DOI:
10.1039/C5SC03905C
(Edge Article)
Chem. Sci., 2016,
7, 2229-2238
A diversity-oriented synthesis of bioactive benzanilides via a regioselective C(sp2)–H hydroxylation strategy†
Received
15th October 2015
, Accepted 3rd December 2015
First published on 3rd December 2015
Abstract
A diversity-oriented synthesis of bioactive benzanilides via C(sp2)–H hydroxylation has been studied. Different regioselectivity was observed with Ru(II) and Pd(II) catalysts. The reaction demonstrates excellent regioselectivity, good tolerance of functional groups, and high yields. A wide range of ortho-hydroxylated-benzanilides can be readily synthesized with excellent regioselectivity via this new synthetic strategy. Computational investigations revealed that the regioselectivity was controlled mainly by both steric and electronic factors. Steric effects determine the regioselective outcomes in the Ru-catalyzed reaction, while electronic effects are dominant in the Pd-catalyzed reaction.
Introduction
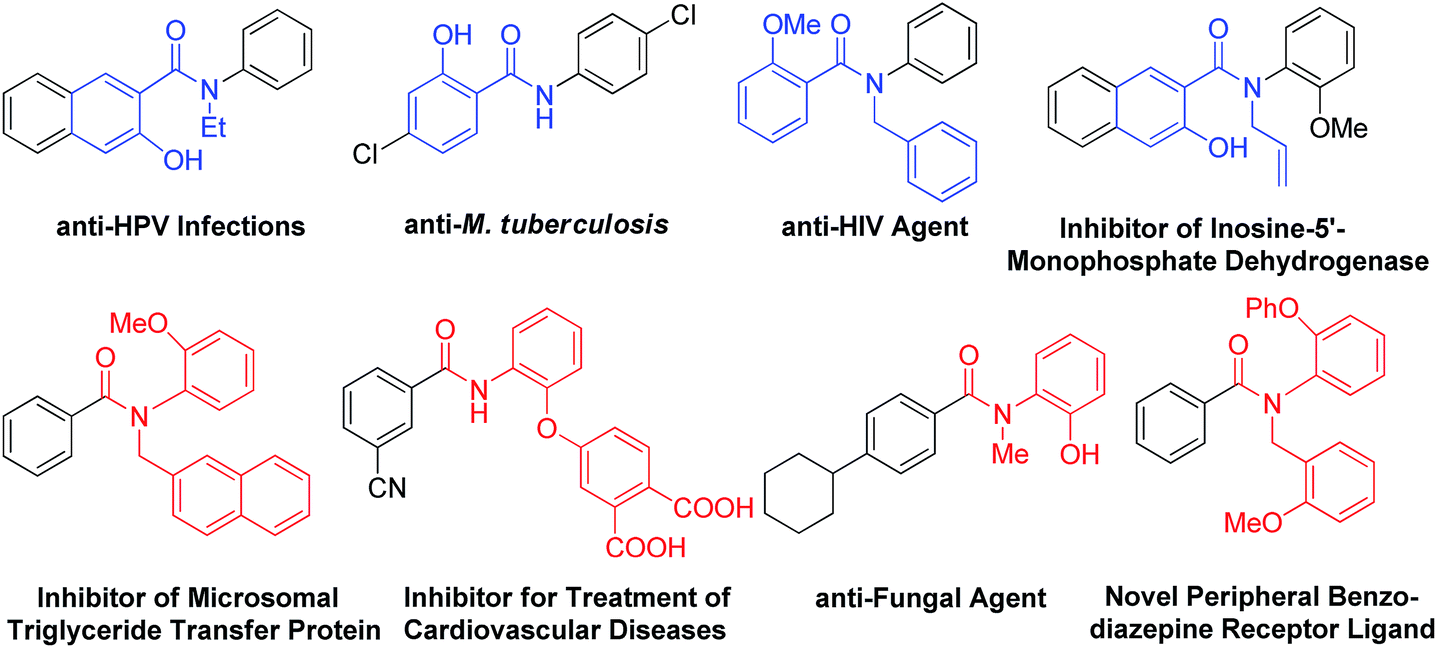
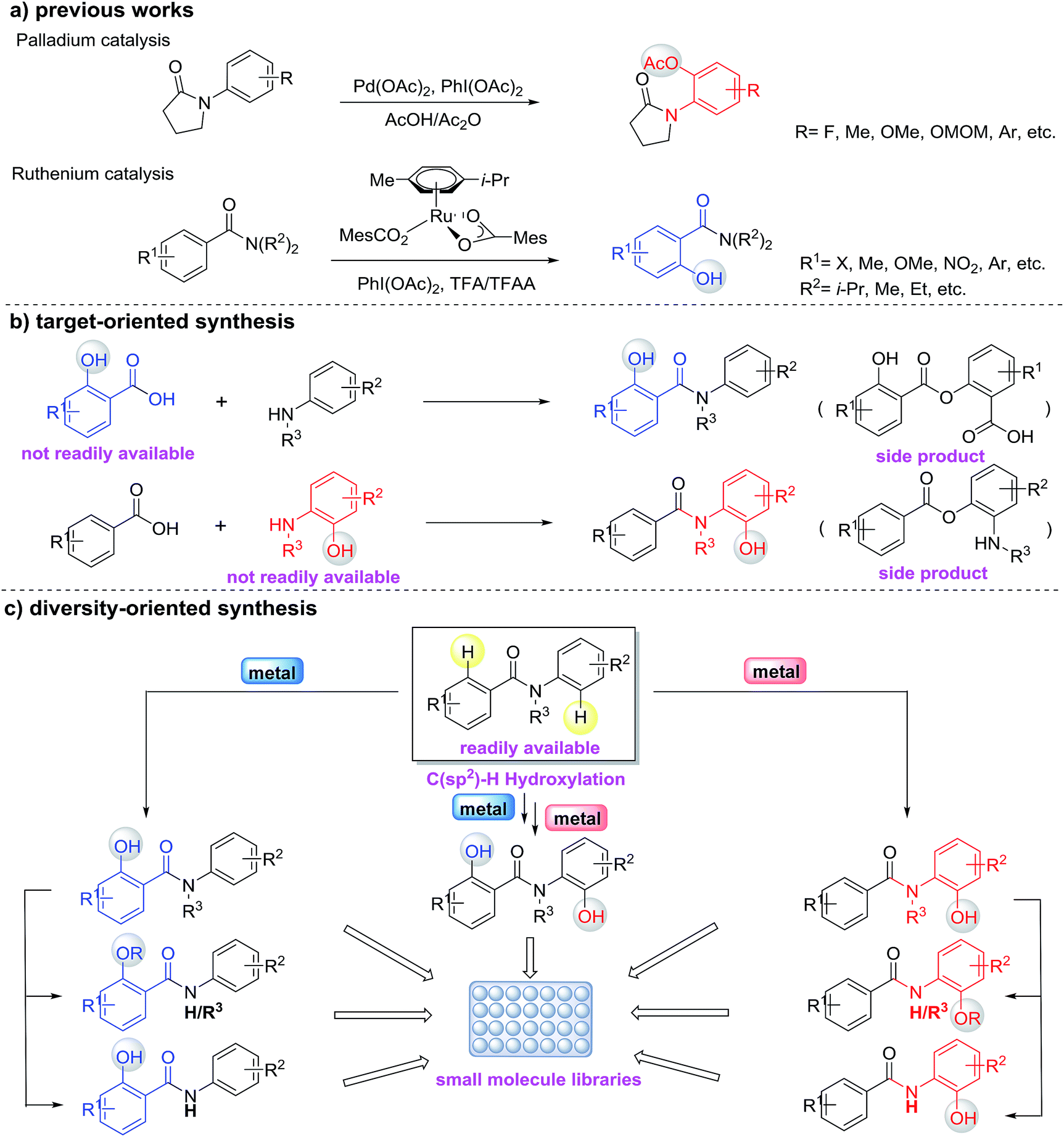
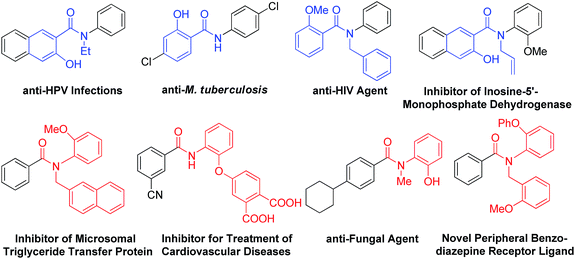
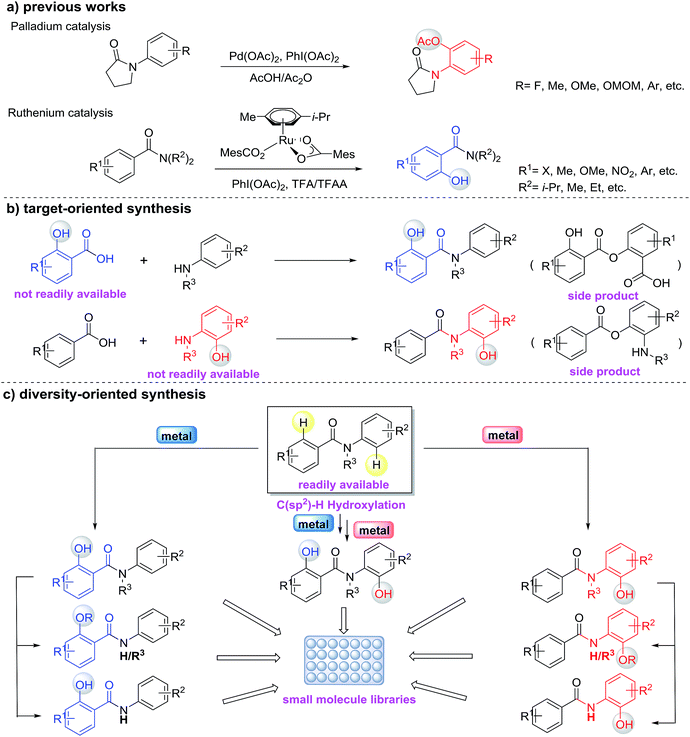
Hydroxylated benzanilides are an important class of chemicals used frequently in pharmaceuticals and agrochemicals (Fig. 1).1 The position of the hydroxyl group, i.e. at the ortho-position in either the benzoyl or the aniline moiety, may determine dramatically different bio-activities. For example, in a DNA-PK inhibitor the hydroxyl group is on the benzoyl segment, but it is on the aniline ring in a rotamase inhibitor. Synthesis and screening of small molecule libraries containing diverse hydroxylated benzanilides will be a highly valuable means of identifying potent hits in early drug discovery. With target-oriented synthesis, hydroxylated benzanilides can be prepared via the condensation of carboxylic acids with amines, but this strategy usually suffers from limitations such as poor availability of starting materials, difficult reaction procedures, low yields and the formation of side products (Scheme 1b).2 For example, when salicylic acid reacts directly with aniline, interaction of the carboxyl and phenolic hydroxyl groups could lead to a significant side reaction. Diversity-oriented synthesis (DOS) is a strategy for rapid access to relatively small libraries of organic molecules with an emphasis on skeletal diversity.3 Structurally diverse compound libraries are highly valuable for drug discovery; to date, many important small molecule modulators of challenging biological targets have been identified from DOS-derived compound libraries.
 |
| | Fig. 1 Biologically important molecules containing hydroxylated benzanilides. | |
 |
| | Scheme 1 A diversity-oriented synthesis for accessing hydroxylated benzanilides via C(sp2)–H functionalization. | |
Due to the challenges in reactivity and selectivity however, the use of C–H functionalization in DOS is still uncommon.4 In the last decade, direct functionalization of sp2 C–H bonds by transition-metals emerged as a powerful method for generating new C–O bonds.5,6 Among these studies, the ruthenium(II)-catalyzed C–H oxygenation of benzamides5n,6b and the palladium(II)-catalyzed C–H oxygenation of anilides5s,5t,6a are the most relevant (Scheme 1a). Although the impressive progress of C–H hydroxylation and other oxidations have provided a toolbox for the functionalization of benzanilides, there are challenges that remain for directed C–H hydroxylation in terms of regioselectivity, scope and practicality of these protocols. For instance, the knowledge about regioselective C–H hydroxylation of substrates containing two or more arenes and its corresponding applicable utility in the preparation of biologically important molecules is still surprisingly limited. Inspired by the diversification of bioactive molecules via the controlled chemical or enzymatic oxidation of C–H bonds reported by Baran,7 we envisioned that the diversity-oriented synthesis via regioselective C–H hydroxylation for bioactive benzanilides could be achieved using different metal catalysts (Scheme 1c). In this paper, we report the development of Pd(II)- or Ru(II)-catalyzed regioselective C–H hydroxylation of benzanilides, which provides a convenient method for the preparation of diverse ortho-hydroxylated benzanilide derivatives. This regioselective strategy as a means of accessing different types of hydroxylated benzanilides directly from benzanilide substrates is particularly attractive and depends highly on the practicality of such chemical processes. Our computational studies revealed that the distinct regioselectivity outcomes are dictated by both steric and electronic factors, and highlighted the importance of the coordination geometries that metal ions prefer for regioselectivity.
Results and discussion
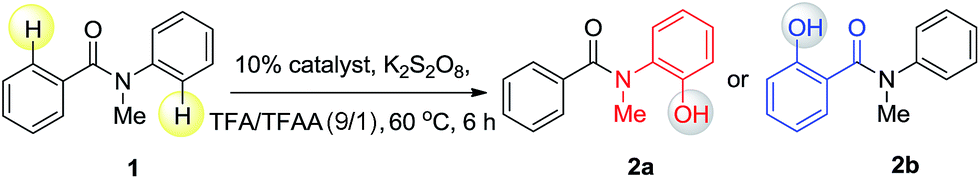
Since different metals form various cyclic metal intermediates, sometimes via different mechanisms,8,9 we speculated that regioselective C–H activation of benzanilides could occur with metal catalysts by attacking distinct ortho protons. In such cases, regioselective C–H hydroxylation may be realized with suitable reagents and oxidants. We started our investigation with substrates containing two arenes. As shown in Table 1, to test this hypothesis, we initiated a model study with N-methyl-benzanilide (1) in the presence of various transition metal catalysts in a TFA (trifluoroacetic acid)/TFAA (trifluoroacetic anhydride) cosolvent system with different oxidants, including PhI(OAc)2, NaIO4, HIO3, selectfluor, Na2S2O8, K2S2O8, H2O2, etc. (for details, please see the ESI†). We discovered that among the screened catalysts, only rhodium, ruthenium and palladium can provide significant amounts of phenol products. In contrast, other metal catalysts including Cu, Fe, Ni, Ag, Au and Co, failed to promote the transformations under the same reaction conditions (entries 1–13). Further analysis of the experimental results revealed an interesting phenomenon that rhodium and ruthenium catalysts always gave the same regioselective products. However, different regioselectivity outcomes were observed under palladium catalysis. For example, when a Ru(II) or Rh(II) catalyst was used, hydroxylation occurred ortho to the carbonyl group to give compound 2b (entries 17–18). In the presence of a Pd(II) catalyst, hydroxylation occurred ortho to the acylated amine group to afford the hydroxylated product 2a (entry 21). In terms of the efficiency of metal catalysis, Pd(II) and Ru(II) were found to be better than Rh(II). Various Pd(II), Rh(II) and Ru(II) catalysts were examined in this reaction, and a consistent regioselectivity with varying product yields was observed (entries 14–23). Among the tested oxidants, K2S2O8 was noted to be superior over others. Generally, 0.1 equiv. of the Ru(II) or Pd(II) catalyst was enough to effectively promote the reaction. Typically the reaction will proceed to completion in TFA/TFAA within 10 hours, in a clean manner, with 2.0 to 3.0 equivalents of the oxidant.
Table 1 Regioselectivity with different metals on N-methyl-benzanilides
|
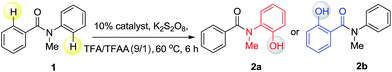
|
| Entry |
Catalyst |
2a yielda (%) |
2b yielda (%) |
|
Conversion ratio.
Isolated yield.
0.5 h.
|
| 1 |
FeBr3 |
<5 |
<5 |
| 2 |
Fe(OAc)2 |
<5 |
<5 |
| 3 |
Co(OAc)2 |
<5 |
<5 |
| 4 |
CoF3 |
<5 |
<5 |
| 5 |
Bis(1,5-cyclooctadiene)nickel |
<5 |
<5 |
| 6 |
Bis(triphenylphosphine)nickel dichloride |
<5 |
<5 |
| 7 |
CuI |
<5 |
<5 |
| 8 |
[μ-Bis(diphenylphosphino)methane]Au2Cl2 |
<5 |
<5 |
| 9 |
AuCl |
<5 |
<5 |
| 10 |
Mn(OAc)2 |
<5 |
<5 |
| 11 |
NbCl5 |
<5 |
<5 |
| 12 |
(1,5-Cyclooctadiene)(methoxy)iridium |
<5 |
<5 |
| 13 |
Ce(SO4)2·4H2O |
<5 |
<5 |
| 14 |
Rh2(TFA)4 |
<5 |
43 |
| 15 |
[(C6H5)3P]3RhCl |
<5 |
8 |
| 16 |
[Cp*RhCl2]2 |
<5 |
<5 |
| 17 |
Rh2(OAc)4 |
<5 |
37 |
|
18
|
[Ru(p-cymene)Cl
2
]
2
|
<5
|
93
|
| 19 |
[(C6H5)3P]3Ru(CO)(Cl)H |
<5 |
73 |
| 20 |
[(C6H5)3P]3RuCl2 |
<5 |
60 |
|
21
|
Pd(OAc)
2
|
70
|
<5
|
| 22c |
Pd(acac)2 |
67 |
<5 |
| 23c |
Pd(TFA)2 |
57 |
<5 |
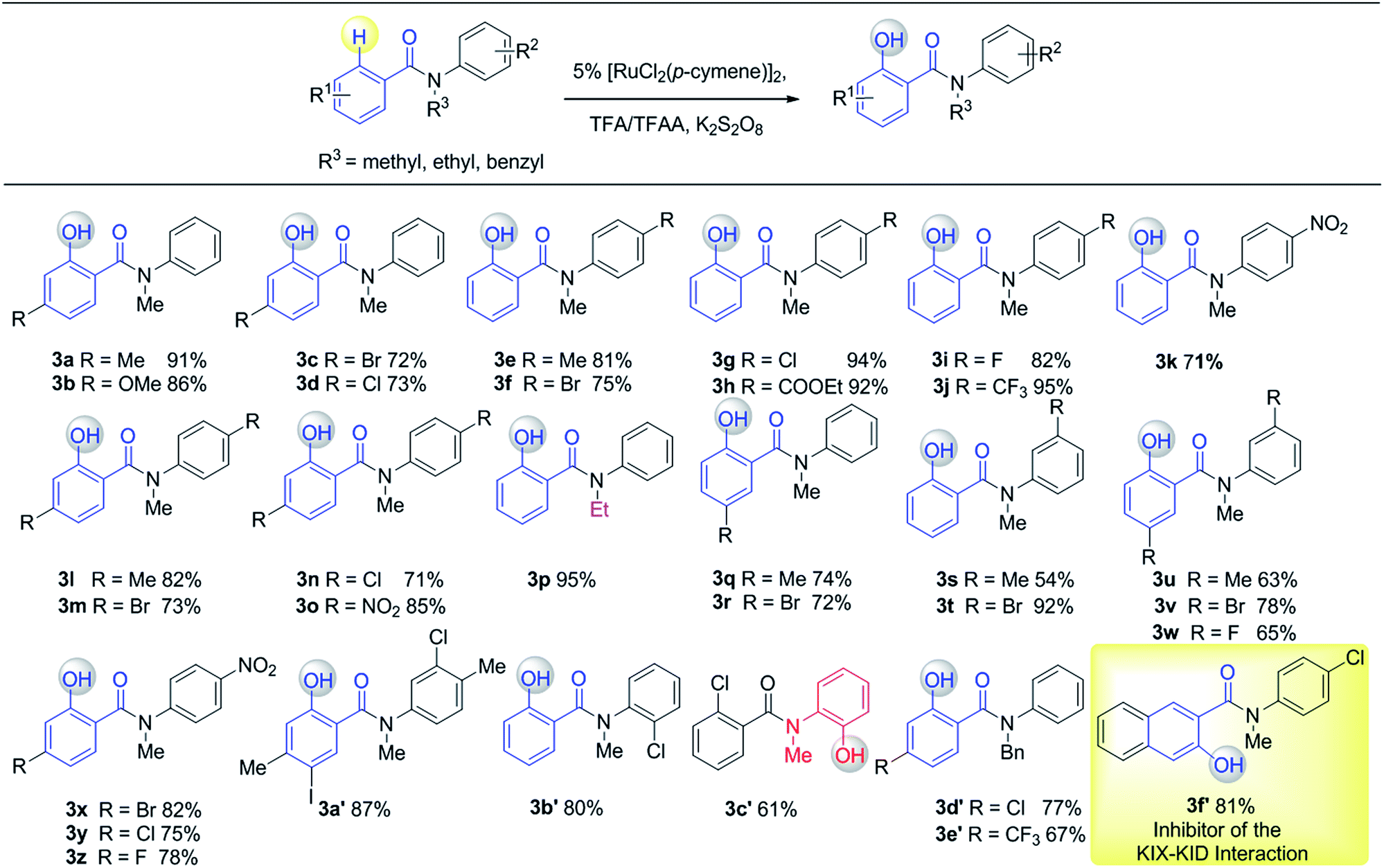
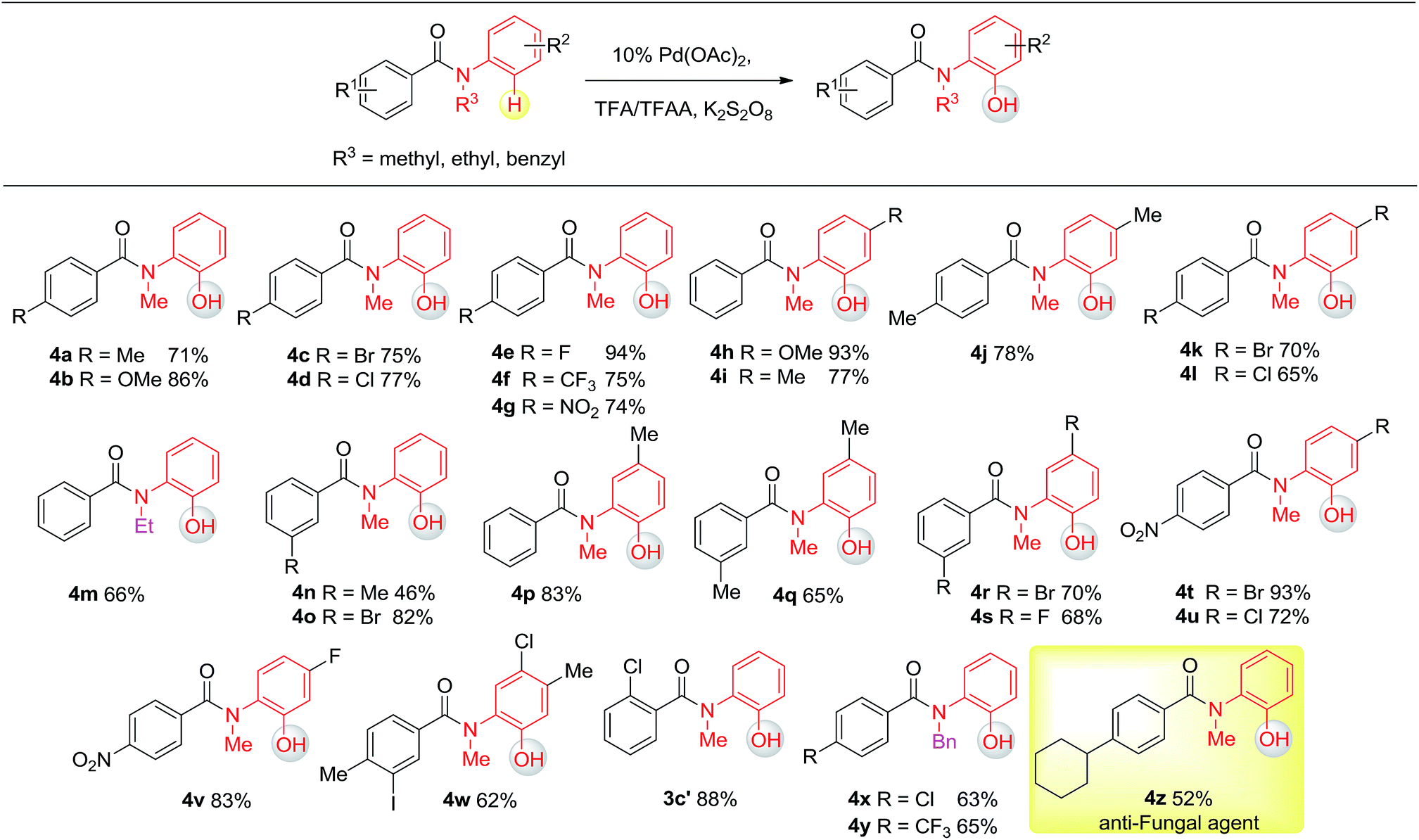
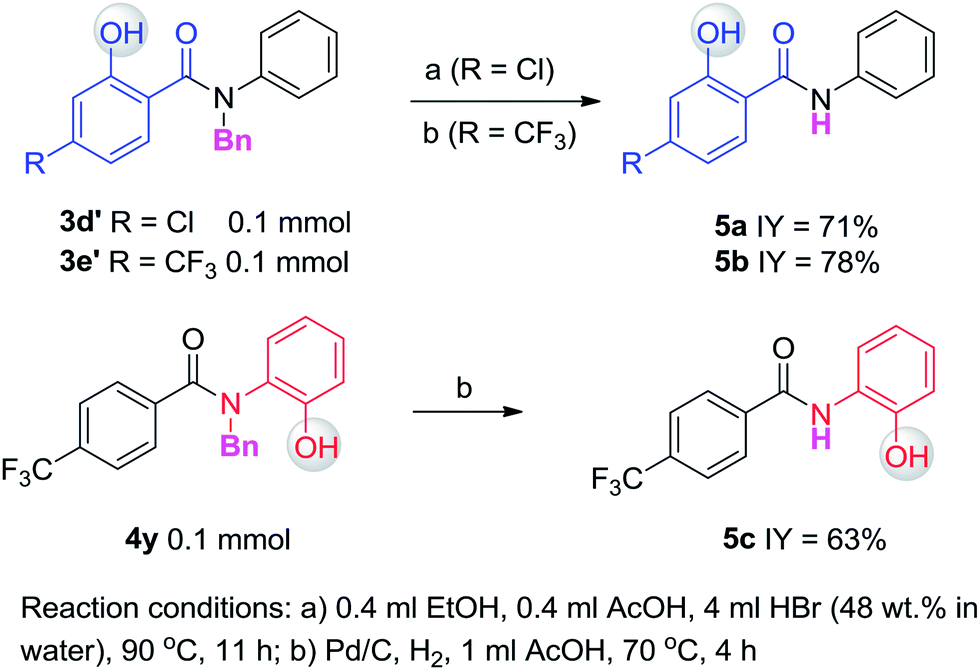
Having established the experimental studies of the regioselectivity results using Ru(II) or Pd(II), we further examined the substrate scope and robustness of this method. As illustrated in Fig. 2 and 3, a broad range of phenol derivatives (3a–3f′, 4a–4z) were prepared in good to excellent yields. The ortho, meta and para-substituted aryl groups, as well as the electron withdrawing and electron donating functional groups (halide, methyl, ester, CF3, and nitro groups) are well tolerated and only specific regioselective phenol products were obtained. As a result of steric effects, meta-substituted substrates always gave only one regioisomer formed by hydroxylation from the less hindered side. In all cases of ruthenium and palladium catalysis, the corresponding regioselective phenol products can be consistently obtained. A special example is about 2-chloro-N-methyl-N-phenylbenzamide. When it was used as the substrate, hydroxylation took place at the ortho-position of the acetylamino group in the presence of either a ruthenium or palladium catalyst to afford the hydroxylated product 3c′. The steric hindrance from the ortho-substituted chlorine group may account for this result. To our delight, N-ethyl-benzanilide and N-benzyl-benzanilides can be used in place of N-methyl-benzanilides as well and provide the desired phenol products (3p, 3d′, 3e′, 4m, 4x, 4y) with excellent reactivity and regioselectivity similar to those of the methyl counterparts. Ortho-hydroxylated benzanilides with unprotected N–H can be readily prepared by regioselective deprotection of the corresponding ortho-hydroxy-N-alkyl-benzanilides (Scheme 2). Thus, our “hydroxylation and deprotection” strategy can readily afford two kinds of ortho-hydroxylated benzanilides, which also represent an important class of bioactive molecules.10 As shown in Scheme 2, a one-step deprotection procedure involving acidic or hydrogenation conditions readily removes the N-benzyl group to furnish the ortho-hydroxylated-N–H-benzanilide product 5a, 5b and 5c respectively.
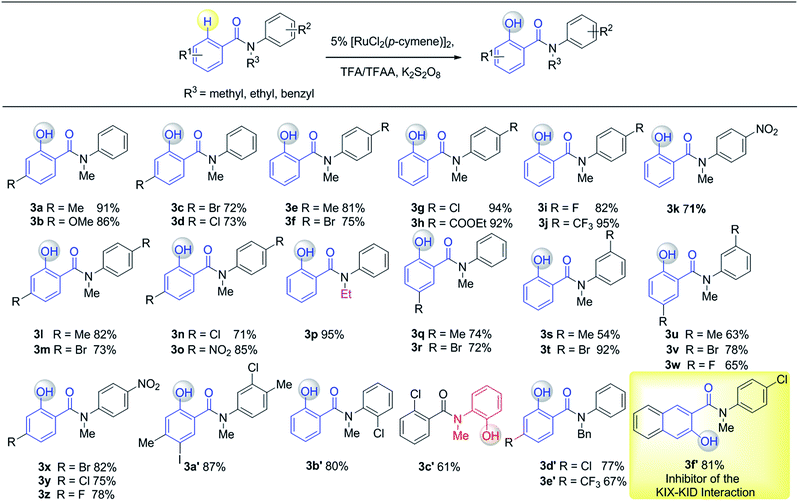 |
| | Fig. 2 Substrate scope of Ru(II) in C(sp2)–H hydroxylation (isolated yield). | |
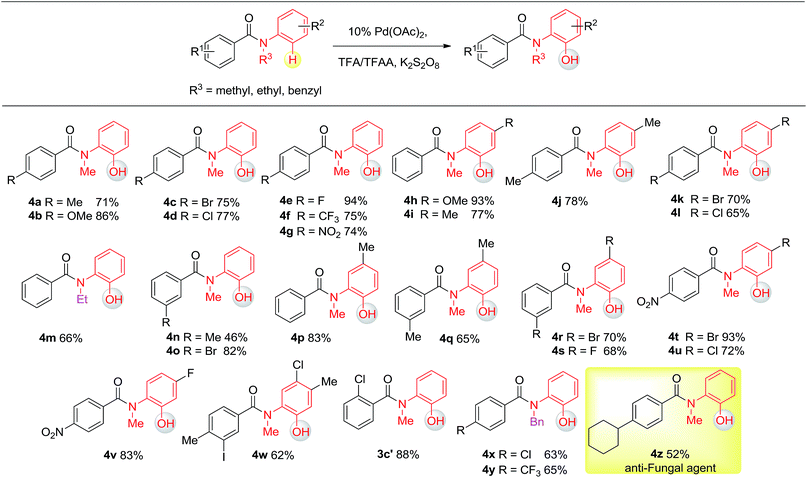 |
| | Fig. 3 Substrate scope of Pd(II) in C(sp2)–H hydroxylation (isolated yield). | |
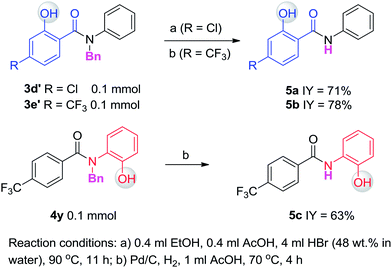 |
| | Scheme 2 Deprotection of ortho-hydroxylated N-benzyl-benzanilides. | |
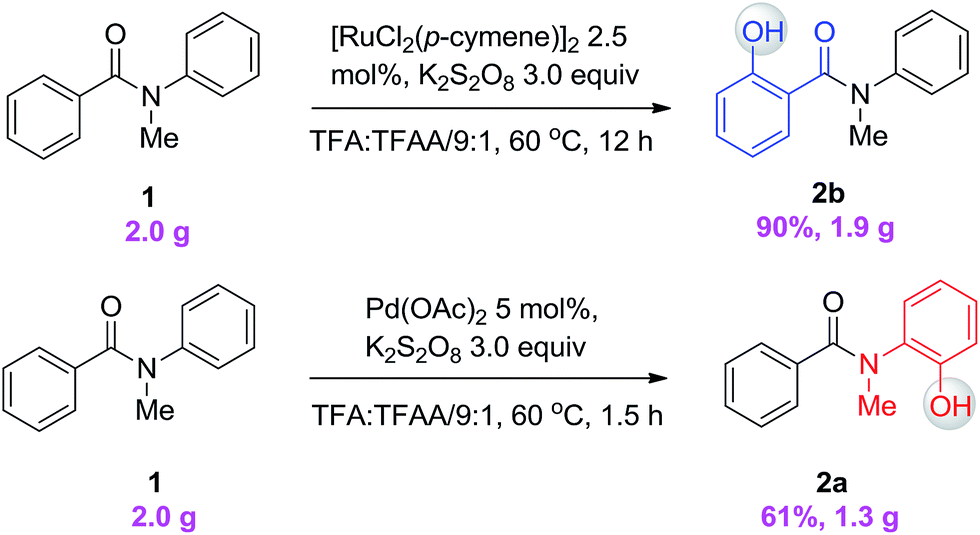
To demonstrate both the practicality and effectiveness of this method for large-scale synthesis, we prepared 2b and 2a on a gram scale under the optimized reaction conditions with only 2.5 mol% [RuCl2(p-cymene)]2 or 5 mol% Pd(OAc)2 catalyst (Scheme 3). In comparison to conventional approaches requiring 2-(alkylamino) phenol derivatives or substituted benzoyl peroxides, which are generally relatively expensive, our method, with its operational simplicity and ready availability of starting materials, is advantageous for rapid access to these molecules. The applicability of this reaction was further demonstrated in the synthesis of biologically important molecules. As exemplified in Fig. 2 and 3, Ru(II)-catalyzed C–H hydroxylation can smoothly transform the substrate, N-(4-chlorophenyl)-N-methyl-2-naphthamide, in a good yield into the hydroxylated product 3f′, which represents a class of effective inhibitors of the KIX–KID interaction.11a KID is the kinase-inducible domain in the cyclic AMP response element binding protein (CREB). KIX is the KID-interacting domain in CREB-binding protein (CBP). This type of compound has been identified as a cell-permeable inhibitor of the KIX–KID interaction which directly binds to KIX. Our method provides a convenient way by which to synthesize this type of naphthol derivative with excellent chemoselectivity and this synthetic strategy might present a new opportunity to develop novel transcription-based cancer therapeutic agents. Similarly, a one-step protocol via a Pd(II)-catalyzed C–H hydroxylation could easily convert 4-cyclohexyl-N-methyl-N-phenylbenzamide into the fungicide1g4z. The preparation of this type of molecule with traditional methods has always suffered from poor selectivity and low yields.
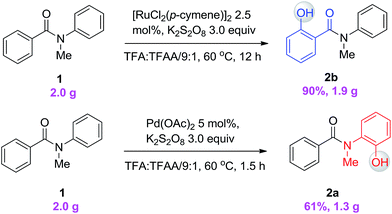 |
| | Scheme 3 Gram-scale synthesis of ortho-hydroxylated N-methyl-benzanilides. | |
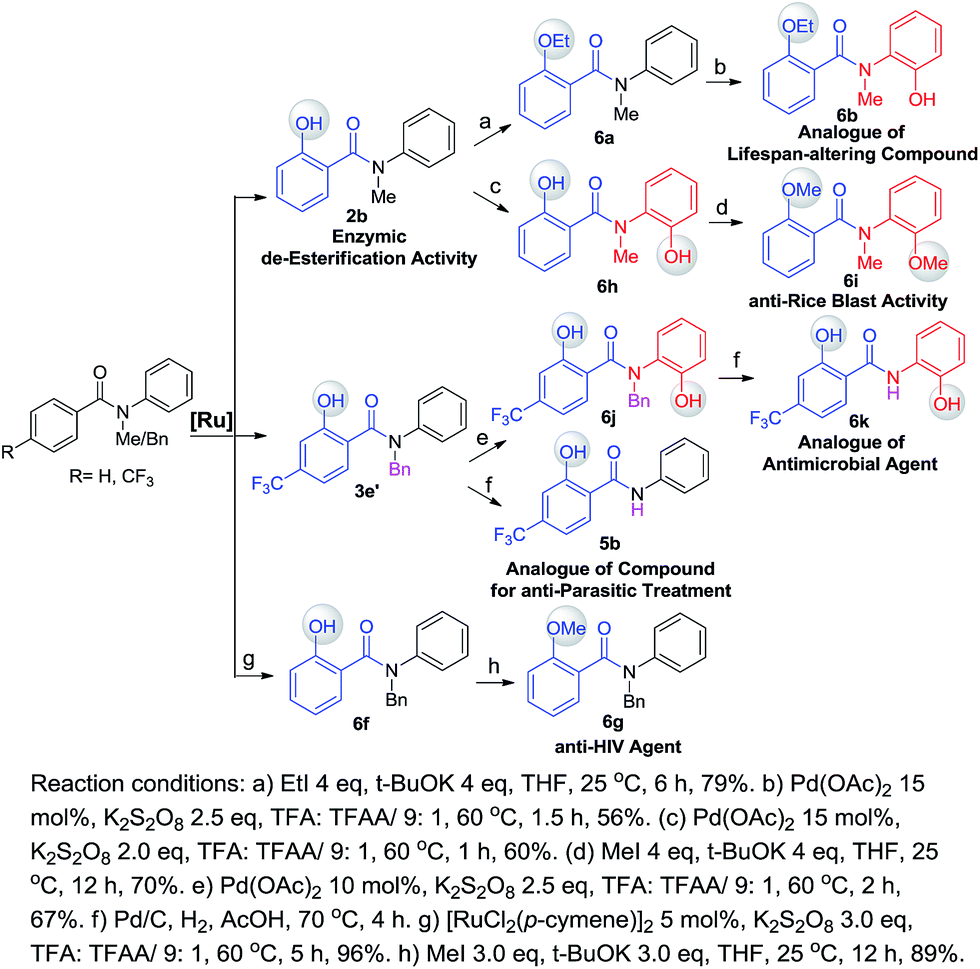
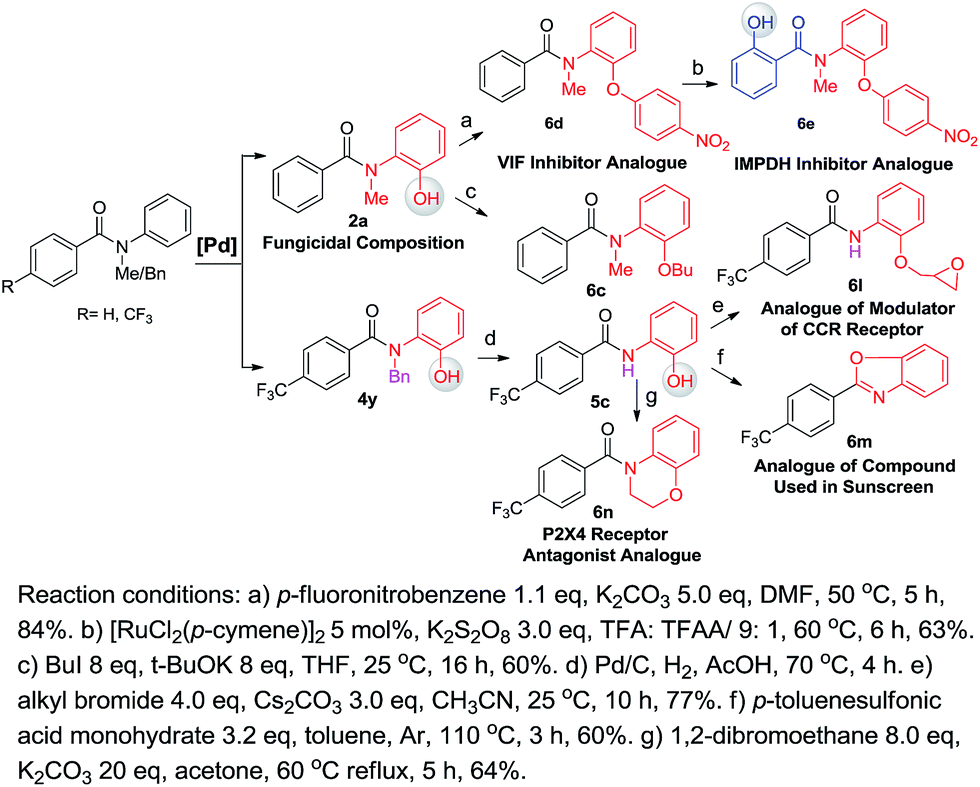
One of our long-term goals is to synthesize a diverse collection of ortho-hydroxylated benzanilide derivatives via a diversity-oriented synthesis strategy, which can be further screened in different biological assays to facilitate early drug discovery. To illustrate the utility of our new synthetic method for the preparation of a focused small molecule library of benzanilides, 20 different benzanilides can be rapidly constructed from two common precursors within 3–4 steps. As exemplified in Scheme 4 and 5, a wide variety of bioactive benzanilides can be tolerated and prepared under our hydroxylation conditions. For instance, the basic structures of 2b and 2a prepared according to our method have been demonstrated to have enzymatic de-esterification activity11b and anti-fungal activity.11c The ortho-hydroxylated N–H-benzanilide product 5b is an analogue of a compound for anti-parasitic treatment.11d The hydroxyl group can be further transformed into an alkyl ether (6a, 6c, 6i), aryl ether (6d, 6e) or derivatives of morpholine (6n) or oxazole (6m). These molecules have different reported bioactivities in the literature. For example, the ether compounds 6b, 6d and 6e are analogues of a lifespan-altering compound,11e a virion infectivity factor (VIF) inhibitor that blocks viral replication11f and an inosine-5′-monophosphate dehydrogenase (IMPDH) inhibitor,1d respectively. When the proton on the amide nitrogen of an ortho-hydroxylated benzanilide is substituted by a benzyl group, and the hydroxyl group is transformed to a methoxyl group, the corresponding benzanilide becomes an anti-HIV agent1c (6g). This type of structure serves as a central scaffold in the study of non-nucleoside reverse transcriptase inhibitors. The synthesis of 6g can be started from a simple benzanilide through a sequential benzylation–hydroxylation–methylation transformation. Remarkably, as shown in Scheme 4, dihydroxylated products can be easily obtained via a two-step hydroxylation manipulation using two different catalysts (Pd and Ru). Among them, the di-methoxylated product 6i has anti-rice blast activity11g and the di-hydroxylated N–H-benzanilide product 6k is an analogue of an antimicrobial agent.11h Besides N-alkyl-benzanilides, useful analogues of N–H-benzanilide compounds, can also be readily prepared by this method. We smoothly obtained three types of analogues from one precursor, N-(2-hydroxyphenyl)-4-(trifluoromethyl)benzamide (5c). Among them, one is a modulator of the CCR-receptor (6l). As is known, chemokines play an important role in immune and inflammatory responses in various diseases and disorders, including asthma and allergic diseases; CCR receptors represent good targets for drug development since agents which modulate these receptors would be useful in the treatment of these disorders and diseases.1i Using the same starting material, an analogue of a compound used in sunscreen, 2-(4-(trifluoromethyl)phenyl)benzo[d]oxazole1j (6m), was readily prepared via an intramolecular dehydration reaction. The third obtained benzamide is a P2X4 receptor antagonist analogue (6n), which is a derivative of morpholine. It inhibits the ATP-induced calcium influx in 1321N1 astrocytoma cells stably transfected with the human P2X4 receptor, and it has potential as a drug for the treatment of neuropathic pain and neurodegenerative diseases.1k To our pleasure, all these C–H hydroxylation reactions demonstrated excellent regio- and chemoselectivity, as well as reactivity.
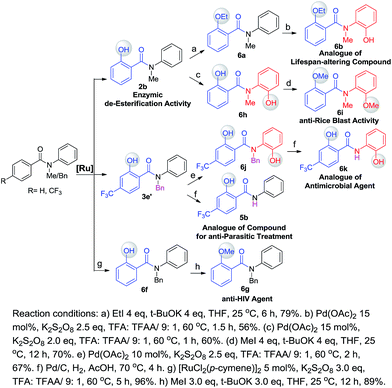 |
| | Scheme 4 A useful compound analogue library prepared via diversity-oriented synthesis (with [Ru] as a catalyst). | |
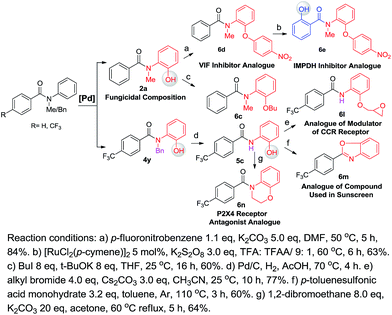 |
| | Scheme 5 A useful compound analogue library prepared via diversity-oriented synthesis (with [Pd] as a catalyst). | |
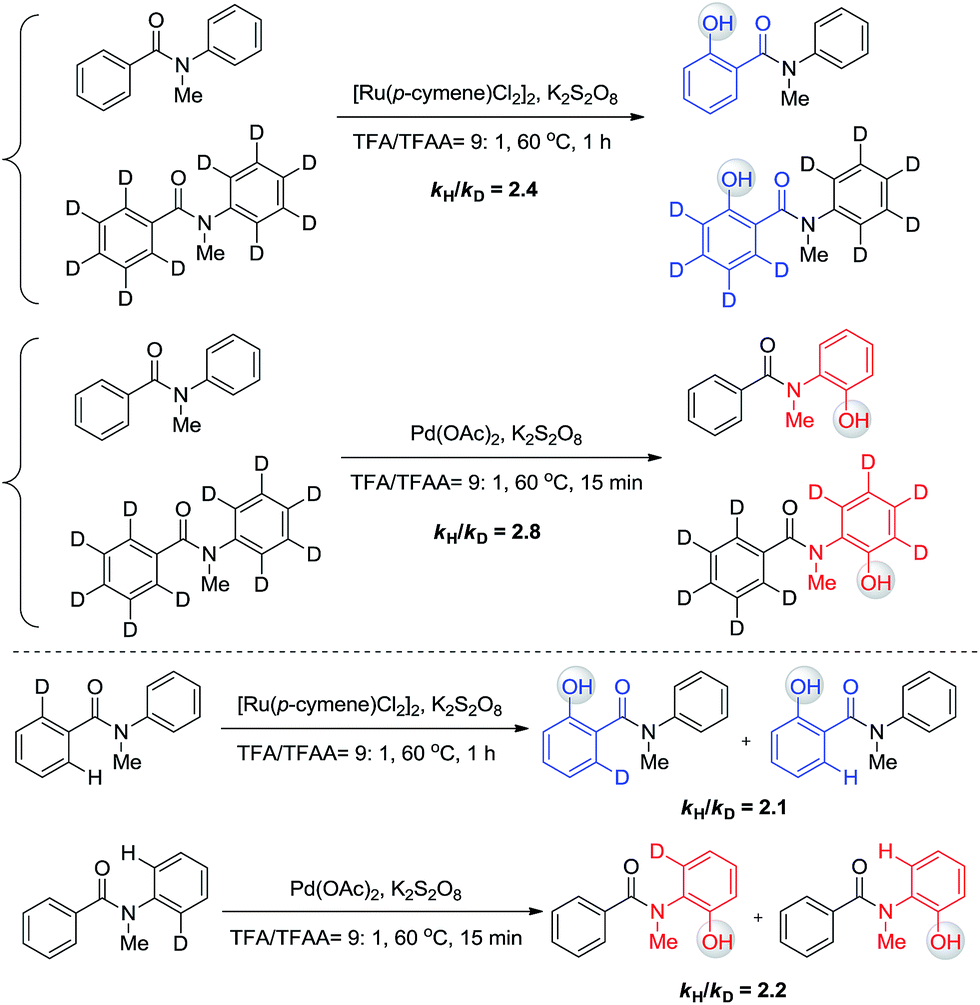
The phenomenon that Ru and Pd have distinct regioselectivity is of great interest. Mechanistic studies were then performed to reveal the origin of the selectivity. Our previous kinetic isotope effect (KIE) studies of similar reactions revealed that C–H activation is involved in the rate-determining step.6 To examine whether C–H activation is also the critical step to determine the selectivity for this work, inter- and intramolecular isotope effect measurements were conducted in parallel (Scheme 6). All KIE values are greater than 2, confirming that C–H activation is involved in the rate-limiting step of these two transformations. No H/D exchange was observed for these substrates, indicating that C–H bond activation is irreversible and determines the selectivity.
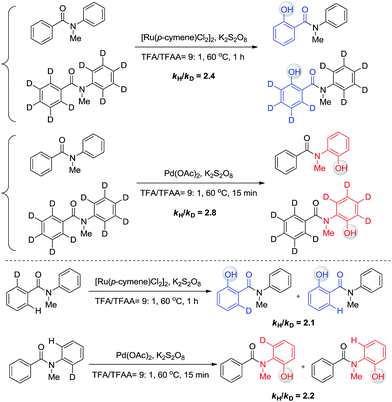 |
| | Scheme 6 Kinetic isotope effect. | |
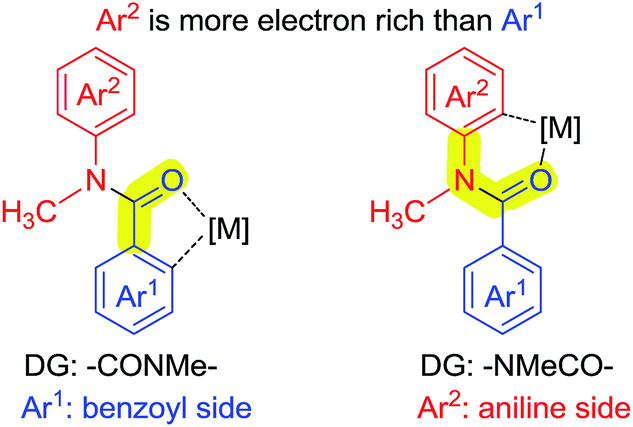
As shown in Scheme 7, N-methyl-benzanilide has two types of directing groups (DGs), amide (–CONMeR) and acylated amine (–NMeCOR). The transition states of the two types of DGs involve a 5-membered ring and a 6-membered ring, respectively. According to our previous studies of the priority order of various DGs,12 the acylated amine group (–NR1COR2) has a higher priority than the amide group (–CONR1R2) for Pd catalysts. This would lead to the hydroxylation on the aniline side and our present experimental data is consistent with this finding. However, the priority ordering of these two DGs is contrary for the Ru catalyst, which causes the different regioselectivity for C–H activation. In light of these results, a density functional theory (DFT) study was conducted.13
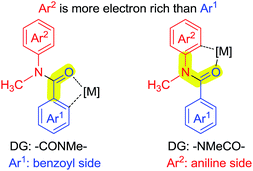 |
| | Scheme 7 Two types of DGs of N-methyl-benzanilide. | |
Recent mechanistic studies on carboxylate-assisted transition-metal catalyzed C–H activation suggested that the concerted metalation–deprotonation (CMD) mechanism with an inner- or outer-sphere base was the most favorable mechanism.14
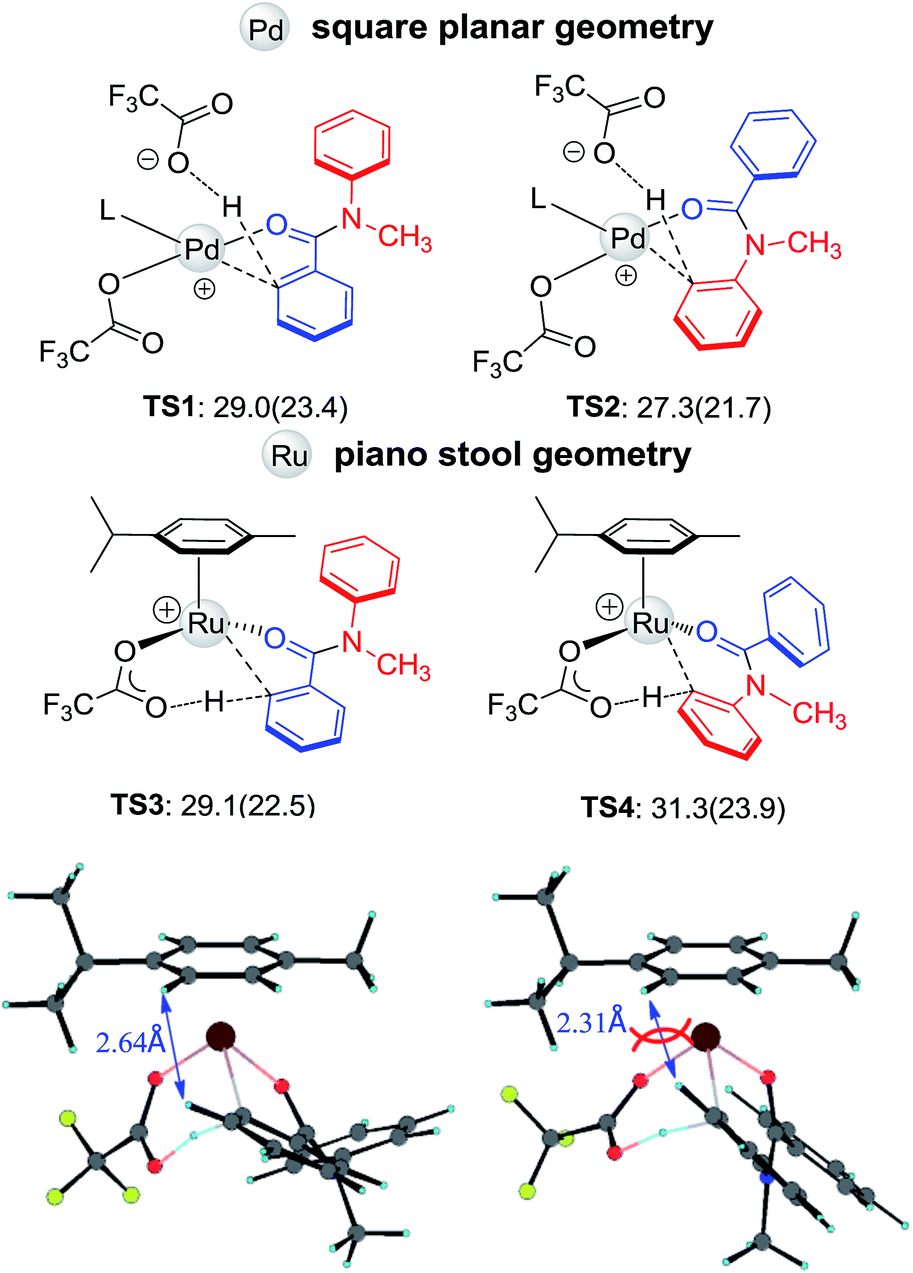
The initial steps of forming active catalysts are shown in Fig. S1 and S2.† Given the fact that the C–H activation step determines the regioselectivity, here we focus on the C–H activation step. Four critical transition structures, TS1–TS4, are depicted in Fig. 4. The calculation is consistent with the experimentally observed regioselectivity. Early kinetic and computational studies demonstrated that C–H activation catalyzed by Pd(OAc)2 is electrophilic in character.15 When reacting in trifluoroacetic acid (TFA), the dissociation of one TFA ligand makes the Pd center even more electrophilic. Therefore, electron-donating substituents on the arene group facilitate the reaction.1,15 For N-methyl-benzanilide, the more electron rich Ar2 is expected to undergo C–H activation preferentially. Indeed, for the Pd catalyst, the regioselectivity is dominated by the electronic effect (see Fig. 4, TS1 and TS2), which is also consistent with the priority ordering of the DGs,12 and using the dissociated acid ligand as an outer-sphere base to deprotonate the C–H bond might be the most favorable mechanism (see ESI†).16 However, the use of a Ru catalyst results in the hydroxylation at Ar1. Inspection of the geometries illustrates that steric factors may be controlling the regioselectivity, overwhelming the electronic effect in this reaction with Ru. Unlike the square planar geometry in the case of Pd(II), Ru(II) adopts a “piano stool” geometry, which is more crowded. Due to the steric repulsion from the N-methyl group, Ar1 and Ar2 both twist away from the amide plane. In TS4, the constraint of the newly formed metallocycle and the repulsion of the N-methyl group force Ar2 to be closer (2.31 Å) to the p-cymene cap and as a result, TS4 is less favorable than TS3 by 2.2 kcal mol−1.
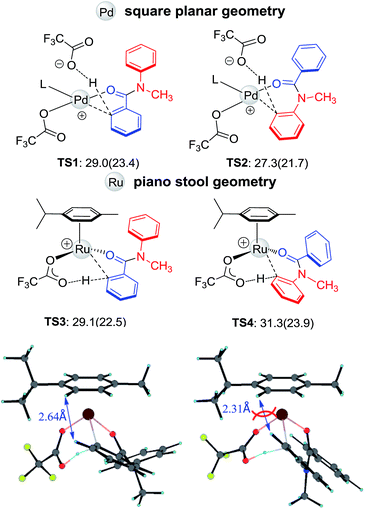 |
| | Fig. 4 The structures and free energies of activation of the Ru- and Pd-catalyzed C–H activations on Ar1 and Ar2 sides for N-methyl-benzanilide; electronic activation energies (in kcal mol−1) are in parentheses (L = N-methyl-benzanilides). | |
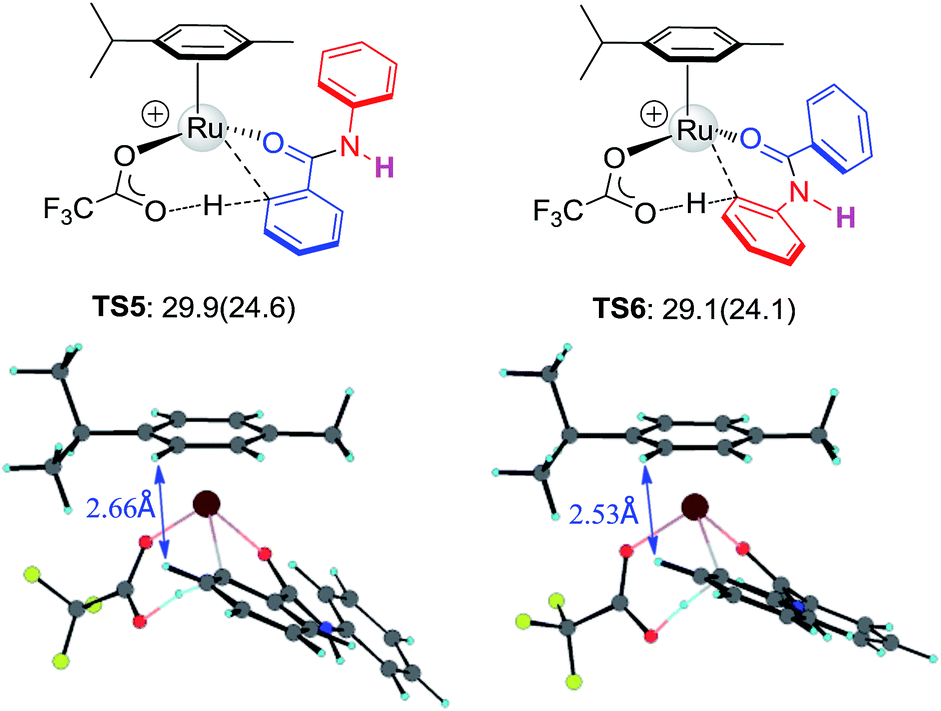
To further examine the hypothesis that the steric effect overrides the electronic effect in the Ru-catalysis transition state, we replaced the N-methyl group with H to reduce the steric repulsion. As illustrated in Fig. 5, the steric constraint in TS6 is largely diminished in the absence of the N-methyl group and preferred activation of Ar1viaTS5 was predicted.
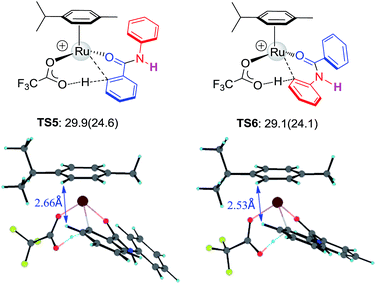 |
| | Fig. 5 The structures and free energies of activation of Ru catalyzed C–H activation on Ar1 and Ar2 sides for N–H-benzanilide; electronic activation energies (in kcal mol−1) are in parentheses. | |
Experiments with benzanilides were then conducted to verify the computational prediction. Hydroxylation indeed occurs on the ortho-position of the aniline (Ar1) with either the Pd(II) or Ru(II) catalyst (Scheme 8). However, the yield of these reactions with N-benzanilides is unsatisfactory under the current conditions. Accordingly, we recommend the protocol in Scheme 2 for the synthesis of ortho-hydroxylated N–H-benzanilides.
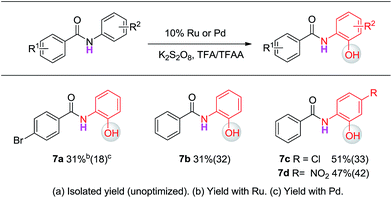 |
| | Scheme 8
Ortho-hydroxylation of N–H-benzanilides.a | |
In a further test to verify the role of repulsion between the substrate and the p-cymene cap, additional model calculations were performed. The p-cymene cap was replaced by smaller ligands or deleted to eliminate the steric factor and the corresponding energies were calculated. In these cases, the regioselectivity is reversed (see Fig. S3† for more details), and consequently, the steric repulsion between the cap and the aryl ring is thought to play an important role in the regioselectivity. This highlights the importance of considering the coordination geometries preferred by a metal center when performing carboxylate-assisted transition-metal catalyzed C–H activation reactions.17
Conclusions
We have demonstrated the regioselective ortho-hydroxylation of N-alkyl-benzanilides catalyzed by Pd(II) or Ru(II) to access two distinct ortho-hydroxylated regio-isomers. When Pd(II) is employed as the catalyst, the hydroxylation occurs on the phenyl ring of aniline. In contrast, a reversed regioselective product will be obtained in the presence of the Ru(II) catalyst. This method has been found to be generally useful for the preparation of a broad range of ortho-hydroxylated-N-alkyl-benzanilides with excellent regio-selectivity. Related ortho-hydroxylated benzanilides can be easily obtained by subsequent deprotection. These two types of compounds have been widely used in the initial stages of drug discovery. The reaction demonstrates excellent regioselectivity and good functional group tolerance, and proceeds with high yields. Its utility has been well exemplified in further synthetic applications of bioactive molecules. Further computational studies revealed that both steric and electronic factors account for the origin of the different regioselectivity between Ru(II) and Pd(II). Moreover, the coordination geometries that a metal center prefers are important for the regioselectivity. Further studies into the scope and the application of this reaction are currently in progress in our laboratory.
Acknowledgements
This work was supported by the national ‘973’ grant from the Ministry of Science and Technology (grant # 2011CB965300, 2013CB911501), National Natural Science Foundation of China (grant # 21302106, 81573277, 21133002, 21232001 and 21302006), Nanshan (KC2014ZDZJ0026A) and Tsinghua University Initiative Scientific Research Program.
Notes and references
-
(a) E. J. Androphy and J. D. Baleja, U. S. Pat. Appl. Publ., 2005 Search PubMed , 20050250855;
(b) M. Kratky, J. Vinsova and J. Stolarikova, Molecules, 2012, 17, 12812 CrossRef CAS PubMed;
(c) R. W. Buckheit Jr, T. L. Kinjerski, V. Fliakas-Boltz, J. D. Russell, T. L. Stup, L. A. Pallansch, W. G. Brouwer, D. C. Dao, W. A. Harrison, R. J. Schultz, J. P. Bander and S. S. Yang, Antimicrob. Agents Chemother., 1995, 39, 2718 CrossRef CAS;
(d) J. Saunders, D. Elbaum, P. Novak, D. Naegele, S. Bethiel, S. Ronkin, M. Badia, C. Frank, D. Stamos, W. Walters and D. Pearlman, PCT Int. Appl., 1999 Search PubMed , 9955663;
(e) R. J. Booth, H. T. Lee, J. K. Pontrello, R. R. Ramharack and B. D. Roth, U. S. Pat. Appl. Publ., 2002 Search PubMed , 20020156281;
(f) K. A. Rytved, N. Dragsted, N. C. B. Nyborg, L. Iversen and M. Kristiansen, U. S. Pat. Appl. Publ., 2004 Search PubMed , 20040082641;
(g) Y. Suda, Y. Kojima, K. Hirakawa and K. Nakamura, Jpn. Kokai Tokkyo Koho, 1981 Search PubMed , 56158705;
(h) T. Okubo, R. Yoshikawa, S. Chaki, S. Okuyama and A. Nakazato, Bioorg. Med. Chem., 2004, 12, 423 CrossRef CAS PubMed;
(i) T. Eriksson and K. Lawitz, PCT Int. Appl., 2002 Search PubMed , 2002076457;
(j) L. E. Cohen, U. S. Pat. Appl. Publ., 2014 Search PubMed , 20140127275;
(k) V. Hernandez-Olmos, A. Abdelrahman, A. El-Tayeb, D. Freudendahl, S. Weinhausen and C. E. Mueller, J. Med. Chem., 2012, 55, 9576 CrossRef CAS PubMed.
-
(a) S. Y. Shang, D. Zhang-Negrerie, Y. F. Du and K. Zhao, Angew. Chem., Int. Ed., 2014, 53, 6216 CrossRef CAS PubMed;
(b) Z.-J. Zhang, X.-J. Quan, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 3292 CrossRef CAS PubMed.
-
(a) S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed;
(b) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef PubMed;
(c) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed;
(d) T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48 CrossRef CAS PubMed;
(e) J. Zhang, J. Wu, B. Hong, W. Ai, X. Wang, H. Li and X. Lei, Nat. Commun., 2014, 5, 4614 CAS;
(f) S. Suzuki, Y. Segawa, K. Itami and J. Yamaguchi, Nat. Chem., 2015, 7, 227 CrossRef CAS PubMed;
(g) R. A. Bauer, T. A. Wenderski and D. S. Tan, Nat. Chem. Biol., 2012, 9, 21 CrossRef PubMed;
(h) F. Kopp, C. F. Stratton, L. B. Akella and D. S. Tan, Nat. Chem. Biol., 2012, 8, 358 CrossRef CAS PubMed;
(i) B. M. Ibbeson, L. Laraia, E. Alza, C. J. O'Connor, Y. S. Tan, H. M. L. Davies, G. McKenzie, A. R. Venkitaraman and D. R. Spring, Nat. Commun., 2014, 5, 3155 Search PubMed;
(j) H. S. G. Beckmann, F. Nie, C. E. Hagerman, H. Johansson, Y. S. Tan, D. Wilcke and D. R. Spring, Nat. Chem., 2013, 5, 861 CrossRef CAS PubMed;
(k) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 Search PubMed.
-
(a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed;
(b) J. Li, J. S. Cisar, C.-Y. Zhou, B. Vera, H. Williams, A. D. Rodríguez, B. F. Cravatt and D. Romo, Nat. Chem., 2013, 5, 510 CrossRef CAS PubMed.
-
(a) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed;
(b) X. Li, Y.-H. Liu, W.-J. Gu, B. Li, F.-J. Chen and B.-F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed;
(c) Y. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827 CrossRef CAS PubMed;
(d) T. Yamaguchi, E. Yamaguchi, N. Tada and A. Itoh, Adv. Synth. Catal., 2015, 357, 2017 CrossRef CAS;
(e) H.-Y. Zhang, H.-M. Yi, G.-W. Wang, B. Yang and S.-D. Yang, Org. Lett., 2013, 15, 6186 CrossRef CAS PubMed;
(f) P. Y. Choy and F. Y. Kwong, Org. Lett., 2013, 15, 270 CrossRef CAS PubMed;
(g) K. Seth, M. Nautiyal, P. Purohit, N. Parikh and A. K. Chakraborti, Chem. Commun., 2015, 51, 191 RSC;
(h) J. Dong, P. Liu and P. Sun, J. Org. Chem., 2015, 80, 2925 CrossRef CAS;
(i) A. Banerjee, A. Bera, S. Guin, S. K. Rout and B. K. Patel, Tetrahedron, 2013, 69, 2175 CrossRef CAS;
(j) V. S. Thirunavukkarasu and L. Ackermann, Org. Lett., 2012, 14, 6206 CrossRef CAS PubMed;
(k) Y. Yang, Y. Lin and Y. Rao, Org. Lett., 2012, 14, 2874 CrossRef CAS PubMed;
(l) F.-Z. Yang and L. Ackermann, Org. Lett., 2013, 15, 718 CrossRef CAS PubMed;
(m) X. Yang, G. Shan and Y. Rao, Org. Lett., 2013, 15, 2334 CrossRef CAS PubMed;
(n) V. S. Thirunavukkarasu, J. Hubrich and L. Ackermann, Org. Lett., 2012, 14, 4210 CrossRef CAS PubMed;
(o) F. Yang, F. Song, W. Li, J. Lan and J. You, RSC Adv., 2013, 3, 9649 RSC;
(p) G. Wang, T. Yuan and X. Wu, J. Org. Chem., 2008, 73, 4717 CrossRef CAS PubMed;
(q) K. Padala and M. Jeganmohan, Chem. Commun., 2013, 49, 9651 RSC;
(r) K. Padala and M. Jeganmohan, Chem.–Eur. J., 2014, 20, 4092 CrossRef CAS PubMed;
(s) D. Kalyani and M. S. Sanford, Org. Lett., 2005, 7, 4149 CrossRef CAS PubMed;
(t) L. V. Desai, K. J. Stowers and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 13285 CrossRef CAS PubMed;
(u) V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29 RSC;
(v) F. Yang, K. Rauch, K. Kettelhoit and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 11285 CrossRef CAS PubMed;
(w) W. Liu and L. Ackermann, Org. Lett., 2013, 15, 3484 CrossRef CAS PubMed;
(x) T. Yoneyama and R. H. Crabtree, J. Mol. Catal. A: Chem., 1996, 108, 35 CrossRef CAS;
(y) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS PubMed.
-
(a) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070 CrossRef CAS PubMed;
(b) G. Shan, X. Han, Y. Lin and Y. Rao, Org. Biomol. Chem., 2013, 11, 2318 RSC.
-
(a) T. Brueckl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed;
(b) H. Renata, Q. Zhou and P. S. Baran, Science, 2013, 339, 59–63 CrossRef CAS PubMed;
(c) N. C. Wilde, M. Isomura, A. Mendoza and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4909–4912 CrossRef CAS PubMed;
(d) Q. Michaudel, G. Journot, A. Regueiro-Ren, A. Goswami, Z. Guo, T. P. Tully, L. Zou, R. O. Ramabhadran, K. N. Houk and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 12091 CrossRef CAS PubMed.
-
(a) L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032 CrossRef CAS PubMed;
(b) E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161 CrossRef CAS PubMed;
(c) A. G. Algarra, W. B. Cross, D. L. Davies, Q. Khamker, S. A. Macgregor, C. L. McMullin and K. Singh, J. Org. Chem., 2014, 79, 1954 CrossRef CAS PubMed.
- R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS PubMed.
-
(a) F. Hirayama, H. Koshio, T. Ishihara, S. Hachiya, K. Sugasawa, Y. Koga, N. Seki, R. Shiraki, T. Shigenaga, Y. Iwatsuki, Y. Moritani, K. Mori, T. Kadokura, T. Kawasaki, Y. Matsumoto, S. Sakamoto and S.-i. Tsukamoto, J. Med. Chem., 2011, 54, 8051 CrossRef CAS PubMed;
(b) J. Wang, L. Chen, S. H. Sinha, Z. Liang, H. Chai, S. Muniyan, Y.-W. Chou, C. Yang, L. Yan, Y. Feng, K. Kathy Li, M.-F. Lin, H. Jiang, Y. George Zheng and C. Luo, J. Med. Chem., 2012, 55, 7978 CrossRef CAS PubMed;
(c) M. Gooyit, N. Tricoche, S. Lustigman and K. D. Janda, J. Med. Chem., 2014, 57, 5792 CrossRef CAS PubMed;
(d) C.-L. Chen, F.-L. Liu, C.-C. Lee, T.-C. Chen, A. A. Ahmed Ali, H.-K. Sytwu, D.-M. Chang and H.-S. Huang, J. Med. Chem., 2014, 57, 8072 CrossRef CAS PubMed.
-
(a) B. X. Li and X. S. Xiao, ChemBioChem, 2009, 10, 2721 CrossRef CAS PubMed;
(b) V. S. Parmar, A. Kumar, A. K. Prasad, R. Kumar, K. S. Bisht, Poonam, S. C. Jain and C. E. Olsen, J. Indian Chem. Soc., 1998, 75, 810 CAS;
(c)
Y. Suda, Y. Kojima, K. Hirakawa and K. Nakamura, Jpn. Kokai Tokkyo Koho, 1981, 56145206 Search PubMed;
(d)
E. Hodgkins, Eur. Pat. Appl., 2015 Search PubMed, 2875726;
(e)
D. S. Goldfarb, U. S. Pat. Appl. Publ., 2009 Search PubMed, 20090163545;
(f) A. Ali, J. Wang, R. S. Nathans, H. Cao, N. Sharova, M. Stevenson and T. M. Rana, ChemMedChem, 2012, 7, 1217 CrossRef CAS PubMed;
(g) S. Ito, Y. Kojima, K. Fujimori, K. Matsunari, I. Shimazaki and Y. Suda, J. Pestic. Sci., 1986, 11, 579 CrossRef CAS;
(h) F. A. Pasha, M. Muddassar, C. Lee and S. J. Cho, Environ. Toxicol. Pharmacol., 2008, 26, 128 CrossRef CAS PubMed.
- X. Sun, G. Shan, Y. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440 CrossRef CAS PubMed.
- Geometries were optimized with B3LYP and LANL2DZ+f (1.472 for Pd and 1.235 for Ru) basis sets for metals and 6-31G(d) for other atoms. Single point energies were calculated with M06 and SDD for Pd and Ru and 6-311++G(d,p) for other atoms with the SMD solvation model. All calculations were performed with Gaussian 09, M. J. Frisch, et al., Gaussian, Inc., Wallingford CT, 2009. More details about the computational methods are included in the ESI†.
-
(a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS PubMed;
(b) Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820 RSC;
(c) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, 658 CrossRef CAS PubMed;
(d) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed;
(e) D. G. Musaev, A. Kaledin, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 1690 CrossRef CAS PubMed;
(f) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed;
(g) G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136, 894 CrossRef CAS PubMed.
-
(a) A. D. Ryabov, I. K. Sakodinskaya and A. K. Yatsimirsky, J. Chem. Soc., Dalton Trans., 1985, 2629 RSC;
(b) A. E. Shilov and G. B. Shuípin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed;
(c) C. G. Jia, D. G. Piao, J. Z. Oyamada, W. J. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS PubMed;
(d) B. Biswas, M. Sugimoto and S. Sakaki, Organometallics, 2000, 19, 3895 CrossRef CAS;
(e) K. J. Stowers and M. S. Sanford, Org. Lett., 2009, 11, 4584 CrossRef CAS PubMed;
(f) B. Xiao, T.-J. Gong, J. Xu, Z.-J. Liu and L. Liu, J. Am. Chem. Soc., 2011, 133, 1466 CrossRef CAS PubMed;
(g) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed;
(h) S. Zhang, L. Shi and Y. Ding, J. Am. Chem. Soc., 2011, 133, 20218 CrossRef CAS PubMed.
-
(a) M. Ahlquist, R. A. Periana and W. A. Goddard, Chem. Commun., 2009, 2373 RSC;
(b) D. H. Ess, T. B. Gunnoe, T. R. Cundari, W. A. Goddard and R. A. Periana, Organometallics, 2010, 29, 6801 CrossRef CAS;
(c) D. H. Ess, W. A. Goddard and R. A. Periana, Organometallics, 2010, 29, 6459 CrossRef CAS;
(d) D. Munz, D. Meyer and T. Strassner, Organometallics, 2013, 32, 3469 CrossRef CAS.
- S. Mazumder, D. W. Crandell, R. L. Lord and M.-H. Baik, J. Am. Chem. Soc., 2014, 136, 9414 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds and computational details. See DOI: 10.1039/c5sc03905c |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Tian-Yu
Sun‡
b,
Yun-Dong
Wu
bc,
Xinhao
Zhang
a,
Tian-Yu
Sun‡
b,
Yun-Dong
Wu
bc,
Xinhao
Zhang