
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new Pseudomonas quinolone signal (PQS) binding partner: MexG†
James T.
Hodgkinson
a,
Jeremy
Gross
b,
Ysobel R.
Baker
a,
David R.
Spring
a and
M.
Welch
 *b
*b
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
bDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge, CB2 1GA, UK. E-mail: mw240@cam.ac.uk
First published on 20th January 2016
Abstract
The opportunistic pathogen Pseudomonas aeruginosa utilises the cell–cell signalling mechanism known as quorum sensing to regulate virulence. P. aeruginosa produces two quinolone-based quorum sensing signalling molecules; the Pseudomonas quinolone signal (PQS) and its biosynthetic precursor 2-heptyl-4(1H)-quinolone (HHQ). To date, only one receptor (the PqsR protein) has been identified that is capable of binding PQS and HHQ. Here, we report on the synthesis of PQS and HHQ affinity probes for chemical proteomic studies. The PQS affinity probe very effectively captured PqsR in vitro. In addition, we also identified an interaction between PQS and the “orphan” RND efflux pump protein, MexG. The PQS–MexG interaction was further confirmed by purifying MexG and characterizing its ability to bind PQS and HHQ in vitro. Our findings suggest that PQS may have multiple binding partners in the cell and provide important new tools for studying quinolone signalling in P. aeruginosa and other organisms.
Introduction
Bacterial cells within a given population can act collectively to coordinate gene expression via a cell-population density manner.1 This natural phenomenon termed quorum sensing (QS) is achieved by the release of small signalling molecules which can diffuse freely between bacterial cells and can be detected by specific receptor proteins, localised within the cell cytoplasm or cell membrane. The most common class of signalling molecules utilised by Gram-negative bacterial species are N-acyl-homoserine lactones (AHLs). The receptor proteins for AHLs are LuxR homologues.2 However, aside from the AHL class of signalling molecule, other chemically distinct classes of QS signal have been discovered. These include; quinolones, boron diesters, and cyclic peptides.3The opportunistic pathogen Pseudomonas aeruginosa produces three QS signalling molecules. These are; (S)-3-oxo-dodecanoyl-homoserine lactone (OdDHL), (S)-butyl-homoserine lactone (BHL) and 2-heptyl-3-hydroxy-4(1H)-quinolone, also known as the Pseudomonas Quinolone Signal (PQS).4 LasR is the principal receptor protein for OdDHL and RhlR is the principal receptor for BHL, although another LuxR-type protein, QscR, is also known to bind OdDHL and may form heterodimers with LasR/RhlR.5 PqsR (also known as MvfR) is currently the only known receptor for PQS.6 However, 2-heptyl-4(1H)-quinolone (HHQ) – the biosynthetic precursor to PQS – is also considered a fourth signalling molecule and also binds PqsR, albeit with lower affinity.6a PQS is considered to be a multifunctional signalling molecule and has been reported to be involved in the regulation (via PqsR) of several important virulence factors including elastase, pyocyanin, and LecA.7 It has also been linked to other functions independent of PqsR such as membrane vesicle formation, autolysis, efflux pump regulation and siderophore (pyoverdine) production.7b,8 HHQ has also been shown to play an important role in quinolone signalling.6a
Since the discovery of quinolone signalling over a decade ago, the field has received copious interest from the chemical biology community.6–8 Most studies have focused on using genetic approaches to dissect the signalling pathway(s) of PQS and HHQ, and the effect(s) of these molecules on the phenotype and fitness of the organism. However, fewer studies have investigated the action of these molecules at a molecular level. Indeed, and aside from the PqsR, no other PQS receptor proteins have been identified. In many ways, this is unexpected, especially considering the large number of phenotypes affected by PQS (many of which appear to be regulated independent of PqsR). Furthermore, we note that alkyl quinolone signalling in a related genus, Burkholderia, presumably involves different types of receptor molecule, since the Burkholderia sp. genome sequences do not encode any obvious PqsR homologues.9a During our structure–activity analyses (SAR) of the PQS molecule, we became interested in identifying potentially novel PQS receptors.10 Therefore, and to further investigate the mode of action of this important signalling molecule we set out to design and synthesise quinolone signalling affinity probes, with the aim of carrying out a chemical-proteomic analysis of potential PQS and HHQ binding partners.
One approach used in chemical proteomics involves tethering a chemically-modified version of the small molecule of interest via a linker to biotin or a biocompatible resin.11 This resin or biotin-tagged small molecule (affinity probe) is then incubated with a cell-free lysate of the organism of interest. Proteins bound with high-affinity to the capture agent can be separated from non-bound or weakly-bound proteins simply by washing the resin. Proteins bound with high affinity to the resin can then be eluted, resolved by SDS-PAGE and identified. Although we initially set out to use the PQS and HHQ affinity probes to identify novel or additional protein–ligand interactions in P. aeruginosa cell lysates, the probes have wider potential applications in other research areas involving quinolone signalling, e.g., to study inter-species “cross-talk” in Burkholderia and Altermonas spp which also produce alkyl-quinolones.9 Intriguingly, both Burkholderia sp. and P. aeruginosa, are major pathogens in cystic fibrosis patients and form mixed biofilms.12 PQS and HHQ have also been found to influence a number of phenotypes in other bacterial species, such as biofilm formation, bacterial growth and motility.13 Such studies have led to the hypothesis that in the bacterial world PQS and HHQ may play an underlying role in gene regulation and inter-species modulation in complex multi-bacterial communities.14 In addition, PQS is known to have immune-modulatory effects suggesting possible interaction with eukaryotic targets as well, so the affinity probe could be used to identify these as well.15
Results
Design & synthesis of PQS and HHQ affinity probes
The first step in the design of the PQS affinity probe was to strategically attach the linker moiety at a position that minimized the impact of immobilization on the biological activity of PQS. From our previous SAR studies we decided to attach the linker to the 5 position of the quinolone ring as modifications at this position resulted in only minor loss of biological activity in both PqsR-dependent and PqsR-independent phenotypes.10 However, strong electron-donating substituents in the 5 and 6 positions resulted in a large decrease in biological activity. Therefore, in the design of our PQS affinity probe we refrained from attaching strongly electron-donating groups to the quinolone core. PQS analogue 3 with the iodine group in the 5 position was furnished utilising our one-pot microwave irradiation procedure (Scheme 1).16 We hypothesised that the iodine in analogue 3 could be used as a “handle” for palladium-catalyzed cross-coupling reactions with a suitable linker moiety. Analogue 3 was protected as masked bis benzyl derivative to give quinoline 4. The linker moiety 5 was prepared in two steps from commercially available starting material (see ESI†). Coupling of the prepared linker moiety and quinoline 4 under Heck coupling reaction conditions proceeded in moderate yield to furnish the two regioisomers 6 and 7. In one pot, under standard hydrogenation conditions, the azide and alkene functionalities were reduced and the benzyl protecting groups were removed to unmask the quinolone tautomer of PQS intermediate 8 in good yield.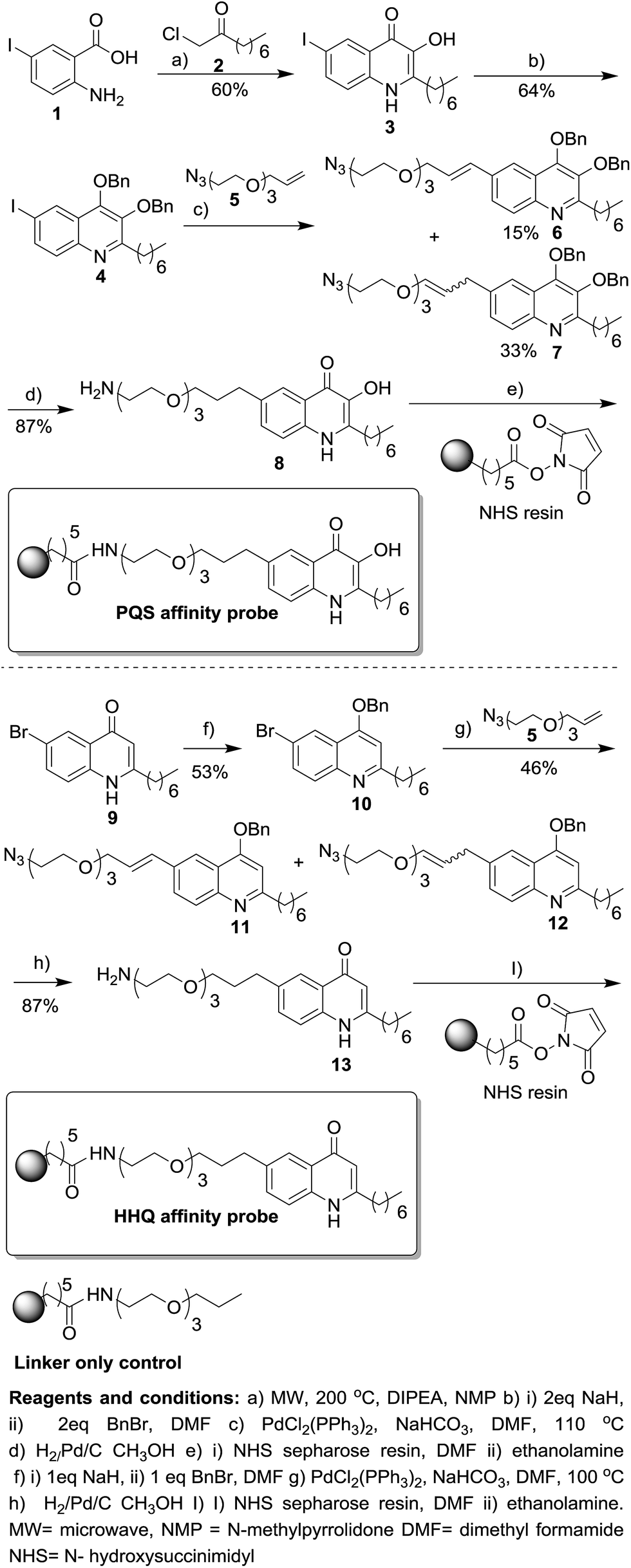 | ||
| Scheme 1 Synthesis of HHQ and PQS affinity probes. For full experimental details on the preparation of intermediates and the linker only control see ESI.† | ||
Intermediate 8 was coupled to N-hydroxysuccinimidyl (NHS) sepharose beads completing the synthesis of the PQS affinity probe. Although the biological activity of HHQ is not as well studied as PQS we also decided to attach the linker moiety to the same 5 position of the quinolone ring in HHQ. Thus, the two affinity probes could be used in chemical proteomic studies for the direct comparison of HHQ and PQS interactions with proteins. The 5-bromine HHQ analogue 9 was prepared in two steps (see ESI†). With analogue 9 in hand the subsequent steps that were used for the synthesis of the PQS affinity probe were also utilised for the generation of the HHQ affinity probe, with similar yields being obtained in both cases. As a control, we also synthesised a “linker-only” resin to assess non-specific protein interactions with the linker/resin.
The PQS affinity probe binds PqsR and iron in vitro
We next sought to determine if the immobilized PQS retained biological activity. PQS is known to have at least two important activities in the cell; it binds PqsR and it also binds/captures iron. To investigate the former, we first tried to over-express and purify full-length PqsR from recombinant Escherichia coli with a view to carrying out binding experiments. Initial attempts at over-expressing hexa-histidine-tagged full-length PqsR protein, or the hexa-histidine-tagged ligand-binding domain of PqsR6a failed because the expressed protein formed insoluble inclusion bodies that did not yield a soluble product after refolding (data not shown). However, we were able to over-express the ligand-binding domain (LBD) of PqsR in soluble form and in high yield as a maltose-binding protein (MBP) fusion.17 With purified PqsRLBD-MBP in hand we measured the ability of the fusion protein to bind to the PQS affinity probe. The purified PqsRLBD-MBP was incubated with the PQS affinity probe or with linker-only control resin for 30 minutes.The resins were then washed and the remaining protein on the resins was released by boiling with SDS sample buffer. The released protein was resolved by SDS-PAGE. A large quantity of the PqsRLBD-MBP was retained on the PQS affinity beads following the wash steps. In contrast, very little PqsRLBD-MBP was captured and retained on the control beads (Fig. 1A). PQS has been reported to chelate iron and form a blood-red metal complex.18,8c Addition of FeCl3 to the PQS affinity probe caused the resin to immediately turn deep red, indicating the formation of the PQS–iron complex on the sepharose beads (Fig. 1B). We conclude that the immobilized PQS is able to bind to PqsR and also retains its ability to chelate iron.
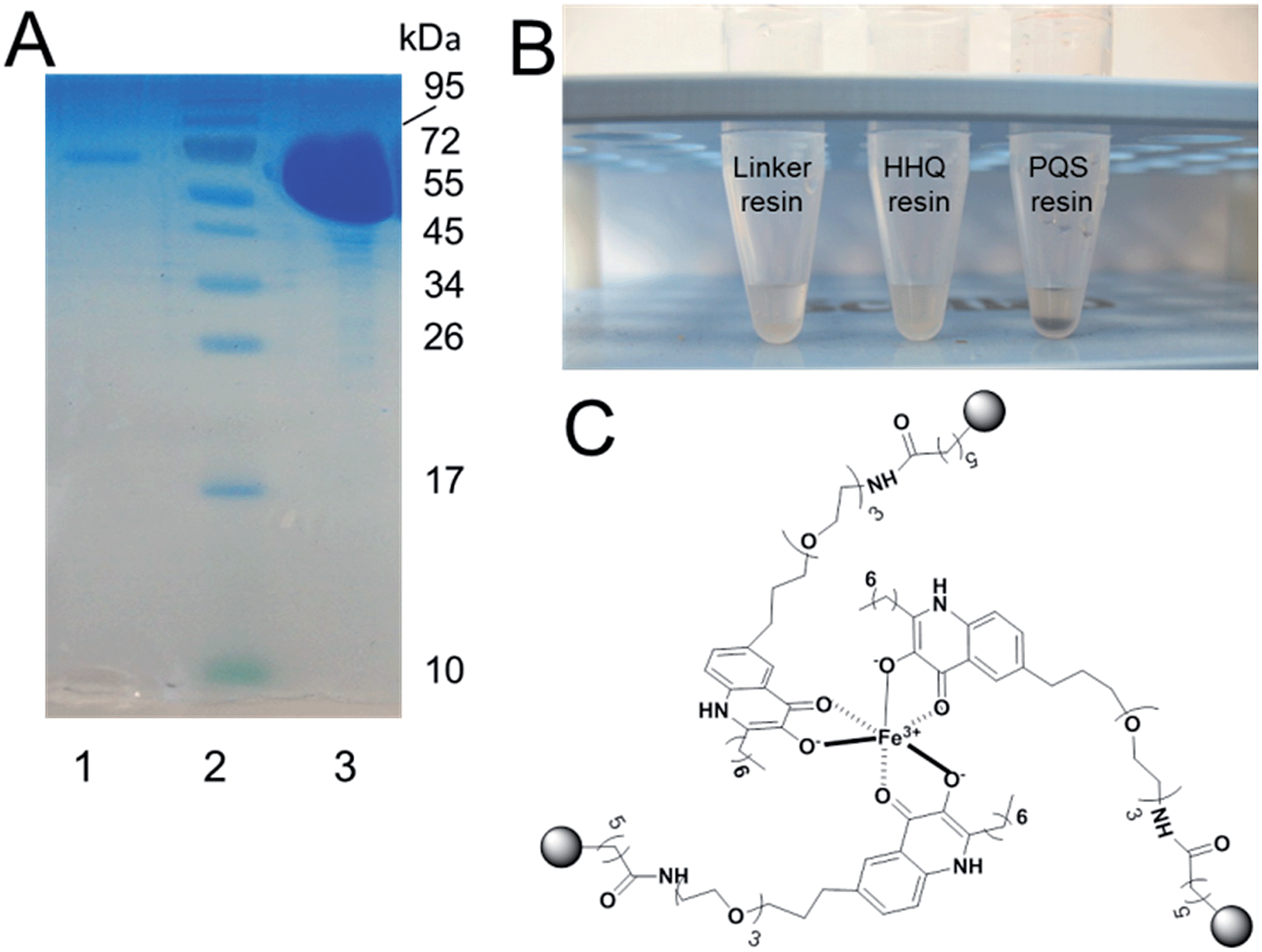 | ||
| Fig. 1 The PQS affinity probe retains affinity for PqsRLBD and Fe3+. (A) Purified PqsR ligand binding domain (PqsRLBD) binds strongly to immobilized PQS but not to the control linker resin. Purified MBP-tagged PqsRLBD was incubated with aliquots of the control or PQS-derivatized resin, see ESI.† The resin samples were extensively washed and the bound protein was eluted by boiling the washed beads in SDS sample buffer. The panel shows a Coomassie Brilliant Blue-stained SDS-PAGE gel of the protein eluting from control beads (lane 1) or PQS-derivatized beads (lane 3). Molecular mass markers are shown in lane 2. (B) Aliquots (50 μL) of control beads, HHQ beads or PQS beads (as indicated) were mixed with 1 mM FeCl3. The PQS beads immediately turned deep red due to the formation of the PQS–iron complex. (C) Proposed structure of the PQS–Fe complex on probe resulting in dark red colouration. | ||
Identification of MgtA and MexG as potential binding partners of PQS
The immobilized PQS probe was used to capture potential PQS-interacting proteins from cell lysates of P. aeruginosa. Cell lysates were prepared from stationary phase cultures of a wild-type strain of P. aeruginosa (PA14) and from an isogenic (alkyl quinolone deficient) pqsB mutant for comparison. The cytoplasmic fraction of cells yielded a high background level of binding to both the PQS and control resins (data not shown) so instead, we focussed on capturing proteins from the detergent-solubilized membrane fraction. This choice was partly driven by the fact that PQS is known to partition into the cell membrane; indeed, PQS is thought to move between cells enclosed within membrane vesicles.8d Two proteins were repeatedly captured from an extract of Triton X100-solubilized PA14 membranes by the PQS resin and not by the control resin. These proteins were resolved by SDS-PAGE and identified by MALDI-MS (Fig. 3A). The protein band with the highest molecular mass was identified as PA4825, also known as MgtA. This is a P-type ATPase involved in Mg2+ transport with a predicted molecular mass of 100 kDa. This is not the first time quinolone signalling has been linked to magnesium homoeostasis.19The second protein that was repeatedly captured by the PQS affinity probe had a molecular mass of approximately 15 kDa and was identified as a predicted inner membrane protein, PA4205, or MexG (Fig. 2). Commensurate with the small size of MexG and noting that integral membrane proteins such as this are rarely ever expressed at high levels, only small amounts of MexG were routinely captured. MexG is encoded by the mexGHI-opmD operon, and is a component of a resistance-nodulation-cell division (RND) efflux pump. These are large protein complexes that traverse both the inner and outer bacterial membrane, allowing active transport of substrates, including many antibiotics and biocides. RND pumps are found in many Gram-negative bacteria and almost invariably have a tripartite structure that includes an inner membrane “pump” component, a periplasmic “membrane fusion” protein and an outer membrane channel (MexI, MexH and OpmD, respectively). However, the MexGHI-OpmD pump is unusual because it contains a fourth protein, MexG, which is not found in most other RND-type complexes. The role of MexG is currently not clear, although it is a member of the DoxX superfamily. The DoxX domain (E = 2.6 × 10−15) in MexG spans residues 15–95 (Fig. 2). It has been previously suggested that the annotated start codon (in PseudoCAP) for mexG may be incorrect, and that the ORF actually encodes an 11.2 kDa protein.20 However, our data suggest that the annotated start codon is indeed correct and that the full-length protein is 15 kDa in size. Interestingly, previous reports have shown that mexI and opmD mutants are deficient in the production of PQS-regulated virulence factors such as pyocyanin, pyoverdine, elastase and rhamnolipid.8a Furthermore, it has been previously reported in an alkyl quinolone deficient pqsR mutant, expression of mexG is severely down regulated.21 Consistent with this, no MexG was captured from lysates prepared from an alkyl quinolone deficient pqsB mutant compared with the wild-type (Fig. 2). The established link between PQS signalling and mexG made it a logical candidate for further analysis.
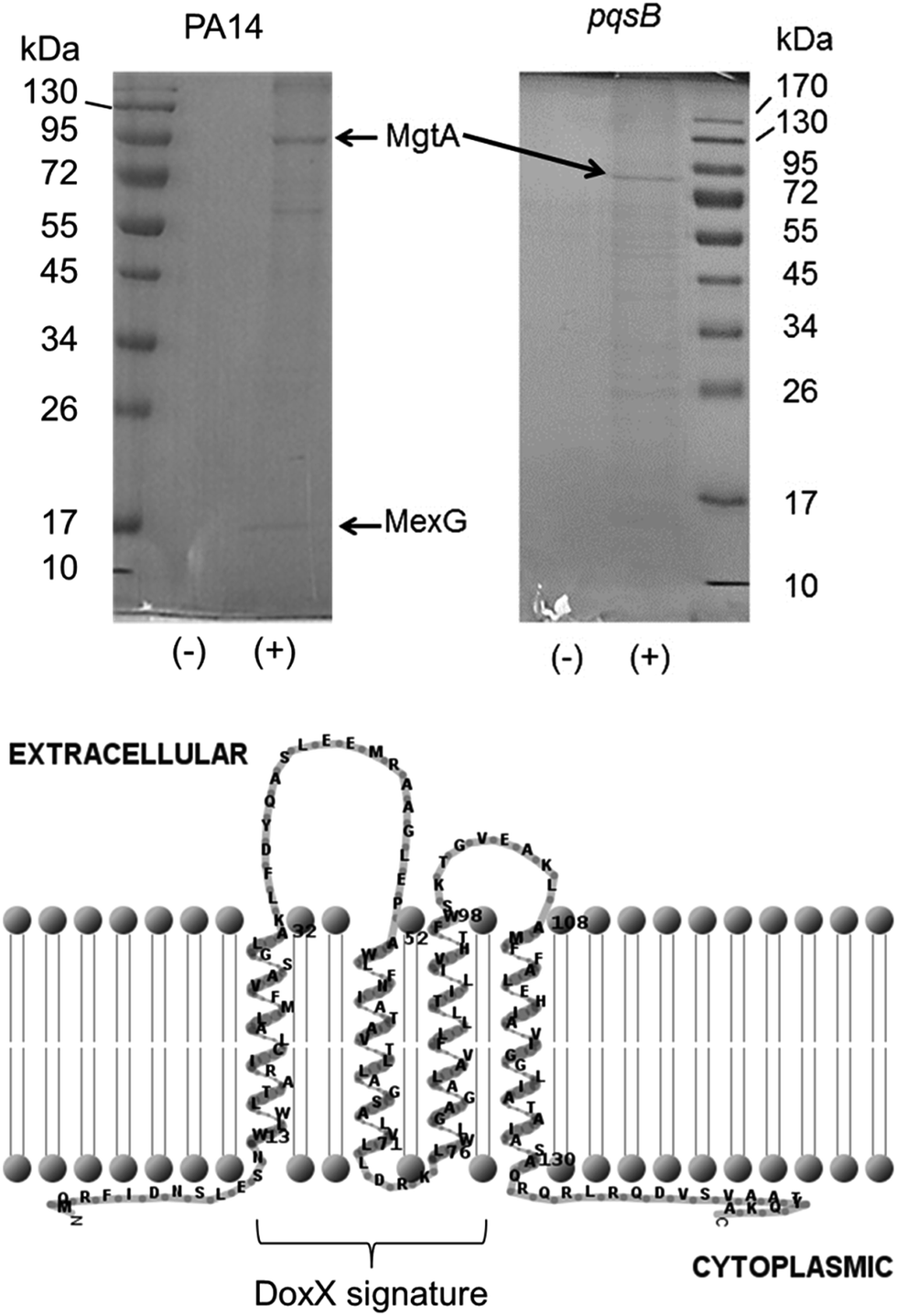 | ||
| Fig. 2 The PQS affinity probe captures MgtA and MexG from the detergent-solubilized cell membrane fraction. Detergent-solubilized cell membranes from stationary phase cultures of wild-type PA14 or an isogenic pqsB mutant, as indicated, were mixed with aliquots of either control resin (−) or PQS resin (+). Following extensive washing of the resins, bound proteins were eluted by boiling the resin samples in SDS sample buffer. The eluted proteins were resolved by SDS-PAGE and an image of the Coomassie Brilliant Blue-stained gel is shown. Following staining of the gel, a selection of bands were excised and identified by MALDI-MS. The MgtA and MexG-containing bands are indicated. Predicted membrane topology of MexG. The DoxX domain (residues 15–95, encompassing most of helices 1–3) signature is indicated. The MexG amino acid sequence was analysed by TMHMM and the output was depicted using TMRPRES2D. | ||
Deletion of the DoxX domain abolishes the ability of MexG to bind PQS in vivo
To further investigate the MexG–PQS interaction, we generated an in-frame mexG mutant in which amino acids 3–108 were deleted, removing the predicted periplasmic loops and the first three transmembrane domains (which collectively, comprise the DoxX domain sequence signature in MexG). Detergent-solubilized membranes from the ΔmexG mutant and from the progenitor wild-type strain (PAO1) were incubated with control or PQS-derivatized resin. Following washing, the bound protein(s) were resolved by SDS-PAGE (Fig. 3). MexG was captured by the PQS resin from extracts of wild-type membranes, but was absent from the corresponding mexG mutant membranes.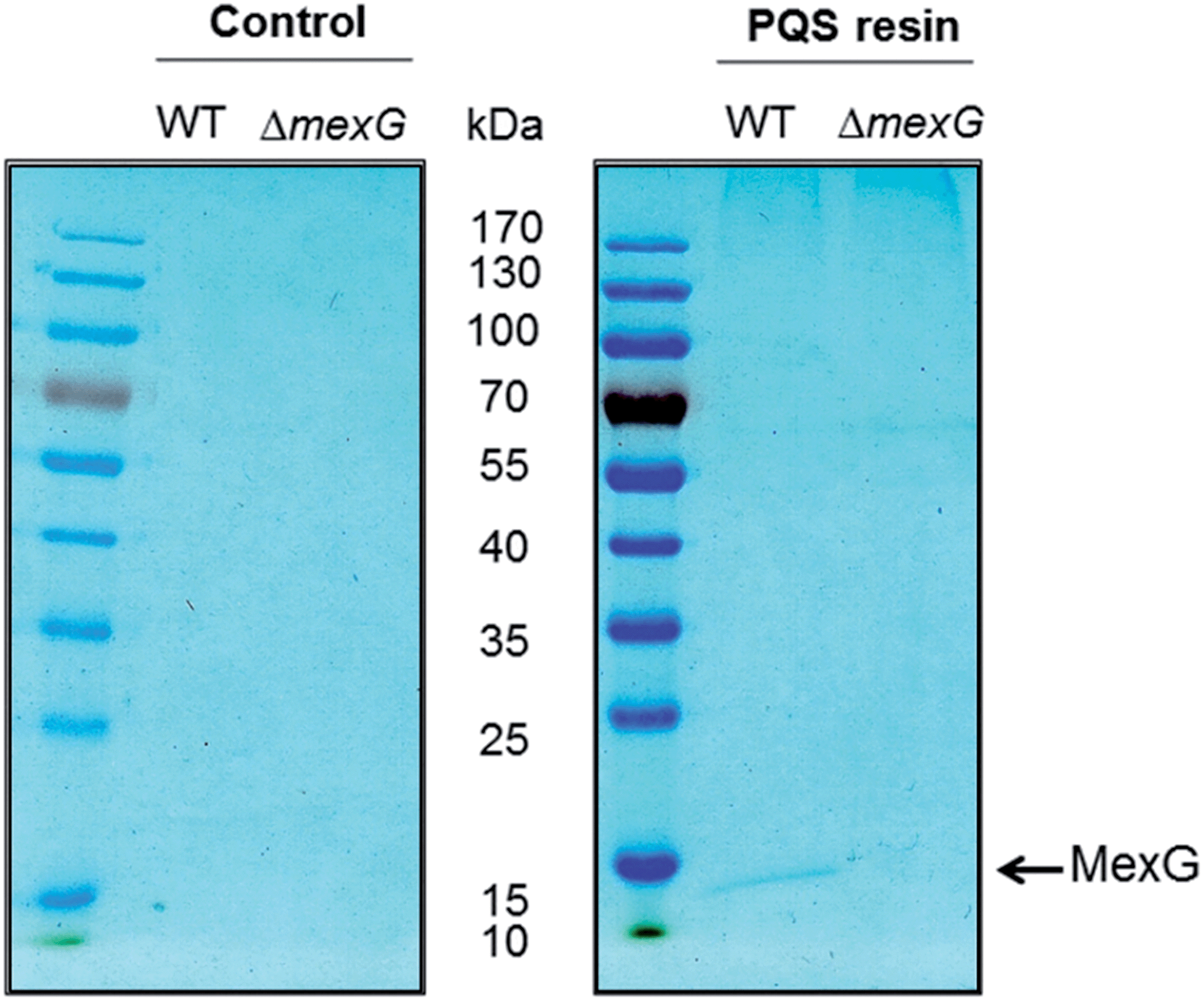 | ||
| Fig. 3 Deletion of the DoxX motif abolishes MexG binding to PQS. Detergent-solubilized membranes from either wild-type PAO1 or from an in-frame mexG deletion mutant (as indicated) were mixed with control resin or PQS-derivatized resin and incubated to allow binding. The resins were washed extensively and the bound proteins were eluted by boiling in SDS sample buffer. The figure shows a Coomassie Brilliant Blue-stained gel of the proteins bound to control resin (left panel) and PQS resin (right panel). Note that essentially the only protein detectable in this experiment was MexG from the wild-type extract bound to the PQS resin. Control = linker only resin. | ||
Purified MexG binds to the PQS and HHQ affinity probes in vitro
To further investigate the interaction of MexG with PQS, we over-expressed hexa-histidine-tagged MexG in E. coli. N- and C-terminal His6 tags were tested; the N-terminally tagged protein was expressed in higher quantities (ca. 0.5 mg L−1 culture) and partitioned exclusively into the Triton-X100 soluble membrane fraction. We found that MexG could also be solubilized from the membrane fraction using DDM (dodecyl maltoside, 1% w/v), and since (unlike Triton X-100) this detergent does not absorb light at 280 nm, it was used for subsequent extraction/purification steps. Purified DDM-solubilized MexG was incubated with the linker-only (control) resin, PQS resin and HHQ resin. Following washing of the resins, SDS-PAGE revealed that MexG bound to both PQS and HHQ affinity probes but not to the linker only resin (Fig. 4A). To show that these resins are not simply binding to any protein at high concentrations via potential hydrophobic interactions, the experiment was repeated with bovine serum albumin (BSA). Serum albumin is to known to have an affinity for fatty acids, so this protein would be expected to bind non-specifically to hydrophobic matrices. No BSA could be recovered from PQS or HHQ probes after washing (Fig. 4B).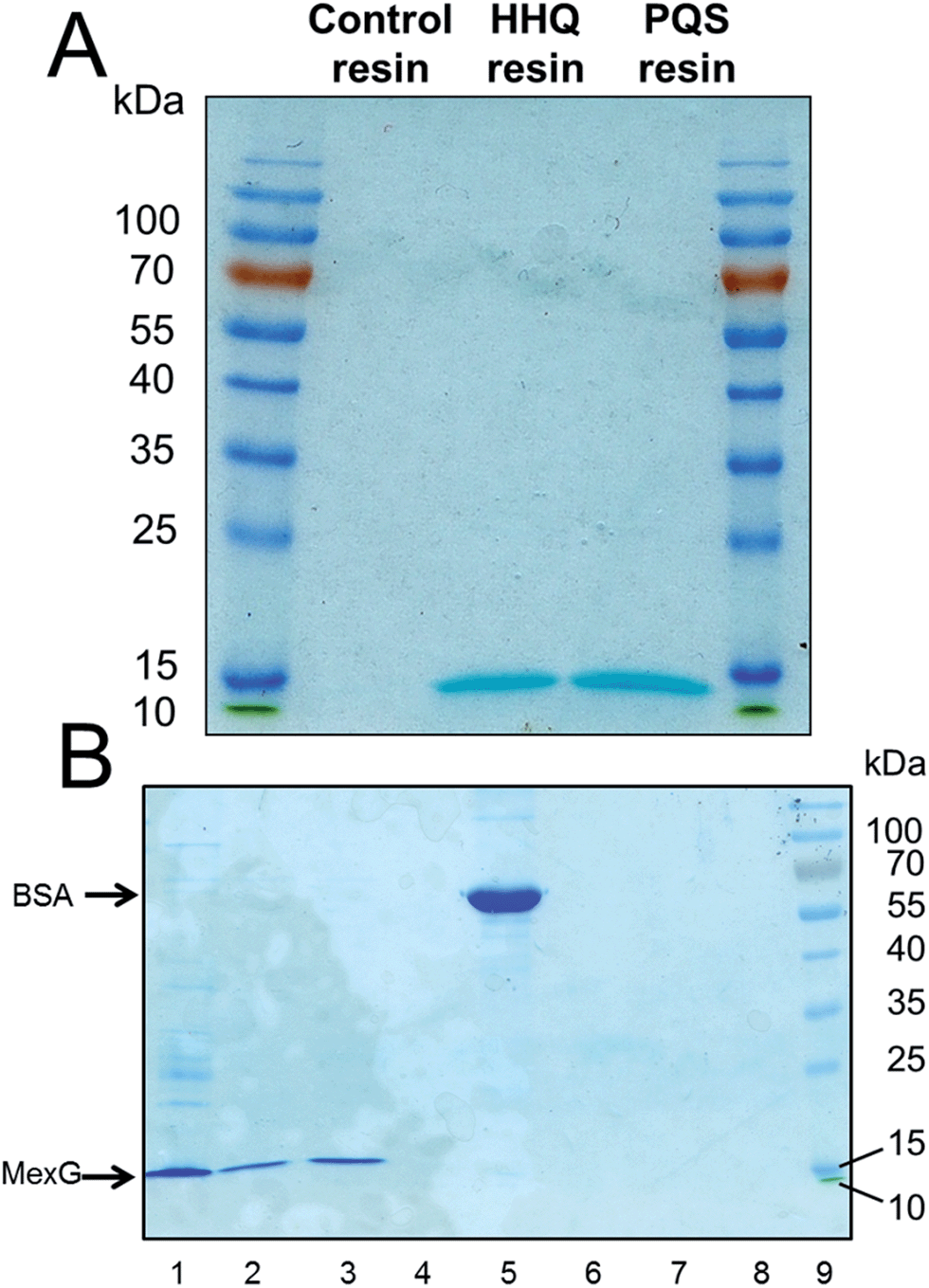 | ||
| Fig. 4 Purified MexG binds to immobilized PQS and immobilized HHQ in vitro. (A) Purified MexG was incubated with either linker resin, HHQ or PQS affinity probes (as indicated) for 1 h. After extensive washing of the resins, the bound protein was eluted in SDS sample buffer and resolved by SDS-PAGE. The resulting Commassie Brilliant Blue-stained gel is shown. (B) The experiment in (A) was repeated using BSA in place of MexG. Lane 1; an aliquot of the MexG sample that was loaded onto the resin, lane 2; MexG bound to PQS resin, lane 3; MexG bound to HHQ resin, lane 4; MexG bound to control resin, lane 5; BSA alone, lane 6; BSA bound to PQS resin, lane 7; BSA bound to HHQ resin, lane 8; BSA bound to control resin, lane 9; molecular mass markers. | ||
Soluble PQS outcompetes MexG binding to the PQS affinity probe
MexG binding to the PQS-derivatized resin could be outcompeted in the presence of exogenously-added synthetic PQS (Fig. 5A). The binding of MexG to the PQS affinity resin was inhibited when the nominal “free” concentration of PQS was >10 mM. However, the actual concentration of soluble “free” PQS is likely much lower than this, since PQS is only sparingly soluble in aqueous solutions, even in the presence of detergent (which was added to the binding buffer to maintain MexG solubility). This notwithstanding, we did note that the amount of soluble PQS (assayed by monitoring the formation of red coloured Fe3+–PQS complex in the supernatants from the binding assay) increased in proportion with the total amount of PQS present, even after PQS was observed to precipitate (Fig. 5A). We also examined whether Fe3+ affected MexG binding to the HHQ and PQS resins. It did not (Fig. 5B).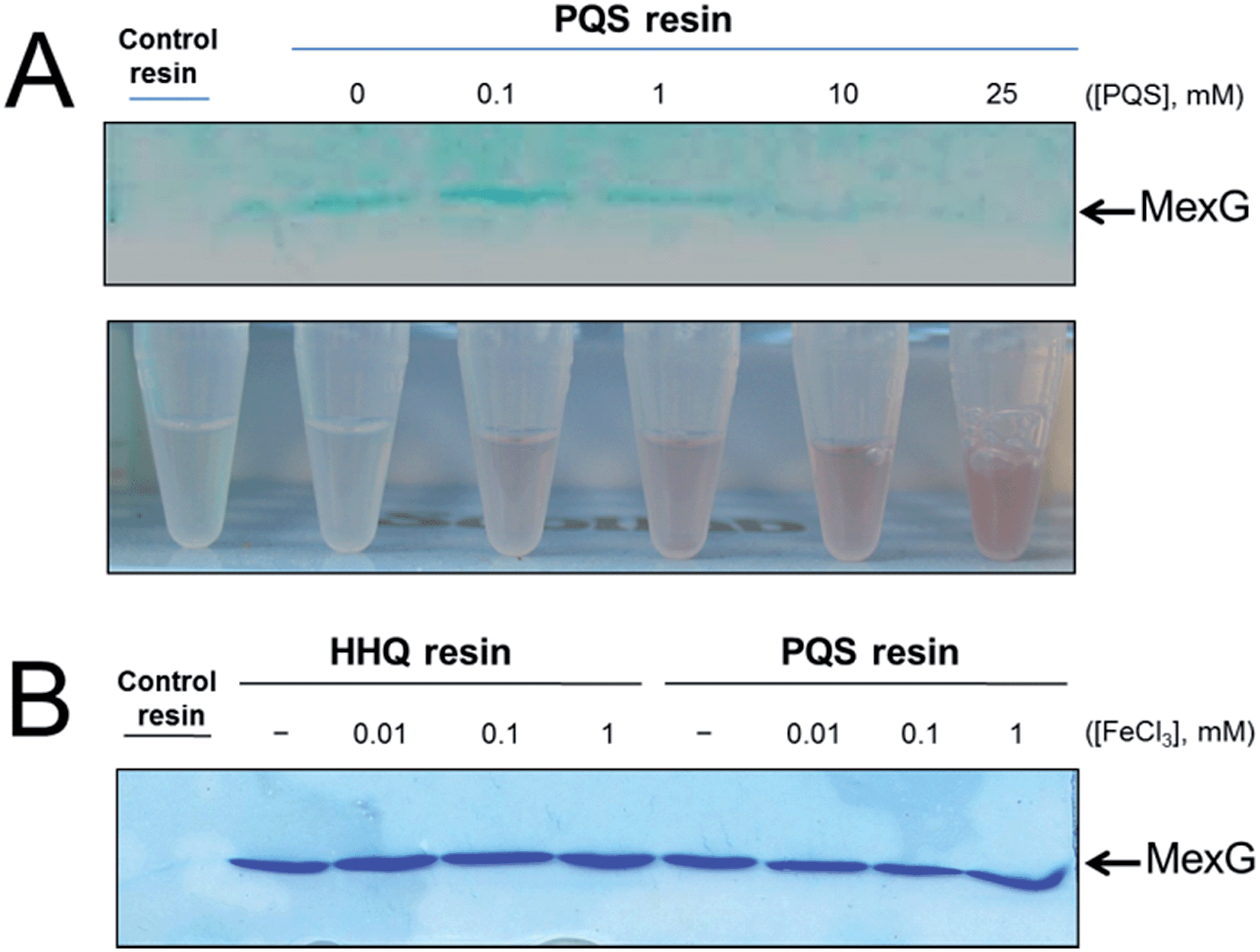 | ||
| Fig. 5 Soluble PQS outcompetes MexG bound to the PQS affinity probe. (A) Purified MexG was incubated with PQS resin or control resin (as indicated) in the presence of increasing concentrations of “free” PQS. The bound protein remaining after extensive washing of the resins was visualised by SDS-PAGE. The upper panel shows the Commassie Brilliant Blue-stained SDS-PAGE gel. The lower panel shows Eppendorf tubes containing the unbound material after incubating with the resin (but before the wash steps) mixed with FeCl3. Note the increased intensity of red Fe3+–PQS complex formed at higher “free” PQS concentrations correlates with a reduction in the amount of MexG bound to the resin after washing. (B) Purified MexG was inclubated with control resin, HHQ resin or PQS resin (as indicated) in the presence of the indicated concentrations of FeCl3. Bound proteins were resolved by SDS-PAGE and the image shows the resulting Coomassie Brilliant Blue-stained gel. | ||
PQS alters the fluorescence spectrum of MexG
The PQS–MexG interaction was further analysed using fluorescence spectroscopy. Excitation of detergent-solubilized MexG (which contains 5 Trp residues, three of which (W15, W53 and W77) are located in the DoxX domain) at 295 nm led to an emission peak at 330 nm (Fig. 6A). This suggests that the Trp residues are primarily located in a hydrophobic environment, consistent with the predicted transmembrane location of MexG. In contrast, excitation of PQS at 295 nm gave rise to an emission peak at 450 nm (Fig. 6Ainset). When DMSO alone was titrated into purified MexG, no changes in the emission spectrum were observed (Fig. 6A). However, when PQS dissolved in DMSO was titrated into purified MexG, quenching of MexG fluorescence (330 nm) was observed with a concomitant increase in emission at 450 nm (Fig. 6B). These data suggest that PQS physically binds MexG and elicits a change in the environment of one or more of the Trp fluorophores. Since all of the Trp residues reside in the transmembrane segments, this suggests that the PQS binding site may be associated with these segments, or may drive a conformational change that affects these segments. To further quantify the PQS:MexG interaction, we titrated known amounts of PQS into a MexG sample of known concentration and monitored the fluorescence quenching at 330 nm. High concentrations of added PQS led to quenching of >80% of the fluorescence (Fig. 6C (inset)). Scatchard analysis of the data yielded distinctly biphasic binding behaviour, indicative of tight binding of MexG to PQS (characterized by Kd = 1 μM) as well as weaker binding (Kd = 5 μM). The weaker binding only became apparent at high concentrations of PQS (Fig. 6C).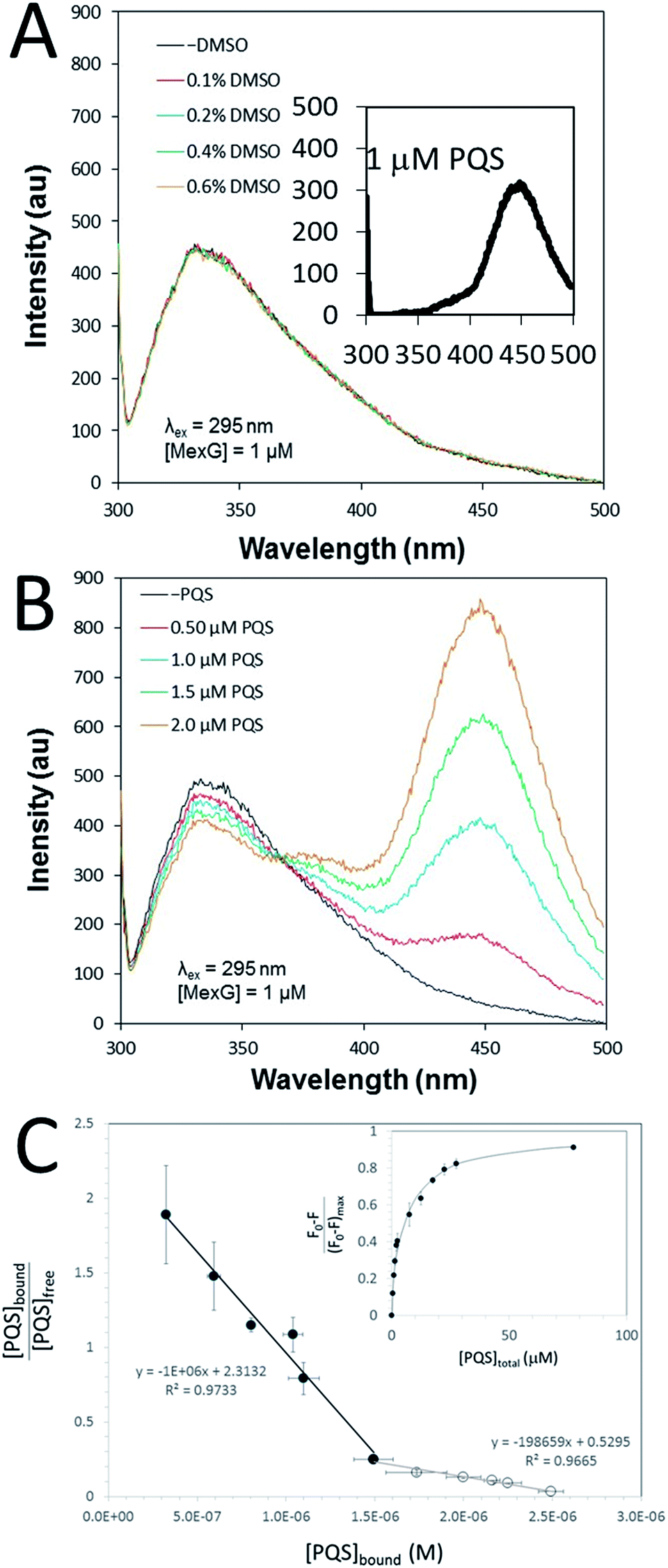 | ||
| Fig. 6 Fluorescence emission spectra of MexG and PQS. (A) The fluorescence emission spectra of purified MexG (1 μM final concentration) was measured in the presence of the indicated amounts of added DMSO (DMSO was used as a solvent for PQS). The excitation wavelength was 295 nm. The highest final concentration of DMSO tested (0.6% v/v) was greater than the highest DMSO concentration achieved during the titration of PQS. Note that the addition of DMSO does not change the emission spectrum of MexG. Inset. Emission spectrum of 1 μM PQS excited at 295 nm. (B) Emission spectrum of purified MexG in the presence of increasing concentrations of PQS. The excitation wavelength was 295 nm. Note how increasing PQS concentrations lead to an increase in emission at ca. 450 nm and a concomitant decrease (quenching) of emission at ca. 340 nm. (C) Quantitation of PQS binding to MexG. Scatchard plot of the binding data derived from fluorescence quenching experiments. Note the biphasic binding behaviour of the MexG:PQS interaction, indicative of tight (filled symbols) and weaker (open symbols) binding interactions. The slope of each linear regression yields −1/Kd. Inset. The figure shows the fraction of protein fluorescence that was quenched at different total concentrations of added PQS. | ||
Discussion
In this work we have synthesized and characterized novel affinity probes designed to assist in identifying potentially novel binding partners involved in alkyl quinolone signalling in the opportunistic human pathogen, P. aeruginosa. Using these tools, we have been able to identify at least two potential additional binding partners for HHQ/PQS, and we have characterized one of these interactions in some detail. Given that for practical reasons – we restricted our pull down analysis to the detergent-solubilized membrane fraction, it seems probable that we have only identified a subset of the possible alkyl quinolone interacting partners in the cell. Additionally, it must be borne in mind that although the design of our affinity probes was informed by previous SAR analyses, it is possible that for steric reasons, the immobilized ligand may not retain an ability to interact with the full spectrum of binding partners in the cell. In this regard, it is noteworthy that we did not pull-down the only other known PQS receptor, PqsR, especially since we demonstrated that the purified ligand-binding domain of PqsR binds robustly to the PQS probe in vitro. Moreover, GST-tagged PqsR (also known as MvfR) has been previously-reported to associate with the P. aeruginosa membrane fraction.6b Presumably, either (i) the concentration of membrane-associated PqsR is too low to detect using our pull-downs, (ii) the conformation of the membrane-associated PqsR prevents it from binding PQS, (iii) expression of the tagged protein from a medium copy vector leads to spurious sub-cellular localization, or (iv) the conformation of full-length PqsR is subtly different to that of the purified PqsR LBD, thereby preventing the linker-modified PQS from being able to access the LBD in the native protein. We cannot comment further on which of these possibilities, if any, is correct.A key finding of the current work was that the PQS affinity probe was able to capture MexG from crude cell extracts. We were further able to reconstitute this interaction using purified MexG in vitro. The purified MexG bound strongly to immobilized PQS and immobilized HHQ, and this binding could be out-competed in the presence of excess ligand. Iron had no apparent effect on the interaction, in spite of the fact that this metal was shown to bind to the PQS (but not HHQ) resin. Deletion of the DoxD domain abolished the binding of MexG to the PQS resin, as did mutation of the PQS biosynthetic cluster. The latter was expected because pqs biosynthetic mutants display pronounced down-regulation of genes dependent on PQS for their expression, including MexG (see below). Finally, using intrinsic Trp fluorescence as a reporter, we confirmed that PQS alters the emission characteristics of purified MexG in vitro. The observed change in the protein fluorescence spectrum in the presence of PQS may reflect (i) a PQS-induced change in the conformation of the protein, (ii) the consequence of direct juxtaposition of bound PQS adjacent to one or more of the protein Trp residues (without any associated conformational change), or (iii) a degree of resonance energy transfer from the Trp residues to the bound PQS. The latter has been observed experimentally in PqsR.17 We cannot currently discriminate between these three possibilities, but note that all are indicative of a direct interaction between PQS and the purified MexG. Quantitative analysis of the MexG:PQS interaction using this approach yielded a Kd of 1 μM, which is very similar to that (1.2 μM) measured for PQS binding to PqsR.17 A small proportion of the purified protein yielded a higher Kd (5 μM), which may indicate the presence of a second, weaker binding site. To put these values into context, the concentration of extracellular PQS in bacterial cultures has been reported to range from 6 μM to 25 μM.17 These results suggest that MexG is easily capable of binding PQS in vivo, and furthermore, can potentially compete with PqsR for PQS binding. The MexG–PQS interaction was unexpected, although given that the MexGHI-OpmD efflux pump has already been reported to be involved in virulence regulation, QS and quinolone signalling molecule homeostasis, we suggest that the interaction may have functional importance.8a Currently, the function of MexG remains unclear; as previously noted, MexG is unique in that it is not conserved in other RND pumps in P. aeruginosa. However, MexG does have sequence similarity to a subunit of a known quinolone oxidase, DoxD.21 Earlier workers have noted links between quinolone signalling and MexG (although none have identified a direct interaction between this protein and the signalling molecule). For example, based on microarray analyses, Déziel and co-workers reported that mexG transcription was severely down-regulated in a pqsR mutant (which is unable to make alkyl quinolones) and β-galactosidase activity from a mexG:lacZ reporter was essentially abolished in the same mutant.21 These observations are consistent with our own finding that much less MexG is captured in cell extracts of a pqsB mutant compared with the wild-type (like the pqsR mutant, a pqsB mutant is unable to make alkyl quinolones). In addition, Aendekerk et al. showed that virulence, quinolone biosynthesis and growth rate are all severely impaired when insertions are introduced into mexI or opmD. These observations were partly attributed to the intracellular accumulation (due to presumed impaired efflux) of an alkyl quinolone precursor, anthranilate.8a However, it is unlikely that MexG is involved in anthranilate export since the in-frame mexG mutant displayed wild-type growth kinetics, suggesting that it was not accumulating cytotoxic precursors.
In addition to MexG, we also identified a P-type ATPase, MgtA, as a potential PQS interaction partner. MgtA expression is stimulated when Mg2+ concentrations become low, perhaps to compensate for the PhoPQ-dependent down-regulation of corA expression (CorA is the preferred Mg2+ transporter when Mg2+ is not limiting). PQS signalling has previously been linked to magnesium homeostasis and under Mg2+ limiting conditions the expression of lipopolysaccharide-modifying enzymes and alkyl quinolone biosynthesis enzymes is known to increase, as does PQS production.19 In the light of this, one possibility is that PQS acts to allosterically stimulate MgtA and it is tempting to speculate that one reason why PQS biosynthesis is up-regulated under Mg2+ limiting conditions is to enhance magnesium uptake into the cell. It should be noted that our data suggest that PQS does not appear to affect mgtA expression; the amount of MgtA that was pulled down in the wild-type and pqsB mutant extracts was roughly comparable (Fig. 2A). Further studies will be required to determine the exact role played by PQS in magnesium homeostasis. Moreover, it should be noted that unlike MexG, we did not independently verify the PQS–MgtA interaction through e.g., reconstitution of the binding reaction in vitro using purified components. Until such confirmatory studies are carried out, the PQS–MgtA interaction has to be considered as speculative.
Conclusions
Quorum sensing and quinolone signalling has attracted significant interest from the chemical biology community. Indeed, quorum sensing interference has been long posited as a potential supplement to conventional antibiotic therapy. Here, we report on the design and chemical synthesis of novel affinity probes to investigate binding partners of the Pseudomonas Quinolone Signal (PQS). Using these novel molecular tools, we identified and confirmed a new PQS binding partner – MexG – a component of an RND-type efflux pump that has been previously implicated in PQS-dependent signalling, but hitherto not thought to interact with PQS. Furthermore, we have provided evidence that PQS may also interact with the alternative Mg2+ transporter, MgtA. Our studies suggest that alkyl quinolones not only affect the cell by altering the transcriptional profiles of genes; they also bind directly to protein partners in the cell. Given the attention currently being paid to quorum sensing blockers, a more in-depth understanding of these multi-functional signalling molecules will be important if we are to develop such molecules as potential therapeutic agents in the future.Experimental
PQS, HHQ and HQNO synthesis
PQS was prepared as previously reported.16 HHQ was prepared as previously reported.22PQS, HHQ affinity probe synthesis and linker control
Full experimental details and characterisation data can be found in the ESI.†Purification of PqsR ligand binding domain (LBD)
Purification of the PqsRLBD-maltose binding protein chimera was carried out as previously reported.17PqsRLBD binding to the PQS affinity probe
The PQS affinity resin (100 μL, isopropanol suspension) was added to a 1.5 mL Eppendorf tube. Resin buffer (20 mM Tris–HCl, 200 mM NaCl, pH 7.4), 1 mL was added to the beads and the resin suspension was centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 2 min, 4 °C). The remaining solution (0.9 mL) was carefully removed and discarded so as not to disturb the pelleted resin. This procedure was repeated a further two times to pre-equilibrate the resin. The purified PqsRLBD (200 μL, 6 mg mL−1) was incubated separately with the washed/equilibrated PQS affinity probe resin and the linker-only resin, in 1.5 mL Eppendorf tubes with gentle mixing at 4 °C for 35 min. The resin was then pelleted (10000 × g, 2 min, 4 °C) and the overburden of supernatant was removed. The resin pellets were then washed five times in resin buffer (1 mL) to remove non-bound proteins. After the last wash as much buffer as possible was removed without disturbing the resin pellet. Denaturing buffer 50 μL (50 mM Tris–HCl, 2% sodium dodecyl sulphate, 0.1% bromophenol blue, 10% glycerol, 10 mM dithiothreitol) was then added to the resin suspension and the samples were heated at 95 °C for 10 min. The resin was then briefly pelleted and the eluted proteins were analysed by SDS polyacrylamide gel electrophoresis (12% gel).
000 × g, 2 min, 4 °C). The remaining solution (0.9 mL) was carefully removed and discarded so as not to disturb the pelleted resin. This procedure was repeated a further two times to pre-equilibrate the resin. The purified PqsRLBD (200 μL, 6 mg mL−1) was incubated separately with the washed/equilibrated PQS affinity probe resin and the linker-only resin, in 1.5 mL Eppendorf tubes with gentle mixing at 4 °C for 35 min. The resin was then pelleted (10000 × g, 2 min, 4 °C) and the overburden of supernatant was removed. The resin pellets were then washed five times in resin buffer (1 mL) to remove non-bound proteins. After the last wash as much buffer as possible was removed without disturbing the resin pellet. Denaturing buffer 50 μL (50 mM Tris–HCl, 2% sodium dodecyl sulphate, 0.1% bromophenol blue, 10% glycerol, 10 mM dithiothreitol) was then added to the resin suspension and the samples were heated at 95 °C for 10 min. The resin was then briefly pelleted and the eluted proteins were analysed by SDS polyacrylamide gel electrophoresis (12% gel).
Preparation of P. aeruginosa cell lysate enriched in membrane proteins
PA14 (UCBPP-PA14) was obtained from Dr S. Diggle (Nottingham University, UK). DH125 (a PA14-derived pqsB:TnphoA mutant) was generated by Deziel et al.21. Due to its relative ease of genetic manipulation, PAO1 was used to generate the ΔmexG mutant. Fresh LB media (100 mL) was inoculated to an initial OD600 of 0.05 with an overnight culture of wild-type PA14 or DH125. The cultures were grown at 37 °C for 10 h with good aeration (250 rpm). The OD600 was measured and cultures were harvested by sedimentation at (7400 × g, 4 °C, 30 min). The cell pellet was resuspended in resin buffer (10 mL) containing a protease inhibitor cocktail tablet (Roche). The cells were then lysed by ultrasonication on ice. The crude lysate was clarified by centrifugation (15000 × g, 4 °C, 30 min) and aliquots (1 mL) of the supernatant were collected. The supernatant aliquots were then centrifuged (365000 × g, 4 °C, 30 min) to pellet the membrane fraction and the supernatant (soluble fraction) was discarded. The pelleted membranes were washed once with resin buffer (1 mL) and re-centrifuged (365000 × g, 4 °C, 30 min) to remove residual contaminating soluble proteins. The supernatant was discarded and the pellet was resuspended in buffer (1 mL) containing 0.5% Triton X-100. The suspension was clarified once again by centrifugation (365000 × g, 4 °C, 30 min) and aliquots (1 mL) of the supernatant were then stored at −80 °C until further use.
Binding of proteins from P. aeruginosa cell lysate to the affinity probes
The same procedure was followed as described above to measure PqsRLBD binding to the affinity probe, with minor modifications. Smaller volumes of the affinity probe (50 μL) and linker-only resin (50 μL) were used, and the solubilized membrane fraction (0.5 mL) was added in place of purified PqsRLBD. The reaction incubation time was increased to 1 h. In the wash steps 0.1% Triton-X-100 was present in the resin buffer.Cloning and expression of mexG
Full-length mexG was PCR-amplified from PAO1 genomic DNA using primers TAGTGGATCCCAGCGCTTCATCGATAACTC and TAGTAAGCTTTCAGGCCTTCTGGTA GGTGG. The PCR product was digested with BamHI and HindIII and cloned into similarly-digested pQE80 (Qiagen). The plasmid was introduced into E. coli ER2556 by CaCl2 transformation. Overnight cultures of ER2556 (pQE80mexG) were inoculated 1:100 into 3 × 1 L LB containing 50 μg mL−1 carbenicillin. Cultures were incubated at 37 °C with shaking at 250 rpm until they reached an OD600 of 0.5. IPTG was then added (1 mM final concentration) and the cultures were incubated with shaking (250 rpm) at 25 °C for 16 h. Cells were harvested by centrifugation (6000 × g, 30 min, 4 °C) and resuspended in 5 mL lysis buffer (50 mM Tris–HCl, 100 mM NaCl pH 8.0) per gram of wet cell pellet. The cells were lysed by sonication on ice. Lysates were clarified by low-speed centrifugation (4000 × g, 30 min, 4 °C). The supernatant was then centrifuged at higher speed (136000 × g (SW28 rotor), 2 h, 4 °C) to collect membrane pellets. The supernatant was discarded and each pellet was washed once with 30 mL lysis buffer. The membrane pellets were then suspended in 50 mM Tris–HCl, 100 mM NaCl, 1% n-dodecyl-β-D-maltoside, pH 8.0 and incubated with gentle mixing at 4 °C for 1 h. The detergent-solubilised membranes were then centrifuged (136000 × g, 1 h, 4 °C) to remove insoluble debris and the supernatants were applied at a flow rate of 0.5 mL min−1 to an Ni-NTA agarose column (2 mL packed bed volume) pre-equilibrated with 50 mM Tris–HCl, 100 mM NaCl, 0.03% n-dodecyl-β-D-maltoside, pH 8.0. The column was washed overnight with 1 L of equilibration buffer and bound proteins were eluted in 50 mM Tris–HCl, 100 mM NaCl, 0.03% n-dodecyl-β-D-maltoside, 300 mM imidazole pH 8.0. The purified protein was concentrated by ultrafiltration (Millipore, molecular mass cutoff 10 kDa) and stored short-term at 4 °C. For certain downstream applications, MexG was further purified using a Superdex 75 gel filtration column.
MexG binding to the affinity probes
Affinity resin (25 μl) was dispensed into 1.5 mL microcentrifuge tubes. To equilibrate the resin, 1 mL of resin buffer (20 mM Tris–Cl, 200 mM NaCl, pH 7.4) containing 1% Triton X-100 was added and the tubes were mixed by inversion. The resin was pelleted by centrifugation (10000 × g, 2 min, 4 °C) and the supernatant was discarded. This equilibration process was repeated three times. Purified His6-MexG was then added to the resin and tubes were mixed by inversion at 4 °C for 1 h. The resin was collected (10000 × g, 2 min, 4 °C) and the supernatant was discarded. To wash away unbound proteins, 1 mL resin buffer containing 1% Triton X-100 was added and the tubes were mixed vigorously. The resin was collected (10000 × g, 2 min, 4 °C) and the supernatant was removed. This wash process was repeated six times. Bound proteins were eluted and analysed as described above for the PqsRLBD capture experiments.
Fluorescence analyses
Fluorescence spectra were recorded using an LS-50 Perkin Elmer spectrofluorimeter. Samples were excited at 295 nm in 50 mM Tris–HCl, 100 mM NaCl, 0.03% n-dodecyl-β-D-maltoside, pH 8.0. The excitation and emission slit widths were 2.5 nm and 5 nm respectively. The scan speed was 50 nm s−1 with a range from 300–500 nm. All experiments were performed at 25 °C. For quantitative analysis of PQS binding to MexG, the fluorescence signal (F0) of a known concentration of MexG was measured at 330 nm. PQS was subsequently titrated into the sample and the fluorescence (F) was measured after mixing. A plot of 1/(F0 − F) vs. 1/[PQS] was used to estimate (F0 − F) at saturating PQS concentrations (i.e., (F0 − F)max). Assuming a MexG:PQS stoichiometry of 1 (which, given the small size of PQS, seems reasonable), then the value of [MexG] × (F0 − F)/(F0 − F)max yields [PQS]bound. It follows that [PQS]free is [PQS]total – [PQS]bound. A plot of [PQS]bound/[PQS]freevs. [PQS]bound (i.e., a Scatchard plot) has a slope of −1/Kd. The data in Fig. 6C represent the results of 3 independent experiments.Construction of in-frame mexG deletion mutant
An 870 bp fragment corresponding to the 5′ flanking region of mexG was PCR-amplified from PAO1 genomic DNA using primers GAAT![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) CCAGTTGCGTTTCGTCGAACG and GGCACTGCATGGGTCGTTCCTTGTGCTG (HindIII and XbaI sites underlined). The fragment was directionally cloned into pUCP20 digested with HindIII and XbaI to generate pUCP20mexG5′. In parallel, a 742 bp fragment corresponding to the 3′ region of mexG was PCR-amplified using primers GGTAATGTTCTTCGCCCTCGAACACATC and CAGTGAGAAATTGCTTTTCAGGGTCCGC (XbaI and SacI sites underlined). This PCR product was cloned between the XbaI and SacI sites on pUCP20mexG5′ to generate pUCP20ΔmexG. The sequence between the HindIII and SacI sites was excised and then cloned into pEX18Tc, yielding pEX18TcΔmexG. The pEX18TcΔmexG was mobilized into PAO1 from E. coli β2163 by conjugation. Mutants were obtained by sacB-based allele exchange. Putative mutants were confirmed by PCR-amplifying the mexG region and sequencing.
CCAGTTGCGTTTCGTCGAACG and GGCACTGCATGGGTCGTTCCTTGTGCTG (HindIII and XbaI sites underlined). The fragment was directionally cloned into pUCP20 digested with HindIII and XbaI to generate pUCP20mexG5′. In parallel, a 742 bp fragment corresponding to the 3′ region of mexG was PCR-amplified using primers GGTAATGTTCTTCGCCCTCGAACACATC and CAGTGAGAAATTGCTTTTCAGGGTCCGC (XbaI and SacI sites underlined). This PCR product was cloned between the XbaI and SacI sites on pUCP20mexG5′ to generate pUCP20ΔmexG. The sequence between the HindIII and SacI sites was excised and then cloned into pEX18Tc, yielding pEX18TcΔmexG. The pEX18TcΔmexG was mobilized into PAO1 from E. coli β2163 by conjugation. Mutants were obtained by sacB-based allele exchange. Putative mutants were confirmed by PCR-amplifying the mexG region and sequencing.
Acknowledgements
Work in the MW lab is supported by the BBSRC (BB/M019411/1), MRC and EU (Marie Curie ETN). Work in the DRS lab is supported by EPSRC, ERC and the Wellcome Trust. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no [279337/DOS]. JTH was supported by Trinity College Cambridge and a studentship from the MRC. YRB is an EPSRC-funded PhD student.Notes and references
- (a) C. M. Waters and B. L. Bassler, Annu. Rev. Cell Dev. Biol., 2005, 21, 319 CrossRef CAS PubMed; (b) G. D. Geske, J. C. O'Neil and H. E. Blackwell, Chem. Soc. Rev., 2008, 37, 1432 RSC; (c) N. A. Whitehead, A. M. Barnard, H. Slater, N. J. Simpson and G. P. Salmond, FEMS Microbiol. Rev., 2001, 25, 365 CrossRef CAS PubMed.
- (a) W. R. D. J. Galloway, J. T. Hodgkinson, S. D. Bowden, M. Welch and D. R. Spring, Chem. Rev., 2011, 111, 28 CrossRef CAS PubMed; (b) J. S. Dickschat, Nat. Prod. Rep., 2010, 27, 343 RSC.
- (a) S. Atkinson and P. Williams, J. R. Soc., Interface, 2009, 6, 959 CrossRef CAS PubMed; (b) L. C. Antunes, R. B. Ferreria, M. M. Buckner and B. B. Finlay, Microbiology, 2010, 156, 2271 CrossRef CAS PubMed.
- (a) P. N. Jimenez, G. Koch, J. A. Thompson, K. B. Xavier, R. H. Cool and W. J. Quax, Microbiol. Mol. Biol. Rev., 2012, 76, 46 CrossRef CAS PubMed; (b) G. Laverty, S. P. Gorman and B. F. Gilmore, Pathogens, 2014, 3, 596 CrossRef CAS PubMed.
- M. J. Lintz, K. Oinuma, C. L. Wysoczynski, E. P. Greenberg and M. E. Churchill, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15763 CrossRef CAS PubMed.
- (a) G. Xiao, E. Déziel, J. He, F. Lépine, B. Lesic, M. H. Castonguay, S. Milot, A. P. Tampakaki, S. E. Stachel and L. G. Rahme, Mol. Microbiol., 2006, 62, 1689 CrossRef CAS PubMed; (b) H. Cao, G. Krishnan, B. Goumnerov, J. Tsongalis, R. Tompkins and L. G. Rahme, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14613 CrossRef CAS PubMed.
- (a) E. C. Pesci, J. B. Milbank, J. P. Pearson, S. McKnight, A. S. Kende, E. P. Greenberg and B. H. Iglewski, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11229 CrossRef CAS PubMed; (b) S. P. Diggle, K. Winzer, S. R. Chhabra, K. E. Worrall, M. Cámara and P. Williams, Mol. Microbiol., 2003, 50, 29 CrossRef CAS PubMed; (c) L. A. Gallagher, S. L. McKnight, M. S. Kuznetsova, E. C. Pesci and C. Manoil, J. Bacteriol., 2002, 184, 6472 CrossRef CAS PubMed.
- (a) S. Aendekerk, S. P. Diggle, Z. Song, N. Høiby, P. Cornelis, P. Williams and M. Cámara, Microbiology, 2005, 151, 1113 CrossRef CAS PubMed; (b) D. A. D'Argenio, M. W. Calfee, P. B. Rainey and E. C. Pesci, J. Bacteriol., 2002, 184, 6481 CrossRef; (c) S. P. Diggle, S. Matthijs, V. J. Wright, M. P. Fletcher, S. R. Chhabra, I. L. Lamont, X. Kong, R. C. Hider, P. Cornelis, M. Cámara and P. Williams, Chem. Biol., 2007, 14, 87 CrossRef CAS PubMed; (d) L. Mashburn-Warren, J. Howe, K. Brandenburg and M. Whiteley, J. Bacteriol., 2009, 191, 3411 CrossRef CAS PubMed.
- (a) L. Vial, F. Lépine, S. Milot, M. C. Groleau, V. Dekimpe, D. E. Woods and E. Déziel, J. Bacteriol., 2008, 190, 5339 CrossRef CAS PubMed; (b) S. P. Diggle, P. Lumjiaktase, F. Dipilato, K. Winzer, M. Kunakorn, D. A. Barrett, S. R. Chhabra, M. Cámara and P. Williams, Chem. Biol., 2006, 13, 701 CrossRef CAS PubMed; (c) J. F. Dubern and S. P. Diggle, Mol. BioSyst., 2008, 4, 882 RSC.
- J. Hodgkinson, S. D. Bowden, W. R. D. J. Galloway, D. R. Spring and M. Welch, J. Bacteriol., 2010, 192, 3833 CrossRef CAS PubMed.
- (a) S. Ziegler, V. Pries, C. Hedberg and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 2744 CrossRef CAS PubMed; (b) M. Radia, Curr. Opin. Chem. Biol., 2011, 15, 570 CrossRef PubMed; (c) U. Rix and G. Superti-Furga, Nat. Chem. Biol., 2009, 5, 616 CrossRef CAS PubMed; (d) L. Margarucci, M. C. Monti, B. Fontanella, R. Riccio and A. Casapullo, Mol. BioSyst., 2011, 7, 480 RSC.
- L. Eberl and B. Tümmler, Int. J. Med. Microbiol., 2004, 294, 123 CrossRef CAS PubMed.
- (a) F. J. Reen, M. J. Mooij, L. J. Holcombe, C. M. McSweeney, G. P. McGlacken, J. P. Morrissey and F. O'Gara, FEMS Microbiol. Ecol., 2011, 77, 413 CrossRef CAS PubMed; (b) T. Inaba, H. Oura, K. Moringana, M. Toyofuku and N. Nomura, Microbes Environ., 2015, 30, 189 CrossRef PubMed.
- (a) S. P. Diggle, P. Cornelis, P. Williams and M. Cámara, Int. J. Med. Microbiol., 2006, 296, 83–91 CrossRef CAS PubMed; (b) Y. Tashiro, Y. Yawata, M. Toyofuku, H. Uchiyama and N. Nomura, Microbes Environ., 2013, 28, 13 CrossRef PubMed.
- (a) D. S. Hooi, B. W. Bycroft, S. R. Chhabra, P. Williams and D. I. Pritchard, Infect. Immun., 2004, 72, 6463 CrossRef CAS PubMed; (b) M. E. Skindersoe, L. H. Zeuthen, S. Brix, L. N. Fink, J. Lazenby, C. Whittall, P. Williams, S. P. Diggle, H. Froekiaer, M. Cooley and M. Giskov, FEMS Immunol. Med. Microbiol., 2009, 55, 335 CrossRef CAS PubMed.
- (a) J. T. Hodgkinson, W. R. D. J. Galloway, S. Saraf, I. R. Baxendale, S. V. Ley, M. Ladlow, M. Welch and D. R. Spring, Org. Biomol. Chem., 2011, 9, 57 RSC; (b) J. T. Hodgkinson, W. R. D. J. Galloway, M. Welch and D. R. Spring, Nat. Protoc., 2012, 7, 1184 CrossRef CAS PubMed.
- M. Welch, J. T. Hodgkinson, J. Gross, D. R. Spring and T. Sams, Biochemistry, 2013, 52, 4433 CrossRef CAS PubMed.
- (a) A. Zaborin, K. Romanowski, S. Gerdes, C. Holbrook, F. Lepine, J. Long, V. Poroyko, S. P. Diggle, A. Wilke, K. Righetti, I. Morozova, T. Babrowski, D. C. Liu, O. Zaborina and J. C. Alverdy, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6327 CrossRef CAS PubMed; (b) F. Bredenbruch, R. Geffers, M. Nimtz, J. Buer and S. Häussler, Environ. Microbiol., 2006, 8, 1318 CrossRef CAS PubMed.
- (a) T. Guina, M. Wu, S. I. Miller, S. O. Purvine, E. C. Yi, J. Eng, D. R. Goodlett, R. Aebersold, K. R. Ernst and K. A. Lee, J. Am. Soc. Mass Spectrom., 2003, 14, 742 CrossRef CAS PubMed; (b) T. Guina, S. O. Purvine, E. C. Yi, J. Eng, D. R. Goodlett, R. Aebersold and S. I. Miller, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2771 CrossRef CAS PubMed; (c) Y. Tashiro, S. Ichikawa, T. Nakajima-Kambe, H. Uchiyama and N. Nomura, Microbes Environ., 2010, 25, 120 CrossRef PubMed.
- S. Aendekerk, B. Ghysels, P. Cornelis and C. Baysse, Microbiology, 2002, 148, 2371 CrossRef CAS PubMed.
- E. Déziel, S. Gopalan, A. P. Tampakaki, F. Lépine, K. E. Padfield, M. Saucier, G. Xiao and L. G. Rahme, Mol. Microbiol., 2005, 55, 998 CrossRef PubMed.
- A. Woschek, M. Mahout, K. Mereiter and F. Hammerschmidt, Synthesis, 2007, 10, 1517 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental protocols and characterisation data. See DOI: 10.1039/c5sc04197j |
| This journal is © The Royal Society of Chemistry 2016 |