tetrahedron, 2000, 56, 1725 CrossRef; (c) A. Caballero, A. Prieto, M. M. Díaz-Requejo and P. J. Pérez, Eur. J. Inorg. Chem., 2009, 2009, 1137 CrossRef.
- (a) L. T. Scott and G. J. DeCicco, J. Am. Chem. Soc., 1974, 96, 322 CrossRef CAS; (b) M. M. Díaz-Requejo, T. R. Belderraín, M. C. Nicasio, S. Trofimenko and P. J. Pérez, J. Am. Chem. Soc., 2002, 124, 896 CrossRef; (c) H. V. R. Dias, R. G. Browning, S. A. Richey and C. J. Lovely, Organometallics, 2004, 23, 1200 CrossRef CAS; (d) For seminal studies see A. Demonceau, A. F. Noels, A. J. Hubert and P. Teyssié, J. Chem. Soc., Chem. Commun., 1981, 688 RSC; (e) For a recent review see A. Caballero, M. M. Díaz-Requejo, M. R. Fructos, A. Olmow, J. Urbano and P. J. Pérez, Dalton Trans., 2015, 44, 20295 RSC.
- For comparison, the energy barrier for methyl phenyldiazoacetate insertion into cyclopentane was calculated to be 17.4 kcal mol−1: J. Hansen, J. Autschbach and H. M. L. Davies, J. Org. Chem., 2009, 74, 6555 CrossRef CAS PubMed.
- Select examples and reviews: (a) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831 RSC; (b) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899 RSC; (c) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed; (d) J. Kim and S. Chang, Angew. Chem., Int. Ed., 2014, 53, 2203 CrossRef CAS PubMed; (e) A. G. Schafer and S. B. Blakey, Chem. Soc. Rev., 2015, 44, 5969 RSC.
- (a) S. E. Denmark, R. A. Stavenger, A.-M. Faucher and J. P. Edwards, J. Org. Chem., 1997, 62, 3375 CrossRef CAS PubMed; (b) Y. Motoyama, N. Makihara, Y. Mikami, K. Aoki and H. Nishiyama, Chem. Lett., 1997, 26, 951 CrossRef; (c) M. A. Stark and C. J. Richards, Tetrahedron Lett., 1997, 38, 5881 CrossRef CAS; (d) J.-i. Ito, T. Shiomi and H. Nishiyama, Adv. Synth. Catal., 2006, 348, 1235 CrossRef CAS; (e) H. Nishiyama, Chem. Soc. Rev., 2007, 36, 1133 RSC; (f) C. P. Owens, A. Varela-Alvarez, V. Boyarskikh, D. G. Musaev, H. M. L. Davies and S. B. Blakey, Chem. Sci., 2013, 4, 2590 RSC.
- Select examples of phebim complexes: (a) X.-Q. Hao, J.-F. Gong, C.-X. Du, L.-Y. Wu, Y.-J. Wu and M. P. Song, Tetrahedron Lett., 2006, 47, 5033 CrossRef CAS; (b) K. Ma and J. You, Chem.–Eur. J., 2007, 13, 1863 CrossRef CAS PubMed; (c) K. Hyodo, S. Nakamura and N. Shibata, Angew. Chem., Int. Ed., 2012, 51, 10337 CrossRef CAS PubMed; (d) X.-Q. Hao, Y.-W. Zhao, J.-J. Yang, J.-L. Niu, J.-F. Gong and M. P. Song, Organometallics, 2014, 33, 1801 CrossRef CAS; (e) T. Wang, X.-Q. Hao, J.-J. Huang, J.-L. Niu, J.-F. Gong and M.-P. Song, J. Org. Chem., 2013, 78, 8712 CrossRef CAS PubMed; (f) E. Milczek, N. Boudet and S. Blakey, Angew. Chem., Int. Ed., 2008, 47, 6825 CrossRef CAS PubMed.
- C. Hansch and A. Leo, Exploring QSAR: Fundamentals and Applications in Chemistry and Biology, American Chemical Society, Washington, DC, 1995 Search PubMed.
- (a) J. J. Miller and M. S. Sigman, Angew. Chem., Int. Ed., 2008, 47, 771 CrossRef CAS PubMed; (b) M. S. Sigman and J. J. Miller, J. Org. Chem., 2009, 74, 7633 CrossRef CAS PubMed; (c) K. C. Harper, E. N. Bess and M. S. Sigman, Nat. Chem., 2012, 4, 366 CrossRef CAS PubMed; (d) K. C. Harper, S. C. Vilardi and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 2482 CrossRef CAS PubMed.
- W. Karcher and J. Devillers, Practical Applications of Quantitative Structure–Activity Relationships (QSAR) in Environmental Chemistry and Toxicology, Springer, Netherlands, 1990 Search PubMed.
- (a) D. M. Guptill, C. M. Cohen and H. M. L. Davies, Org. Lett., 2013, 15, 6120 CrossRef CAS PubMed; (b) D. M. Guptill and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 17718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and computational details are provided. CCDC 1408224. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00190d |
| This journal is © The Royal Society of Chemistry 2016 |