
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitchable π-electronic network of bis(α-oligothienyl)-substituted hexaphyrins between helical versus rectangular circuit†
Juwon
Oh
a,
Hirotaka
Mori
b,
Young Mo
Sung
a,
Woojae
Kim
a,
Atsuhiro
Osuka
*b and
Dongho
Kim
*a
aSpectroscopy Laboratory for Functional π-Electronic Systems, Department of Chemistry, Yonsei University, 50 Yonsei-ro, Seoul 120-749, Korea. E-mail: dongho@yonsei.ac.kr
bDepartment of Chemistry, Graduated School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: osuka@kuchem.kyoto-u.ac.jp
First published on 8th December 2015
Abstract
The switching phenomena of conformation with π-electronic network through deprotonation–protonation processes were investigated by employing a series of 5,20-bis(α-oligothienyl) substituted hexaphyrins(1.1.1.1.1.1). They showed significant changes in the absorption and emission spectra with deprotonation, and returned to the initial state with protonation. Through NMR measurements and single crystal X-ray diffraction analysis, we found that the 5,20-bis(α-oligothienyl) substituted hexaphyrins, which possess acyclic, helical electronic networks involving oligothienyl chains in dumbbell conformations (type-I) in a neutral form, underwent effective deprotonation upon treatment with tetrabutylammonium fluoride (TBAF) to generate the corresponding dianions, which display cyclic electronic networks with enhanced aromaticity in rectangular conformations (type-II). Our quantum calculation results provide an unambiguous description for the switchable conformation and π-conjugation, which revealed that a deprotonation-induced enhanced aromatic conjugation pathway is involved in the switchable π-electronic network.
Introduction
π-Conjugation in molecular systems, one of the most interesting subjects in chemical, physical and materials science, not only determines molecular properties but also has promising potential applications in nanotechnology, such as for optoelectronic and photovoltaic devices. Since the π-conjugated pathway is largely governed by molecular structures, there are numerous approaches towards π-conjugated molecular systems with well-defined structures in order to understand the structure–property relationship, which ultimately leads to the control of molecular properties.1Among various π-conjugated molecular systems, expanded porphyrins have recently emerged as promising functional dyes with potential applications as anion sensors, near-infrared (NIR) dyes and nonlinear optical (NLO) materials, because they possess fully conjugated cyclic electronic networks as evinced by strong and red-shifted absorption spectra.2 Versatile electronic states have been realized for expanded porphyrins, which include Hückel aromatic and antiaromatic species, Möbius aromatic3 and antiaromatic species,4 and stable radicals.5 As another attribute, expanded porphyrins become increasingly conformationally flexible with an increase in ring size. Since pyrrole and meso-methine carbon constituents of expanded porphyrins afford molecular flexibility and a highly conjugated electronic system, an intricate interplay of the ring size, connectivity, constitutional heterocycle, intramolecular steric congestion, and intramolecular hydrogen bonding determines the conformations of expanded porphyrins. Importantly the electronic properties of expanded porphyrins are heavily dependent upon their conformations, and thus the control of conformations of expanded porphyrins is vital in the realization of expanded porphyrins with the desired properties. As an external stimulus, protonation has often been employed to control the conformations of expanded porphyrins by disrupting intramolecular hydrogen bonding interactions.6 In contrast, deprotonation has rarely been used for the same purpose.7
Among the series of meso-pentafluorophenyl-substituted expanded porphyrins, [26]hexaphyrin 1 is a benchmark molecule in light of the strong aromaticity and roughly planar rectangular conformation (type-II, Chart 1).7 It has been shown that in [26]hexaphyrins dumbbell conformations (type-I) are intrinsically more stable as compared with type-II conformations owing to their more effective intramolecular hydrogen bonding interactions, where the type-II conformation of 1 is formed due to the steric congestion between the two meso-pentafluorophenyl substituents at the 5- and 20-positions.8,9 This has been supported by the type-I conformations of 2.5a
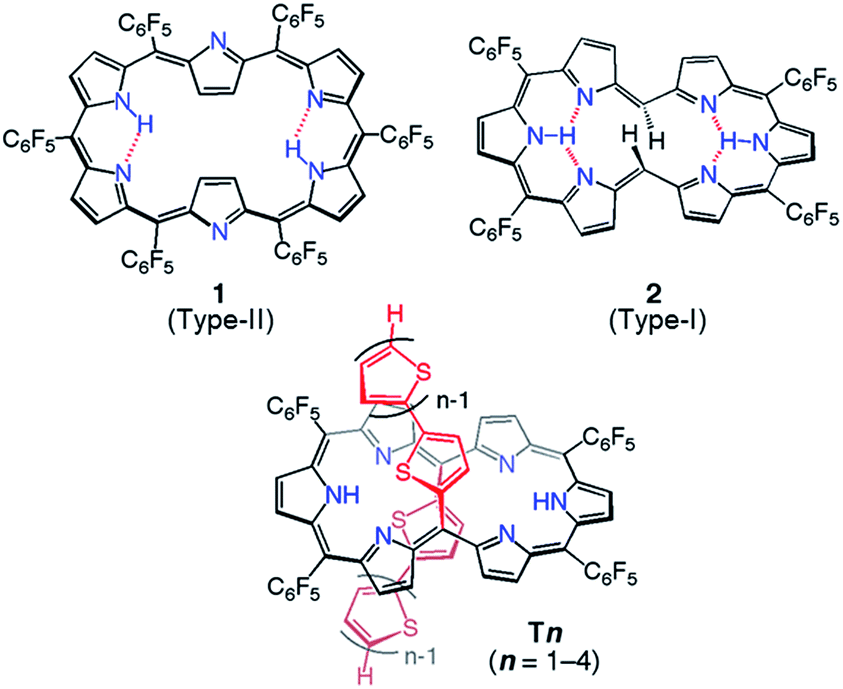 | ||
| Chart 1 Molecular structures of type-I and type-II [26]hexaphryins and the series of 5,20-bis(α-oligothienyl) substituted [26]hexaphyrins, T1–T4. | ||
Since such structural flexibility and effective π-conjugation of expanded porphyrins are desirable for the modification of their electronic structures, they can be regarded as ideal candidates for multiple π-electronic networks, such as dual electronic conjugation, three dimensional aromaticity or electronic network switching. For this purpose, there have been persistent efforts for an elaborate modulation of electronic structures of expanded porphyrins, such as core-modification, appending internal bridges and changing the meso-substituent.3,5 In the same line, we recently reported a set of 5,20-bis(α-oligothienyl) substituted hexaphyrins(1.1.1.1.1.1) (Chart 1, T1–T4) with type-I conformations due to the small size of oligo(2-thienyl) substituent.9,10 In these structures, the oligothienyl chains are located at the hinge positions of the hexaphyrin (5- and 20-positions) with small dihedral angles with the hexaphyrin core, effectively perturbing the electronic structure of the [26]hexaphyrin core. This perturbation becomes increasingly larger upon elongation of the oligothienyl chain, and has been ascribed to the significant contribution of an acyclic, helical π-conjugated network.
Here, because the core molecular systems of T1–T4 are composed of flexible hexaphyrins, the possibility of their forming multiple π-conjugated networks still remains an intriguing question. The hexaphyrins characteristically possess a highly conjugated flexible structure, where an intramolecular hydrogen-bonding network, due to the regularly arranged six pyrrole rings with meso-methine carbons, significantly affects the molecular geometry.11 As a result, the core π-conjugated system of hexaphyrins is readily modified by various approaches such as protonation, deprotonation, metallation and redox processes,7,12 which enables hexaphyrins to adopt various conformations such as type-I, type-II and even doubly-twisted conformations with Hückel and Möbius aromatic and antiaromatic electronic states. In this paper, we report the deprotonation of T1–T4 with tetrabutylammonium fluoride (TBAF) as an effective means to switch the type-I conformations of T1–T4 to the corresponding type-II conformations with a concurrent change in the electronic system from an acyclic, helical network to a cyclic, aromatic network.
Results and discussion
Steady-state absorption and emission
[26]Hexaphyrin 1 shows an absorption spectrum with a sharp and strong Soret-like band at 566 and Q-bands at 711, 768, 881, and 1019 nm in CH2Cl2, and undergoes smooth deprotonation upon treatment with TBAF to afford 12−, which takes a similar rectangular shape (type II; Scheme 1a) and displays an exceedingly enhanced and sharp Soret-like band at 601 nm.7a,13 Along with these spectral changes, 12− shows a longer S1-state lifetime and intensified fluorescence, all of which indicate an enhancement of the aromaticity of the hexaphyrin network. The absorption spectra of T1–T4 in CH2Cl2 are radically different from that of 1 due to the perturbed electronic structures by the oligothienyl chains, showing ill-defined broad bands as shown in Fig. 1.10 Upon addition of TBAF, the absorption spectra of T1–T4 exhibited drastic changes, becoming quite similar to that of 12−. The Soret-like bands are observed at 616, 643, 653, and 658 nm and the Q-bands are observed at 1014, 1023, 1030, and 1034 nm for T12−–T42−, respectively. Compared to the weak fluorescence of T1–T4, dianions T12−–T42− emit significantly intensified fluorescence at 1030, 1054, 1054, and 1056 nm with distinct vibronic structures (Fig. S1†). These results strongly suggest that the formed dianions, T12−–T42−, take rectangular aromatic conformations. Furthermore, the observed small Stokes shifts of T12−–T42− suggest certain structural rigidities of their S1-states. As indicated by the absorption and fluorescence spectra (Fig. S2†), the protonation of T12−–T42− with trifluoroacetic acid (TFA) gave rise to the restoration of T1–T4, indicating a nice reversibility of the deprotonation and protonation sequence.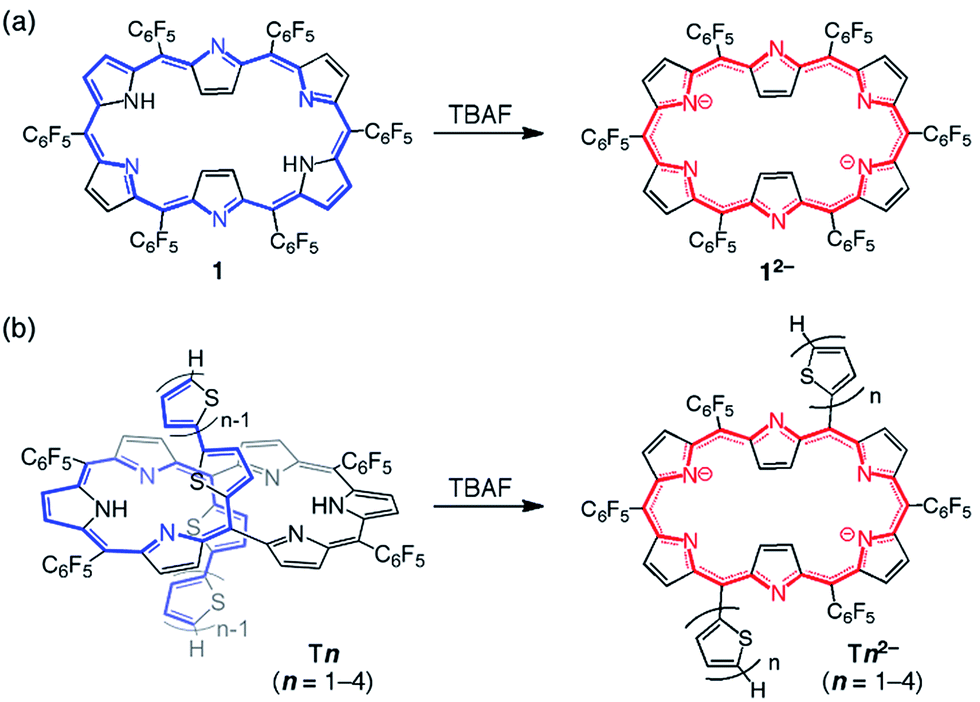 | ||
| Scheme 1 Deprotonation of [26]hexaphyrins (a): 1 and (b): Tn. | ||
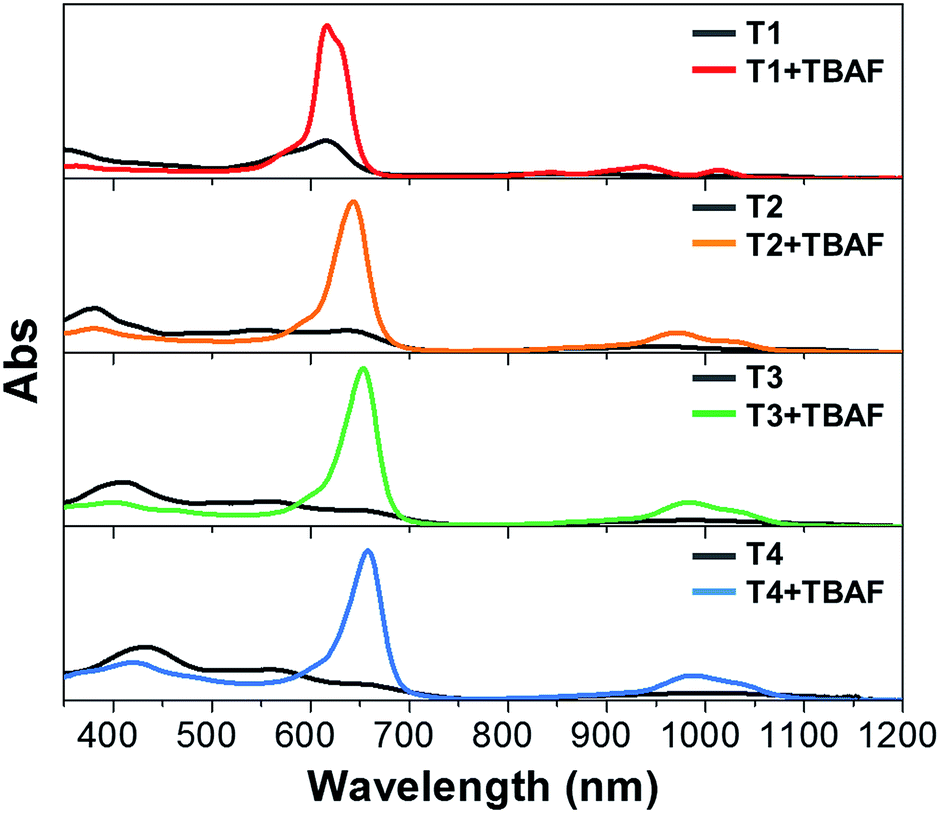 | ||
| Fig. 1 Steady-state absorption spectra of T1–T4 in CH2Cl2 with and without TBAF. | ||
NMR spectra and X-ray crystal structure
The structures of dianions T12−–T42− have been investigated by 1H NMR spectroscopy (Fig. 2). The 1H NMR spectra of T2–T4 are very broad at room temperature owing to their fast conformational dynamics and a low temperature (213 K) is necessary to obtain their clear 1H NMR spectra (Fig. S3–S6†).10 Notably, the signals of the two inner NH protons of T2–T4 were observed in the range −8.4 to −8.2 and −5.5 to −5.3 ppm. These data indicate that the helical π-conjugated network of the oligothienyl chains results in an asymmetric π-electronic environment on the hexaphyrin cores. In contrast, the 1H NMR spectra of T12−–T42− in THF-d8 are sharp even at 25 °C, displaying a set of sharp signals, showing four doublets due to the outer β protons in the range of 9.4–8.6 ppm, signals due to the oligothienyl substituents in the range of 8.0–7.0 ppm, and a characteristic two doublets due to the inner β protons at around −3.1 ppm (Fig. S7–S10†). Only a type-II rectangular conformation bearing the oligothienyl chains on the long side (Scheme 1b) in solution is consistent with these spectral data, clearly indicating that the deprotonation induces a conformational change from the type-I to the type-II conformation with an inversion of the oligothienyl chains and pyrrole rings. Moreover, the large Δδ values of 12.5–12.1 ppm signify an intense diatropic ring current,14 demonstrating that T12−–T42− possess a cyclic aromatic π-conjugated network.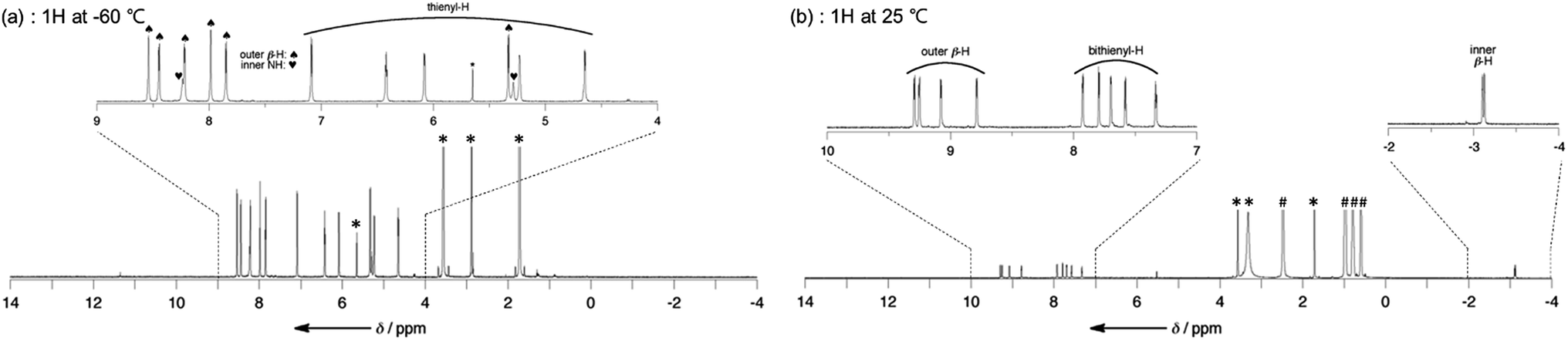 | ||
| Fig. 2 1H NMR spectra of T2 in THF-d8 in the (a) absence of TBAF and (b) presence of an excess amount of TBAF. * and # represent peaks due to residual solvent and the tetrabutylammonium cation, respectively. | ||
We attempted to get crystals of the dianions. Single crystals of salt T22−(TBA+)2 were obtained by vapor diffusion of diethyl ether into a solution of T2 in CH2Cl2 in the presence of an excess amount of TBAF. In the crystal, the structure of T22− was not rectangular but was a triangular shape (Fig. S11†). Similar triangular conformations were observed in diprotonated [28]hexaphyrins, where coulombic repulsion between the two positive charges plays a key role in producing a triangular shape.15 On the other hand, in contrast to a compactly packed solid-state condition, negative charges of T22− can be shielded by the surrounding solvent and countercations in the solution-state. Moreover, T22− has a different charge position (negative charges on the pyrrolic nitrogens) from that of hexaphyrin cations. These features can reduce coulombic repulsion in T22−, leading to the rectangular shape of typical [26]hexaphyrins. Collectively, the triangular structure of T22− in the solid-state may be arising from the crystal packing environment and delicate coulombic interactions. Such a discrepancy between structures in solution and in the solid-state has been observed for several expanded porphyrins.3c,12
Importantly, although T22− showed a triangular conformation in the solid state, this crystal structure strongly suggests that the deprotonation triggers a conformational change of the hexaphyrin cores of T1–T4, involving the inversion of the pyrrole rings and oligothienyl chains, with a concurrent change in the π-electronic networks from an acyclic, helical conjugation to a rectangular aromatic conjugation; while the oligothienyl chains oriented inward in T2 favor the extension of the π-conjugation between one tripyrrodimethene and the oligothienyl chains, the planar structure and outward-pointing oligothienyl chains of T22− are advantageous for the aromatic cyclic [26]π-conjugation pathway.
Excited-state dynamics
The excited-state dynamics of T12−–T42− were studied by femtosecond transient absorption (fs-TA) measurements. Our previous study revealed that the S1-state lifetime of T1 was 35.4 ps, being considerably shorter than those of strongly aromatic [26]hexaphyrins 1 (98 ps) and 2 (138 ps), and those of T2–T4 became shortened with an increase of the oligothienyl chains (15.2, 10, and 8.5 ps, respectively), indicating an increasing electronic perturbation of the meso-oligothienyl chains and hence an increasing importance of the helical conjugation.10 In contrast, the TA spectra of T12−–T42− displayed strong ground state bleaching (GSB) signals at around 600–650 nm and weak excited state absorption (ESA) signals at both sides of the GSB signals (Fig. 3), which are quite similar to those of 1 and 12−. Moreover, the TA spectra of T12−–T42− showed beautiful single exponential decays with dramatically elongated S1-state lifetimes (820, 420, 320, and 310 ps for T12−–T42−, respectively) along with decays of longer lifetimes due to their triplet state (Table S1†). The observed exponential decays of T12−–T42− were even much longer than that of 1, being comparable to 12−. These excited-state dynamics of T12−–T42− are concluded to indicate their enhanced aromaticities.7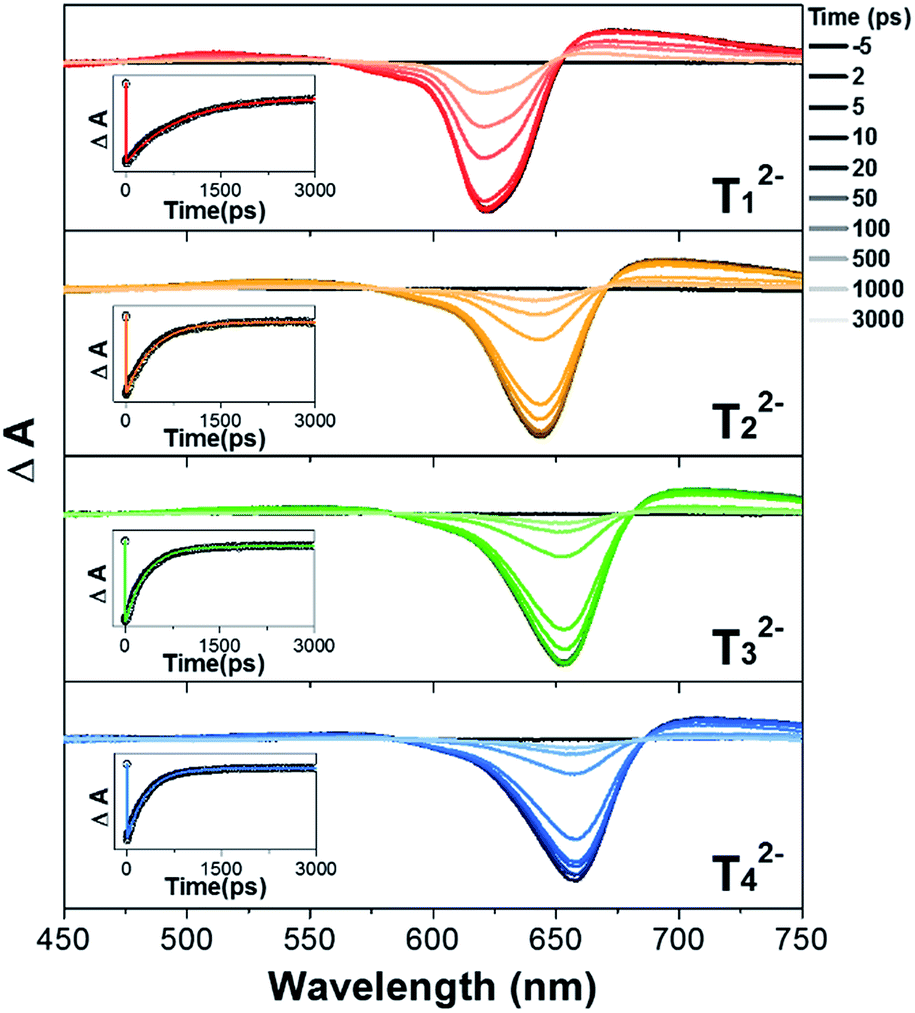 | ||
| Fig. 3 Femtosecond time-resolved transient absorption spectra and decay profiles (inset) of T12−–T42− in CH2Cl2. | ||
Theoretical calculations
To obtain more insights into the switchable conformational and conjugated features of T1–T4, density functional theory (DFT) calculations have been performed by use of the Gaussian 09 program at the CAM-B3LYP/6-31G** level. Firstly, the relative energies of the dumbbell, type-I, versus rectangular, type-II, conformations of T1–T4 have been calculated to show that, except for T1, the former is more stable than the latter by ca. 11–14 kcal mol−1 (Table S2†). The optimized structures of T2–T4 exhibited an asymmetric arrangement of the oligothienyl chains, where one tripyrrodimethene of the hexaphyrin core was helically aligned with the oligothienyl chains (Fig. S12†). The nucleus independent chemical shift (NICS)16 values of T1–T4 at the center of the internal left and right pockets have been calculated to be −12.9 and −12.9 ppm for T1, −1.34 and −3.61 ppm for T2, −1.46 and −3.87 ppm for T3, and −1.53 and −2.14 ppm for T4, respectively (Fig. S13†), being consistent with their weakly aromatic characters as observed in their 1H NMR spectra. Here, along with the 1H NMR spectra of T2–T4, the asymmetric NICS values, −1.53 to −1.34 and −3.87 to −3.14 ppm at the left and right pockets, respectively, represent the asymmetric π-electronic environment in their hexaphyrin cores, resulting from the major helical π-conjugated network. Furthermore, the harmonic oscillator model of aromaticity (HOMA)17 values are also well matched with their weak aromaticity arising from the 26π hexaphyrin circuits (0.819 for T1, 0.585 for T2, 0.586 for T3, and 0.585 for T4; Fig. S14†). In a similar fashion, the molecular orbital (MO) structures of T2–T4 displayed the electron densities distributed on the acyclic helical pathways in HOMO and HOMO−1 (Fig. 4 and S15†). These results provide support for a decreasing aromaticity with elongation of the oligothienyl chains, and thus the significant contribution of the helix-like conjugations.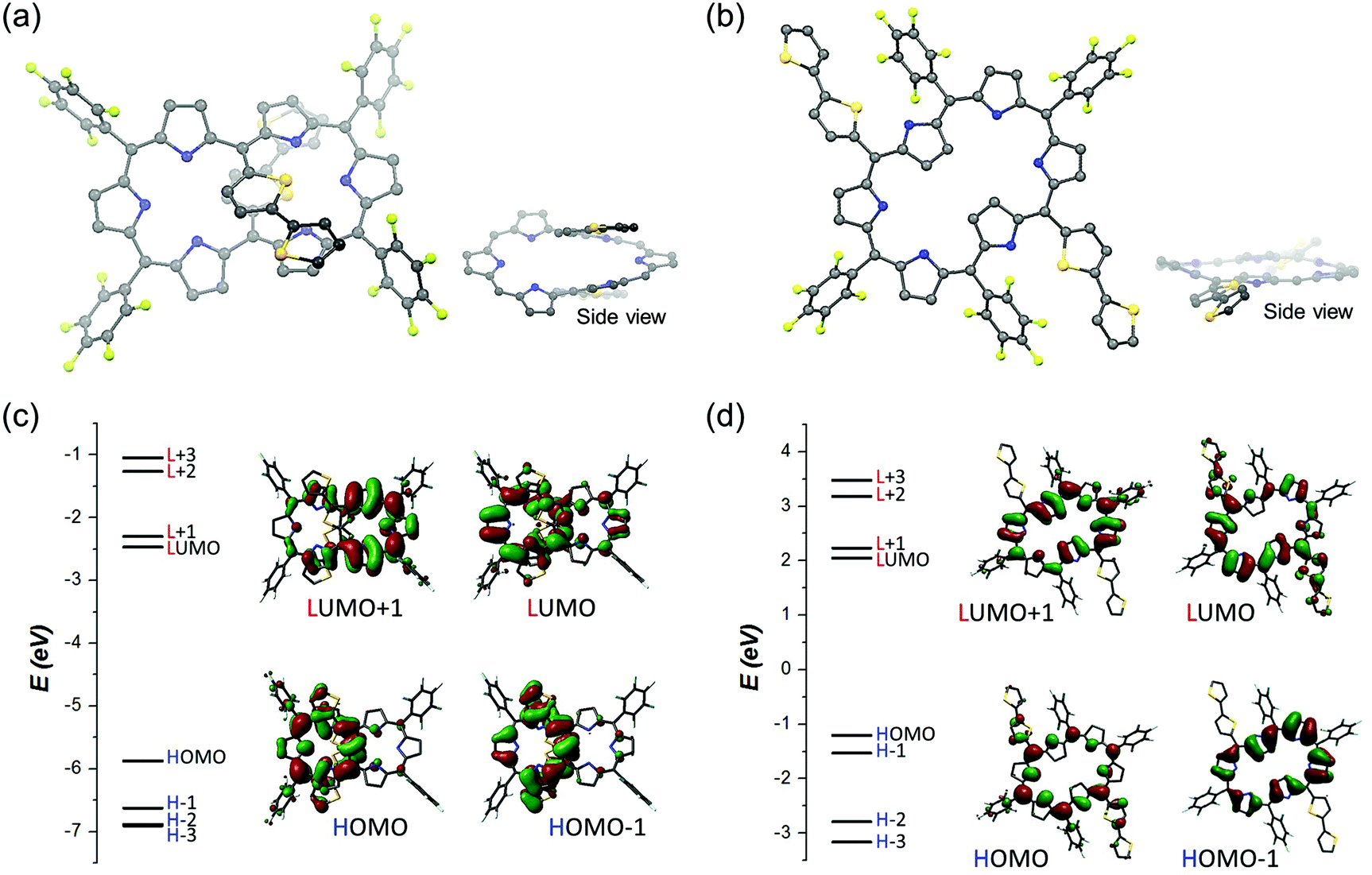 | ||
| Fig. 4 Optimized structures of (a) T2 and (b) T22− from the top and side view and energy level diagrams with molecular orbitals of (c) T2 and (d) T22−. meso-Pentafluorophenyl groups are omitted for clarity in the side view figures. | ||
Next, the geometrical optimizations of the dianions of T12−–T42− have been conducted on the basis of the rectangular conformations observed in their 1H NMR spectra (Fig. S16†). In contrast to their neutral forms, the type-II conformations have been calculated to be more stable than the type-I conformation by 17–29 kcal mol−1 for the dianions T12−–T42−, in line with the observed experimental results (Table S3†). The type-II conformations of T12−–T42− with the outward-pointing oligothienyl chains exhibited their planar structures for effective π-conjugation on the hexaphyrin cores. The NICS values at the center of the macrocycle of T12−–T42− have been calculated to be ca. −9.2 ppm (Fig. S17†), representing the aromatic nature of T12−–T42− due to the major cyclic π-conjugation pathways. Moreover, the HOMA values depict the aromatic π-conjugation pathways (Fig. S18†). Furthermore, the frontier MOs (FMOs) showed the degenerated HOMO−1/HOMO and LUMO/LUMO+1 states, following Gouterman's four-orbital model (Fig. S19†).18 The TD-DFT calculation indicated that their lowest four transitions resulted from the configurational interactions between four FMOs, which is characteristic of aromatic porphyrinoids, having intense B-like transitions with weak Q-like transitions.18 Collectively, these computational results reproduce the experimental results well and suggest that the 26π aromaticity of T1–T4 was effectively enhanced by deprotonation accompanied by large structural changes.
Effect of deprotonation–protonation on the switchable conformation and π-electronic network
These switchable conformations with the π-electronic network of T1–T4 by deprotonation–protonation are attributable to two features: deprotonation-induced (1) conformational change and (2) alternative cyclic conjugation pathway. In hexaphyrins, the regularly aligned pyrrole rings provide intramolecular hydrogen-bonding interactions, which has a significant effect on their conformations.19 With small meso-substituents, such as thienylene and pyrrylene, the effective intramolecular hydrogen-bonding seizes the pyrrole subunits, leading to the type-I conformation. In this geometry, the small meso-aryl substituents can appreciably participate in the π-electronic networks with small dihedral angles.9,20 However, the larger meso-substituents, such as phenylene derivatives, hinder the intramolecular hydrogen-bonding due to steric congestion, resulting in the compromised type-II conformation. Here, since T1–T4 contain oligothienyl derivatives at the meso-positions, their effective hydrogen-bonding enforces the hexaphyrin cores to adopt a type-I conformation. In this geometry, inward-pointing 5,20-oligothienyl chains induced the small dihedral angle between thiophene and pyrrole in the range 15–20° and the large dihedral angle between pyrroles in the range 40–45° (Fig. S12†). This structural feature enables an effective conjugative interaction between the tripyrrodimethane and oligothienyl chains. As a result, T1–T4 possess helically π-conjugated systems with elongation of the oligothienyl chains. Here, the removal of the inner NH protons by deprotonation effectively cuts off the intramolecular hydrogen-bonding interactions of T1–T4, which gives rise to the conformational change from the type-I to type-II conformation in the solution-state.20 Such a conformational change involves the inversion of the pyrrole rings and oligothienyl chains, which entirely alters the geometry of hexaphyrin core; while the dihedral angles between thiophene and pyrrole rings are estimated to be 51–55°, those between the pyrrole rings are significantly reduced to ∼10°. Such geometry leads to π-electron delocalization on the planar hexaphyrin core. Consequently, the breaking of intramolecular hydrogen-bonding interactions by deprotonation leads to the type-II conformation with aromatic π-conjugation and restoring of the hydrogen-bonding interactions by protonation gives rise to a return to the type-I conformation with a major helical π-conjugation in T1–T4 (Scheme 2).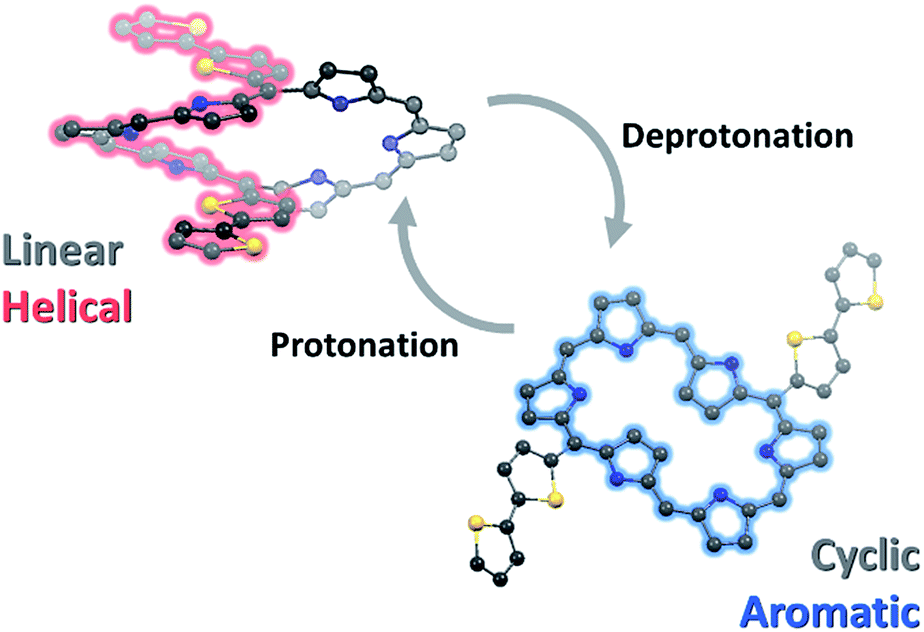 | ||
| Scheme 2 Switchable conformation and π-electronic network of Tn by deprotonation–protonation. | ||
In addition to the conformational change, it is expected that the deprotonation process induces an alternative cyclic conjugation pathway, a dianionic hexa-aza[24]annulene pathway, for T12−–T42− (Scheme 1b), resulting in an enhancement of the aromaticity and a distinct switchable π-conjugated network. In our previous study, while 1 showed π-conjugation on the tetra-aza[26]annulene pathway, the deprotonation process to 12− induced an alternative hexa-aza[24] π-conjugation pathway containing all six nitrogen atoms, leading to a more effective 26π-aromaticity (Scheme 1a).7a In accordance with these previous results, the enhanced aromatic π-conjugation on the dianionic hexa-aza[24]annulene pathway is considered in T12−–T42−. The NICS values at the center of the pyrrole rings showed contrasting features based on the hexa-aza[24]annulene circuit; in contrast to the small NICS values outside the circuit (at the D, F, G, and I positions), those of −10.6 to −12.5 ppm inside the circuit (at the E, and H positions) reflect the alternative [24]annulene π-conjugation pathway. Furthermore, the anisotropy of the induced current density (ACID) plots of T12−–T42− shows that their electron density is distributed only along the 24-membered circuit (Fig. S20†), which obviously depicts the hexa-aza[24] π-conjugation.21 The HOMA values were calculated on the two kinds of the electronic conjugation pathways; the dianionic hexa-aza[24]annulene circuit and the tetra-aza[26]annulene pathway (Fig. S18†). The HOMA values of the dianionic hexa-aza[24]annulene pathway of T12−–T42− were much larger (ca. 0.82) than those of the latter (ca. 0.61). A comparative analysis of the absorption spectra of protonated and deprotonated species (Fig. S21†) clearly demonstrates the effect of deprotonation-induced enhanced aromaticity on the switchable features, where protonated T1–T4 showed spectral changes but which wer not distinct as those of the deprotonated ones. These results strongly suggest that the deprotonation intensifies the aromatic nature of T12−–T42− and induces the conformational changes. As a result, T1–T4 show a distinct switchable π-electronic network between the linear helical and cyclic aromatic pathways.
During the course of our continuing studies on the relationship between the structural factors and aromaticity of expanded porphyrins, protonation of the interior imine nitrogens has been found to effectively cut off the intramolecular hydrogen-bonding interactions and smoothly release the structural restraints of expanded porphyrins, leading to a favorable conformation to realize more stable electronic conjugations. Although the deprotonation of expanded porphyrins has not been well studied, the present results imply that the deprotonation strategy is promising in the effective realization of novel switchable π-conjugated networks and enhancing aromaticity.
Conclusions
The switchable conformation and π-electronic network in T1–T4 by deprotonation–protonation were investigated. The breaking (or recovery) of intramolecular hydrogen-bonding interactions of hexaphyrin cores by deprotonation (or protonation) gave rise to the conformational changes in T1–T4, which effectively controlled the π-electronic network between the linear helical and cyclic aromatic π-conjugations. Our finding implies that deprotonation is another effective approach to switch π-conjugated networks and enhance the aromaticity in addition to protonation.The control of the molecular structure and π-conjugation pathway will provide useful information on the potential for designing supramolecular multiply π-conjugated systems and the development of molecular electronics. Moreover, the elaborate modulation of π-conjugation pathways suggests that the highly conjugated and flexible molecular systems, such as expanded porphyrins, are the ideal platform for controlling the π-delocalized electronic nature.
Acknowledgements
This research at Yonsei University supported by the Global Research Laboratory Program (2013K1A1A2A02050183) funded by the Ministry of Science, ICT & Future, Korea. The work at Kyoto was supported by JSPS KAKENHI Grant Numbers (25220802 and 25620031). H. M. acknowledges a JSPS Fellowship for Young Scientists. The quantum calculations were performed using the supercomputing resources of the Korea Institute of Science and Technology Information (KISTI).Notes and references
- (a) S. Shaik, A. Shurki, D. Danovich and P. C. Hiberty, Chem. Rev., 2001, 101, 1501 CrossRef CAS; (b) T. M. Krygowshi, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383 CrossRef PubMed; (c) Y. M. Sung, M.-C. Yoon, J. M. Lim, H. Rath, K. Naoda, A. Osuka and D. Kim, Nat. Chem., 2015, 7, 418 CrossRef CAS PubMed.
- (a) T. K. Chandrashekar and S. Venkatraman, Acc. Chem. Res., 2003, 36, 676 CrossRef CAS PubMed; (b) J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS PubMed; (c) M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288 CrossRef PubMed; (d) S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342 CrossRef CAS PubMed.
- (a) M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz and L. Szterenberg, Angew. Chem., Int. Ed., 2007, 46, 7869 CrossRef PubMed; (b) Y. Tanaka, S. Saito, S. Mori, N. Aratani, H. Shinokubo, N. Shibata, Y. Higuchi, Z. S. Yoon, K. S. Kim, S. B. Noh, J. K. Park, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 681 CrossRef CAS PubMed; (c) J. Sankar, S. Mori, S. Saito, H. Rath, M. Suzuki, Y. Inokuma, H. Shinokubo, K. S. Kim, Z. S. Yoon, J.-Y. Shin, J. M. Lim, Y. Matsuzaki, O. Matsushita, A. Muranaka, N. Kobayashi, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 13568 CrossRef CAS PubMed; (d) Z. S. Yoon, A. Osuka and D. Kim, Nat. Chem., 2009, 1, 113 CrossRef CAS PubMed.
- (a) E. Pacholska-Dudziak, J. Skonieczny, M. Pawlicki, L. Szterenberg, Z. Ciunik and L. Latos-Grażyński, J. Am. Chem. Soc., 2008, 130, 6182 CrossRef CAS PubMed; (b) T. Higashino, J. M. Lim, Y. Miura, S. Saito, J.-Y. Shin, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 4950 CrossRef CAS PubMed; (c) T. Higashino, B. S. Lee, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2012, 51, 13105 CrossRef CAS PubMed.
- (a) T. Koide, G. Kashiwazaki, M. Suzuki, K. Furukawa, M.-C. Yoon, S. Cho, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 9661 CrossRef CAS PubMed; (b) H. Rath, S. Tokuji, N. Aratani, K. Furukawa, J. M. Lim, D. Kim, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 1489 CrossRef CAS PubMed; (c) T. Koide, K. Furukawa, H. Shinokubo, J.-Y. Shin, K. S. Kim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2010, 132, 7246 CrossRef CAS PubMed; (d) T. Y. Gopalakrishna, J. S. Reddy and V. G. Anand, Angew. Chem., Int. Ed., 2014, 53, 10984 CrossRef CAS PubMed; (e) Y. Tanaka, T. Yoneda, K. Furukawa, T. Koide, H. Mori, T. Tanaka, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10908 CrossRef CAS PubMed; (f) Y. Hisamune, K. Nishimura, K. Isakari, M. Ishida, S. Mori, S. Karasawa, T. Kato, S. Lee, D. Kim and H. Furuta, Angew. Chem., Int. Ed., 2015, 54, 7323 CrossRef CAS PubMed.
- (a) S. Saito, J.-Y. Shin, J. M. Lim, K. S. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 9657 CrossRef CAS PubMed; (b) M. Stępień, B. Szyszko and L. Latos-Grażyński, J. Am. Chem. Soc., 2010, 132, 3140 CrossRef PubMed; (c) J. M. Lim, J.-Y. Shin, Y. Tanaka, S. Saito, A. Osuka and D. Kim, J. Am. Chem. Soc., 2010, 132, 3105 CrossRef CAS PubMed; (d) T. Koide, K. Youfu, S. Saito and A. Osuka, Chem. Commun., 2009, 6047 RSC; (e) S. Ishida, T. Higashino, S. Mori, H. Mori, N. Aratani, T. Tanaka, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 3427 CrossRef CAS PubMed; (f) Z. Zhang, W.-Y. Cha, N. J. William, E. L. Rush, M. Ishida, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 7591 CrossRef CAS PubMed; (g) T. Soya, W. Kim, D. Kim and A. Osuka, Chem.–Eur. J., 2015, 21, 8341 CrossRef CAS PubMed.
- (a) W.-Y. Cha, J. M. Lim, M.-C. Yoon, Y. M. Sung, B. S. Lee, S. Katsumata, M. Suzuki, H. Mori, Y. Ikawa, H. Furuta, A. Osuka and D. Kim, Chem.–Eur. J., 2012, 18, 15838 CrossRef CAS PubMed; (b) W.-Y. Cha, T. Yoneda, S. Lee, J. M. Lim, A. Osuka and D. Kim, Chem. Commun., 2014, 50, 548 RSC.
- (a) M. G. P. M. S. Neves, R. M. Martins, A. C. Tomé, A. J. D. Silvestre, A. M. S. Silva, V. Félix, M. G. B. Drew and J. A. S. Cavaleiro, Chem. Commun., 1999, 385 RSC; (b) J.-Y. Shin, H. Furuta, K. Yoza, S. Igarashi and A. Osuka, J. Am. Chem. Soc., 2001, 123, 7190 CrossRef CAS PubMed.
- M. Suzuki and A. Osuka, Chem.–Eur. J., 2007, 13, 196 CrossRef CAS PubMed.
- H. Mori, M. Suzuki, W. Kim, J. M. Lim, D. Kim and A. Osuka, Chem. Sci., 2015, 6, 1696 RSC.
- A. Osuka and S. Saito, Chem. Commun., 2011, 47, 4330 RSC.
- (a) H. Rath, S. Tokuji, N. Aratani, K. Furukawa, J. M. Lim, D. Kim, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 1489 CrossRef CAS PubMed; (b) M.-C. Yoon, P. Kim, H. Yoo, T. Koide, S. Tokuji, S. Saito, A. Osuka and D. Kim, J. Phys. Chem. B, 2011, 115, 14928 CrossRef CAS PubMed; (c) M.-C. Yoon, J.-Y. Shin, J. M. Lim, S. Saito, T. Yoneda, A. Osuka and D. Kim, Chem.–Eur. J., 2011, 17, 6707 CrossRef CAS PubMed; (d) T. Higashino and A. Osuka, Chem. Sci., 2013, 4, 1087 RSC.
- M. Suzuki and A. Osuka, Chem. Commun., 2005, 3685 RSC.
- J. K. Park, Z. S. Yoon, K. S. Kim, S. Mori, J.-Y. Shin, A. Osuka and D. Kim, J. Am. Chem. Soc., 2008, 130, 824 CrossRef PubMed.
- S. Ishida, T. Higashino, S. Mori, H. Mori, N. Aratani, T. Tanaka, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., 2014, 126, 3495 CrossRef.
- T. M. Krygowski and K. M. Cryanski, Chem. Rev., 2001, 101, 1385 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- (a) M.-C. Yoon, S. Cho, M. Suzuki, A. Osuka and D. Kim, J. Am. Chem. Soc., 2009, 131, 7360 CrossRef CAS PubMed; (b) J.-Y. Shin, K. S. Kim, M.-C. Yoon, J. M. Lim, Z. S. Yoon, A. Osuka and D. Kim, Chem. Soc. Rev., 2010, 39, 2751 RSC.
- (a) H. Mori, Y. M. Sung, B. S. Lee, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2012, 51, 12459 CrossRef CAS PubMed; (b) K. Naoda, H. Mori, J. Oh, K. H. Park, D. Kim and A. Osuka, J. Org. Chem., 2015, 80, 11726 CrossRef CAS PubMed.
- K. Naoda, Y. M. Sung, J. M. Lim, D. Kim and A. Osuka, Chem.–Eur. J., 2014, 20, 7698 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1425255. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04263a |
| This journal is © The Royal Society of Chemistry 2016 |